

Abstract
Complex signaling cross‐talks between different growth factor cascades orchestrate the primary brain cancer development. Among the frequent deregulated oncogenic pathways, the ligand‐activated wild‐type epidermal growth factor receptor (EGFR), constitutively activated EGFRvIII mutant and sonic hedgehog pathways have attracted much attention because of their pivotal roles in pediatric medulloblastomas and adult glioblastoma multiformes (GBM) brain tumors. The enhanced expression levels and activation of EGFR, EGFRvIII mutant and hedgehog signaling elements can provide key roles for the sustained growth, migration and local invasion of brain tumor‐initiating cells (BTICs) and their progenies, resistance to current therapies and disease relapse. These tumorigenic cascades also can cooperate with Wnt/β‐catenin, Notch, platelet‐derived growth factor (PDGF)/PDGF receptors (PDGFRs), hepatocyte growth factor (HGF)/c‐Met receptor and vascular endothelial growth factor (VEGF)/VEGF receptors (VEGFRs) for the acquisition of a more malignant behavior and survival advantages by brain tumor cells during disease progression. Therefore, the simultaneous targeting of these oncogenic signaling components including wild‐type EGFR, EGFRvIII mutant and hedgehog pathways may constitute a potential therapeutic approach of great clinical interest to eradicate BTICs and improve the efficacy of current clinical treatments by radiation and/or chemotherapy against aggressive and recurrent medulloblastomas and GBMs.
Keywords: brain tumor‐initiating cells, carcinogenesis, combination therapy, EGFR, EGFRvIII, glioblastoma multiforme, hedgehog, medulloblastomas, molecular targeting, receptor tyrosine kinases
Abbreviations:
- ABC
ATP‐binding cassette
- AURKA
aurora kinase A
- ASPM
protein, abnormal spindle‐like microcephaly associated protein
- BMP
bone morphogenic protein
- BTICs
brain tumor‐initiating cells
- CDK
cyclin‐dependent kinase
- CNS
central nervous system
- CXCR4
CXC chemokine receptor 4
- EGF
epidermal growth factor
- EGFR
epidermal growth factor receptor
- GBM
glioblastoma multiforme
- GCPs
granule cell precursors
- GFAP
glial fibrillary acidic protein
- GNP
granule neuron precursors
- HGF
hepatocyte growth factor
- JAK2
Janus‐activated kinase 2
- MAPKs
mitogen‐activated protein kinases
- MARCKS
myristoylated alanine‐rich protein kinase C substrate
- miRNA
microRNAs
- MMPs
matrix metalloproteinases
- NGF
nerve growth factor
- NF‐κB
nuclear factor‐kappa B
- NPCs
neural progenitor cells
- NSCs
neural stem cells
- PDGF
platelet‐derived growth factor
- PDGFR
platelet‐derived growth factor receptor
- C; PI3K
phosphatidylinositol‐3′ kinase
- PIP2
phosphatidylinositol (3,4)‐bisphosphate
- PIP3
phosphatidylinositol (3,4,5)‐trisphosphate
- PKC
protein kinase
- PTEN
phosphatase tensin homolog deleted on chromosome 10
- RTKs
receptor tyrosine kinases
- SDF‐1
stromal cell‐derived factor‐1
- SHH
sonic hedgehog ligand
- shRNA
small hairpin RNA
- siRNA
small interference RNA
- SMO
smoothened
- SREBP‐1
sterol regulatory element‐binding protein 1
- STAT3
signal transducers and activators of transcription 3
- SUFU
suppressor of fused
- TBP
TATA‐binding protein
- TF
tissue factor
- TGF‐β
transforming growth factor‐β
- uPA
urokinase‐type plasminogen activator
- VEGF
vascular endothelial growth factor
- VEGFR
vascular endothelial growth factor receptor
INTRODUCTION
Malignant primary brain tumors, including medulloblastomas and gliomas, such as high‐grade astrocytomas and glioblastoma multiforme (GBM) brain tumors, also designated as glioblastomas, are among the most frequent, rapidly growing and lethal tumors of the central nervous system (CNS) in children and adults, respectively (Figure 1) 66, 106, 207. The image‐guided complete surgical excision of infiltrating pediatric medulloblastomas and adult GBM tumors, which can invade the surrounding tissues and spread to different locations in the brain and/or spine through their diffusion into the brain parenchyma and cerebrospinal fluid is typically ineffective and leads to local relapse of the disease 66, 106. Also, highly aggressive medulloblastomas and GBMs are usually refractory to current clinical therapies by high‐dose external beam radiotherapy, targeted brachytherapy and/or adjuvant chemotherapeutic treatments with diverse DNA‐alkylating agents such as temozolomide, nitrosoureas and/or cisplatin 66, 106, 207. Especially, the GBM patients treated with radiotherapy plus adjuvant temozolomide, which represents the standard of care in several clinics, have a poor median survival time of about 14.6 months after diagnosis (207). The inefficacy of current therapies by radiation and diverse chemotherapeutic regimens for treating the patients with locally advanced and disseminated medulloblastomas and GBMs has been associated with an enhanced expression of different oncogenic products and the rapid development of resistance mechanisms by brain cancer cells 66, 106, 207. Moreover, the use of high‐dose ionizing radiation and/or chemotherapeutic drugs might cause systemic and neurological toxicity, such as temozolomide‐induced CD4+ lymphopenia and severe long‐term side effects 66, 106, 207.
Figure 1.
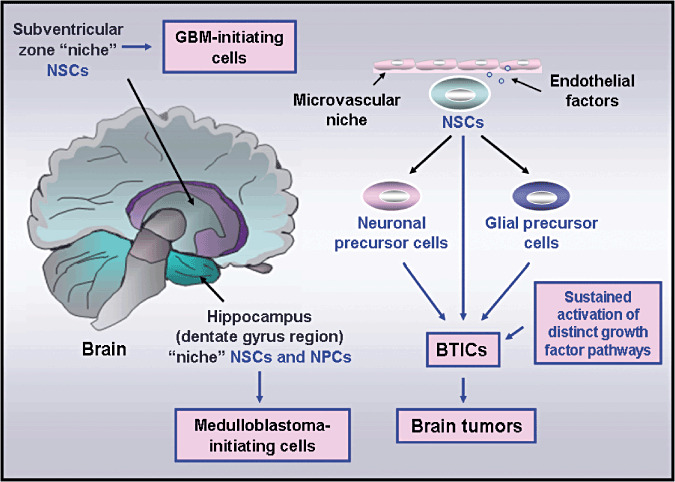
Scheme showing the anatomic localization of the niches of neural stem/progenitor cells in the human adult brain as well as their malignant transformation into brain tumor‐initiating cells. The occurrence of genetic alterations leading to a sustained activation of distinct developmental signaling pathways, including wild‐type epidermal growth factor receptor (EGFR), EGFRvIII mutant and/or sonic hedgehog pathways, in neural stem/progenitor cells (NSCs or NPCs) is indicated. These molecular changes in NSCs or NPCs may culminate in their malignant transformation into brain tumor‐initiating cells (BTICs) and brain tumor development, including medulloblastomas or glioblastoma multiforme (GBM) brain tumors.
In general, the initiation and progression of primary brain tumors, including medulloblastomas and gliomas, are accompanied by the accumulation of distinct genetic alterations in diverse tumor suppressor gene products such as phosphatase tensin homolog deleted on chromosome 10 (PTEN), p53, p16INK4A and/or p19ARF and oncogenic products 100, 104, 106, 156, 181, 217, 219. The genetic alterations resulting in a sustained activation of different tumorigenic cascades mediated through different receptor tyrosine kinases (RTKs) and their downstream signaling elements, phosphatidylinositol‐3′ kinase (PI3K)/Akt and Ras/mitogen‐activated protein kinases (MAPKs) can cooperate for the acquisition of more aggressive phenotypes and survival advantages by brain cancer cells 100, 104, 106, 135, 217, 246. Among the frequent genetic alterations that might contribute to primary brain tumor initiation and progression, the wild‐type EGFR receptor tyrosine kinase (erbB1 or HER1) is amplified, overexpressed and/or mutated in approximately 30%–60% of GBM patients (2, 3) 1, 5, 24, 66, 92, 156, 197, 214, 224. It has also been noticed that the EGFR amplification is often accompanied by an upregulated expression of the deletion mutant EGFR variant III form, also designated as EGFRvIII or ΔEGFR, as well as the PTEN loss‐induced Akt activation, and associated with a poor overall survival of GBM patients 5, 59, 66, 197, 214, 234. More specifically, the immunohistochemical and reverse transcriptase‐polymerase chain reaction (RT‐PCR) analyses have revealed that an increase of constitutively active EGFRvIII mutant expression frequently occurs in approximately 30%–60% of GBM patients while no expression of this mutant is observed in the normal adult brain and any other tissues (Figure 2) 5, 151, 166, 197, 234, 244. Moreover, the activation of the PI3K/Akt pathway is frequently detected in up to about 77%–87% of the tumor tissue samples from GBM patients 1, 38, 56, 66. In addition, a persistent activation of the sonic hedgehog cascade often occurs during medulloblastoma and GBM initiation and progression, and may provide critical functions for the acquisition of more malignant phenotypes by brain tumor cells (Figure 3) 8, 11, 15, 35, 37, 39, 40, 51, 89, 109, 130, 145, 156, 183, 187, 193, 238, 241, 242. Also, other molecular pathways that are frequently deregulated in medulloblastoma and GBM cells during the progression to advanced disease stages include Wnt/β‐catenin, Notch, platelet‐derived growth factor (PDGF)/PDGF receptors (PDGFRs), hepatocyte growth factor (HGF)/c‐Met receptor, stem cell factor (SCF) receptor c‐KIT, vascular endothelial growth factor (VEGF)/VEGF receptors (VEGFRs) and erbB2 (HER2) in the case of medulloblastomas (Figure 4) 3, 4, 7, 21, 23, 24, 35, 39, 67, 71, 78, 104, 112, 135, 156, 192, 203, 211, 219, 224. These tumorigenic cascades may contribute in cooperation with the EGFR/EGFRvIII and hedgehog signaling networks to treatment resistance and a poor survival of brain cancer patients.
Figure 2.
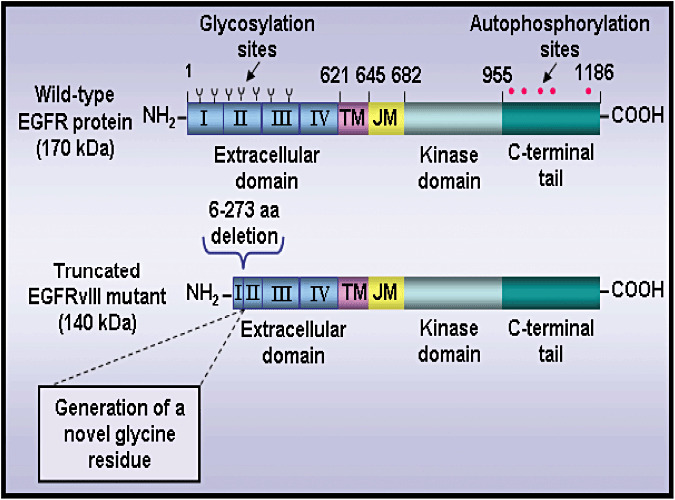
Schematic structures of human wild‐type EGFR protein and truncated EGFRvIII mutant. The scheme shows the extracellular domains divided in four subdomains I–IV, transmembrane region (TM), juxtamembrane segment (JM), tyrosine kinase domain and C‐terminal tail. The residue number corresponds to the human epidermal growth factor receptor (EGFR) amino acid (aa) sequence excluding the signal peptide sequence. The position of potential glycosylation sites and tyrosine phosphorylation sites are indicated. Moreover, the in‐frame deletion of the 6–273 aa segment in the extracellular domain of the truncated mutant epidermal growth factor receptor variant III (EGFRvIII mutant), which results in the generation of a novel glycine residue at the fusion junction between aa at the positions 5 and 274 that is specific to the mutant EGFRvIII receptor is also indicated.
Figure 3.
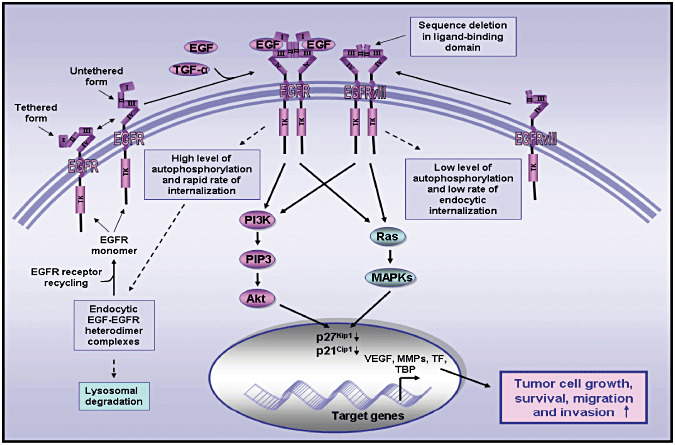
Schematic representations showing the structural and functional differences between the wild‐type EGFR and truncated EGFRvIII mutant in brain cancer cells. This scheme shows the activation of the wild‐type EGFR through a homodimerization induced by the binding of its ligand EGF or TGF‐α and ligand‐independent activation of constitutively activated EGFRvIII mutant which is characterized by a sequence deletion in the 6–273 amino acid region within the ligand‐binding domain. The high level of autophosphorylation of wild‐type EGFR and rapid endocytic internalization of EGF–EGFR complexes that might result in either the receptor recycling or lysosomal degradation as compared with a low level of autophosphorylation and low rate of endocytic internalization of the truncated EGFRVIII mutant are also illustrated. The oncogenic effects induced through the sustained activation of EGF/EGFR and constitutively activated EGFRvIII mutant in brain cancer cells is also indicated. EGF, epidermal growth factor; EGFR, epidermal growth factor receptor; EGFRvIII, mutant epidermal growth factor receptor variant III; TGF‐α, transforming growth factor‐α.
Figure 4.
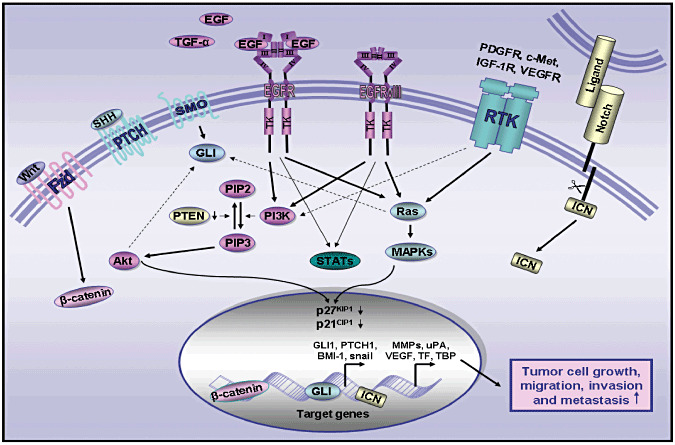
Schematic representation showing the oncogenic signaling elements induced through the sustained activation of distinct growth factor pathways involved in the malignant behavior of brain cancer cells. This scheme shows the stimulatory effect induced through the activation of wild‐type EGFR, constitutively activated EGFRvIII mutant, sonic hedgehog, Wnt/β‐catenin, Notch and other RTKs such as c‐MET, PDGFR and IGF‐R1 on the Ras/mitogen activated protein kinases (MAPKs) and phosphatidylinositol 3′ kinase (PI3K)/Akt and potential signaling cross‐talks between these cascades. The downregulation of phosphatase tensin homolog deleted on chromosome 10 (PTEN), which may promote the accumulation of phosphatidylinositol (3,4,5)‐trisphosphate (PIP3) induced by PI3K, and activation of Akt cascade is also indicated. Moreover, the stimulation of these developmental pathways, which may result in an inhibition of cyclin‐dependent kinase inhibitors p27KIP1 and p21CIP1 and enhanced expression of diverse target gene products, is also illustrated. EGF, epidermal growth factor; EGFR, epidermal growth factor receptor; EGFRvIII, mutant epidermal growth factor receptor variant III; Fzd, frizzled receptor; ICN, intracellular domain of Notch; MMPs, matrix metalloproteinases; PTCH, pathched receptor; RTK, receptor tyrosine kinase; SHH, sonic hedgehog; SMO, smoothened coreceptor, TBP, TATA‐binding protein; STAT, signal transducer and activator of transcription; TF, tissue factor; TGF‐α, transforming growth factor‐α; uPA, urokinase type‐plasminogen activator; VEGF, vascular endothelial growth factor; Wnt, Wingless ligand.
Importantly, recent accumulating lines of experimental evidence also suggest that certain primary malignant CNS tumors, including medulloblastomas and GBMs, could derive from the malignant transformation of neural stem/progenitor cells (NSCs/NPCs) or their more committed progenies into brain cancer stem/progenitor cells, also designated as brain tumor‐initiating cells (BTICs) in postnatal and adult life 69, 76, 90, 154, 199, 221, 247. It has been shown that BTICs endowed with a high self‐renewal capacity but an aberrant differentiation ability, were able to give rise ex vivo and in vivo to the total tumor cell mass containing a heterogeneous population of cancer cells consisting of a mixture of the astrocytes, oligodendrocytes and/or ependymal cell‐like cells in different proportions that recapitulated the architecture and phenotypic features of the original patient's brain tumors (Figure 1) 76, 90, 99, 199. It has also been reported that the wild‐type EGFR, EGFRvIII mutant and hedgehog cascades, in cooperation with other genetic alterations, can play critical roles for the malignant transformation of NSCs/NPCs into BTICs during medulloblastoma and GBM development, treatment resistance and disease relapse (3, 4) 11, 14, 32, 37, 41, 59, 97, 119, 130, 140, 151, 238. Consequently, the multitargeted strategies of wild‐type EGFR, EGFRvIII mutant, hedgehog and other oncogenic products with the current clinical treatments by radiation and/or chemotherapy, might represent more promising therapies as monotherapies for treating the patients diagnosed with aggressive and recurrent primary brain tumors (Figure 5). In regard with this, we review the most recent advancements on the key oncogenic functions supplied by the wild‐type EGFR, truncated EGFRvIII mutant, sonic hedgehog and downstream signaling elements such as PI3K/Akt and cross‐talks with other tumorigenic cascades in BTICs and their progenies during the primary brain tumor development. Of great clinical interest, recent studies supporting the therapeutic benefit to target wild‐type EGFR/EGFRvIII mutant, hedgehog and other oncogenic signaling elements to eradicate BTICs and their progenies and thereby improve the current clinical treatments and develop a novel effective combination therapy against highly aggressive and recurrent medulloblastomas and GBMs are also discussed.
Figure 5.
