

Abstract
Helicobacter pylori survives and grows at low pHs via acid acclimation mechanisms that enable periplasmic pH homeostasis. Important components include a cytoplasmic urease; a pH-gated urea channel, UreI; and periplasmic α-carbonic anhydrase. To allow the rapid adjustment of periplasmic pH, acid acclimation components are recruited to the inner membrane in acid. The ArsRS two-component system, in an acid-responsive manner, controls the transcription of the urease gene cluster and α-carbonic anhydrase. The aim of this study is to determine the role of ArsS in protein trafficking as a component of acid acclimation. H. pylori wild-type and ΔarsS bacteria were incubated at acidic and neutral pHs. Intact bacteria, purified membranes, and total protein were analyzed by Western blotting and urease activity measurements. The total urease activity level was decreased in the ΔarsS strain, but the acid activation of UreI was unaffected. A 30-min acid exposure increased the level and activity of urease proteins at the membrane in the wild type but not in the ΔarsS strain. The urease levels and activity of the ΔarsS strain after a 90-min acid exposure were similar to those of the wild type. ArsS, in addition to its role in urease gene transcription, is also involved in the recruitment of urease proteins to the inner membrane to augment acid acclimation during acute acid exposure. Urease membrane recruitment following prolonged acid exposure in the absence of ArsS was similar to that of the wild type, suggesting a compensatory mechanism, possibly regulated by FlgS, underscoring the importance of urease membrane recruitment and activation in periplasmic pH homeostasis.
INTRODUCTION
Helicobacter pylori is a Gram-negative, neutralophilic bacterium that colonizes the normal acid-secreting human stomach (17). H. pylori is bioenergetically classified as a neutralophile based on the inability to maintain a proton motive force outside a range of pH 4 to 8 in the absence of urea (21). In the presence of physiologic urea concentrations, the bacteria require acid for survival (6). The presence of urea restores membrane potential and proton motive force at an acidic pH (35). Given the distinct site of colonization and the requirements for acidity and urea, an understanding of mechanisms that allow the bacteria to sense and adapt to the environment is important for developing a more complete picture of acid acclimation mechanisms and for unveiling new targets for eradication.
Bacteria sense and respond to their environment via two-component systems (TCSs). A TCS is typically composed of a sensor histidine kinase and a response regulator. H. pylori expresses two known pH-sensing histidine kinases, ArsS, an integral inner membrane protein, and soluble, cytoplasmic FlgS. We previously described the effect of the deletion of FlgS, where urease is no longer activated in acidic media and neither urease nor its accessory proteins assemble at the membrane in association with the urea channel, UreI (34). We now investigate the complementary role of ArsS in the regulation of urease activity, activation, and membrane assembly and develop a model where the acid responsiveness of these two kinases enables a response to the broad range of environmental pHs to which the organism is exposed.
Neither urease nor ureI deletion mutants are able to colonize the stomach (9, 22, 36). In acid, the urease-catalyzed breakdown of urea restores membrane potential and proton motive force by raising the periplasmic pH to ∼6.1, allowing gastric colonization without the need for a large-scale pH increase of the gastric environment (35). The intragastric pH varies between ∼pH 5.0 in the digestive phase and pH ∼1.0 in the absence of food (38). The organism must therefore adapt to this variable-acid-stress environment; thus, it not only must respond rapidly to a fall in the periplasmic pH but also must respond to a fall in the cytoplasmic pH.
Although almost all bacteria have TCSs, the numbers differ, often related to the variability of the environment(s) which they inhabit. Bacteria average about 52 TCSs, with many regulating virulence determinants (7). Pseudomonas aeruginosa, which is found in many different environments, has about 64 histidine kinases and 72 response regulators (11). Besides the chemotaxis proteins CheAY2 (HP0392), CheY1 (HP1067), CheV1 (HP0019), CheV2 (HP0616), and CheV3 (HP0393), H. pylori contains only three histidine kinases and five response regulators (2, 39). ArsRS, among other roles, is responsible for the pH-responsive regulation of genes involved in acid acclimation (14). FlgS is the only cytoplasmic histidine kinase in H. pylori (2, 39) and has an acid-responsive regulon distinct from the flagellar genes controlled in conjunction with the response regulator FlgR (45). FlgRS is involved in the expression of the class 2 flagellar genes (24). CrdRS (HP1365/HP1364) responds to increased concentrations of copper ions via the positive regulation of the copper resistance determinant CrdAB-CzcAB (41). The histidine kinase CrdS was reported previously to be required at pH 5 in strain J99 (15), a finding later determined to be strain specific. CrdS is not involved in acid-responsive gene regulation in strain G27 or 26695 (31). The orphan response regulator HP1021 is required for normal bacterial growth and is thought to be involved in acetone metabolism (4, 18, 27). The orphan response regulator HP1043 is essential for viability (18, 32) and may play a role in regulating growth (8).
The ArsRS TCS mediates the pH-responsive transcriptional control of the urease gene cluster, genes involved in acid acclimation and pH homeostasis, as well as several other genes (5, 20, 29, 44). ArsS is considered to be a pH sensor since the pH-dependent induction of transcription of target genes is abolished in an ΔarsS strain (28). Indeed, point mutations of histidine residues in the periplasmic input domain of ArsS show that H94 is crucial for acid sensing in H. pylori (23). Its response regulator, arsR, is essential, while the histidine kinase gene arsS can be deleted without an effect on in vitro growth (4, 18). The replacement of arsR with a gene containing a point mutation at the phosphorylation site (D52N) results in a viable strain, suggesting that the regulon consists of genes controlled by both unphosphorylated and phosphorylated ArsR (32). Therefore, ArsR is essential for growth, independent of phosphorylation, whereas gene regulation in response to an acidic pH requires the phosphorylation of ArsR (29). There are two main aspects of the acidic-pH response of the ArsRS TCS. One is the transcriptional regulation of various pH-homeostatic genes (29, 44). The other, the major focus of this study, is the regulation of the trafficking of the soluble products of the urease gene cluster to the inner membrane in response to an acidic-pH challenge. We previously described the role of the cytoplasmic histidine kinase, FlgS, in urease membrane recruitment at pH <3.0 (34) and now show that the membrane-bound histidine kinase, ArsS, is also involved in urease membrane recruitment at pH 4.5. Thus, acidic stress is counteracted by cooperation between ArsS and FlgS for urease membrane recruitment. A recruitment response to the cytoplasmic pH, mediated by FlgS, and, as shown in this study, a response to the periplasmic pH, mediated by ArsS, allow a graded response to the gastric luminal pH (34).