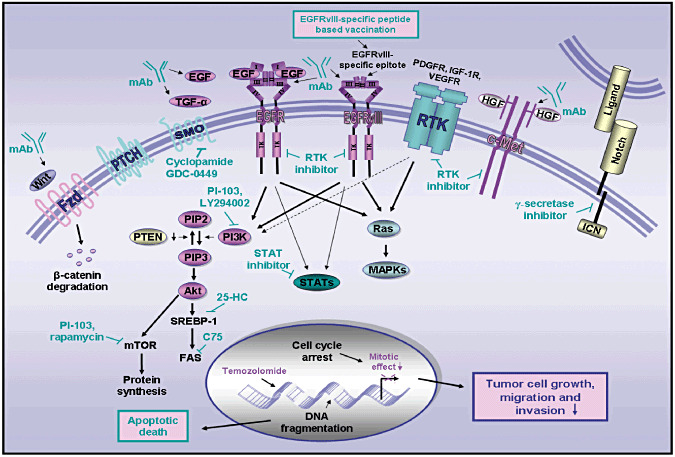
Novel multitargeted strategies against highly aggressive and invasive medulloblastomas and glioblastoma multiforme (GBM) brain tumors. The scheme shows the potential inhibitory effects induced by diverse pharmacological agents targeting wild‐type EGFR, EGFRvIII mutant, sonic hedgehog, Wnt/β‐carenin, HGF/c‐Met receptor and Notch/ligand as well as current chemotherapeutic drug, temozolomide on the growth, survival, invasion and metastasis of brain tumor cells. The potent anticarcinogenic agents include a monoclonal antibody (mAb) or immunotoxins directed against EGF or TGF‐α ligand, wild‐type EGFR or EGFRvIII mutant, a mAb directed against Wnt and HGF as well as a selective inhibitor of EGFR/EGFRvIII tyrosine kinase activity (gefitinib and erlotinib), smoothened coreceptor (SMO) activity (cyclopamine or GDC‐0449), or Notch (γ‐secretase inhibitor). The molecular targeting of downstream signaling elements induced through the activation of these growth factor pathways and phosphatase tensin homolog deleted on chromosome 10 (PTEN) downregulation‐promoted PI3K/Akt activation is also indicated. The pharmacological agents include a specific inhibitor of JAK/STATs, PI3K (LY294002), mTOR (rapamycin), dual inhibitor of PI3K/mTOR (PI‐103), SREBP‐1 (25‐HC) and FAS (C75). The vaccination based on the use of EGFRvIII‐specific peptide is also indicated. EGF, epidermal growth factor; EGFR, epidermal growth factor receptor; EGFRvIII, mutant epidermal growth factor receptor variant III; 25‐HC, 25‐hydroxycholesterol; HGF, hepatocyte growth factor; ICN, intracellular domain of Notch; JAK2, Janus‐activated kinase; MAPKs, mitogen‐activated protein kinases; c‐MET, hepatocyte growth factor receptor; mTOR, mammalian target of rapamycin; PI3K, phosphatidylinositol 3′ kinase; PIP2, phosphatidylinositol (3,4)‐bisphosphate; PIP3, phosphatidylinositol (3,4,5)‐trisphosphate; STAT3, signal transducer and activator of transcription 3; SREBP‐1, sterol regulatory element‐binding protein‐1; TGF‐α, transforming growth factor‐α; Wnt, Wingless ligand.
IMPLICATION OF THE MALIGNANT TRANSFORMATION OF NSCs/NPCs INTO BTICs IN PRIMARY BRAIN CANCER DEVELOPMENT
Phenotypic and functional features of NSCs/NPCs
Adult neurogenesis, astrogliogenesis and tissue repair in central and peripheral nervous tissues may occur through the activation of adult NSCs/NPCs 13, 28, 144, 201, 210, 220, 252. The NSCs/ NPCs have been identified within two specific germinal regions of the brain: the subventricular zone bordering lateral ventricle in the forebrain and the dentate gyrus in the hippocampus (Figure 1) 13, 28, 127, 128, 201, 230, 252. Multipotent NSC/NPCs localized in the germinal subraventricular zone, which express different stem cell‐like markers such as CD133 and/or nestin and possess a high self‐renewal potential, can give rise to three principal cell lineages, including mature neurons and glial cells, astrocytes and oligodentrocytes 13, 28, 201, 220, 230, 252. NSCs/NPCs endowed with a multilineage differentiation potential and regenerative capacity can generate the progenitor cells that migrate along the blood vessels at distant damaged areas of the brain and participate to regenerate and repair the injured tissues by generating further differentiated and functional progenies. Moreover, NSCs/NPCs, including NPCs designated as neural precursor cells, found in the subgranular cell layer of the hippocampus, can generate the granule cell projection neurons that integrate into existing neuronal circuitry (Figure 1) 28, 144, 230. In addition, multipotent adult stem/progenitor cells expressing the glial markers that are able to give rise to the dopaminergic glomus cells have also been identified in the peripheral nervous system within a germinal center termed the carotid body (161).
The stringent regulation of NSC/NPC behavior during neurogenesis and astrogliogenesis in developing and adult CNS is accomplished through the stimulation of diverse developmental signaling cascades initiated by different growth factors that display neurogenic, neuroprotective and/or neurorestorative effects. Among them, there are EGF/EGFR, sonic hedgehog, Wnt/β‐catenin, Notch, insulin‐like growth factor‐receptor type 1 (IGF‐1R), PDGF/PDGFRs, bone morphogenic protein (BMP), nerve growth factor (NGF), transforming growth factor‐β (TGF‐β) and stromal cell‐derived growth factor‐1 (SDF‐1)/CXC chemokine receptor 4 (CXCR4) pathways 6, 13, 28, 43, 60, 72, 77, 116, 145, 159, 172, 200, 201, 208, 210, 220, 230, 231, 252. The interplay of these growth factor cascades is involved in the regulation of the balance between the quiescence, proliferation, differentiation and migration of NSCs/NPCs and their progenies. Moreover, the immature NSCs/NPCs are localized within a specialized local microenvironment called the microvascular niche consisting of the neighboring endothelial cells that tightly regulate their functions through the direct interactions and the release of specific soluble factors (Figure 1) 42, 62, 73, 113, 195, 196, 209, 230, 252. Importantly, a growing body of evidence has revealed that the deregulation in these developmental growth factor cascades and their local microenvironment might lead to the development of primary brain cancers, including medulloblastomas and GBMs (Figure 1).
Cellular origin and heterogeneity of primary brain tumors
The pediatric and adult brain tumors are heterogeneous malignancies that include distinct cancer subtypes with different cellular origins, which may be associated with specific inherited disorders and genetic alterations occurring in primitive NSCs/NPCs or more committed cell lineage progenies with stem cell‐like properties in the developing brain or adult CNS as well as the changes in their local microenvironment, niche (Figure 1) 16, 24, 35, 70, 72, 129, 148, 154, 164, 192, 218, 224. Numerous genetic and proteomic analyses combined with the gain‐ and loss‐function studies and development of animal models of medulloblastoma and GBM have led to their molecular classification in different biological and clinical subtypes characterized by particular genetic alterations as well as the establishment of their cellular origin 16, 24, 31, 39, 48, 70, 72, 115, 122, 129, 157, 162, 164, 192, 193, 218, 224. Among the primary brain tumors, gliomas are classified in four grades, Grades I–IV, including Grade IV GBMs, which is the most aggressive grade. The glioma have been classified into different subgroups including at least three different GBM subtypes that comprise classical, proneural and neural subtypes that are characterized by frequent genetic aberrations in EGFR, PDGFR or neurofibromatosis 1, respectively 15, 24, 39, 111, 122, 224. It has been reported that the gliomas might originate from the malignant transformation of NSCs, glial stem cells or their progenitors within the ventricular zone germinal layer into BTICs in postnatal and adult life 69, 76, 90, 148, 154, 186, 199, 221, 225, 247. Moreover, it has also been shown that the PDGF‐B transfer to more committed glial progenitor cells, including oligodendrocyte progenitor cells, can give rise to Grade II gliomas with an incidence of 33% (129).
In addition, the medulloblastomas have also been classified in four to six different subgroups characterized by the frequent genetic aberrations in specific signaling pathways including Wnt/β‐catenin and sonic hedgehog subgroups 31, 48, 60, 72, 115, 144, 157, 162, 193, 218, 254. The sonic hedgehog subgroup of medulloblastoma, which is frequent in the desmoplastic and nodular subtype, is often associated with the genetic deregulations, including inactivating mutations in tumor suppressor genes patched receptor 1 (PTCH1) and suppressor of fused (SUFU) that lead to a sustained activation of sonic hedgehog ligand (SHH)/glioma‐associated oncogene (GLI) cascade 48, 60, 72, 115, 144, 157, 162, 193, 202, 218, 254. The hedgehog subgroup may arise in children of the malignant transformation of neuronal lineage committed progenitors, designated as cerebellar granule cell precursors (GCPs), located in the external granular layer within the cerebellum into BTICs (Figure 1) 72, 144. Moreover, the classic histological subtype of medullublastoma, which often harbors the genetic alterations in Wnt/β‐catenin pathway and is generally associated with a more good prognosis, may derive outside the cerebellum from the malignant transformation of cells of the dorsal brainstrem 31, 48, 70, 115, 157, 162, 218.
Phenotypic and functional features of BTICs
The subpopulation of BTICs detected in medulloblastomas or GBMs typically express the stem cell‐like markers, such as CD133, nestin, CD44 and/or Oct‐3/4 and possess a high self‐renewal and aberrant multilineage differentiation potential, including the ability to form nonadherent spheres in vitro and serially transplantable tumors in animal models in vivo 76, 90, 99, 121, 199. It has been shown that highly tumorigenic CD133+ BTICs isolated from medulloblastomas or GBMs were able to give rise ex vivo and in vivo to a heterogeneous population of differentiated CD133‐ brain tumor cells expressing the neuronal (β‐III tubulin) and astrocytic glial fibrillary acidic protein (GFAP) markers in proportions that reflected the cellular heterogeneity of the original patient's brain tumors 76, 90, 99, 121, 199.
Importantly, CD133+ BTICs have also been reported to contribute to the resistance to current radiation and chemotherapeutic treatments and disease relapse 10, 22, 64, 96, 130, 131, 136, 146, 147. For instance, it has been observed that CD133+ BTICs detected in three primary cell lines established from glioblastoma patients, expressed high mRNA levels of CXCR4, O6‐methylguanine DNA methyltransferase (MGMT), brain cancer‐related protein (BCRP)/ABCG2 multidrug transporter, anti‐apoptotic factors such as Bcl‐2, survivin, inhibitor of apoptosis proteins (IAPs), hedgehog signaling elements and the transcriptional repressor of the E‐cadherin, snail (130). These BTICs were also more resistant to chemotherapeutic agents, such as temozolomide, carboplatin, etoposide and paclitaxel, as compared with the CD133‐ cell fraction 64, 130. Particularly, it has been shown that the development of treatment‐refractory infiltrating brain tumors can be promoted by CD133+ GBM cells endowed with neural and mesenchymal properties and which are capable of forming spheres in culture (64). Moreover, the enhanced expression of CD133 marker and sphere forming‐capacity of BTICs has been reported to be the independent predictors of the clinical outcome of the glioma patients 160, 177, 249. In this regard, it has also been observed that a high number of CD133+/nestin+ BTICs was detected in N29 and N32 rat tumor glioma cell lines maintained under stem cell and differentiation culture conditions in vitro as well as after intracranial implantation of tumor cells into syngeneic hosts (19). Hence, it appears that the number of highly tumorigenic BTICs may vary with the glioma subtype.
Recent accumulating lines of evidence have also indicated that poorly differentiated CD133‐ glioma progenitor cells expressing other neural and glial stem/progenitor cell‐like markers such as nestin, stage‐specific embryonic antigen 1 (SSEA‐1)/CD15 and A2B5 endowed with a high self‐renewal potential were able to give rise to CD133+ and CD133‐ glioma cells 18, 30, 36, 65, 85, 102, 158, 173, 204, 213. The immature CD133‐ BTICs could initiate sphere growth in vitro and tumor formation in vivo with a high frequency comparable with CD133+ BTICs 18, 30, 36, 65, 85, 102, 158, 173, 204. In contrast, moderately differentiated and self‐renewal CD133‐ cells only given rise to more differentiated CD133‐ glioma cells with no self‐renewal ability 30, 65, 173. Overall, these observations suggest that different brain tumor subtypes may arise from distinct self‐renewing BTICs, which can exhibit different phenotypic markers and functional features. Moreover, these data also indicate the possibility that the self‐renewing BTICs detected within the same primary brain tumors may be organized under form a lineage hierarchy. The cellular hierarchy could consist of self‐renewing BTIC subpopulations characterized by specific tumorigenic and aberrant multilineage differentiation capacities as observed in other cancer types such as the leukemias and epithelial cancers 30, 65, 146, 147, 158, 173, 227. However, future studies are required to more precisely establish the hierarchical organization, specific stem cell‐like markers and functions of self‐renewing CD133+ and CD133‐ BTIC subpopulations detected within the same or distinct primary brain tumors from patients at different stages of the disease progression.
In addition, the malignant transformation of BTICs and their progenies has also been associated with the occurrence of distinct genetic alterations in these immature cancer cells leading to a sustained activation of diverse developmental cascades that are involved in the stringent regulation of the behavior of their normal counterpart, NSCs/NPCs in the postnatally developing cerebellum and adult brain 8, 27, 35, 72, 102, 159. The growth factor pathways frequently deregulated in BTICs and their progenies include EGFR, hedgehog, Wnt/β‐catenin, Notch, PDGF/PDGFR, HGF/c‐Met, BMP and TGF‐β cascades and downstream signaling element PI3K/Akt 8, 27, 35, 121. In this matter, we are reporting the specific functions of wild‐type EGFR, constitutively activated EGFRvIII mutant and sonic hedgehog cascades and potential interactive signaling cross‐talks with other tumorigenic pathways that can contribute to the malignant transformation of BTICs and their progenies during primary brain cancer development, including medulloblastomas and GBMs.
Functions of the wild‐type EGFR and EGFRvIII mutant during primary brain cancer development
Numerous studies have revealed that a sustained activation of the wild‐type EGFR and truncated EGFRvIII mutant can cooperate for the malignant transformation of brain cancer cells, angiogenic process and tumor development 14, 106, 121, 217. The enhanced expression and activation of wild‐type EGFR in conjunction with the EGFRvIII mutant have been shown to contribute to the acquisition of a more malignant behavior by BTICs and their progenies during primary brain cancer initiation and progression (Figure 3) 20, 76, 99. For instance, the primary GBMs, which are aggressive brain cancers that typically progress rapidly without evidence of a transitory step of lower‐grade tumor, are frequently accompanied by an overexpression of the wild‐type EGFR and/or EGFRvIII mutant 69, 154, 221, 247. In the same way, it has also been reported that all tumorigenic CD133+/nestin+ BTICs populations isolated from seven patients with GBMs expressed the wild‐type EGFR (76). Moreover, the results from the histoimmunostaining analyses of GBM cells detected in tumor tissues from patients have indicated that the EGFRvIII mutant was expressed in CD133+ GBM cells (249). The results of RT‐PCR and Western blot analyses of tumor spheres established from primary brain tumors have also revealed that eight out of 11 GBMs, one out of two medulloblastomas and one out of one ependymoma were positive for the EGFRvIII mutant (235). The EGFRvIII mutant has also been observed to be co‐expressed with the stem cell‐like marker CD133+ in a high fraction (70 ± 8%, n = 8) of the GBM cell population detected by fluorescence‐activated cell sorting (235). Importantly, although the EGFRvIII mutant is rapidly lost when the brain tumor cells are maintained in the culture conditions promoting their differentiation, the EGFRvIII expression has been detected for up to 3 months in the tumor spheres endowed with a high self‐renewal ability maintained in serum‐free stem cell growth media ex vivo (235).
The ligand‐activated wild‐type EGFR and constitutively activated EGFRvIII mutant can stimulate similar intracellular pathways as well as display particular functional properties and activate specific downstream signaling effectors in a brain cancer cell type‐dependent manner 4, 8, 66, 143, 144, 148. More specifically, the activation of the wild‐type EGFR by its secreted ligands, including EGF and TGF‐α, might occur through an autocrine loop or in a juxtacrine or a paracrine manner, and leads to the stimulation of diverse intracellular signaling elements in primary brain cancer cells including BTICs (Figure 4) 106, 132, 214, 248. These intracellular signaling components include PI3K/Akt, Ras/MAPKs, Janus‐activated kinase 2 (JAK2)/signal transducer and activators of transcription 3 (STAT3) and phospholipase C‐γ (PLC‐γ) (Figure 4) 96, 106, 132, 214, 248. The activation of these downstream signaling elements might result in the inhibition of nuclear cyclin‐dependent kinase (CDK) inhibitors, p21CIP1 and p27KIP1 and the induction of mitotic effects, including an enhanced expression of metalloproteinases (MMPs), urokinase‐type plasminogen activator (uPA) and VEGF. Thereby, the EGFR activation may promote the growth, survival, migration and local invasion of brain cancer cells, including medulloblastoma‐ and GBM‐initiating cells, and the angiogenic process.
In the case of the somatically mutated EGFRvIII receptor, which lacks an N‐terminal 267‐amino acid portion of the full‐length EGFR's extracellular ligand‐binding domain, it has been shown that this truncated RTK protein can act as a constitutively autophosphorylated and activated receptor that mediates its oncogenic effects in the absence of a ligand (2, 3, 4) 14, 106. The constitutively activated EGFRvIII typically exhibits a 10‐fold lower level of autophosphorylation relative to the ligand‐activated wild‐type EGFR 75, 84, 97, 189, 248. This difference between EGFRvIII and wild‐type EGFR phosphorylation status may influence their cell‐surface stability, functional properties and rate of internalization in cancer cells (Figure 3). More specifically, the downregulation of the cell‐surface EGFRvIII mutant through its internalization by endocytosis and degradation in the lysosomes is low compared with the wild‐type EGFR complexes that generally undergo a rapid internalization through clathrin‐coated endocytic vesicles followed by the receptor recycling at the cell surface or lysosomal degradation (Figure 3) 75, 84, 97, 226. Hence, the enhanced stability of the constitutively activated EGFRvIII mutant at the plasma membrane can promote its malignant transforming effects on brain tumor cells and contribute at least in part to the functional divergences noticed between the EGFRvIII mutant and wild‐type EGFR, including their differential ability to activate different signaling elements such as MAPKs and target genes 63, 97, 151, 189, 215. Particularly, the constitutively activated EGFRvIII mutant can persistently stimulate diverse intracellular cascades and upregulate the expression of numerous target genes such as MMPs, tissue factor (TF) and TATA‐binding protein (TBP). In turn, these gene products may contribute to the malignant behavior of brain tumor cells including BTICs 20, 44, 56, 63, 94, 98, 106, 117, 142, 151, 153, 155, 174, 182, 228. The downstream signaling elements that might be induced through the EGFRvIII mutant in a brain cancer cell type‐dependent manner include PI3K/Akt, Ras/MAPK, JAK2/STAT3, protein kinase C‐α (PKC‐α)/myristoylated alanine‐rich PKC substrate (MARCKS) and abnormal spindle‐like microcephaly associated (ASPM) protein (Figure 3) 20, 44, 56, 63, 94, 106, 117, 142, 151, 153, 155, 174, 182, 228. Moreover, the expression of the EGFRvIII mutant in the GBM patients has also been associated with a decreased expression level of collapsing response mediator protein 1 (CRMP1), which in turn may promote the invasion of EGFRvIII‐expressing GBM cells (153). Consequently, the EGFRvIII mutant can play critical roles in the proliferation and migration of GBM cells, and enhanced tumorigenicity and angiogenic process, and cooperate with wild‐type EGFR and other oncogenic products for primary brain cancer tumor development. In addition, the co‐expression and heterodimerization of the wild‐type EGFR and EGFRvIII mutant or the formation of heterodimer between these receptors and erbB2 and interactive cross‐talks with hedgehog and other tumorigenic cascades in medulloblastoma and GBM cells including BTICs also can contribute to their acquisition of a more malignant behavior and treatment resistance (Figure 4) 7, 24, 35, 95, 101, 126, 134, 137, 152, 163, 191, 205.