MATERIALS AND METHODS
Bacterial strains and culture conditions.
H. pylori strain ATCC 43504 was used. A nonpolar ATCC 43504 ΔarsS deletion mutant was constructed by allelic exchange, as previously described (43) and as summarized below. Bacteria were grown under microaerobic conditions (5% O2, 10% CO2, and 85% N2) either on Trypticase soy agar (TSA) plates supplemented with 5% sheep blood (Gibco) or in brain heart infusion (BHI) medium (Difco) supplemented with 7% horse serum (Gibco) and 0.25% yeast extract (Difco). Bacteria in media were grown in the presence of Dent selective supplement (Oxoid), and the ΔarsS strain was grown with 20 μg/ml kanamycin (Sigma).
Construction of the ΔarsS strain.
A genomic knockout of arsS was constructed by homologous recombination. The gene was first sequenced in H. pylori strain ATCC 43504. As the gene was originally sequenced in strain 26695 as 2 gene locus tags (HP0164 and HP0165) with 2 different reading frames, it was necessary to delineate the true start and stop sites and overlap site. Sequencing primers were designed upstream and downstream of the start and stop sites (5′-TGAAAGCATTGCGATTGAGA-3′ and 5′-CCTTCATTCCGCTCACTTCT-3′). Sequencing was carried out at the Keck facility. The sequence was obtained and analyzed by using Vector NTI software, and the construction of the knockout proceeded by using this sequence. pBluescript (Stratagene) containing a kanamycin resistance gene in the multicloning site flanked by SalI (5′) and BglII (3′) was used to generate the knockout plasmid. Primers were designed to flank the regions approximately 600 bp upstream of the 5′ end of the gene and 400 bp downstream from the 3′ end. The 600-bp upstream segment was amplified with a 5′ primer containing a site for digestion by XbaI (5′-CATGTAACCAATCTAGATGAGCCATATACCGGC-3′) and a 3′ primer containing a site for digestion by SalI (5′-CTTTAAAAAAGATAGAGGTCGACATAACCCCTTAACTCC-3′). The 400-bp downstream segment was amplified with a 5′ primer containing a site for digestion by BglII (5′-CCCGAAAATTTGAGATCTGTGAGCGGAATGAAGGGG-3′) and a 3′ primer containing a site for digestion by Acc65I (5′-GCCCATGGTCGGTACCTTCACAAAAACACAAATCCGC-3′). The purified PCR products were sequentially ligated into pBluescript around the kanamycin resistance gene. The construct was introduced into H. pylori strain ATCC 43504 by natural transformation, and colonies were selected in the presence of 40 μg/ml kanamycin. The higher kanamycin concentration was used only for selection; subsequent culture conditions were the same as those described above. Knockouts were confirmed by a series of PCRs.
The expression of the downstream gene HP0163 in the knockout mutant was confirmed by reverse transcriptase PCR (RT-PCR). RNA was isolated by using a combination of TRIzol (Invitrogen) and the RNeasy minikit (Qiagen), as described previously (46). cDNA was constructed by using Omniscript enzyme (Qiagen) and random primers (Invitrogen) with 6 μg RNA as the template. One microliter of cDNA was then used in a PCR with the following primers for HP0163: sense primer 5′-GCCATGCGTTAAACAAGGAT-3′ and antisense primer 5′-ATATCCACACCGCTTTCAGC-3′. A single band of the appropriate size was generated, which was identical to the genomic DNA (gDNA) control (data not shown), confirming that downstream gene expression was not affected by the deletion of arsS. PCR analysis confirming the absence of the arsS gene and the presence of the replacement kanamycin cassette as well as the normal expression of downstream genes confirmed the specificity of the gene deletion and the nonpolar nature of the knockout.
Preparation of lysates and purified membranes.
Cells of wild-type (wt) H. pylori or the ΔarsS strain were incubated in 10 ml BHI medium at pH 4.5 (pH set with HCl) or pH 7.4 for 30 or 90 min with shaking under microaerobic conditions and then harvested by centrifugation (1,500 × g for 10 min at 4°C). Pellets were resuspended in 3 ml ice-cold 25 mM phosphate buffer (PB) with 5 mM EDTA and 150 μl bacterial protease inhibitor (Sigma) and lysed by 3 passes through a French press at 20,000 lb/in2. Unbroken bacteria were removed by a 10-min spin at 4°C at 1,500 × g. The resulting supernatant was centrifuged (10,000 × g for 10 min at 4°C). Membrane proteins were pelleted by centrifugation (100,000 × g for 1 h at 4°C). To purify the proteins, the membrane pellet was resuspended and layered onto 5 ml 25 mM PB with 5 mM EDTA and 20% sucrose and then spun at 100,000 × g at 4°C for 1 h. The spin through sucrose was repeated two times, and the membrane pellet was then resuspended in 200 μl 25 mM PB with 5 mM EDTA. SDS (0.1%) was added to the membranes to help resuspend the proteins. This procedure removes all unattached proteins form the membrane fraction, as previously described (40). The protein concentration was determined by the bicinchoninic acid (BCA) method (Pierce).
Urease activity. (i) Intact bacteria.