Functions of the sonic hedgehog cascade during primary brain tumor development
The sustained activation of canonical sonic hedgehog cascade consisting of SHH ligand/PTCH1/smoothened (SMO) co‐receptor/GLI transcription factors, which can cooperate with other developmental pathways such as EGFR, Wnt/β‐catenin and Notch to induce the proliferation of NSCs/NPCs in the postnatal development and adult brain, is also deregulated in a subset of primary brain tumors, including medulloblastomas and GBMs 8, 11, 37, 60, 100, 159, 169, 187, 191, 193, 205, 254. The inactivation mutations in tumor suppressor genes, PTCH1 receptor and SUFU, or activating mutations in oncogenic SMO co‐receptor of the hedgehog cascade is prevalent in about 15% of all the cases of sporadic medulloblastomas 176, 178, 216, 254. Also, an upregulation of the expression levels and activities of hedgehog signaling elements might occur in medulloblastoma and GBM cells including BTICs during the brain cancer progression to locally advanced and disseminated stages 11, 35, 37, 40, 51, 89, 95, 109, 183, 187, 191, 193, 238, 241, 242. The persistent activation of sonic hedgehog cascade may lead to an enhanced expression of GLI‐targeted genes such as GLI‐1, cyclin DI and D2, Bmi‐1 polycomb ring finger oncogene, snail, Bcl‐2, insulin receptor substrate 1 (IRS1) and adenosine triphosphate (ATP)‐binding cassette (ABC) multidrug transporters. Thereby, the activated hedgehog pathway may contribute to the acquisition of a more malignant behavior and survival advantages by medulloblastoma‐ and GBM‐initiating cells and their progenies (Figure 4) 11, 35, 37, 40, 51, 89, 95, 109, 145, 183, 187, 206, 238, 241, 242. More specifically, it has been shown that the sustained activation of SHH/PTCH1/GLI‐1 can promote the self‐renewal and sphere‐forming ability of BTICs from GBM patients (238).
In addition, several signaling cross‐talks between the hedgehog signaling network and other tumorigenic cascades have been proposed to cooperate for the malignant transformation of medulloblastoma and/or glioma cells 95, 145, 191, 206. For instance, it has been shown that the noncanonical hedgehog pathway activated through RTKs such as EGFR may lead to the stimulation of Ras and Akt, that in turn can activate the GLI‐targeted gene expression and GLI‐mediated cellular responses in glioma cells 145, 206. Moreover, the activation of hedgehog in glioma‐initiating cells may also result in the GLI‐1‐induced IRS1 expression that in turn can stimulate the IGF‐I‐mediated MAPK activation (95). Additionally, the characterization of different animal models, in which the wild‐type EGFR and/or EGFRvIII mutant were overexpressed or the hedgehog signaling elements were altered, has also indicated the critical importance of these tumorigenic cascades for the malignant transformation of NSCs/NPCs into highly tumorigenic BTICs during brain cancer development 8, 9, 46, 92, 125.
In vivo animal models of primary brain tumor development
Numerous investigations carried out using animal models, including transgenic mice, have indicated that the enhanced expression level and/or activity of wild‐type EGFR, truncated EGFRvIII mutant and their downstream signaling elements, such as Ras and Akt and/or a persistent activation of hedgehog cascade in NSCs or more committed neuronal or glial precursor cells, can cooperate with other genetic alterations to induce primary brain cancer initiation and progression 8, 9, 46, 83, 92, 93, 100, 125, 232. For instance, it has been shown that the loss of both p16INK4A and p19ARF tumor suppressor genes combined with a constitutive expression of the wild‐type EGFR in NSCs or astrocytes resulted in their acquisition of malignant features, suggesting that these cells can serve as the cell‐of‐origin for certain high‐grade malignant gliomas (9). In the same way, the overexpression of the EGFRvIII mutant in PTEN−/− neural precursor cells also resulted in an enhanced activation of Ras/Erk and PI3K/Akt pathways as compared with PTEN+/+, PTEN+/+/EGFRvIII or PTEN−/− neural precursor cells (125). More specifically, the transformed EGFRvIII‐expressing PTEN−/− neural precursor cells were characterized by an increase in the expression level of CD133 stem cell‐like marker and enhanced centrosome amplification, in vitro proliferation and colony formation in soft‐agar as well as a higher resistance to oxidative stress and ionizing radiation relative to nonmalignant neural precursor cells (125). The EGFRvIII‐expressing PTEN−/− neural precursor cells also formed highly mitotic tumors characterized by a nuclear pleomorphism, necrotic areas and the expression of glioblastoma markers (125). Moreover, it has been observed that the intracranial injection of the EGFRvIII‐expressing PTEN−/− neural precursor cells isolated from these primary tumors in mice led to the development of invasive, GFAP‐ and nestin‐positive tumors (125).
In addition, the results from another study performed using avian retroviral vector to transfer the mutant EGFRvIII gene to the glial precursors and astrocytes in transgenic mice expressing tv‐a, which is a gene encoding the retrovirus receptor under the control of the brain cell type‐specific promoters, have indicated that the expression of the EGFRvIII mutant induced lesions resembling human glioma tumors (92). It has been noticed that these brain lesions occurred more frequently when the gene transfer to mice was made into the progenitor‐specific nestin promoter as compared with the astrocyte‐specific GFAP promoter, suggesting that these lesions can arise more efficiently in immature brain stem/progenitor cells than their differentiated progenies (92). The experiments consisting of simultaneously infecting tv‐a transgenic mice with vectors carrying CDK4 and EGFRvIII or by infecting tv‐a transgenic mice bearing a disrupted INK4a‐ARF locus with the EGFRvIII‐carrying vector alone, have also revealed that these combined genetic alterations resulted in glioma formation (92).
On the other hand, numerous genetically engineered mouse models of primary brain tumors have been generated by altering the hedgehog signaling elements, alone or in combination with different tumor suppressor genes such as p53 8, 86, 100, 143, 205, 241. It has been shown that the PTCH knockout mice, SMOA1/SMOA1 or SMOA1;Bmi‐1+/+ and SMOA1;Bmi‐1+/− mice expressing SMO and Bmi‐1 spontaneously developed typical medulloblastoma subtypes arising from the expansion of the cerebellar granule neuron precursors (GNP) within the external germinal layer relevant to human disease 86, 100, 143, 241. Particularly, 94% of the homozygous SMOA1/SMOA1 mouse model under the control of the GNP promoter ND2 developed medulloblastoma subtypes by 2 months of age that exhibited leptomeningeal dissemination of neoplastic cells to the brain and spine, as observed in many cases of the patients diagnosed with locally metastatic medulloblastomas (86).
In addition, numerous other animal models of primary brain tumors have also been engineered that support the critical implication of the other growth factor cascades such as PDGF/PDGFRs and HGF/c‐Met in malignant transformation of NSCs/NPCs and/or their more committed progenies in medulloblastoma and GBM development 2, 70, 82, 83, 87, 129, 213. For instance, an animal model of human GBM tumors has been generated by retroviral expression of a constitutively active mutant K‐RasG12V construct that acts downstream of the EGFR in cultured neurospheres composed of NSCs/NPCs derived from the subraventricular zone of brains of neonatal p16INK4A−/−;p19ARF−/− transgenic mice follows by the injection of the infected neurospheres into the basal ganglia of wild‐type recipient mice (213). The immunohistochemical analyses of this mouse model of aggressive GBM, which has been combined with a reporter system termed NS‐GFP where the nucleostemin (NS) promoter driving a high expression of green fluorescent protein (GFP) expression in normal and malignant NSCs/NPCs, has revealed that that c‐Met was phosphorylated in NS‐GFP+ BTICs expressing stem cell‐like marker nestin at the invasion front of the tumors. Moreover, the activation of c‐Met cascade by its ligand HGF in isolated NS‐GFP+ BTICs also increased their invasion capacity in vitro supporting the potential role of HGF/c‐Met in GBM development and the therapeutic benefit of its targeting to eradicate invasive GBM‐initiating cells (213).
Hence, on the basis of these observations, it appears that the occurrence in neural or glial stem/progenitor cells of distinct genetic alterations in the specific tumor suppressor genes and oncogenes, including wild‐type EGFR, EGFRvIII mutant and hedgehog signaling elements, like those prevailing in certain cases of human medulloblastomas and GBMs, are required to induce primary brain tumor formation and disease progression. Therefore, the simultaneous molecular targeting of the wild‐type EGFR, EGFRvIII mutant, hedgehog and other gene products such as PDGFRs and c‐Met that are frequently deregulated during human primary brain cancer development might constitute a more promising therapeutic strategy as monotherapies to eradicate the total brain tumor cell mass and prevent disease relapse. In this matter, we are reporting the data from recent investigations supporting the therapeutic interest of targeting wild‐type EGFR, EGFRvIII mutant, hedgehog and other oncogenic signaling elements, alone or in combination with current therapies, for overcoming the treatment resistance of BTICs and their progenies, eradicating the total tumor cell mass and preventing disease recurrence.
Current clinical therapies against primary brain cancers
The therapeutic treatment of the patients with primary brain tumors in the clinics largely varies with the cancer subtype, its anatomic localization and grade at the time of diagnosis and may include surgical excision of tumor, radiotherapy, chemotherapy and stem cell‐based transplantation therapy, alone or in combination 66, 106, 148, 207. The chemotherapeutic drugs such as temozolomide or nitrosourea agents, including 1,3‐bis(2‐chloroethyl)‐1‐nitrosourea (BCNU carmustine), 1‐(2‐chloroethyl)‐3‐cyclohexyl‐1‐nitrosourea (CCNU, lomustine) and cisplatin are often used for treating the patients with aggressive primary brain cancers (Figure 5) 66, 106, 207. Many efforts have been made to develop new and effective combination therapies for treating patients with primary brain cancers in considering the frequent genetic alterations and complex signaling cross‐talks between the growth factors involved in the disease progression. More particularly, new drug classes that are able to penetrate the blood–brain barrier and specifically target the deregulated gene products in brain tumor cells may represent an alternative therapeutic option for the patients with disseminated and recurrent brain tumors that are resistant to conventional therapies. More importantly, numerous recent studies have also revealed that BTICs, which are more resistant than their differentiated progenies to radiation treatment and chemotherapies including temozolomide, may be responsible for the tumor regrowth after treatment initiation and disease recurrence 10, 22, 64, 96, 130, 131, 136, 146, 147. Consequently, the targeting of the total tumor cell mass including highly tumorigenic BTICs is of immense importance for preventing the disease relapse. We review here the data from recent studies supporting the potential benefit of targeting distinct developmental signaling cascades such as RTKs including EGFR/EGFRvIII and sonic hedgehog signaling pathways using the specific inhibitors, alone or in combination, to eradicate BTICs and their progenies, and thereby improve the current therapies 10, 37, 146, 148.
Molecular targeting of the wild‐type EGFR and EGFRvIII mutant signaling pathways
Diverse therapeutic strategies targeting wild‐type EGFR and/or EGFRvIII mutant, including the use of tyrosine kinase activity inhibitors, monoclonal antibodies (mAbs) directed against these RTKs or EGF and TGF‐α ligands, immunotoxins and peptide‐based vaccination have given promising results in preclinical and clinical trials, alone or in combination with the current treatments by radiotherapy and/or chemotherapies (Figure 5) 103, 106, 119, 139, 144, 146, 148, 167, 214. Moreover, the inhibition of the downstream signaling elements, such as PI3K/Akt/mammalian target of rapamycin (mTOR) or sterol regulatory element‐binding protein 1 (SREBP‐1), JAK2/STAT3, MARCKS, ASPM and KLHDC8 referred to as a substitute for ΔEGFR expression‐1 (SΔE) might also constitute an alternative therapeutic strategy for improving the antitumoral efficacy induced through the EGFR and EGFRvIII blockade 44, 80, 94, 106, 132, 150, 153.
Different mAbs specifically recognizing the wild‐type EGFR and/or EGFRvIII mutant, including mouse mAb 528, 806 and 3C10 and 225, the chimeric mouse/human version, mAb C225 (cetuximab or erbitux) and humanized mAb 425 (EMD72000 or matuzumab) have been shown to induce the antitumoral effects on GBM cells in vitro and/or in vivo (Figure 5) 54, 105, 126, 137, 149, 163, 168, 236. It has been proposed that the cetuximab or mAb528 treatment of GBM cells might prevent the heterodimer formation between the wild‐type EGFR and EGFRvIII molecules by sterically impairing the adoption of an extended conformation by these receptors, which is necessary for their dimerization and activation (Figure 3) 126, 137, 163. For instance, the intracranial delivery of cetuximab has been reported to induce the tumor growth inhibitory, anti‐invasive and apoptotic effects on three of seven cases of diffusely invasive xenografts established from different human GBM spheroids exhibiting EGFR amplification and EGFRvIII expression, which have been implanted after short‐term culture into the brains of nude mice (137). In contrast, all the unresponsive GBM tumors lacked amplified and/or mutated EGFR expression (137). Importantly, the co‐targeting of tumor‐specific EGFRvIII mutant and stem cell‐like marker CD133 (AC133) by using a recombinant bispecific antibody (BsAb) directed against mutant EGFRvIII receptor and CD133 protein was also effective to eradicate BTICs while the normal neurospheres expressing only CD133 were insensitive to this BsAb treatment (236). Hence, this therapeutic approach is of great therapeutic interest to specifically target CD133+/EGFRvIII+ BTICs, and thereby prevents the secondary effects on the normal CD133+ NSCs/NPCs.
In addition, diverse specific inhibitors of EGFR/EGFRvIII tyrosine kinase activity such as orally active gefitinib (Iressa) and erlotinib (OSI‐774), dual EGFR/erbB2‐TKIs such as PK1166, lapatinib (GW572017), and pan and irreversible TKI of all four erbB family members, including EGFR (erbBb1)/erbB2/erbB3/erbB4 (CI‐1033 and EKB569) have also been shown to induce the anti‐proliferative and cytotoxic effects on primary brain tumor cells (Figure 5) 29, 76, 106, 117, 123, 132, 139, 144, 146, 148, 150, 165. More specifically, it has been reported that gefitinib induced an anti‐proliferative effect on three medulloblastoma cell lines (D283, D341 and Daoy) and four primary cultures established from medulloblastomas, with apoptosis only in D283 and D341 cell lines as well as a growth inhibitory effect on D341 and Daoy cell‐derived xenografts (139). It has also been noted that the ectotopic overexpression of the erbB2 (HER2) in Daoy cells markedly enhanced their sensitivity to antitumoral effects induced by gefitinib treatment in vivo (139). These data indicate that gefitinib can inhibit the EGFR‐induced transactivation of erbB2 receptor molecule which is overexpressed in about 40% of medulloblastomas and associated with a poor outcome of the patients 67, 74. Importantly, it has been observed that the gefitinib or erlotinib induced the anti‐proliferative and cytotoxic effects on EGFR+/CD133+ BTICs from five patients with gliomablastomas, while two cases of GBMs with a high level of Akt activation in BTICs were insensitive to both drugs or only sensitive to high concentrations of erlotinib (76). These observations suggest that a combination of a specific inhibitor of wild‐type EGFR and EGFRvIII mutant activities with the pharmacological agents inhibiting downstream signaling elements, including PI3K/Akt, could be more effective in certain GBM patients than the single drugs to eradicate the total tumor cell mass. In addition, several clinical trials have also been undertaken to investigate the therapeutic interest of targeting wild‐type EGFR and/or truncated EGFRvIII mutant for improving the efficacy of current therapeutic treatment of brain cancer patients 81, 170, 171, 180.
Clinical trials with the wild‐type EGFR and truncated EGFRvIII mutant inhibitors
The results from clinical trials have revealed the therapeutic interest of using the RTK inhibitors such as gefitinib and erlotinib or mAbs directed against wild‐type EGFR and truncated EGFRvIII mutant for the treatment of GBM patients, and more particularly in combination with radiation therapy or chemotherapy 81, 170, 171, 180. In general, it has been noticed that the response of the patients to the RTK inhibitors was positively associated with the expression of wild‐type EGFR/EGFRvIII mutant and PTEN in certain cases of GBM patients 81, 106, 138, 140, 188. For instance, the data from a recent phase II study have indicated that the inclusion of erlotinib plus temozolomide during and after radiation therapy of newly diagnosed GBM patients improved the median survival to 19.3 months as compared with 14.1 months observed for a treatment consisting of radiation plus temozolomide without erlotinib (171). The results from the comparative analyses of GBM patient's tissue specimens obtained pre‐ and posttreatment with a dual EGFR/erbB2 inhibitor, lapatinib have also revealed that this agent inhibited the EGFR tyrosine kinase activity and downstream signaling elements including PI3K/Akt‐induced the nuclear translocation of SREBP that may contribute to the resistance of GBM patients to mTOR inhibitor, rapamycin (80).