Bacteria were added to 100 mM PB containing 5 mM KCl, 138 mM NaCl, 0.5 mM MgCl2, 1 mM CaCl2, 10 mM glucose, 1 mM glutamine, and 5 mM [14C]urea. The buffer pH range was 3 to 8. The pH of the buffer during the course of the experiment did not change by more than 0.1 pH units.
(ii) Total urease activity.
Lysates and purified membranes were isolated as described above. Proteins were added to 100 mM PB (pH 7.4) containing 5 mM KCl, 138 mM NaCl, 0.5 mM MgCl2, 1 mM CaCl2, 10 mM glucose, 1 mM glutamine, and 5 mM [14C]urea with a specific activity of 10 mCi/mmol. The pH of the buffer during the course of the experiment did not change by more than 0.1 pH units.
(iii) Urease activity over time.
Cells of wt H. pylori strain ATCC 43504 and the ΔarsS strain grown on TSA plates were suspended in 500 μl of BHI medium (pH 7.4). Two hundred twenty microliters of the suspension was added to 15 ml of BHI medium at pH 7.4, 5.5, or 4.5 and incubated at 37°C under microaerobic conditions. Aliquots (1.5 ml) were obtained at 0, 30, 90, and 180 min and immediately centrifuged at 17,900 × g for 5 min at 4°C. The pellets were resuspended in 25 mM ice-cold PB (pH 7.0) and centrifuged at 17,900 × g for 5 min at 4°C. The pellet was resuspended in 150 μl of 25 mM PB (pH 7.0) containing 0.01% polyoxyethylene-8-lauryl ether (C12E8; Sigma) to permeabilize the bacterial membranes without a disruption of the bacteria (42).
(iv) Measurement of urease activity.
Urease activity was measured radiometrically (19, 35). Ten microliters of bacteria or bacterial protein was added to the buffer mixture containing 5 mM [14C]urea with a specific activity of 10 mCi/mmol, as described above. Plastic wells containing 500 mM KOH-soaked filter paper hung from rubber stoppers were used to collect the total [14C]O2 from the hydrolysis of urea. Urease activity was measured for 30 min at 37°C with constant agitation. The reaction was terminated with 5 N H2SO4 to release all the generated CO2, and the mixture was incubated for 30 min at 37°C. The wells were placed into a scintillation cocktail (HiIonicFluor; Packard Instruments), and the radioactivity was measured by scintillation counting (1216 RackBeta; LKB Instruments). The protein concentration was determined by the BCA method (Pierce). Results are expressed in μmol/min/mg protein.
SDS-PAGE and Western blotting.
Total lysates, supernatants, and purified membranes prepared as described above were size fractionated by SDS-PAGE using 4 to 12% NuPAGE Bis-Tris gradient gels (Invitrogen) and transferred onto nitrocellulose (Bio-Rad). Western blot analysis was performed. Monoclonal UreA and UreB antibodies (Austral Biologics) were used at dilutions of 1:1,000 and 1:20,000, respectively. Immunolabeling was detected by using the Supersignal West Pico chemiluminescence system (Pierce).
RESULTS
Effect of medium pH on urease activity in the isogenic ΔarsS strain.
The urease activity was measured in intact bacteria over a range of medium pHs between 3.0 and 8.0 in strong buffer to avoid a pH increase due to urea hydrolysis. In the wild type, the urease activity increased 5-fold between pH 8.0 and 3.0, with half-maximal activation at pH 5.9. The ΔarsS strain demonstrated a lower level of activity at a neutral pH and a steep increase in activity with a decrease in the pH below 6.5, with maximal activity at pH 5.0 and a decline between pH 4.0 and 3.0. The decrease in activity at pH 3.0 is most easily attributed to a decline in the cytoplasmic pH in the absence of a proper increase of the periplasmic pH, leading to the decreased activity of the neutral-pH-optimum urease (Fig. 1A). These data also show that the proton-gated urea channel, UreI, is functional in the ΔarsS strain, as shown by the increase in the level of urease activity at pH <6.0. The lower maximal activity level in the ΔarsS strain is consistent with a decrease in the levels of production and activation of urease in the absence of this pH sensor kinase (44). In agreement with these results, the total urease activity level (determined by using lysed bacteria, bypassing UreI) was decreased in ΔarsS cells by about 50% (Fig. 1B).
Fig 1.
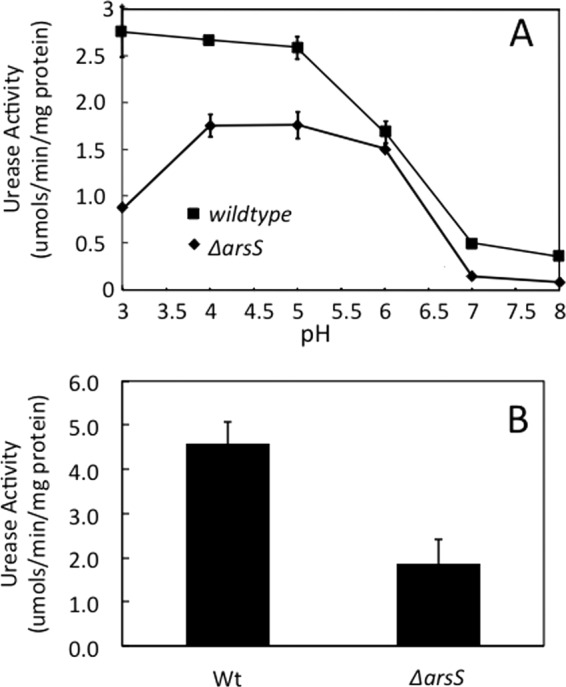
Urease activity in intact organisms and total urease activity. (A) H. pylori wild type and ΔarsS were incubated for 30 min in strong buffers of various pHs with [14C]urea, and the liberation of [14C]O2 was measured (n = 3). Like the wild type, the ΔarsS strain shows acid activation of cytoplasmic urease mediated by UreI, with minimal activity at a neutral pH and increasing activity with decreasing medium pHs, with maximal activity below pH 5.5. (B) The urease activity of H. pylori wild-type and ΔarsS lysates was measured in strong buffer at pH 7.4. In agreement with the curve shown in panel A, the level of total urease activity is lower in the absence of ArsS (n = 3).
Levels of urease proteins.
The deletion of arsS resulted in decreases in the levels of UreA and UreB, with levels of UreA falling by 40% and levels of UreB falling by 20% compared to the wild type, as shown by Western blot analysis of the total bacterial lysate (Fig. 2). This reflects the decrease in ureA and ureB mRNA levels seen in the absence of arsS (44).
Fig 2.

Total production of urease proteins. (A) Lysates were obtained from wild-type and ΔarsS bacteria grown overnight on TSA plates supplemented with 5% sheep blood at pH 7.4. SDS-PAGE using 10 μg loaded protein and Western blot analysis were done, and densitometry was performed. There was a decrease in the total level of production of urease protein in the absence of the pH-sensing histidine kinase ArsS compared to wild-type bacteria (n = 3) (means ± standard errors of the means). (B) Representative Western blot. MW, molecular weight marker (in thousands).
Time course of pH-dependent urease activation.
The time course of urease activation was determined to investigate whether the deletion of arsS had an effect on the acid-dependent activation of apourease. The total urease activity was measured at various time points after incubation at an acidic or neutral pH. The level of urease activity of the wild-type strain increased >3-fold during the first 90 min of incubation at pH 5.5 compared to that at pH 7.4, and that level of activity was maintained for the remainder of the incubation period (Fig. 3). It was previously shown that this acid-induced increase in the level of urease activity was independent of protein synthesis but dependent on nickel and UreI expression (33). In contrast, there was a 30-min delay in the initiation of urease activation in the ΔarsS strain in acid, with the eventual attainment of levels close to those of the wild type only after 180 min of incubation (Fig. 3). No change in activity was seen at pH 7.4 for either strain. With the deletion of arsS, the delay in the acid-induced increase in the urease activity level suggests an alternative compensatory mechanism for the increase, underscoring the importance of this signaling system in ensuring a rapid response to acidity.
Fig 3.
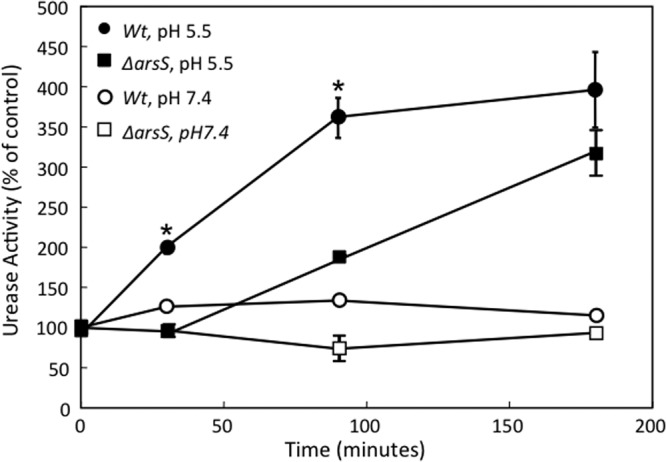
Time course of urease activation of H. pylori. H. pylori wild-type and ΔarsS strains were incubated in BHI medium at pH 5.5 or 7.4 for 30 to 180 min, and the total urease activity was measured in the presence of the nonionic detergent C12E8 to bypass UreI. In the wild-type strain, there was a >3-fold increase in the level of urease activity at pH 5.5 compared to that at pH 7.4 after 30 min, and it remained elevated for 3 h. The absence of ArsS had no effect on urease activity at pH 7.4 but delayed the increase in the urease activity level at an acidic pH seen with the wild-type strain. Activity approached wild-type levels after 180 min (n = 3) (means ± standard errors of the means) (∗, P < 0.05 by t test).
Membrane recruitment of cytoplasmic urease proteins.
To determine whether urease is assembled at the membrane in association with UreI and whether this urease-membrane association is regulated by ArsS in response to an acidic pH, Western blot analysis was performed on different cellular fractions of H. pylori. Antibodies against UreA and UreB were used for the immunodetection of these proteins from purified membrane fractions of H. pylori wild-type and ΔarsS strains exposed to pH 4.5, 5.5, and 7.4. Densitometry was performed by averaging the results of 3 Western blots of 3 independent membrane preparations at each pH. As shown in Fig. 4, the incubation of H. pylori at pH 4.5 for 30 min, compared to that at pH 7.4, resulted in an increase in the levels of membrane-associated UreA and UreB. The arsS deletion abolished this pH-induced increase in the levels of membrane-associated UreA and UreB (Fig. 4). Hence, the membrane recruitment of the urease subunits and the activation of the apoenzyme are dependent on the presence of this inner membrane sensor kinase. The ratios of band densities at pH 4.5/pH 7.4 were ≤1 for the ΔarsS strain, which may be explained by protein degradation. Similar trends were seen when the experiments were performed at pH 5.5 (data not shown). These data also show that the activation of the apoenzyme occurs only when assembled with UreI at the inner membrane.
Fig 4.