In addition, several anti‐EGFRvIII antibodies and EGFRvIII mutant‐specific peptides have also been developed, which may serve as potential carriers for radioconjugate‐ and immunotoxin‐based treatments and therapeutic tools for vaccination of patients diagnosed with primary brain tumors overexpressing the truncated EGFRvIII mutant 33, 45, 106, 133, 179, 185, 234. In fact, the EGFRvIII mutant, which is not expressed in normal tissues, constitutes an ideal and attractive tumor‐specific target for cellular or humoral immunotherapy in glioma patients. Indeed, the unique extracellular epitope of truncated EGFRvIII mutant, which is formed subsequently to an in‐frame genomic deletion that splits a codon and creates a novel glycine residue at the fusion junction, can serve as a unique antigenic site that might be targeted using antibody‐based antitumor vaccines (2, 3, 4, 5) 17, 33, 185, 240. For instance, a number of clinical studies have been performed by using an EGFRvIII mutant‐based cancer vaccine designated as PEPvIII‐KLH. The PEPvIII‐KLH construct consists of an EGFRvIII‐specific peptide PEPvIII (LEEKKGNYVVTDHC), which corresponds to a 13‐amino acid sequence that spans the EGFRvIII fusion junction combined with an additional cysteine residue to facilitate the chemical conjugation to a keyhole limpet hemocyanin (KLH) (2, 3). It has been reported that PEPvIII‐KLH can generate EGFRvIII‐specific antibodies in the patients with high‐grade gliomas 33, 185, 190. Moreover, the brain tumor resection and sequential treatment of GBM patients with radiation plus DNA‐damaging alkylator temozolomide followed by intradermal injections of the cancer vaccine PEPvIII‐KLH was also accompanied by a specific T‐ and B‐cell‐induced immune response and elimination of tumor cells expressing the EGFRvIII mutant 33, 88. The overall survival of newly diagnosed GBM patients overexpresing the EGFRvIII mutant after this treatment type was significantly enhanced to about 26 months as compared with 15 months observed for patients treated with radiation plus temozolomide alone 88, 185. It has also been noticed that the temozolomide‐induced lymphopenia associated with this treatment improved the efficacy of the peptide vaccination by inhibiting regulatory T cells or their delayed recovery 33, 88. Similarly, the results from a phase II multicenter trial with 22 cases of newly diagnosed GBM patients overexpressing EGFRvIII consisting of external beam radiation therapy followed by vaccinations with PEPvIII‐KLH and granulocyte macrophage‐colony stimulatory factor (GM‐CSF) have indicated that the humoral and immune responses were manifested in these patients. The median time‐to‐progression of the GBM patients was of 14.2 months, which is superior to only 7.1 months observed for the GBM patients who have not been treated with temozolomide (185). Although these results are interesting, it is noteworthy that a downregulation of the EGFRvIII mutant and stimulation of diverse tumorigenic cascades might occur in certain GBM patients during disease progression, and contribute to the disease recurrence (185). These observations suggest that it is necessary to also include other potential tumor‐specific antigens to immunotherapy such as interleukin‐13 receptor α2 in addition to the EGFRvIII mutant‐specific peptide in the future investigations in order to prevent treatment resistance (184). In this regard, we are reporting recent studies supporting the clinical interest to also target the hedgehog cascade to eradicate the medulloblastoma‐ and GBM‐initiating cells and their progenies.
Molecular targeting of the hedgehog signaling cascade
Based on the observations indicating that the hedgehog cascade can provide critical functions for the survival and treatment resistance of BTICs and their progenies, numerous studies have been made to identify and validate novel pharmacological agents that are able to inhibit this tumorigenic cascade for killing the total brain tumor cell mass (4, 5) 11, 35, 37, 40, 41, 51, 95, 109, 145, 183, 187, 238, 241, 242. It has been reported that the treatment with a selective inhibitor of the SMO coreceptor of hedgehog pathway, cyclopamine or GLI‐1 knockdown eradicated the glioma cells from GBM patients endowed with a high self‐renewal ability and expressing the stem cell‐like markers in vitro and inhibited the intracranial growth of tumors derived from these immature glioma cells in vivo 11, 37, 95, 187, 238. More specifically, a long‐term cyclopamine treatment of CD133+ gliomasphere cells expressing high expression levels of the stemness genes (PTCH1, GLI‐1 transcription factor, Bmi‐1, Nanog, Oct‐3/4, SRY‐box containing gene 2 “SOX2” and proliferating cell nuclear antigen “PCNA”) was effective to eradicate all of these BTICs in culture, and induce the regression of glioma tumors established from the gliomasphere cells in nude mice in vivo, without systemic toxicity (37). Importantly, the analyses of 30 glioma samples from the patients before and after treatment with temozolomide or nimustine (ACNU) have revealed that the overexpression of the GLI1 transcriptional effector of sonic hedgehog correlated with glioma recurrence after chemotherapy (41). Of therapeutic interest, it has also been reported that the use of cyclopamine at a lower concentration in combination with the current therapeutic drug, temozolomide induced the additive or synergistic anti‐proliferative and apoptotic effects on the gliomasphere cells in vitro (37). In the same way, the suppression of GLI‐1 activity by small hairpin RNA (shRNA) also inhibited IGF‐1‐induced proliferation, clonogenicity and invasion of glioma‐initiating cells (95). The blockade of the hedgehog with a GLI antagonist GANT or inhibition of IGF‐1R or MAPK by using AG1024 or PD98059 was also effective to improve the cytotoxic effects induced by temozolomide or irradiation on glioma‐initiating cells (95). In addition, the use of small, chemically synthesized SMO antagonists, SANT‐1 and SANT‐2, or natural compounds that can counteract the SMO translocation to the primary cilium, and thereby inhibit hedgehog activation in brain cancer cells, also may constitute another potential therapeutic strategy 40, 109.
In the case of medulloblastomas, it has been shown that the specific products of the sterol biosynthesis, including cholesterol and oxysterols, can provide key roles for the SMO translocation to the ciliary structure and stimulation of the hedgehog pathway (40). Interestingly, the blockade of the sterol synthesis using specific inhibitors that impair the translocation of SMO molecules to the primary cilium has been observed to reduce the SHH‐mediated cell proliferation in medulloblastoma cells (40). It has also been shown that a systemic delivery of antifungal agent designated itraconazole, which acts as a noncompetitive inhibitor of hedgehog pathway, prevented the SHH‐induced SMO accumulation in the primary cilium and suppressed the growth of subcutaneously implanted primary medulloblastoma cells from a PTCH+/−;p53−/− mouse (109). Itraconazole also potentiated the growth inhibitory effect induced by low doses of a potent SMO antagonist, 3‐Keto‐N‐(aminoethyl‐aminocaproyl‐dihydrocinnamoyl)‐cyclopamine (KAAD‐cyclopamine) derivative (109). Interestingly, a natural and nontoxic plant compound, curcumin has also been reported to inhibit the hedgehog cascade by reducing the expression levels of SHH protein and GLI target gene products including Bcl‐2, β‐catenin and the activated/phosphorylated form of Akt and NF‐κB in medulloblastoma cells (53). These molecular events induced by curcumin were accompanied by a cell cycle arrest and apoptotic death of medulloblastoma cells (53). Moreover, curcumin potentiated the cytotoxic effects induced by cisplatin or ionizing radiation on medulloblastoma cells (53). Of particular clinical interest, the results from a case report about a patient diagnosed with a systemic metastatic medulloblastoma associated with inactivating mutation in PTCH1, that was refractory to multiple prior clinical treatments, have also indicated that a rapid tumor regression and reduction of symptoms occurred in this patient after a treatment with SMO antagonist GDC‐0449 (183). The occurrence of a mutation in the SMO protein impairing the binding of GDC‐0449 to the SMO molecules, however, had led to GDC‐0449‐resistance, tumor regrowth evident about 3 months after GDC‐0449 treatment initiation at some sites and the death of the cancer patient 183, 242. Thus, additional studies are necessary to establish the effects induced through the hedgehog blockade in medulloblastoma‐initiating cells vs. their progenies as well as the mechanisms that are associated with their intrinsic and/or acquired resistance to a specific inhibitor of the hedgehog cascade after a long‐term exposure to the drug. In this regard, as the hedgehog and EGFR/EGFRvIII cascades also can cooperate with other tumorigenic pathways for the acquisition of more malignant phenotypes and survival advantage by BTICs and their progenies during disease progression, the targeting of distinct oncogenic products may be more effective for developing new combination therapies against highly aggressive and recurrent medulloblastomas and GBMs.
Other molecular targets for eradicating BTICs and their progenies and reversing drug resistance
Other gene products often altered in BTICs and their progenies during medulloblastoma and GBM formation and progression may also be targeted, alone or in combination therapies, for eradicating the total brain tumor cell mass and reversing drug resistance. Among them, there are telomerase, Wnt/β‐catenin, Notch, erbB2, PDGF/PDGFRs, HGF/c‐Met, c‐KIT, CD44, VEGF/VEGFR, ABC multidrug transporters and/or downstream signaling elements such as PI3K/Akt, STATs, NF‐kB, aurora kinase A (AURKA), MYCN, insulin‐like growth factor binding protein 2 (IGFB2), neuronal adhesion molecule L1CAM and snail (4, 5) 3, 4, 7, 21, 24, 34, 58, 78, 91, 96, 112, 114, 135, 136, 146, 192, 203, 211, 212, 213, 233, 239, 246. It has been reported that the blockade of these oncogenic signaling components by using a specific inhibitor or antagonist, mAb, antisense oligonucleotide or small siRNA, alone or in combination therapy, led to a growth inhibition, apoptotic cell death and/or a reduction of invasiveness and dissemination of medulloblastoma and GBM cells in vitro or in animal models in vivo (Figure 5) 3, 4, 52, 55, 58, 96, 108, 132, 136, 212, 251. More specifically, the treatment of GBM‐initiating cells with telomerase antagonist, imetelstat has been observed to result in the telomere shortening and induce the antiproliferative effect on these immature tumor cells in vitro and in GBM subcutaneous xenograft tumors in vivo (136). A combination of imetelstat with temozolomide also induced the additive cytotoxic effects on GBM‐initiating cells in vitro at least in part by activating DNA damage response pathway (136). Moreover, recent studies have also revealed the therapeutic interest of targeting the small miRNAs, which are often deregulated during etiopathogenesis and the progression of medulloblastomas and GBMs 68, 79, 223. For instance, it has been observed that the transfection of miRNA‐124, miRNA‐137 or miRNA‐451 induced the differentiation of human GBM‐derived stem cells and a growth arrest of GBM cells 68, 198. Moreover, the upregulation of the expression levels of different miRNAs such as miRNA‐34a, miRNA‐128a, or miRNA‐129 suppressor genes in medulloblastoma or GBM cells was also effective at inhibiting the cell growth, survival and invasion, at least in part, by downregulating different oncogenes such as Notch, c‐Met and Bmi‐1 79, 223, 237. In this matter, we review recent data supporting the therapeutic interest to also target Notch, PDGFR and c‐Met pathways and PI3K/Akt signaling elements for eradicating the medulloblastoma and GBM cells and improving the anticarcinogenic efficacy of the specific inhibitors of EGFR/EGFRvIII and hedgehog pathways, and current chemotherapeutic drugs.
Targeting Notch, PDGFR and c‐Met tumorigenic signaling pathways
The molecular targeting of Notch and other RTK cascades such as PDGF/PDGFR receptors and HGF/c‐Met receptor signaling elements, which are frequently deregulated in BTICs and their progenies during the development of medulloblastomas and GBMs, including the brain tumor subtypes overexpressing EGFR/EGFRvIII and/or hedgehog signaling elements, also represent potential strategies against aggressive primary brain tumors (4, 5) 3, 4, 7, 21, 23, 24, 35, 71, 104, 112, 114, 118, 139, 168, 175, 191, 203, 222, 250. For instance, it has been observed that the GBM subtypes characterized by a high level of activated EGFR, are also characterized by the Notch pathway activation as indicated by elevated expression levels of Notch ligands, cleaved Notch receptor, and downstream target gene product Hes1 (Figure 4) (24). Interestingly, it has been reported that the inhibition of Notch receptor activity by using γ‐secretase inhibitor, N‐[N‐(3,5‐difluorophenacetyl‐L‐alanyl)]‐S‐phenylglycine t‐butyl ester (DAPT) or the downregulation of its ligands Delta‐like‐1 and Jagged by siRNA reduced the proliferation and survival of diverse human glioma cell lines in vitro and in vivo at least in part by decreasing the EGFR expression level (Figure 5) (175). More importantly, the Notch blockade by γ‐secretase inhibitors has also reported to preferentially induce the antiproliferative and apoptotic effects on the self‐renewing and tumorigenic CD133+/nestin+ medulloblastoma‐ and glioblastoma‐initiating cells as compared with their differentiated progenies 57, 58. More specifically, it has been shown that the treatment with the γ‐secretase inhibitors reduced HSR‐GBM1 neurosphere growth and clonogenicity in vitro as well as the tumor growth of intracranial HSR‐GBM1 neurosphere xenografts in nude mice by depleting the CD133+/nestin+ glioblastoma‐initiating cells (58). It has also been noticed that the growth inhibitory and apoptotic effects induced by Notch blockade on CD133+/nestin+ glioblastoma‐initiating cells may be mediated at least in part through a decrease of Akt and STAT3 phosphorylation (58). In the same way, a combination of γ‐secretase inhibitor MRK‐003 with hedgehog inhibitor cyclopamine was also more effective at inducing apoptosis, decreasing cell growth, and inhibiting colony‐forming ability of the medulloblastoma and GBM cell lines, including low‐passage neurospheres isolated from freshly resected human GBMs relative to single drugs (191). Moreover, the inhibition of Notch and sonic hedgehog signaling elements, which are overexpressed in CD133+ GMB cells, with GSI‐1 and cyclopamine, also sensitized the chemoresistant CD133+ GBM cell fraction from GMB cell lines to the cytotoxic effects induced by current chemotherapeutic drug, temozolomide (222).
In addition, a combination of different orally active small molecules that act as the specific inhibitors of different RTKs such as PDGFR, EGFR, c‐Met, RET, c‐KIT and/or VEGFR‐2 or use of pan‐RTK inhibitor has been shown to abrogate the primary brain tumor cell proliferation, survival and tumor angiogenesis 3, 4, 251. For instance, it has been reported that imatinib mesylate (STI571 or Gleevec), which may inhibited PDGFR, abrogated the migration and invasion of human Daoy and D556 medulloblastoma cells and induced the Daoy cell apoptosis in vitro by inhibiting the PDGFB‐induced Erk and Akt signaling pathways and EGFR transactivation concomitant with an upregulation of PTEN expression and activity (4). Furthermore, the pan‐RTK inhibitor, sunitinib, which possesses the ability to cross the blood–brain barrier, also inhibited PDGFR‐signaling cascade and EGFR transactivation and induced anti‐migratory effects on Daoy and D556 medulloblastoma cell lines and primary murine SMO/SMO medulloblastoma cells from the homozygous SMO/SMO mouse model (3).
On the other hand, it has been observed that the overexpression of EGFRvIII in U87MG GBM cells was accompanied by a coactivation of several RTKs, including c‐Met supporting the interest to co‐target these two RTKs (168). In support with this, it has been shown that a combination of an anti‐HGF antibody, L2G7 with erlotinib synergistically reduced the tumor growth and increased the apoptosis of glioma cells in PTENnull/HGF+/c‐Met+/EGFRvIII+ U87MG glioma xenografts in mice in vivo (Figure 5) (117). Also, a combination of an anti‐EGFR antibody (panitumumab) with an anti‐HGF antibody (AMG102) substantially inhibited the tumor growth of EGFRvIII‐expressing U87MG glioma xenografts in mice through an apoptotic mechanism as compared with single agents (168). Future preclinical and clinical studies are however required to ascertain the cytotoxic effects induced by these pharmacological agents targeting different RTKs, alone or in combination therapy, on isolated BTICs with the stem cell‐like properties.
Targeting PI3K/Akt signaling pathway
The stimulation of the PI3K/Akt and their downstream effectors such as mTOR or SREBP‐1, which are the frequent oncogenic events occurring during medulloblastoma and GBM development, might be induced in part through the activation of the RTKs such as wild‐type EGFR, EGFRvIII mutant, PDGFRs, IGF‐R1 and other growth factor signaling cascades as well as the PTEN inactivation (3, 4) 1, 25, 38, 56, 66, 214, 229. More specifically, the tumor suppressor protein PTEN is a phosphatase that can dephosphorylate the key lipid second messager phosphatidylinositol 3, 4, 5‐trisphosphate (PIP3) on the 3 position, generating phosphatidylinositol 3, 5‐bisphosphate (PIP2) and thereby antagonize the function of PI3K (4, 5). Consequently, the PTEN downregulation or loss during brain cancer development may promote the PIP3 accumulation induced through the PI3K activated by RTKs and stimulate its downstream effectors including the serine‐threonine kinase Akt (4, 5). In this matter, several investigations have revealed the therapeutic interest of targeting the PI3K/Akt/mTOR or SREBP‐1 signaling components, alone or in combination therapy, to improve the cytotoxic effects induced by different antitumoral agents including the wild‐type EGFR and truncated EGFRvIII mutant inhibitors, and reduce the growth and induce the apoptotic death of GBM cells in vitro and in vivo (Figure 5) 55, 56, 72, 80, 124, 228. More specifically, it has been reported that the molecular targeting of PI3K/Akt/mTOR by using a specific inhibitor of PI3K (LY294002), Akt (Akt inhibitor III/SH‐6) or mTOR (rapamycin) was more effective at inducing a growth inhibition and apoptosis of CD133+ BTICs relative to their differentiated CD133‐ progenies from primary glioblastoma tissues in vitro and in vivo (55). The targeting of IGFBP2, which is overexpressed in most GBM patients, by shRNA or using a IGFBP2 neutralizing antibody (aBP2) has also been reported to inhibit the neurosphere formation of single GBM cells from patients (96). Interestingly, the aBP2 treatment was also effective in enhancing the sensibility of glioma stem cells to cytotoxic effects induced by an inhibitor of EGFR (AG1478), PDGFR (AG1296) or IGF‐1R (AG1024), as well as the etoposide (VP‐16) and temozolomide at least in part by inhibiting Akt activation‐induced drug resistance in these immature tumor cells (96). In the same way, the analyses of animal models of medulloblastoma resistant to SMO coreceptor hedgehog inhibitor have revealed that a chromosomal amplification of GLI‐2 and upregulation of IGF‐1R/PI3K pathway, and more rarely the mutations in the SMO protein may lead to a reactivation of hedgehog cascade and tumor regrowth after treatment with a SMO antagonist NVP‐LDE225 (25). Of clinical interest, a combination of PI3K inhibitor NVP‐BKM120 or the dual PI3K‐mTOR (mammalian target of rapamycin) inhibitor NVP‐BEZ235 plus NVP‐LDE225 markedly delayed the tumor growth and development of resistance in PTCH+/−;p53−/− tumors subcutaneously implanted in nude mice supporting the interest to co‐target hedgehog pathway and PI3K to prevent the resistance of medulloblastomas to SMO antagonist (25).
In addition, the activation of PI3K/Akt might also result in the activation of SREBP‐1/FAS signaling elements and thereby enhance the expression of fatty acids in response to cholesterol and fatty acid deprivation following the EGFR/EGFRvIII‐mediated cell division (80). Of therapeutic interest, the inhibition of this pro‐survival metabolic pathway mediated through activated EGFR/EGFRvIII has been observed to induce a massive rate of apoptotic death in glioma cell lines expressing a high EGFR level (80). The therapeutic strategies that may be used to block this metabolic pathway include the inhibition of the fatty acid synthesis by using 25‐hydroxycholesterol (25‐HC), which acts as a specific inhibitor of SREBP‐1 processing, hydroxymethylglutaryl coenzyme A reductase (HMG‐CoA) inhibitor, atorvastatin and a FAS inhibitor C75, alone or in combination (Figure 5) (80). For instance, it has been reported that the inhibition of this fatty acid biosynthetic pathway in U87MG‐EGFRvIII glioma cells with a combination of atorvastatin and C75 resulted in a significant reduction of the in vitro tumor growth of xenografts derived from these cancer cells (80).