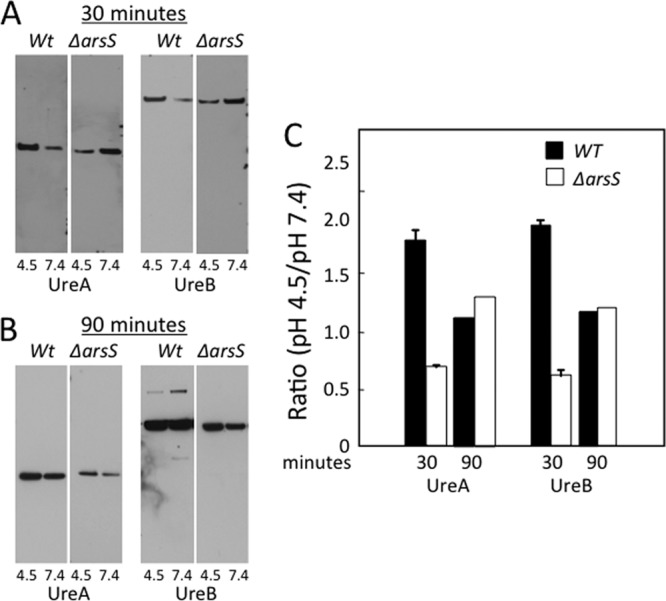
Membrane recruitment of urease proteins. Wild-type and ΔarsS bacteria were incubated at pH 4.5 or 7.4 for 30 or 90 min, membranes were isolated and purified, and 10 μg protein was used for SDS-PAGE. (A and B) Western blot analysis was performed by using antibodies against the cytoplasmic proteins UreA and UreB. Representative Western blots are shown for UreA and UreB. (C) An apparent increase in the level of each of the urease proteins in the membrane fraction in acid in the wild-type bacteria was confirmed by densitometry analysis. This effect was abolished in ΔarsS bacteria. After 90 min at an acidic pH, the membrane recruitment of urease proteins in ΔarsS bacteria approached wild-type levels (B and C) (n = 3).
As shown in Fig. 3, in the absence of ArsS, prolonged acid exposure (>1 h) was required for an increase in the level of urease activity. To determine if there was a similar increase in the level of urease proteins associated with the membrane even in the absence of ArsS, cells of the wild type and knockout mutants were incubated at pH 4.5 for 90 min. There was an increase in the levels of UreA and UreB in both the wild-type and knockout strains at pH 4.5, but the increase required 90 min of incubation for the deletion mutant, rather than the 30 min seen for the wild-type strain (Fig. 4B and C). This finding suggests that ArsS is required for an acute (<30-min) response to acidity, while the mechanism for the membrane recruitment of urease proteins triggered by prolonged acid exposure may be regulated by the cytoplasmic kinase, FlgS, activated when impaired or incomplete periplasmic alkalization ultimately leads to a decrease in the cytoplasmic pH (26, 34, 45).
Urease activity of membrane fractions.
The incubation of isolated H. pylori membranes at pH 4.5 compared to pH 7.4 for 30 min resulted in an increase in the total urease activity level of the membrane fraction by approximately 2-fold (Fig. 5). The deletion of arsS prevented this acidic-pH-dependent increase in the urease activity level in the membrane fraction. This is in agreement with the Western blot data showing the ArsS-dependent recruitment of cytoplasmic urease proteins to the membrane in acid after 30 min of incubation.
Fig 5.
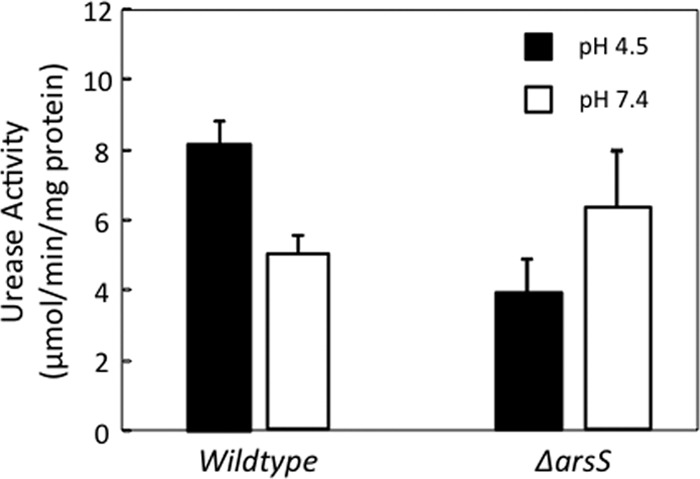
Urease activity of purified membranes. Wild-type and ΔarsS bacteria were incubated at pH 4.5 or 7.4 for 30 min. Bacteria were lysed, and membranes were isolated and purified. The total urease activity of the membrane fractions was measured by quantifying the release of [14C]O2 from [14C]urea. There was an increase in the level of urease activity in the membrane fraction of the wild-type bacteria after incubation in acid that was abolished in ΔarsS bacteria, in agreement with the Western blot data obtained from identical incubation and membrane isolation experiments (n = 3).
DISCUSSION
H. pylori is a neutralophile since it grows in vitro over a narrow pH range between 6.0 and 8.0 and optimally at pH 7.4. However, in its natural habitat, the gastric mucosa, H. pylori is often exposed to pH 3.0 and below (3, 12). Therefore, it has developed the mechanism of acid acclimation to combat the various acidities at its site of colonization.
Acid acclimation contrasts with acid resistance or acid tolerance utilized by other neutralophiles to maintain a viable cytoplasmic pH between 4.0 and 5.0, allowing survival but not growth (10, 37). Acid acclimation is the ability of H. pylori to buffer its periplasm to near neutrality in acid, thus maintaining sufficiently high cytoplasmic pH and transmembrane potential to allow for continued metabolism and growth, allowing gastric colonization. This unique property requires the activity of a neutral-pH-optimum cytoplasmic urease, a cytoplasmic β-carbonic anhydrase, a proton-gated urea channel, and a membrane-anchored periplasmic α-carbonic anhydrase (16, 33).
Although H. pylori shares some of the cytoplasmic pH-homeostatic systems found in other neutralophilic bacteria, it does not express the genes of the acid fitness island of Escherichia coli (2, 39). On the other hand, H. pylori expresses a variety of genes that contribute to the buffering of the periplasm and cytoplasm, generating a relatively neutral pH in its periplasm in spite of the acidity in its gastric environment (46). Both the cytoplasmic pH and inner membrane potential of H. pylori in acid are compatible with not only survival but also growth and, hence, colonization (21, 33).