In considering the fact that the activation of RTKs such as EGFR and downstream signaling elements PI3K/Akt and Ras may lead to the stimulation of a noncanonical hedgehog pathway in brain cancer cells, the targeting of these signaling effectors also might constitute another therapeutic approach to reduce the GLI‐targeted gene expression and oncogenic response 145, 206. In support with this, it has been reported that a specific inhibitor of mitogen‐activated protein kinase kinase (MEK) (U0126) or Akt activity (Akt inhibitor III also designated SH‐6) in combination with cyclopamine synergistically inhibited the growth of U87 human glioma cells in vitro (Figure 5) (206). The enhanced colony formation of U87 glioma cells induced by oncogenic Akt1 and GLI1 was also reduced after a treatment with GLI3R transcriptional repressor or tumor suppressor protein, SUFU or PTEN (206). Importantly, LY294002, Akt inhibitor VIII or rapamycin was also effective to eradicate the SHH‐dependent CD133+ glioma cells as well as SHH‐independent‐CD133+ glioma cells from glioma patients that did not express the hedgehog components and which may persist after hedgehog blockade (238). All these data support the great therapeutic interest to also target PI3K/Akt signaling elements in combination therapies for eradicating the total brain tumor cell mass and prevent disease relapse. Additionally, it has also been shown that the targeting of the local microenvironment of BTICs and their progenies might also constitute an adjuvant therapy to inhibit angiogenesis, and thereby prevent their intracranial dissemination and disease relapse.
Targeting of the local microenvironment of BTICs and their progenies
The local microenvironment of brain cancer cells, and more particularly the microvascular niche may influence the tumor neoangiogenesis and growth and dissemination of BTICs at distant intracranial sites from the primary brain tumors 26, 62, 73. Reciprocally, BTICs can also contribute to tumor angiogenesis by promoting the local endothelial cell activity and recruitment of bone marrow‐derived circulating endothelial progenitor cells (EPCs) by releasing diverse cytokines such as VEGF and stromal cell‐derived factor 1 (SDF‐1) 62, 194, 243. Therefore, the targeting of microvascular niche components by using the anti‐angiogenic agents targeting different angiogenic factors such as the VEGF/VEGFR system, matrix‐degrading MMPs and hypoxia‐inducible factors (HIFs) may constitute adjuvant treatments alone or in combination with the current radiotherapy or chemotherapies to counteract the primary brain tumor development 7, 12, 21, 26, 61, 62, 112, 203. Interestingly, it has been observed that the treatment of the mice bearing orthotopic U87 glioma cell xenografts with an anti‐VEGF mAb, bevacizumab markedly reduced the microvasculature density and tumor growth of vessel‐associated self‐renewing CD133+/nestin+ BTICs (26). The anticarcinogenic effect of bevacizumab was accompanied by a decrease in the number of vessel‐associated self‐renewing CD133+/nestin+ BTICs (26). In the same way, a combination of a VEGFR‐2 antibody DC101 and cytotoxic agent (cyclophosphamide) was also more effective in reducing the number of gliomasphere cells in C6 glioma xenografts in vivo than individual agents (61). In addition, it has also been observed that the VEGF‐dependant proliferation of medulloblastoma and glioma cells expressing VEGFR‐1 and VEGFR‐2 was reduced in the presence of a VEGF or VEGFR inhibitor suggesting that an autocrine loop mediated through VEGF–VEGFR system may contribute to the malignant behavior of brain tumor cells 112, 203. Importantly, the results from a recent phase II clinical trial have also revealed that the orally active pan‐VEGFR tyrosine kinase inhibitor AZD2171 (cediranib) was effective as a monotherapy and associated with manageable toxicity in a subset of patients with recurrent glioblastomas (12). It has however been noted that the patients with recurrent GBMs typically relapse after an initial response to anti‐VEGFR therapy, and AZD2171‐treated recurrent GBMs express high level of PDGFR and c‐Met supporting the interest to also target diverse RTKs in combination with the anti‐angiogenic therapy (47). In this regard, it has been reported that pan‐RTK inhibitor, ZD6474, which inhibits VEGFR‐2 and EGFR was effective at suppressing the angiogenesis and tumor growth of intracranial xenografts derived from EGFRvIII‐expressing U87MG and short‐term cultured primary GBM8 cells in vivo (243). Moreover, the histidine‐rich glycoprotein (HRG), a plasma protein with antiangiogenic properties that can inhibit endothelial cell adhesion and migration, was effective at inhibiting the development of malignant glioma and completely prevent the occurrence of glioblastoma induced by PDGFB in RCAS/TV‐A mouse model of gliomas (107).
Of particular interest, the use of genetically modified migrating NSCs or other cell types such as bone marrow‐derived stem/progenitor cells, which are able to migrate through the CNS and reach the intracranial neoplastic sites also offers great promise as a delivery vehicle for a specific release of the anti‐angiogenic factors, cytotoxic drugs or antibodies to diverse distant brain tumor sites 110, 120, 141, 245, 253. Indeed, NSCs and other adult stem/progenitor cells possess an inherent tumor tropic property and typically migrate throughout the brain to reach the primary medulloblastoma and GBM and infiltrative brain tumor sites that release diverse chemoattractant factors 110, 253. As a matter of fact, it has been observed that the intracerebral injection of BM‐ or fetal tissue‐derived NSC‐like cells engineered to express interleukin‐12 or tumor necrosis factor‐α (TNF‐α) related apoptosis inducing ligand (TRAIL), resulted in their specific recruitment within intracranial glioma, and the release of therapeutic gene product concomitant with an inhibition of tumor growth 49, 50, 120. Moreover, it has been shown that the intracerebral injection of bone marrow‐derived NSC‐like cells or EPCs engineered to express an anti‐angiogenic factor such as a secreted form of platelet factor 4 impaired tumor angiogenesis and U87MG cell‐derived brain tumor growth in vivo (120). Hence, the combination of these different therapeutic strategies targeting BTICs and their progenies as well as their local microenvironment offers great promise to eradicate the total tumor cell mass and prevent the disease relapse.
CONCLUSIONS AND PERSPECTIVES
Together, these recent investigations have revealed that the enhanced expression and activity of the wild‐type EGFR, EGFRvIII mutant and hedgehog signaling elements represent frequent transforming events occurring during primary brain cancer development, and more particularly during medulloblastoma and GBM progression to aggressive and invasive cancer subtypes. These oncogenic pathways can provide critical roles for the cell proliferation, survival and migration of BTICs and their progenies, and thereby cooperate with other oncogenic events for tumor formation and local invasion, treatment resistance and disease relapse.
Despite these important advancements, future studies are required to more precisely establish the specific intracellular pathways induced through the wild‐type EGFR, truncated EGFRvIII mutant and hedgehog in the BTICs vs. their differentiated progenies during primary brain cancer development. Particularly, it will be important to determine the co‐expression levels of EGFR, EGFRvIII and sonic hedgehog (SHH/PTCH/SMO/GLIs) signaling elements and their colocalization with the stem cell‐like markers such as CD133 on a large panel of medulloblastoma and GBM tissues from patients at different stages of disease progression for further confirmation of their potential implication and cooperation in the acquisition of an aggressive behavior by BTICs and their progenies. Moreover, it is also of great interest to more precisely establish the specific functions supplied by homodimers vs. heterodimers formed by the wild‐type EGFR, truncated EGFRvIII mutant and erBb2 as well as the interactive cross‐talks between these RTKs and other growth factor pathways, including the sonic hedgehog cascade that may cooperate for the progression of primary brain cancer to locally advanced and invasive stages and resistance to current therapies. The establishment of animal models of highly invasive GBM reflecting the genetic alterations occurring during human GBM development, including enhanced activation of wild‐type EGFR, EGFRvIII mutant and hedgehog, is also important to develop more accurate methods of preclinical testing of novel anticarcinogenic agents. Especially, it will be important to develop new animal models to establish the specific functions of sonic hedgehog in GMB development and its interacting oncogenic partners. Also, the implantation of patient's tumor specimens in animal models in vivo may represent the good GBM models to validate the results obtained in conventional non‐invasive orthotopic models using xenografted GBM cell lines.
Hence, these additional studies should lead to the development of more effective multitargeted approaches to inhibit the wild‐type EGFR, EGFRvIII mutant and hedgehog‐mediated tumorigenic pathways and other key signaling elements that cooperate for the acquisition of a more malignant behavior and survival advantages by BTICs and their progenies during disease progression. These multitargeted strategies could be used to improve the current radiation and chemotherapeutic treatments against highly aggressive and lethal primary brain cancers, including pediatric medulloblastomas and adult GBMs, and thereby prevent disease relapse and the death of cancer patients.
ACKNOWLEDGMENTS
The authors on this work are supported in part by U.S. Department of Defense grants (PC074289 and PC074289 and BC074639) and the National Institutes of Health [Grants R01CA78590, EDRN U01CA111294, P50CA127297, R01CA133774, R01CA131944 and R01CA138791]. The authors disclose no potential conflicts of interest.
REFERENCES
- 1. (2008) Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature 455:1061–1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Abel TW, Clark C, Bierie B, Chytil A, Aakre M, Gorska A et al (2009) GFAP‐Cre‐mediated activation of oncogenic K‐ras results in expansion of the subventricular zone and infiltrating glioma. Mol Cancer Res 7:645–653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Abouantoun TJ, MacDonald TJ (2009) Imatinib blocks migration and invasion of medulloblastoma cells by concurrently inhibiting activation of platelet‐derived growth factor receptor and transactivation of epidermal growth factor receptor. Mol Cancer Ther 8:1137–1147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Abouantoun TJ, Castellino RC, MacDonald TJ (2011) Sunitinib induces PTEN expression and inhibits PDGFR signaling and migration of medulloblastoma cells. J Neurooncol 101:215–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Aldape KD, Ballman K, Furth A, Buckner JC, Giannini C, Burger PC et al (2004) Immunohistochemical detection of EGFRvIII in high malignancy grade astrocytomas and evaluation of prognostic significance. J Neuropathol Exp Neurol 63:700–707. [DOI] [PubMed] [Google Scholar]
- 6. Amankulor NM, Hambardzumyan D, Pyonteck SM, Becher OJ, Joyce JA, Holland EC (2009) Sonic hedgehog pathway activation is induced by acute brain injury and regulated by injury‐related inflammation. J Neurosci 29:10299–10308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Andersson U, Schwartzbaum J, Wiklund F, Sjostrom S, Liu Y, Tsavachidis S et al (2010) A comprehensive study of the association between the EGFR and ERBB2 genes and glioma risk. Acta Oncol 49:767–775. [DOI] [PubMed] [Google Scholar]
- 8. Ayrault O, Zhao H, Zindy F, Qu C, Sherr CJ, Roussel MF (2010) Atoh1 inhibits neuronal differentiation and collaborates with Gli1 to generate medulloblastoma‐initiating cells. Cancer Res 70:5618–5627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Bachoo RM, Maher EA, Ligon KL, Sharpless NE, Chan SS, You MJ et al (2002) Epidermal growth factor receptor and Ink4a/Arf: convergent mechanisms governing terminal differentiation and transformation along the neural stem cell to astrocyte axis. Cancer Cell 1:269–277. [DOI] [PubMed] [Google Scholar]
- 10. Bao S, Wu Q, McLendon RE, Hao Y, Shi Q, Hjelmeland AB et al (2006) Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature 444:756–760. [DOI] [PubMed] [Google Scholar]
- 11. Bar EE, Chaudhry A, Lin A, Fan X, Schreck K, Matsui W et al (2007) Cyclopamine‐mediated hedgehog pathway inhibition depletes stem‐like cancer cells in glioblastoma. Stem Cells 25:2524–2533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Batchelor TT, Duda DG, Ancukiewicz M, Plotkin SR, Gerstner E et al (2010) Phase II study of cediranib, an oral pan‐vascular endothelial growth factor receptor tyrosine kinase inhibitor, in patients with recurrent glioblastoma. J Clin Oncol 28:2817–2823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Bath KG, Lee FS (2010) Neurotrophic factor control of adult SVZ neurogenesis. Dev Neurobiol 70:339–349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Batra SK, Castelino‐Prabhu S, Wikstrand CJ, Zhu X, Humphrey PA, Friedman HS et al (1995) Epidermal growth factor ligand‐independent, unregulated, cell‐transforming potential of a naturally occurring human mutant EGFRvIII gene. Cell Growth Differ 6:1251–1259. [PubMed] [Google Scholar]
- 15. Becher OJ, Hambardzumyan D, Fomchenko EI, Momota H, Mainwaring L, Bleau AM et al (2008) Gli activity correlates with tumor grade in platelet‐derived growth factor‐induced gliomas. Cancer Res 68:2241–2249. [DOI] [PubMed] [Google Scholar]
- 16. Becher OJ, Hambardzumyan D, Walker TR, Helmy K, Nazarian J, Albrecht S et al (2010) Preclinical evaluation of radiation and perifosine in a genetically and histologically accurate model of brainstem glioma. Cancer Res 70:2548–2557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Beers R, Chowdhury P, Bigner D, Pastan I (2000) Immunotoxins with increased activity against epidermal growth factor receptor vIII‐expressing cells produced by antibody phage display. Clin Cancer Res 6:2835–2843. [PubMed] [Google Scholar]
- 18. Beier D, Hau P, Proescholdt M, Lohmeier A, Wischhusen J, Oefner PJ et al (2007) CD133(+) and CD133(–) glioblastoma‐derived cancer stem cells show differential growth characteristics and molecular profiles. Cancer Res 67:4010–4015. [DOI] [PubMed] [Google Scholar]
- 19. Bexell D, Gunnarsson S, Siesjo P, Bengzon J, Darabi A (2009) CD133+ and nestin+ tumor‐initiating cells dominate in N29 and N32 experimental gliomas. Int J Cancer 125:15–22. [DOI] [PubMed] [Google Scholar]
- 20. Bikeye SN, Colin C, Marie Y, Vampouille R, Ravassard P et al (2010) ASPM‐associated stem cell proliferation is involved in malignant progression of gliomas and constitutes an attractive therapeutic target. Cancer Cell Int 10:1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Blom T, Roselli A, Hayry V, Tynninen O, Wartiovaara K, Korja M et al (2010) Amplification and overexpression of KIT, PDGFRA, and VEGFR2 in medulloblastomas and primitive neuroectodermal tumors. J Neurooncol 97:217–224. [DOI] [PubMed] [Google Scholar]
- 22. Blough MD, Beauchamp DC, Westgate MR, Kelly JJ, Cairncross JG (2011) Effect of aberrant p53 function on temozolomide sensitivity of glioma cell lines and brain tumor initiating cells from glioblastoma. J Neurooncol 102:1–7. [DOI] [PubMed] [Google Scholar]
- 23. Bodey B, Kaiser HE, Siegel SE (2005) Epidermal growth factor receptor (EGFR) expression in childhood brain tumors. In Vivo 19:931–941. [PubMed] [Google Scholar]
- 24. Brennan C, Momota H, Hambardzumyan D, Ozawa T, Tandon A, Pedraza A et al (2009) Glioblastoma subclasses can be defined by activity among signal transduction pathways and associated genomic alterations. PLoS ONE 4:e7752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Buonamici S, Williams J, Morrissey M, Wang A, Guo R, Vattay A et al (2010) Interfering with resistance to smoothened antagonists by inhibition of the PI3K pathway in medulloblastoma. Sci Transl Med 2:51ra70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Calabrese C, Poppleton H, Kocak M, Hogg TL, Fuller C, Hamner B et al (2007) A perivascular niche for brain tumor stem cells. Cancer Cell 11:69–82. [DOI] [PubMed] [Google Scholar]
- 27. Calzolari F, Malatesta P (2010) Recent insights into PDGF‐induced gliomagenesis. Brain Pathol 20:527–538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Carrillo‐Garcia C, Suh Y, Obernier K, Holzl‐Wenig G, Mandl C, Ciccolini F (2010) Multipotent precursors in the anterior and hippocampal subventricular zone display similar transcription factor signatures but their proliferation and maintenance are differentially regulated. Mol Cell Neurosci 44:318–329. [DOI] [PubMed] [Google Scholar]
- 29. Cemeus C, Zhao TT, Barrett GM, Lorimer IA, Dimitroulakos J (2008) Lovastatin enhances gefitinib activity in glioblastoma cells irrespective of EGFRvIII and PTEN status. J Neurooncol 90:9–17. [DOI] [PubMed] [Google Scholar]
- 30. Chen R, Nishimura MC, Bumbaca SM, Kharbanda S, Forrest WF, Kasman IM et al (2010) A hierarchy of self‐renewing tumor‐initiating cell types in glioblastoma. Cancer Cell 17:362–375. [DOI] [PubMed] [Google Scholar]
- 31. Cho YJ, Tsherniak A, Tamayo P, Santagata S, Ligon A, Greulich H et al (2011) Integrative genomic analysis of medulloblastoma identifies a molecular subgroup that drives poor clinical outcome. J Clin Oncol 29:1424–1430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Choe G, Horvath S, Cloughesy TF, Crosby K, Seligson D, Palotie A et al (2003) Analysis of the phosphatidylinositol 3′‐kinase signaling pathway in glioblastoma patients in vivo . Cancer Res 63:2742–2746. [PubMed] [Google Scholar]
- 33. Choi BD, Archer GE, Mitchell DA, Heimberger AB, McLendon RE, Bigner DD et al (2009) EGFRvIII‐targeted vaccination therapy of malignant glioma. Brain Pathol 19:713–723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Chu SH, Ma YB, Feng DF, Zhang H, Qiu JH, Zhu ZA (2010) c‐Met antisense oligodeoxynucleotides increase sensitivity of human glioma cells to paclitaxel. Oncol Rep 24:189–194. [DOI] [PubMed] [Google Scholar]
- 35. Clark PA, Treisman DM, Ebben J, Kuo JS (2007) Developmental signaling pathways in brain tumor‐derived stem‐like cells. Dev Dyn 236:3297–3308. [DOI] [PubMed] [Google Scholar]
- 37. Clement V, Sanchez P, de Tribolet N, Radovanovic I, Altaba A (2007) HEDGEHOG‐GLI1 signaling regulates human glioma growth, cancer stem cell self‐renewal, and tumorigenicity. Curr Biol 17:165–172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Clement V, Dutoit V, Marino D, Dietrich PY, Radovanovic I (2009) Limits of CD133 as a marker of glioma self‐renewing cells. Int J Cancer 125:244–248. [DOI] [PubMed] [Google Scholar]
- 38. Cloughesy TF, Yoshimoto K, Nghiemphu P, Brown K, Dang J, Zhu S et al (2008) Antitumor activity of rapamycin in a Phase I trial for patients with recurrent PTEN‐deficient glioblastoma. PLoS Med 5:e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Cooper LA, Gutman DA, Long Q, Johnson BA, Cholleti SR, Kurc T et al (2010) The proneural molecular signature is enriched in oligodendrogliomas and predicts improved survival among diffuse gliomas. PLoS ONE 5:e12548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Corcoran RB, Scott MP (2006) Oxysterols stimulate Sonic hedgehog signal transduction and proliferation of medulloblastoma cells. Proc Natl Acad Sci USA 103:8408–8413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Cui D, Xu Q, Wang K, Che X (2010) Gli1 is a potential target for alleviating multidrug resistance of gliomas. J Neurol Sci 288:156–166. [DOI] [PubMed] [Google Scholar]
- 42. Currle DS, Gilbertson RJ (2008) The niche revealed. Cell Stem Cell 3:234–236. [DOI] [PubMed] [Google Scholar]
- 43. Dahmane N, Sanchez P, Gitton Y, Palma V, Sun T, Beyna M et al (2001) The Sonic Hedgehog‐Gli pathway regulates dorsal brain growth and tumorigenesis. Development 128:5201–5212. [DOI] [PubMed] [Google Scholar]
- 44. de la Iglesia N, Konopka G, Puram SV, Chan JA, Bachoo RM, You MJ et al (2008) Identification of a PTEN‐regulated STAT3 brain tumor suppressor pathway. Genes Dev 22:449–462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Denholt CL, Hansen PR, Pedersen N, Poulsen HS, Gillings N, Kjaer A (2009) Identification of novel peptide ligands for the cancer‐specific receptor mutation EFGRvIII using a mixture‐based synthetic combinatorial library. Biopolymers 91:201–206. [DOI] [PubMed] [Google Scholar]
- 46. Ding H, Shannon P, Lau N, Wu X, Roncari L, Baldwin RL et al (2003) Oligodendrogliomas result from the expression of an activated mutant epidermal growth factor receptor in a RAS transgenic mouse astrocytoma model. Cancer Res 63:1106–1113. [PubMed] [Google Scholar]
- 47. diTomaso E, Snuderl M, Kamoun WS, Duda DG, Auluck PK, Fazlollahi L et al (2011) Glioblastoma recurrence after cediranib therapy in patients: lack of “rebound” revascularization as mode of escape. Cancer Res 71:19–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Eberhart CG (2011) Molecular diagnostics in embryonal brain tumors. Brain Pathol 21:96–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Ehtesham M, Kabos P, Gutierrez MA, Chung NH, Griffith TS, Black KL et al (2002) Induction of glioblastoma apoptosis using neural stem cell‐mediated delivery of tumor necrosis factor‐related apoptosis‐inducing ligand. Cancer Res 62:7170–7174. [PubMed] [Google Scholar]
- 50. Ehtesham M, Kabos P, Kabosova A, Neuman T, Black KL, Yu JS (2002) The use of interleukin 12‐secreting neural stem cells for the treatment of intracranial glioma. Cancer Res 62:5657–5663. [PubMed] [Google Scholar]
- 51. Ehtesham M, Sarangi A, Valadez JG, Chanthaphaychith S, Becher MW, Abel TW et al (2007) Ligand‐dependent activation of the hedgehog pathway in glioma progenitor cells. Oncogene 26:5752–5761. [DOI] [PubMed] [Google Scholar]
- 52. El‐Sheikh A, Fan R, Birks D, Donson A, Foreman NK, Vibhakar R (2010) Inhibition of Aurora Kinase A enhances chemosensitivity of medulloblastoma cell lines. Pediatr Blood Cancer 55:35–41. [DOI] [PubMed] [Google Scholar]
- 53. Elamin MH, Shinwari Z, Hendrayani SF, Al‐Hindi H, Al‐Shail E, Khafaga Y et al (2010) Curcumin inhibits the Sonic Hedgehog signaling pathway and triggers apoptosis in medulloblastoma cells. Mol Carcinog 49:302–314. [DOI] [PubMed] [Google Scholar]
- 54. Eller JL, Longo SL, Hicklin DJ, Canute GW (2002) Activity of anti‐epidermal growth factor receptor monoclonal antibody C225 against glioblastoma multiforme. Neurosurgery 51:1005–1013. [DOI] [PubMed] [Google Scholar]
- 55. Eyler CE, Foo WC, LaFiura KM, McLendon RE, Hjelmeland AB, Rich JN (2008) Brain cancer stem cells display preferential sensitivity to Akt inhibition. Stem Cells 26:3027–3036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Fan QW, Knight ZA, Goldenberg DD, Yu W, Mostov KE, Stokoe D et al (2006) A dual PI3 kinase/mTOR inhibitor reveals emergent efficacy in glioma. Cancer Cell 9:341–349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Fan X, Matsui W, Khaki L, Stearns D, Chun J, Li YM et al (2006) Notch pathway inhibition depletes stem‐like cells and blocks engraftment in embryonal brain tumors. Cancer Res 66:7445–7452. [DOI] [PubMed] [Google Scholar]
- 58. Fan X, Khaki L, Zhu TS, Soules ME, Talsma CE, Gul N et al (2010) NOTCH pathway blockade depletes CD133‐positive glioblastoma cells and inhibits growth of tumor neurospheres and xenografts. Stem Cells 28:5–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Feldkamp MM, Lala P, Lau N, Roncari L, Guha A (1999) Expression of activated epidermal growth factor receptors, Ras‐guanosine triphosphate, and mitogen‐activated protein kinase in human glioblastoma multiforme specimens. Neurosurgery 45:1442–1453. [DOI] [PubMed] [Google Scholar]
- 60. Fogarty MP, Kessler JD, Wechsler‐Reya RJ (2005) Morphing into cancer: the role of developmental signaling pathways in brain tumor formation. J Neurobiol 64:458–475. [DOI] [PubMed] [Google Scholar]
- 61. Folkins C, Man S, Xu P, Shaked Y, Hicklin DJ, Kerbel RS (2007) Anticancer therapies combining antiangiogenic and tumor cell cytotoxic effects reduce the tumor stem‐like cell fraction in glioma xenograft tumors. Cancer Res 67:3560–3564. [DOI] [PubMed] [Google Scholar]
- 62. Folkins C, Shaked Y, Man S, Tang T, Lee CR, Zhu Z et al (2009) Glioma tumor stem‐like cells promote tumor angiogenesis and vasculogenesis via vascular endothelial growth factor and stromal‐derived factor 1. Cancer Res 69:7243–7251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Fromm JA, Johnson SA, Johnson DL (2008) Epidermal growth factor receptor 1 (EGFR1) and its variant EGFRvIII regulate TATA‐binding protein expression through distinct pathways. Mol Cell Biol 28:6483–6495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Fu J, Shao CJ, Chen FR, Ng HK, Chen ZP (2010) Autophagy induced by valproic acid is associated with oxidative stress in glioma cell lines. Neuro Oncol 12:328–340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Fukaya R, Ohta S, Yamaguchi M, Fujii H, Kawakami Y, Kawase T et al (2010) Isolation of cancer stem‐like cells from a side population of a human glioblastoma cell line, SK‐MG‐1. Cancer Lett 291:150–157. [DOI] [PubMed] [Google Scholar]
- 66. Furnari FB, Fenton T, Bachoo RM, Mukasa A, Stommel JM, Stegh A et al (2007) Malignant astrocytic glioma: genetics, biology, and paths to treatment. Genes Dev 21:2683–2710. [DOI] [PubMed] [Google Scholar]
- 67. Gajjar A, Hernan R, Kocak M, Fuller C, Lee Y, McKinnon PJ et al (2004) Clinical, histopathologic, and molecular markers of prognosis: toward a new disease risk stratification system for medulloblastoma. J Clin Oncol 22:984–993. [DOI] [PubMed] [Google Scholar]
- 68. Gal H, Pandi G, Kanner AA, Ram Z, Lithwick‐Yanai G, Amariglio N et al (2008) MIR‐451 and Imatinib mesylate inhibit tumor growth of Glioblastoma stem cells. Biochem Biophys Res Commun 376:86–90. [DOI] [PubMed] [Google Scholar]
- 69. Galli R, Binda E, Orfanelli U, Cipelletti B, Gritti A, De Vitis S et al (2004) Isolation and characterization of tumorigenic, stem‐like neural precursors from human glioblastoma. Cancer Res 64:7011–7021. [DOI] [PubMed] [Google Scholar]
- 70. Gibson P, Tong Y, Robinson G, Thompson MC, Currle DS, Eden C et al (2010) Subtypes of medulloblastoma have distinct developmental origins. Nature 468:1095–1099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Gilbertson RJ, Clifford SC (2003) PDGFRB is overexpressed in metastatic medulloblastoma. Nat Genet 35:197–198. [DOI] [PubMed] [Google Scholar]
- 72. Gilbertson RJ, Ellison DW (2008) The origins of medulloblastoma subtypes. Annu Rev Pathol 3:341–365. [DOI] [PubMed] [Google Scholar]
- 73. Gilbertson RJ, Rich JN (2007) Making a tumour's bed: glioblastoma stem cells and the vascular niche. Nat Rev Cancer 7:733–736. [DOI] [PubMed] [Google Scholar]
- 74. Gilbertson RJ, Perry RH, Kelly PJ, Pearson AD, Lunec J (1997) Prognostic significance of HER2 and HER4 coexpression in childhood medulloblastoma. Cancer Res 57:3272–3280. [PubMed] [Google Scholar]
- 75. Grandal MV, Zandi R, Pedersen MW, Willumsen BM, van Deurs DB, Poulsen HS (2007) EGFRvIII escapes down‐regulation due to impaired internalization and sorting to lysosomes. Carcinogenesis 28:1408–1417. [DOI] [PubMed] [Google Scholar]
- 76. Griffero F, Daga A, Marubbi D, Capra MC, Melotti A, Pattarozzi A et al (2009) Different response of human glioma tumor‐initiating cells to EGFR kinase inhibitors. J Biol Chem 284:7138–7148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Grimm I, Messemer N, Stanke M, Gachet C, Zimmermann H (2009) Coordinate pathways for nucleotide and EGF signaling in cultured adult neural progenitor cells. J Cell Sci 122:2524–2533. [DOI] [PubMed] [Google Scholar]
- 78. Guessous F, Zhang Y, diPierro C, Marcinkiewicz L, Sarkaria J, Schiff D et al (2010) An orally bioavailable c‐Met kinase inhibitor potently inhibits brain tumor malignancy and growth. Anticancer Agents Med Chem 10:28–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Guessous F, Zhang Y, Kofman A, Catania A, Li Y, Schiff D et al (2010) microRNA‐34a is tumor suppressive in brain tumors and glioma stem cells. Cell Cycle 9:1031–1036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Guo D, Prins RM, Dang J, Kuga D, Iwanami A, Soto H et al (2009) EGFR signaling through an Akt‐SREBP‐1‐dependent, rapamycin‐resistant pathway sensitizes glioblastomas to antilipogenic therapy. Sci Signal 2:ra82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Halatsch ME, Schmidt U, Behnke‐Mursch J, Unterberg A, Wirtz CR (2006) Epidermal growth factor receptor inhibition for the treatment of glioblastoma multiforme and other malignant brain tumours. Cancer Treat Rev 32:74–89. [DOI] [PubMed] [Google Scholar]
- 82. Hambardzumyan D, Amankulor NM, Helmy KY, Becher OJ, Holland EC (2009) Modeling adult gliomas using RCAS/t‐va technology. Transl Oncol 2:89–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Hambardzumyan D, Parada LF, Holland EC, Charest A (2011) Genetic modeling of gliomas in mice: New tools to tackle old problems. Glia [Epub ahead of print; doi: 10.1002/glia.21142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Han W, Zhang T, Yu H, Foulke JG, Tang CK (2006) Hypophosphorylation of residue Y1045 leads to defective downregulation of EGFRvIII. Cancer Biol Ther 5:1361–1368. [DOI] [PubMed] [Google Scholar]
- 85. Harris MA, Yang H, Low BE, Mukherjee J, Guha A, Bronson RT et al (2008) Cancer stem cells are enriched in the side population cells in a mouse model of glioma. Cancer Res 68:10051–10059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Hatton BA, Villavicencio EH, Tsuchiya KD, Pritchard JI, Ditzler S, Pullar B et al (2008) The Smo/Smo model: hedgehog‐induced medulloblastoma with 90% incidence and leptomeningeal spread. Cancer Res 68:1768–1776. [DOI] [PubMed] [Google Scholar]
- 87. Hede SM, Hansson I, Afink GB, Eriksson A, Nazarenko I, Andrae J et al (2009) GFAP promoter driven transgenic expression of PDGFB in the mouse brain leads to glioblastoma in a Trp53 null background. Glia 57:1143–1153. [DOI] [PubMed] [Google Scholar]
- 88. Heimberger AB, Sun W, Hussain SF, Dey M, Crutcher L, Aldape K et al (2008) Immunological responses in a patient with glioblastoma multiforme treated with sequential courses of temozolomide and immunotherapy: case study. Neuro Oncol 10:98–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Heine VM, Priller M, Ling J, Rowitch DH, Schuller U (2010) Dexamethasone destabilizes Nmyc to inhibit the growth of hedgehog‐associated medulloblastoma. Cancer Res 70:5220–5225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Hemmati HD, Nakano I, Lazareff JA, Masterman‐Smith M, Geschwind DH, Bronner‐Fraser M et al (2003) Cancerous stem cells can arise from pediatric brain tumors. Proc Natl Acad Sci USA 100:15178–15183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Hernan R, Fasheh R, Calabrese C, Frank AJ, Maclean KH, Allard D et al (2003) ERBB2 up‐regulates S100A4 and several other prometastatic genes in medulloblastoma. Cancer Res 63:140–148. [PubMed] [Google Scholar]
- 92. Holland EC, Hively WP, DePinho RA, Varmus HE (1998) A constitutively active epidermal growth factor receptor cooperates with disruption of G1 cell‐cycle arrest pathways to induce glioma‐like lesions in mice. Genes Dev 12:3675–3685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Holland EC, Li Y, Celestino J, Dai C, Schaefer L, Sawaya RA et al (2000) Astrocytes give rise to oligodendrogliomas and astrocytomas after gene transfer of polyoma virus middle T antigen in vivo . Am J Pathol 157:1031–1037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Horvath S, Zhang B, Carlson M, Lu KV, Zhu S, Felciano RM et al (2006) Analysis of oncogenic signaling networks in glioblastoma identifies ASPM as a molecular target. Proc Natl Acad Sci USA 103:17402–17407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Hsieh A, Ellsworth R, Hsieh D (2010) GLI1 regulates IRS1 expression to promote IGF dependent malignant behaviors in glioma stem cells. J Cell Physiol 226:1118–1127. [DOI] [PubMed] [Google Scholar]
- 96. Hsieh D, Hsieh A, Stea B, Ellsworth R (2010) IGFBP2 promotes glioma tumor stem cell expansion and survival. Biochem Biophys Res Commun 397:367–372. [DOI] [PubMed] [Google Scholar]
- 97. Huang HS, Nagane M, Klingbeil CK, Lin H, Nishikawa R, Ji XD et al (1997) The enhanced tumorigenic activity of a mutant epidermal growth factor receptor common in human cancers is mediated by threshold levels of constitutive tyrosine phosphorylation and unattenuated signaling. J Biol Chem 272:2927–2935. [DOI] [PubMed] [Google Scholar]
- 98. Huang PH, Mukasa A, Bonavia R, Flynn RA, Brewer ZE, Cavenee WK et al (2007) Quantitative analysis of EGFRvIII cellular signaling networks reveals a combinatorial therapeutic strategy for glioblastoma. Proc Natl Acad Sci USA 104:12867–12872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Huang Q, Zhang QB, Dong J, Wu YY, Shen YT, Zhao YD et al (2008) Glioma stem cells are more aggressive in recurrent tumors with malignant progression than in the primary tumor, and both can be maintained long‐term in vitro . BMC Cancer 8:304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Huse JT, Holland EC (2009) Genetically engineered mouse models of brain cancer and the promise of preclinical testing. Brain Pathol 19:132–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Johns TG, Perera RM, Vernes SC, Vitali AA, Cao DX, Cavenee WK et al (2007) The efficacy of epidermal growth factor receptor‐specific antibodies against glioma xenografts is influenced by receptor levels, activation status, and heterodimerization. Clin Cancer Res 13:1911–1925. [DOI] [PubMed] [Google Scholar]
- 102. Joo KM, Kim SY, Jin X, Song SY, Kong DS, Lee JI et al (2008) Clinical and biological implications of CD133‐positive and CD133‐negative cells in glioblastomas. Lab Invest 88:808–815. [DOI] [PubMed] [Google Scholar]
- 103. Jung J, Solanki A, Memoli KA, Kamei K, Kim H, Drahl MA et al (2010) Selective inhibition of human brain tumor cells through multifunctional quantum‐dot‐based siRNA delivery. Angew Chem Int Ed Engl 49:103–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Kagawa N, Maruno M, Suzuki T, Hashiba T, Hashimoto N, Izumoto S et al (2006) Detection of genetic and chromosomal aberrations in medulloblastomas and primitive neuroectodermal tumors with DNA microarrays. Brain Tumor Pathol 23:41–47. [DOI] [PubMed] [Google Scholar]
- 105. Kamat V, Donaldson JM, Kari C, Quadros MR, Lelkes PI, Chaiken I et al (2008) Enhanced EGFR inhibition and distinct epitope recognition by EGFR antagonistic mAbs C225 and 425. Cancer Biol Ther 7:726–733. [DOI] [PubMed] [Google Scholar]
- 106. Karpel‐Massler G, Schmidt U, Unterberg A, Halatsch ME (2009) Therapeutic inhibition of the epidermal growth factor receptor in high‐grade gliomas: where do we stand? Mol Cancer Res 7:1000–1012. [DOI] [PubMed] [Google Scholar]