The regulation of urease activity occurs at several levels: the regulation of urea access to urease via the proton-gated urea channel, UreI (42); the insertion of Ni2+ and the activation of the apoenzyme; and the biosynthesis of the structural genes. The proton-gated urea channel, UreI, accelerates urea entry severalfold as the medium pH drops from neutrality to pH 5.5. The NH3 can consume acid entering the cytoplasm, but it also diffuses out along with CO2 across UreI into the periplasm, neutralizing protons entering the periplasm (34). However, the organism has to maintain a relatively neutral periplasm to achieve an adequate bioenergetic profile across its inner membrane for growth at even moderately acidic pHs. The buffering ability of the NH3/NH4+ couple is much attenuated at a neutral pH, since its pKa is 9.2, 3 log orders away from the required pH. To achieve this, H. pylori also expresses a periplasmic membrane-bound α-carbonic anhydrase that generates HCO3− from the exiting CO2, with an effective pKa of 6.1. The periplasmic pH is maintained at around pH 6.1 in the presence of urea over a pH range of 2.5 to 6.2, allowing growth over this range of pHs in the presence, but not the absence, of urea (16, 34, 42).
At a neutral pH, two-thirds of the urease is present as inactive apoenzyme and is distributed throughout the cytoplasm. At acidic pHs, apourease traffics to the membrane and associates with the integral membrane protein UreI, where it is activated by Ni2+ insertion by the two accessory protein pairs UreE/G and UreF/H (40). This allows a relatively rapid response to medium acidification and suggests that the site of urease activation is pH dependent. Thus, there is not only a transcriptional/translational control of urease activity but also posttranslational regulation involving membrane recruitment and apourease activation.
One means of regulation of ureA and ureB transcription under acidic conditions is the activation of the two-component histidine kinase system ArsRS (1, 29, 30, 44, 46). ArsS is a major regulator of the acid response of H. pylori. It triggers the phosphorylation of its response regulator, ArsR, which has within its regulon the urease gene cluster and the α-carbonic anhydrase gene HP1186 (30, 43). About 109 additional genes are also dependent on the ArsRS TCS for increased transcription (29, 44). arsS deletion mutants do not infect the mouse stomach (26). Here we report that ArsS is also required for the acute acid-dependent association of apourease and its nickel-incorporating accessory proteins with UreI at the inner membrane. This increase in the level of membrane-associated urease activity to accelerate periplasmic buffering adds another response of the organism to acid stress.
In the absence of ArsS, the urease activity decreased to one-third of the level of activity of the wild type, and this decrease was seen in both intact bacteria under acidic conditions (pH <5.5), where activity is maximal, and total bacterial lysates. This finding is in agreement with data from previous studies and is a result of the decreased transcription of the ureA and ureB genes, which are regulated by the ArsS cognate response regulator ArsR via the activation of the PureA promoter (30). The absence of ArsS had no effect on the activation of UreI when the external pH fell below 6.5. Since ureI and the urease accessory genes are members of the ArsRS regulon that increase transcription via the binding of phosphorylated ArsR to PureI, this suggests that adequate levels of UreI are constitutively produced to provide saturating levels of urea to cytoplasmic urease.
The level of urease activity of H. pylori exposed to acid increases 3- to 4-fold with time and is maximal after 90 min. This increase in activity is independent of protein synthesis but is nickel and UreI dependent, showing that the increased level of activity with time in acid is the result of nickel insertion into apourease and takes place at the membrane, as shown by the requirement for UreI (33). The time course of urease activity of the ΔarsS strain exposed to acid showed a 30-min delay in activation compared to the wild-type organism and reached wild-type levels only after 180 min. Despite this delay, the activation of apourease proceeded, indicating an alternative, compensatory pathway for urease activation.
Immunoelectron microscopy showed an acid- and UreI-dependent reorganization of UreA and UreB from a predominately cytoplasmic location to the membrane, and native PAGE identified a UreA-UreB-UreI membrane complex (13, 40). In the current study, using Western blot analysis, it was found that the transition from a neutral pH to an acidic pH for 30 min resulted in a ∼2-fold increase in the levels of UreA and UreB at the membrane in the wild-type organism. Under the same conditions, in the ΔarsS strain, there was nearly a 2-fold decrease in the levels of UreA and UreB. Therefore, the delay in urease activation seen in the knockout strain is due to the decreased association of UreA and UreB with UreI at the inner membrane in acid. Exposure to acid for 90 min resulted in a 2-fold increase in both UreA and UreB protein levels compared to those at a neutral pH in both the wild-type and knockout strains, although the levels of UreA and UreB in the knockout strain were lower than those in the wild type. The decrease in UreA/UreB levels in the knockout strain is expected with the loss of the acid-activated transcription of these genes in the absence of ArsS.
Recently, a second, cytoplasmically localized acid responsive sensor kinase, FlgS, was identified in H. pylori. FlgS, due to its cognate response regulator, HP0703, is known to regulate flagellar gene transcription. ΔflgS and ΔflgR knockout mutants are aflagellate and nonmotile (25). The survival of ΔflgS mutants exposed to pH 4.5 for 30 min in the absence of urea is equal to that of the wild-type strain. However, a 30-min acid challenge at pH 2.5 even in the presence of 10 mM urea decreased the survival of ΔflgS bacteria by over 7 orders of magnitude compared to the wild-type organism (45). The membrane recruitment of the urease apoenzyme and its activation are a requirement for acid survival. The survival of ΔflgR mutants at pH 4.5 and 2.5 was indistinguishable from that of the wild-type strain, showing that the role of FlgS in acid survival is independent of its role in flagellar gene regulation and FlgR activation (45). The 3-fold increase in the level of urease activity in H. pylori presented with an acid challenge was completely abolished in the ΔflgS mutant, in contrast to just the delay seen for the ΔarsS mutant. Likewise, there was no increase in the level of UreA or UreB protein associated with the membrane, nor was there an increase in the urease activity of the membrane fraction (34). These findings suggest that a decrease in the cytoplasmic pH, exaggerated in the absence of ArsS, may activate FlgS, leading to an increase in urease recruitment and activation at the membrane, in association with UreI. Therefore, the finding that periplasmic pH homeostasis is compromised in the absence of ArsS suggests that, even at a medium pH of 4.5, the cytoplasmic pH decreases to levels that activate FlgS, resulting in eventual urease membrane recruitment and activation, restoring periplasmic pH homeostasis. Hence, the essential membrane recruitment and membrane-located activation of urease are the result of the integrated action of two histidine sensor kinases, the membrane-located ArsS and the cytoplasmic FlgS. The inhibition of either sensor kinase would compromise the ability to colonize the stomach and therefore provide a target for the eradication of H. pylori.
ACKNOWLEDGMENTS
This work was supported by NIH and USVA grants T32 DK07180-34 (E.A.M.), K12HD034610 (E.A.M.), P30 DH41301 (E.A.M.), DK053462 (G.S.), and 1I01BX001006 (G.S.).
Footnotes
Published ahead of print 3 August 2012
REFERENCES
- 1. Akada JK, Shirai M, Takeuchi H, Tsuda M, Nakazawa T. 2000. Identification of the urease operon in Helicobacter pylori and its control by mRNA decay in response to pH. Mol. Microbiol. 36:1071–1084 [DOI] [PubMed] [Google Scholar]
- 2. Alm RA, et al. 1999. Genomic-sequence comparison of two unrelated isolates of the human gastric pathogen Helicobacter pylori. Nature 397:176–180 [DOI] [PubMed] [Google Scholar]
- 3. Baumgartner HK, Montrose MH. 2004. Regulated alkali secretion acts in tandem with unstirred layers to regulate mouse gastric surface pH. Gastroenterology 126:774–783 [DOI] [PubMed] [Google Scholar]
- 4. Beier D, Frank R. 2000. Molecular characterization of two-component systems of Helicobacter pylori. J. Bacteriol. 182:2068–2076 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Bury-Mone S, et al. 2004. Responsiveness to acidity via metal ion regulators mediates virulence in the gastric pathogen Helicobacter pylori. Mol. Microbiol. 53:623–638 [DOI] [PubMed] [Google Scholar]
- 6. Clyne M, Labigne A, Drumm B. 1995. Helicobacter pylori requires an acidic environment to survive in the presence of urea. Infect. Immun. 63:1669–1673 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Cock PJ, Whitworth DE. 2007. Evolution of prokaryotic two-component system signaling pathways: gene fusions and fissions. Mol. Biol. Evol. 24:2355–2357 [DOI] [PubMed] [Google Scholar]
- 8. Delany I, Spohn G, Rappuoli R, Scarlato V. 2002. Growth phase-dependent regulation of target gene promoters for binding of the essential orphan response regulator HP1043 of Helicobacter pylori. J. Bacteriol. 184:4800–4810 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Eaton KA, Krakowka S. 1994. Effect of gastric pH on urease-dependent colonization of gnotobiotic piglets by Helicobacter pylori. Infect. Immun. 62:3604–3607 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Foster JW. 2004. Escherichia coli acid resistance: tales of an amateur acidophile. Nat. Rev. Microbiol. 2:898–907 [DOI] [PubMed] [Google Scholar]
- 11. Gotoh Y, et al. 2010. Two-component signal transduction as potential drug targets in pathogenic bacteria. Curr. Opin. Microbiol. 13:232–239 [DOI] [PubMed] [Google Scholar]
- 12. Henriksnas J, et al. 2006. Impaired mucus-bicarbonate barrier in Helicobacter pylori-infected mice. Am. J. Physiol. 291:G396–G403 doi:10.1152/ajpgi.00017.2006 [DOI] [PubMed] [Google Scholar]
- 13. Hong W, et al. 2003. Medium pH-dependent redistribution of the urease of Helicobacter pylori. J. Med. Microbiol. 52:211–216 [DOI] [PubMed] [Google Scholar]
- 14. Joseph B, Beier D. 2007. Global analysis of two-component gene regulation in H. pylori by mutation analysis and transcriptional profiling. Methods Enzymol. 423:514–530 [DOI] [PubMed] [Google Scholar]
- 15. Loh JT, Cover TL. 2006. Requirement of histidine kinases HP0165 and HP1364 for acid resistance in Helicobacter pylori. Infect. Immun. 74:3052–3059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Marcus EA, Moshfegh AP, Sachs G, Scott DR. 2005. The periplasmic alpha-carbonic anhydrase activity of Helicobacter pylori is essential for acid acclimation. J. Bacteriol. 187:729–738 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Marshall BJ, Warren JR. 1984. Unidentified curved bacilli in the stomach of patients with gastritis and peptic ulceration. Lancet i:1311–1315 [DOI] [PubMed] [Google Scholar]
- 18. McDaniel TK, Dewalt KC, Salama NR, Falkow S. 2001. New approaches for validation of lethal phenotypes and genetic reversion in Helicobacter pylori. Helicobacter 6:15–23 [DOI] [PubMed] [Google Scholar]
- 19. McDonald JA, Speeg KVJ, Campbell JW. 1972. Urease: a sensitive and specific radiometric assay. Enzymologia 42:1–9 [PubMed] [Google Scholar]
- 20. Merrell DS, Goodrich ML, Otto G, Tompkins LS, Falkow S. 2003. pH-regulated gene expression of the gastric pathogen Helicobacter pylori. Infect. Immun. 71:3529–3539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Meyer-Rosberg K, Scott DR, Rex D, Melchers K, Sachs G. 1996. The effect of environmental pH on the proton motive force of Helicobacter pylori. Gastroenterology 111:886–900 [DOI] [PubMed] [Google Scholar]
- 22. Mollenhauer-Rektorschek M, Hanauer G, Sachs G, Melchers K. 2002. Expression of UreI is required for intragastric transit and colonization of gerbil gastric mucosa by Helicobacter pylori. Res. Microbiol. 153:659–666 [DOI] [PubMed] [Google Scholar]
- 23. Muller S, Gotz M, Beier D. 2009. Histidine residue 94 is involved in pH sensing by histidine kinase ArsS of Helicobacter pylori. PLoS One 4:e6930 doi:10.1371/journal.pone.0006930 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Niehus E, et al. 2004. Genome-wide analysis of transcriptional hierarchy and feedback regulation in the flagellar system of Helicobacter pylori. Mol. Microbiol. 52:947–961 [DOI] [PubMed] [Google Scholar]