- 107. Karrlander M, Lindberg N, Olofsson T, Kastemar M, Olsson AK, Uhrbom L (2009) Histidine‐rich glycoprotein can prevent development of mouse experimental glioblastoma. PLoS ONE 4:e8536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Keir ST, Maris JM, Lock R, Kolb EA, Gorlick R, Carol H et al (2010) Initial testing (stage 1) of the multi‐targeted kinase inhibitor sorafenib by the pediatric preclinical testing program. Pediatr Blood Cancer 55:1126–1133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Kim J, Tang JY, Gong R, Kim J, Lee JJ, Clemons KV et al (2010) Itraconazole, a commonly used antifungal that inhibits Hedgehog pathway activity and cancer growth. Cancer Cell 17:388–399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Kim SU (2011) Neural stem cell‐based gene therapy for brain tumors. Stem Cell Rev 7:130–140. [DOI] [PubMed] [Google Scholar]
- 111. Kinzler KW, Bigner SH, Bigner DD, Trent JM, Law ML, O'Brien SJ et al (1987) Identification of an amplified, highly expressed gene in a human glioma. Science 236:70–73. [DOI] [PubMed] [Google Scholar]
- 112. Knizetova P, Ehrmann J, Hlobilkova A, Vancova I, Kalita O, Kolar Z et al (2008) Autocrine regulation of glioblastoma cell cycle progression, viability and radioresistance through the VEGF‐VEGFR2 (KDR) interplay. Cell Cycle 7:2553–2561. [DOI] [PubMed] [Google Scholar]
- 113. Kokovay E, Goderie S, Wang Y, Lotz S, Lin G, Sun Y et al (2010) Adult SVZ lineage cells home to and leave the vascular niche via differential responses to SDF1/CXCR4 signaling. Cell Stem Cell 7:163–173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Kong DS, Song SY, Kim DH, Joo KM, Yoo JS, Koh JS et al (2009) Prognostic significance of c‐Met expression in glioblastomas. Cancer 115:140–148. [DOI] [PubMed] [Google Scholar]
- 115. Kool M, Koster J, Bunt J, Hasselt NE, Lakeman A, van Sluis P et al (2008) Integrated genomics identifies five medulloblastoma subtypes with distinct genetic profiles, pathway signatures and clinicopathological features. PLoS ONE 3:e3088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Krampert M, Chirasani SR, Wachs FP, Aigner R, Bogdahn U, Yingling JM et al (2010) Smad7 regulates the adult neural stem/progenitor cell pool in a transforming growth factor beta‐ and bone morphogenetic protein‐independent manner. Mol Cell Biol 30:3685–3694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Lal A, Glazer CA, Martinson HM, Friedman HS, Archer GE, Sampson JH et al (2002) Mutant epidermal growth factor receptor up‐regulates molecular effectors of tumor invasion. Cancer Res 62:3335–3339. [PubMed] [Google Scholar]
- 118. Lal B, Xia S, Abounader R, Laterra J (2005) Targeting the c‐Met pathway potentiates glioblastoma responses to gamma‐radiation. Clin Cancer Res 11:4479–4486. [DOI] [PubMed] [Google Scholar]
- 119. Lammering G, Hewit TH, Holmes M, Valerie K, Hawkins W, Lin PS et al (2004) Inhibition of the type III epidermal growth factor receptor variant mutant receptor by dominant‐negative EGFR‐CD533 enhances malignant glioma cell radiosensitivity. Clin Cancer Res 10:6732–6743. [DOI] [PubMed] [Google Scholar]
- 120. Lee J, Elkahloun AG, Messina SA, Ferrari N, Xi D, Smith CL et al (2003) Cellular and genetic characterization of human adult bone marrow‐derived neural stem‐like cells: a potential antiglioma cellular vector. Cancer Res 63:8877–8889. [PubMed] [Google Scholar]
- 121. Lee J, Kotliarova S, Kotliarov Y, Li A, Su Q, Donin NM et al (2006) Tumor stem cells derived from glioblastomas cultured in bFGF and EGF more closely mirror the phenotype and genotype of primary tumors than do serum‐cultured cell lines. Cancer Cell 9:391–403. [DOI] [PubMed] [Google Scholar]
- 122. Li A, Walling J, Ahn S, Kotliarov Y, Su Q, Quezado M et al (2009) Unsupervised analysis of transcriptomic profiles reveals six glioma subtypes. Cancer Res 69:2091–2099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Li B, Chang CM, Yuan M, McKenna WG, Shu HK (2003) Resistance to small molecule inhibitors of epidermal growth factor receptor in malignant gliomas. Cancer Res 63:7443–7450. [PubMed] [Google Scholar]
- 124. Li B, Yuan M, Kim IA, Chang CM, Bernhard EJ, Shu HK (2004) Mutant epidermal growth factor receptor displays increased signaling through the phosphatidylinositol‐3 kinase/AKT pathway and promotes radioresistance in cells of astrocytic origin. Oncogene 23:4594–4602. [DOI] [PubMed] [Google Scholar]
- 125. Li L, Dutra A, Pak E, Labrie JE, III , Gerstein RM, Pandolfi PP et al (2009) EGFRvIII expression and PTEN loss synergistically induce chromosomal instability and glial tumors. Neuro Oncol 11:9–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Li S, Schmitz KR, Jeffrey PD, Wiltzius JJ, Kussie P, Ferguson KM (2005) Structural basis for inhibition of the epidermal growth factor receptor by cetuximab. Cancer Cell 7:301–311. [DOI] [PubMed] [Google Scholar]
- 127. Lim DA, varez‐Buylla A (1999) Interaction between astrocytes and adult subventricular zone precursors stimulates neurogenesis. Proc Natl Acad Sci USA 96:7526–7531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Lim DA, Huang YC, Alvarez‐Buylla A (2007) The adult neural stem cell niche: lessons for future neural cell replacement strategies. Neurosurg Clin N Am 18:81–89. [DOI] [PubMed] [Google Scholar]
- 129. Lindberg N, Kastemar M, Olofsson T, Smits A, Uhrbom L (2009) Oligodendrocyte progenitor cells can act as cell of origin for experimental glioma. Oncogene 28:2266–2275. [DOI] [PubMed] [Google Scholar]
- 130. Liu G, Yuan X, Zeng Z, Tunici P, Ng H, Abdulkadir IR et al (2006) Analysis of gene expression and chemoresistance of CD133+ cancer stem cells in glioblastoma. Mol Cancer 5:67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Liu Q, Nguyen DH, Dong Q, Shitaku P, Chung K, Liu OY et al (2009) Molecular properties of CD133+ glioblastoma stem cells derived from treatment‐refractory recurrent brain tumors. J Neurooncol 94:1–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. Lo HW, Cao X, Zhu H, li‐Osman F (2008) Constitutively activated STAT3 frequently coexpresses with epidermal growth factor receptor in high‐grade gliomas and targeting STAT3 sensitizes them to Iressa and alkylators. Clin Cancer Res 14:6042–6054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. Lorimer IA, Wikstrand CJ, Batra SK, Bigner DD, Pastan I (1995) Immunotoxins that target an oncogenic mutant epidermal growth factor receptor expressed in human tumors. Clin Cancer Res 1:859–864. [PubMed] [Google Scholar]
- 134. Luwor RB, Zhu HJ, Walker F, Vitali AA, Perera RM, Burgess AW et al (2004) The tumor‐specific de2‐7 epidermal growth factor receptor (EGFR) promotes cells survival and heterodimerizes with the wild‐type EGFR. Oncogene 23:6095–6104. [DOI] [PubMed] [Google Scholar]
- 135. MacDonald TJ, Brown KM, LaFleur B, Peterson K, Lawlor C, Chen Y et al (2001) Expression profiling of medulloblastoma: PDGFRA and the RAS/MAPK pathway as therapeutic targets for metastatic disease. Nat Genet 29:143–152. [DOI] [PubMed] [Google Scholar]
- 136. Marian CO, Cho SK, McEllin BM, Maher EA, Hatanpaa KJ, Madden CJ et al (2010) The telomerase antagonist, imetelstat, efficiently targets glioblastoma tumor‐initiating cells leading to decreased proliferation and tumor growth. Clin Cancer Res 16:154–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137. Martens T, Laabs Y, Gunther HS, Kemming D, Zhu Z, Witte L et al (2008) Inhibition of glioblastoma growth in a highly invasive nude mouse model can be achieved by targeting epidermal growth factor receptor but not vascular endothelial growth factor receptor‐2. Clin Cancer Res 14:5447–5458. [DOI] [PubMed] [Google Scholar]
- 138. McLendon RE, Turner K, Perkinson K, Rich J (2007) Second messenger systems in human gliomas. Arch Pathol Lab Med 131:1585–1590. [DOI] [PubMed] [Google Scholar]
- 139. Meco D, Servidei T, Riccardi A, Ferlini C, Cusano G, Zannoni GF et al (2009) Antitumor effect in medulloblastoma cells by gefitinib: ectopic HER2 overexpression enhances gefitinib effects in vivo . Neuro Oncol 11:250–259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140. Mellinghoff IK, Wang MY, Vivanco I, Haas‐Kogan DA, Zhu S, Dia EQ et al (2005) Molecular determinants of the response of glioblastomas to EGFR kinase inhibitors. N Engl J Med 353:2012–2024. [DOI] [PubMed] [Google Scholar]
- 141. Mercapide J, Rappa G, Anzanello F, King J, Fodstad O, Lorico A (2010) Primary gene‐engineered neural stem/progenitor cells demonstrate tumor‐selective migration and antitumor effects in glioma. Int J Cancer 126:1206–1215. [DOI] [PubMed] [Google Scholar]
- 142. Micallef J, Taccone M, Mukherjee J, Croul S, Busby J, Moran MF et al (2009) Epidermal growth factor receptor variant III‐induced glioma invasion is mediated through myristoylated alanine‐rich protein kinase C substrate overexpression. Cancer Res 69:7548–7556. [DOI] [PubMed] [Google Scholar]
- 143. Michael LE, Westerman BA, Ermilov AN, Wang A, Ferris J, Liu J et al (2008) Bmi1 is required for Hedgehog pathway‐driven medulloblastoma expansion. Neoplasia 10:1343–1349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144. Mimeault M, Batra SK (2008) Recent progress on tissue‐resident adult stem cell biology and their therapeutic implications. Stem Cell Rev 4:27–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145. Mimeault M, Batra SK (2010) Frequent deregulations in the hedgehog signaling network and cross‐talks with the EGFR pathway involved in cancer progression and targeted therapies. Pharmacol Rev 62:497–524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146. Mimeault M, Batra SK (2010) New promising drug targets in cancer‐ and metastasis‐initiating cells. Drug Discov Today 15:354–364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147. Mimeault M, Batra SK (2010) Novel therapies against aggressive and recurrent epithelial cancers by molecular targeting tumor‐ and metastasis‐initiating cells and their progenies. Anticancer Agents Med Chem 10:137–151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148. Mimeault M, Hauke R, Mehta PP, Batra SK (2007) Recent advances on cancer stem/progenitor cell research: therapeutic implications for overcoming resistance to the most aggressive cancers. J Cell Mol Med 11:981–1011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149. Mishima K, Johns TG, Luwor RB, Scott AM, Stockert E, Jungbluth AA et al (2001) Growth suppression of intracranial xenografted glioblastomas overexpressing mutant epidermal growth factor receptors by systemic administration of monoclonal antibody (mAb) 806, a novel monoclonal antibody directed to the receptor. Cancer Res 61:5349–5354. [PubMed] [Google Scholar]
- 150. Morgillo F, Bareschino MA, Bianco R, Tortora G, Ciardiello F (2007) Primary and acquired resistance to anti‐EGFR targeted drugs in cancer therapy. Differentiation 75:788–799. [DOI] [PubMed] [Google Scholar]
- 151. Moscatello DK, Holgado‐Madruga M, Godwin AK, Ramirez G, Gunn G, Zoltick PW et al (1995) Frequent expression of a mutant epidermal growth factor receptor in multiple human tumors. Cancer Res 55:5536–5539. [PubMed] [Google Scholar]
- 152. Mukasa A, Wykosky J, Ligon KL, Chin L, Cavenee WK, Furnari F (2010) Mutant EGFR is required for maintenance of glioma growth in vivo, and its ablation leads to escape from receptor dependence. Proc Natl Acad Sci USA 107:2616–2621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153. Mukherjee J, DeSouza LV, Micallef J, Karim Z, Croul S, Siu KW et al (2009) Loss of collapsin response mediator Protein1, as detected by iTRAQ analysis, promotes invasion of human gliomas expressing mutant EGFRvIII. Cancer Res 69:8545–8554. [DOI] [PubMed] [Google Scholar]
- 154. Nicolis SK (2007) Cancer stem cells and “stemness” genes in neuro‐oncology. Neurobiol Dis 25:217–229. [DOI] [PubMed] [Google Scholar]
- 155. Nishikawa R, Ji XD, Harmon RC, Lazar CS, Gill GN, Cavenee WK et al (1994) A mutant epidermal growth factor receptor common in human glioma confers enhanced tumorigenicity. Proc Natl Acad Sci USA 91:7727–7731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156. Nobusawa S, Lachuer J, Wierinckx A, Kim YH, Huang J, Legras C et al (2010) Intratumoral patterns of genomic imbalance in glioblastomas. Brain Pathol 20:936–944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157. Northcott PA, Korshunov A, Witt H, Hielscher T, Eberhart CG, Mack S et al (2011) Medulloblastoma comprises four distinct molecular variants. J Clin Oncol 29:1408–1414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158. Ogden AT, Waziri AE, Lochhead RA, Fusco D, Lopez K, Ellis JA et al (2008) Identification of A2B5+C. Neurosurgery 62:505–514. [DOI] [PubMed] [Google Scholar]
- 159. Palma V, Lim DA, Dahmane N, Sanchez P, Brionne TC, Herzberg CD et al (2005) Sonic hedgehog controls stem cell behavior in the postnatal and adult brain. Development 132:335–344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160. Panosyan EH, Laks DR, Masterman‐Smith M, Mottahedeh J, Yong WH, Cloughesy TF et al (2010) Clinical outcome in pediatric glial and embryonal brain tumors correlates with in vitro multi‐passageable neurosphere formation. Pediatr Blood Cancer 55:644–651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161. Pardal R, Ortega‐Saenz P, Duran R, Lopez‐Barneo J (2007) Glia‐like stem cells sustain physiologic neurogenesis in the adult mammalian carotid body. Cell 131:364–377. [DOI] [PubMed] [Google Scholar]
- 162. Parsons DW, Li M, Zhang X, Jones S, Leary RJ, Lin JC et al (2011) The genetic landscape of the childhood cancer medulloblastoma. Science 331:435–439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163. Patel D, Lahiji A, Patel S, Franklin M, Jimenez X, Hicklin DJ et al (2007) Monoclonal antibody cetuximab binds to and down‐regulates constitutively activated epidermal growth factor receptor vIII on the cell surface. Anticancer Res 27:3355–3366. [PubMed] [Google Scholar]
- 164. Paugh BS, Qu C, Jones C, Liu Z, Adamowicz‐Brice M, Zhang J et al (2010) Integrated molecular genetic profiling of pediatric high‐grade gliomas reveals key differences with the adult disease. J Clin Oncol 28:3061–3068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165. Pedersen MW, Pedersen N, Ottesen LH, Poulsen HS (2005) Differential response to gefitinib of cells expressing normal EGFR and the mutant EGFRvIII. Br J Cancer 93:915–923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166. Pelloski CE, Ballman KV, Furth AF, Zhang L, Lin E, Sulman EP et al (2007) Epidermal growth factor receptor variant III status defines clinically distinct subtypes of glioblastoma. J Clin Oncol 25:2288–2294. [DOI] [PubMed] [Google Scholar]
- 167. Piao Y, Jiang H, Alemany R, Krasnykh V, Marini FC, Xu J et al (2009) Oncolytic adenovirus retargeted to Delta‐EGFR induces selective antiglioma activity. Cancer Gene Ther 16:256–265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168. Pillay V, Allaf L, Wilding AL, Donoghue JF, Court NW, Greenall SA et al (2009) The plasticity of oncogene addiction: implications for targeted therapies directed to receptor tyrosine kinases. Neoplasia 11:448–458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169. Pola R, Ling LE, Silver M, Corbley MJ, Kearney M, Blake PR et al (2001) The morphogen Sonic hedgehog is an indirect angiogenic agent upregulating two families of angiogenic growth factors. Nat Med 7:706–711. [DOI] [PubMed] [Google Scholar]
- 170. Prados MD, Lamborn KR, Chang S, Burton E, Butowski N, Malec M et al (2006) Phase 1 study of erlotinib HCl alone and combined with temozolomide in patients with stable or recurrent malignant glioma. Neuro Oncol 8:67–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171. Prados MD, Chang SM, Butowski N, DeBoer R, Parvataneni R, Carliner H et al (2009) Phase II study of erlotinib plus temozolomide during and after radiation therapy in patients with newly diagnosed glioblastoma multiforme or gliosarcoma. J Clin Oncol 27:579–584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172. Prajerova I, Honsa P, Chvatal A, Anderova M (2010) Distinct effects of sonic hedgehog and Wnt‐7a on differentiation of neonatal neural stem/progenitor cells in vitro . Neuroscience 171:693–711. [DOI] [PubMed] [Google Scholar]
- 173. Prestegarden L, Svendsen A, Wang J, Sleire L, Skaftnesmo KO, Bjerkvig R et al (2010) Glioma cell populations grouped by different cell type markers drive brain tumor growth. Cancer Res 70:4274–4279. [DOI] [PubMed] [Google Scholar]
- 174. Prigent SA, Nagane M, Lin H, Huvar I, Boss GR, Feramisco JR et al (1996) Enhanced tumorigenic behavior of glioblastoma cells expressing a truncated epidermal growth factor receptor is mediated through the Ras‐Shc‐Grb2 pathway. J Biol Chem 271:25639–25645. [DOI] [PubMed] [Google Scholar]
- 175. Purow BW, Haque RM, Noel MW, Su Q, Burdick MJ, Lee J et al (2005) Expression of Notch‐1 and its ligands, Delta‐like‐1 and Jagged‐1, is critical for glioma cell survival and proliferation. Cancer Res 65:2353–2363. [DOI] [PubMed] [Google Scholar]
- 176. Raffel C, Jenkins RB, Frederick L, Hebrink D, Alderete B, Fults DW et al (1997) Sporadic medulloblastomas contain PTCH mutations. Cancer Res 57:842–845. [PubMed] [Google Scholar]
- 177. Rebetz J, Tian D, Persson A, Widegren B, Salford LG, Englund E et al (2008) Glial progenitor‐like phenotype in low‐grade glioma and enhanced CD133‐expression and neuronal lineage differentiation potential in high‐grade glioma. PLoS ONE 3:e1936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178. Reifenberger J, Wolter M, Weber RG, Megahed M, Ruzicka T, Lichter P et al (1998) Missense mutations in SMOH in sporadic basal cell carcinomas of the skin and primitive neuroectodermal tumors of the central nervous system. Cancer Res 58:1798–1803. [PubMed] [Google Scholar]