- 25. O'Toole PW, Lane MC, Porwollik S. 2000. Helicobacter pylori motility. Microbes Infect. 2:1207–1214 [DOI] [PubMed] [Google Scholar]
- 26. Panthel K, Dietz P, Haas R, Beier D. 2003. Two-component systems of Helicobacter pylori contribute to virulence in a mouse infection model. Infect. Immun. 71:5381–5385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Pflock M, et al. 2007. The orphan response regulator HP1021 of Helicobacter pylori regulates transcription of a gene cluster presumably involved in acetone metabolism. J. Bacteriol. 189:2339–2349 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Pflock M, Dietz P, Schar J, Beier D. 2004. Genetic evidence for histidine kinase HP165 being an acid sensor of Helicobacter pylori. FEMS Microbiol. Lett. 234:51–61 [DOI] [PubMed] [Google Scholar]
- 29. Pflock M, et al. 2006. Characterization of the ArsRS regulon of Helicobacter pylori, involved in acid adaptation. J. Bacteriol. 188:3449–3462 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Pflock M, Kennard S, Delany I, Scarlato V, Beier D. 2005. Acid-induced activation of the urease promoters is mediated directly by the ArsRS two-component system of Helicobacter pylori. Infect. Immun. 73:6437–6445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Pflock M, Muller S, Beier D. 2007. The CrdRS (HP1365-HP1364) two-component system is not involved in pH-responsive gene regulation in the Helicobacter pylori strains 26695 and G27. Curr. Microbiol. 54:320–324 [DOI] [PubMed] [Google Scholar]
- 32. Schar J, Sickmann A, Beier D. 2005. Phosphorylation-independent activity of atypical response regulators of Helicobacter pylori. J. Bacteriol. 187:3100–3109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Scott DR, Marcus EA, Weeks DL, Sachs G. 2002. Mechanisms of acid resistance due to the urease system of Helicobacter pylori. Gastroenterology 123:187–195 [DOI] [PubMed] [Google Scholar]
- 34. Scott DR, et al. 2010. Cytoplasmic histidine kinase (HP0244)-regulated assembly of urease with UreI, a channel for urea and its metabolites, CO2, NH3, and NH4(+), is necessary for acid survival of Helicobacter pylori. J. Bacteriol. 192:94–103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Scott DR, et al. 1998. The role of internal urease in acid resistance of Helicobacter pylori. Gastroenterology 114:58–70 [DOI] [PubMed] [Google Scholar]
- 36. Skouloubris S, Thiberge JM, Labigne A, De Reuse H. 1998. The Helicobacter pylori UreI protein is not involved in urease activity but is essential for bacterial survival in vivo. Infect. Immun. 66:4517–4521 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Slonczewski JL, Kirkpatrick C. 2002. Proteomic analysis of pH-dependent stress responses in Escherichia coli and Helicobacter pylori using two-dimensional gel electrophoresis. Methods Enzymol. 358:228–242 [DOI] [PubMed] [Google Scholar]
- 38. Teyssen S, Chari ST, Scheid J, Singer MV. 1995. Effect of repeated boluses of intravenous omeprazole and primed infusions of ranitidine on 24-hour intragastric pH in healthy human subjects. Dig. Dis. Sci. 40:247–255 [DOI] [PubMed] [Google Scholar]
- 39. Tomb JF, et al. 1997. The complete genome sequence of the gastric pathogen Helicobacter pylori. Nature 388:539–547 [DOI] [PubMed] [Google Scholar]
- 40. Voland P, et al. 2003. Interactions among the seven Helicobacter pylori proteins encoded by the urease gene cluster. Am. J. Physiol. 284:G96–G106 doi:10.1152/ajpgi.00160.2002 [DOI] [PubMed] [Google Scholar]
- 41. Waidner B, Melchers K, Stahler FN, Kist M, Bereswill S. 2005. The Helicobacter pylori CrdRS two-component regulation system (HP1364/HP1365) is required for copper-mediated induction of the copper resistance determinant CrdA. J. Bacteriol. 187:4683–4688 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Weeks DL, Eskandari S, Scott DR, Sachs G. 2000. A H+-gated urea channel: the link between Helicobacter pylori urease and gastric colonization. Science 287:482–485 [DOI] [PubMed] [Google Scholar]
- 43. Wen Y, Feng J, Scott DR, Marcus EA, Sachs G. 2007. The HP0165-HP0166 two-component system (ArsRS) regulates acid-induced expression of HP1186 alpha-carbonic anhydrase in Helicobacter pylori by activating the pH-dependent promoter. J. Bacteriol. 189:2426–2434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Wen Y, Feng J, Scott DR, Marcus EA, Sachs G. 2006. Involvement of the HP0165-HP0166 two-component system in expression of some acidic-pH-upregulated genes of Helicobacter pylori. J. Bacteriol. 188:1750–1761 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Wen Y, Feng J, Scott DR, Marcus EA, Sachs G. 2009. The pH-responsive regulon of HP0244 (FlgS), the cytoplasmic histidine kinase of Helicobacter pylori. J. Bacteriol. 191:449–460 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Wen Y, et al. 2003. Acid-adaptive genes of Helicobacter pylori. Infect. Immun. 71:5921–5939 [DOI] [PMC free article] [PubMed] [Google Scholar]