- 179. Reist CJ, Archer GE, Kurpad SN, Wikstrand CJ, Vaidyanathan G, Willingham MC et al (1995) Tumor‐specific anti‐epidermal growth factor receptor variant III monoclonal antibodies: use of the tyramine‐cellobiose radioiodination method enhances cellular retention and uptake in tumor xenografts. Cancer Res 55:4375–4382. [PubMed] [Google Scholar]
- 180. Rich JN, Reardon DA, Peery T, Dowell JM, Quinn JA, Penne KL et al (2004) Phase II trial of gefitinib in recurrent glioblastoma. J Clin Oncol 22:133–142. [DOI] [PubMed] [Google Scholar]
- 181. Rickert CH, Paulus W (2005) Prognosis‐related histomorphological and immunohistochemical markers in central nervous system tumors of childhood and adolescence. Acta Neuropathol 109:69–92. [DOI] [PubMed] [Google Scholar]
- 182. Rong Y, Belozerov VE, Tucker‐Burden C, Chen G, Durden DL, Olson JJ et al (2009) Epidermal growth factor receptor and PTEN modulate tissue factor expression in glioblastoma through JunD/activator protein‐1 transcriptional activity. Cancer Res 69:2540–2549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183. Rudin CM, Hann CL, Laterra J, Yauch RL, Callahan CA, Fu L et al (2009) Treatment of medulloblastoma with hedgehog pathway inhibitor GDC‐0449. N Engl J Med 361:1173–1178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184. Saikali S, Avril T, Collet B, Hamlat A, Bansard JY, Drenou B et al (2007) Expression of nine tumour antigens in a series of human glioblastoma multiforme: interest of EGFRvIII, IL‐13Ralpha2, gp100 and TRP‐2 for immunotherapy. J Neurooncol 81:139–148. [DOI] [PubMed] [Google Scholar]
- 185. Sampson JH, Archer GE, Mitchell DA, Heimberger AB, Bigner DD (2008) Tumor‐specific immunotherapy targeting the EGFRvIII mutation in patients with malignant glioma. Semin Immunol 20:267–275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186. Sanai N, Albarez‐Buylla A, Berger MS (2005) Neural stem cells and the origin of gliomas. N Engl J Med 353:811–822. [DOI] [PubMed] [Google Scholar]
- 187. Sarangi A, Valadez JG, Rush S, Abel TW, Thompson RC, Cooper MK (2009) Targeted inhibition of the Hedgehog pathway in established malignant glioma xenografts enhances survival. Oncogene 28:3468–3476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188. Sarkaria JN, Yang L, Grogan PT, Kitange GJ, Carlson BL, Schroeder MA et al (2007) Identification of molecular characteristics correlated with glioblastoma sensitivity to EGFR kinase inhibition through use of an intracranial xenograft test panel. Mol Cancer Ther 6:1167–1174. [DOI] [PubMed] [Google Scholar]
- 189. Schmidt MH, Furnari FB, Cavenee WK, Bogler O (2003) Epidermal growth factor receptor signaling intensity determines intracellular protein interactions, ubiquitination, and internalization. Proc Natl Acad Sci USA 100:6505–6510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190. Schmittling RJ, Archer GE, Mitchell DA, Heimberger A, Pegram C, Herndon JE et al (2008) Detection of humoral response in patients with glioblastoma receiving EGFRvIII‐KLH vaccines. J Immunol Methods 339:74–81. [DOI] [PubMed] [Google Scholar]
- 191. Schreck KC, Taylor P, Marchionni L, Gopalakrishnan V, Bar EE, Gaiano N et al (2010) The Notch target Hes1 directly modulates Gli1 expression and Hedgehog signaling: a potential mechanism of therapeutic resistance. Clin Cancer Res 16:6060–6070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192. Schwartzbaum JA, Huang K, Lawler S, Ding B, Yu J, Chiocca EA (2010) Allergy and inflammatory transcriptome is predominantly negatively correlated with CD133 expression in glioblastoma. Neuro Oncol 12:320–327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193. Shahi MH, Afzal M, Sinha S, Eberhart CG, Rey JA, Fan X et al (2010) Regulation of sonic hedgehog‐GLI1 downstream target genes PTCH1, Cyclin D2, Plakoglobin, PAX6 and NKX2.2 and their epigenetic status in medulloblastoma and astrocytoma. BMC Cancer 10:614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194. Shaked Y, Henke E, Roodhart JM, Mancuso P, Langenberg MH, Colleoni M et al (2008) Rapid chemotherapy‐induced acute endothelial progenitor cell mobilization: implications for antiangiogenic drugs as chemosensitizing agents. Cancer Cell 14:263–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195. Shen Q, Goderie SK, Jin L, Karanth N, Sun Y, Abramova N et al (2004) Endothelial cells stimulate self‐renewal and expand neurogenesis of neural stem cells. Science 304:1338–1340. [DOI] [PubMed] [Google Scholar]
- 196. Shen Q, Wang Y, Kokovay E, Lin G, Chuang SM, Goderie SK et al (2008) Adult SVZ stem cells lie in a vascular niche: a quantitative analysis of niche cell‐cell interactions. Cell Stem Cell 3:289–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197. Shinojima N, Tada K, Shiraishi S, Kamiryo T, Kochi M, Nakamura H et al (2003) Prognostic value of epidermal growth factor receptor in patients with glioblastoma multiforme. Cancer Res 63:6962–6970. [PubMed] [Google Scholar]
- 198. Silber J, Lim DA, Petritsch C, Persson AI, Maunakea AK, Yu M et al (2008) miR‐124 and miR‐137 inhibit proliferation of glioblastoma multiforme cells and induce differentiation of brain tumor stem cells. BMC Med 6:14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199. Singh SK, Hawkins C, Clarke ID, Squire JA, Bayani J, Hide T et al (2004) Identification of human brain tumour initiating cells. Nature 432:396–401. [DOI] [PubMed] [Google Scholar]
- 200. Sinor‐Anderson A, Lillien L (2011) Akt1 interacts with epidermal growth factor receptors and hedgehog signaling to increase stem/transit amplifying cells in the embryonic mouse cortex. Dev Neurobiol [Epub ahead of print; doi: 10.1002/dneu.20878]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201. Sirko S, von Holst A, Weber A, Wizenmann A, Theocharidis U, Gotz M et al (2010) Chondroitin sulfates are required for fibroblast growth factor‐2‐dependent proliferation and maintenance in neural stem cells and for epidermal growth factor‐dependent migration of their progeny. Stem Cells 28:775–787. [DOI] [PubMed] [Google Scholar]
- 202. Slade I, Murray A, Hanks S, Kumar A, Walker L, Hargrave D et al (2011) Heterogeneity of familial medulloblastoma and contribution of germline PTCH1 and SUFU mutations to sporadic medulloblastoma. Fam Cancer 10:337–342. [DOI] [PubMed] [Google Scholar]
- 203. Slongo ML, Molena B, Brunati AM, Frasson M, Gardiman M, Carli M et al (2007) Functional VEGF and VEGF receptors are expressed in human medulloblastomas. Neuro Oncol 9:384–392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204. Son MJ, Woolard K, Nam DH, Lee J, Fine HA (2009) SSEA‐1 is an enrichment marker for tumor‐initiating cells in human glioblastoma. Cell Stem Cell 4:440–452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205. Stecca B, Altaba A (2009) A GLI1‐p53 inhibitory loop controls neural stem cell and tumour cell numbers. EMBO J 28:663–676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206. Stecca B, Mas C, Clement V, Zbinden M, Correa R, Piguet V et al (2007) Melanomas require HEDGEHOG‐GLI signaling regulated by interactions between GLI1 and the RAS‐MEK/AKT pathways. Proc Natl Acad Sci USA 104:5895–5900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207. Stupp R, Mason WP, van den Bent MJ, Weller M, Fisher B, Taphoorn MJ et al (2005) Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med 352:987–996. [DOI] [PubMed] [Google Scholar]
- 208. Subkhankulova T, Zhang X, Leung C, Marino S (2010) Bmi1 directly represses p21Waf1/Cip1 in Shh‐induced proliferation of cerebellar granule cell progenitors. Mol Cell Neurosci 45:151–162. [DOI] [PubMed] [Google Scholar]
- 209. Sun Y, Goderie SK, Temple S (2005) Asymmetric distribution of EGFR receptor during mitosis generates diverse CNS progenitor cells. Neuron 45:873–886. [DOI] [PubMed] [Google Scholar]
- 210. Suh Y, Obernier K, Holzl‐Wenig G, Mandl C, Herrmann A, Worner K et al (2009) Interaction between DLX2 and EGFR regulates proliferation and neurogenesis of SVZ precursors. Mol Cell Neurosci 42:308–314. [DOI] [PubMed] [Google Scholar]
- 211. Suzuki K, Momota H, Tonooka A, Noguchi H, Yamamoto K, Wanibuchi M et al (2010) Glioblastoma simultaneously present with adjacent meningioma: case report and review of the literature. J Neurooncol 99:147–153. [DOI] [PubMed] [Google Scholar]
- 212. Swartling FJ, Grimmer MR, Hackett CS, Northcott PA, Fan QW, Goldenberg DD et al (2010) Pleiotropic role for MYCN in medulloblastoma. Genes Dev 24:1059–1072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213. Tamase A, Muraguchi T, Naka K, Tanaka S, Kinoshita M, Hoshii T et al (2009) Identification of tumor‐initiating cells in a highly aggressive brain tumor using promoter activity of nucleostemin. Proc Natl Acad Sci USA 106:17163–17168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214. Tang P, Steck PA, Yung WK (1997) The autocrine loop of TGF‐alpha/EGFR and brain tumors. J Neurooncol 35:303–314. [DOI] [PubMed] [Google Scholar]
- 215. Taub N, Teis D, Ebner HL, Hess MW, Huber LA (2007) Late endosomal traffic of the epidermal growth factor receptor ensures spatial and temporal fidelity of mitogen‐activated protein kinase signaling. Mol Biol Cell 18:4698–4471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216. Taylor MD, Liu L, Raffel C, Hui CC, Mainprize TG, Zhang X et al (2002) Mutations in SUFU predispose to medulloblastoma. Nat Genet 31:306–310. [DOI] [PubMed] [Google Scholar]
- 217. Teodorczyk M, Martin‐Villalba A (2010) Sensing invasion: cell surface receptors driving spreading of glioblastoma. J Cell Physiol 222:1–10. [DOI] [PubMed] [Google Scholar]
- 218. Thompson MC, Fuller C, Hogg TL, Dalton J, Finkelstein D, Lau CC et al (2006) Genomics identifies medulloblastoma subgroups that are enriched for specific genetic alterations. J Clin Oncol 24:1924–1931. [DOI] [PubMed] [Google Scholar]
- 219. Tong CY, Hui AB, Yin XL, Pang JC, Zhu XL, Poon WS et al (2004) Detection of oncogene amplifications in medulloblastomas by comparative genomic hybridization and array‐based comparative genomic hybridization. J Neurosurg 100:187–193. [DOI] [PubMed] [Google Scholar]
- 220. Traiffort E, Angot E, Ruat M (2010) Sonic Hedgehog signaling in the mammalian brain. J Neurochem 113:576–590. [DOI] [PubMed] [Google Scholar]
- 221. Tso CL, Shintaku P, Chen J, Liu Q, Liu J, Chen Z et al (2006) Primary glioblastomas express mesenchymal stem‐like properties. Mol Cancer Res 4:607–619. [DOI] [PubMed] [Google Scholar]
- 222. Ulasov IV, Nandi S, Dey M, Sonabend AM, Lesniak MS (2011) Inhibition of Sonic hedgehog and Notch pathways enhances sensitivity of CD133(+) glioma stem cells to temozolomide therapy. Mol Med 17:103–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223. Venkataraman S, Alimova I, Fan R, Harris P, Foreman N, Vibhakar R (2010) MicroRNA 128a increases intracellular ROS level by targeting Bmi‐1 and inhibits medulloblastoma cancer cell growth by promoting senescence. PLoS ONE 5:e10748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224. Verhaak RG, Hoadley KA, Purdom E, Wang V, Qi Y, Wilkerson MD et al (2010) Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell 17:98–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225. Vescovi AL, Galli R, Reynolds BA (2006) Brain tumour stem cells. Nat Rev Cancer 6:425–436. [DOI] [PubMed] [Google Scholar]
- 226. Vieira AV, Lamaze C, Schmid SL (1996) Control of EGF receptor signaling by clathrin‐mediated endocytosis. Science 274:2086–2089. [DOI] [PubMed] [Google Scholar]
- 227. Wang J, Sakariassen PO, Tsinkalovsky O, Immervoll H, Boe SO, Svendsen A et al (2008) CD133 negative glioma cells form tumors in nude rats and give rise to CD133 positive cells. Int J Cancer 122:761–768. [DOI] [PubMed] [Google Scholar]
- 228. Wang MY, Lu KV, Zhu S, Dia EQ, Vivanco I, Shackleford GM et al (2006) Mammalian target of rapamycin inhibition promotes response to epidermal growth factor receptor kinase inhibitors in PTEN‐deficient and PTEN‐intact glioblastoma cells. Cancer Res 66:7864–7869. [DOI] [PubMed] [Google Scholar]
- 229. Wang SI, Puc J, Li J, Bruce JN, Cairns P, Sidransky D et al (1997) Somatic mutations of PTEN in glioblastoma multiforme. Cancer Res 57:4183–4186. [PubMed] [Google Scholar]
- 230. Watts C, McConkey H, Anderson L, Caldwell M (2005) Anatomical perspectives on adult neural stem cells. J Anat 207:197–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231. Wechsler‐Reya RJ, Scott MP (1999) Control of neuronal precursor proliferation in the cerebellum by Sonic Hedgehog. Neuron 22:103–114. [DOI] [PubMed] [Google Scholar]
- 232. Wei Q, Clarke L, Scheidenhelm DK, Qian B, Tong A, Sabha N et al (2006) High‐grade glioma formation results from postnatal pten loss or mutant epidermal growth factor receptor expression in a transgenic mouse glioma model. Cancer Res 66:7429–7437. [DOI] [PubMed] [Google Scholar]
- 233. Welsh JW, Mahadevan D, Ellsworth R, Cooke L, Bearss D, Stea B (2009) The c‐Met receptor tyrosine kinase inhibitor MP470 radiosensitizes glioblastoma cells. Radiat Oncol 4:69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234. Wikstrand CJ, Hale LP, Batra SK, Hill ML, Humphrey PA, Kurpad SN et al (1995) Monoclonal antibodies against EGFRvIII are tumor specific and react with breast and lung carcinomas and malignant gliomas. Cancer Res 55:3140–3148. [PubMed] [Google Scholar]
- 235. Wong A, Mitra S, Del Vecchio CA, Shirboll S (2008) Expression of EGFRvIII in brain tumor stem cells. J Clin Oncol 26:15s. [Google Scholar]
- 236. Wong A, Mitra S, Gupta P (2009) Targeting brain tumor stem cells using a bispecific antibody directed against CD133+ and EGFRvIII+. J Clin Oncol 27:15s. [Google Scholar]
- 237. Wu J, Qian J, Li C, Kwok L, Cheng F, Liu P et al (2010) miR‐129 regulates cell proliferation by downregulating Cdk6 expression. Cell Cycle 9:1809–1818. [DOI] [PubMed] [Google Scholar]
- 238. Xu Q, Yuan X, Liu G, Black KL, Yu JS (2008) Hedgehog signaling regulates brain tumor‐initiating cell proliferation and portends shorter survival for patients with PTEN‐coexpressing glioblastomas. Stem Cells 26:3018–3026. [DOI] [PubMed] [Google Scholar]
- 239. Xu Y, Stamenkovic I, Yu Q (2010) CD44 attenuates activation of the hippo signaling pathway and is a prime therapeutic target for glioblastoma. Cancer Res 70:2455–2464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240. Yang W, Wu G, Barth RF, Swindall MR, Bandyopadhyaya AK, Tjarks W et al (2008) Molecular targeting and treatment of composite EGFR and EGFRvIII‐positive gliomas using boronated monoclonal antibodies. Clin Cancer Res 14:883–891. [DOI] [PubMed] [Google Scholar]
- 241. Yang ZJ, Ellis T, Markant SL, Read TA, Kessler JD, Bourboulas M et al (2008) Medulloblastoma can be initiated by deletion of Patched in lineage‐restricted progenitors or stem cells. Cancer Cell 14:135–145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242. Yauch RL, Dijkgraaf GJ, Alicke B, Januario T, Ahn CP, Holcomb T et al (2009) Smoothened mutation confers resistance to a Hedgehog pathway inhibitor in medulloblastoma. Science 326:572–574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243. Yiin JJ, Hu B, Schornack PA, Sengar RS, Liu KW, Feng H et al (2010) ZD6474, a multitargeted inhibitor for receptor tyrosine kinases, suppresses growth of gliomas expressing an epidermal growth factor receptor mutant, EGFRvIII, in the brain. Mol Cancer Ther 9:929–941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244. Yoshimoto K, Dang J, Zhu S, Nathanson D, Huang T, Dumont R et al (2008) Development of a real‐time RT‐PCR assay for detecting EGFRvIII in glioblastoma samples. Clin Cancer Res 14:488–493. [DOI] [PubMed] [Google Scholar]
- 245. Yu JJ, Sun X, Yuan X, Lee JW, Snyder EY, Yu JS (2006) Immunomodulatory neural stem cells for brain tumour therapy. Expert Opin Biol Ther 6:1255–1262. [DOI] [PubMed] [Google Scholar]
- 246. Yuan L, Santi M, Rushing EJ, Cornelison R, MacDonald TJ (2010) ERK activation of p21 activated kinase‐1 (Pak1) is critical for medulloblastoma cell migration. Clin Exp Metastasis 27:481–491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 247. Yuan X, Curtin J, Xiong Y, Liu G, Waschsmann‐Hogiu S, Farkas DL et al (2004) Isolation of cancer stem cells from adult glioblastoma multiforme. Oncogene 23:9392–9400. [DOI] [PubMed] [Google Scholar]
- 248. Zandi R, Larsen AB, Andersen P, Stockhausen MT, Poulsen HS (2007) Mechanisms for oncogenic activation of the epidermal growth factor receptor. Cell Signal 19:2013–2023. [DOI] [PubMed] [Google Scholar]
- 249. Zeppernick F, Ahmadi R, Campos B, Dictus C, Helmke BM, Becker N et al (2008) Stem cell marker CD133 affects clinical outcome in glioma patients. Clin Cancer Res 14:123–129. [DOI] [PubMed] [Google Scholar]
- 250. Zhang XP, Zheng G, Zou L, Liu HL, Hou LH, Zhou P et al (2008) Notch activation promotes cell proliferation and the formation of neural stem cell‐like colonies in human glioma cells. Mol Cell Biochem 307:101–108. [DOI] [PubMed] [Google Scholar]
- 251. Zhang Y, Guessous F, Kofman A, Schiff D, Abounader R (2010) XL‐184, a MET, VEGFR‐2 and RET kinase inhibitor for the treatment of thyroid cancer, glioblastoma multiforme and NSCLC. IDrugs 13:112–121. [PMC free article] [PubMed] [Google Scholar]
- 252. Zhao C, Deng W, Gage FH (2008) Mechanisms and functional implications of adult neurogenesis. Cell 132:645–660. [DOI] [PubMed] [Google Scholar]
- 253. Zhao D, Najbauer J, Garcia E, Metz MZ, Gutova M, Glackin CA et al (2008) Neural stem cell tropism to glioma: critical role of tumor hypoxia. Mol Cancer Res 6:1819–1829. [DOI] [PubMed] [Google Scholar]
- 254. Zurawel RH, Allen C, Chiappa S, Cato W, Biegel J, Cogen P et al (2000) Analysis of PTCH/SMO/SHH pathway genes in medulloblastoma. Genes Chromosomes Cancer 27:44–51. [DOI] [PubMed] [Google Scholar]