

SUMMARY
1,3-Dipropyl-8-phenylxanthine, a synthetic analog of theophylline and a potent antagonist of adenosine at A1 and A2-adenosine receptors, has been attached covalently through a functionalized chain to amino acids and oligopeptides. The xanthine conjugates have been studied as competitive inhibitors of the specific binding of [3H]N6-cyclohexyladenosine to A1-receptors of rat cerebral cortical membranes and for inhibition of cyclic AMP accumulation elicited by 2-chloroadenosine in guinea pig brain slices through A2-receptors. A free amino group on the extended chain generally resulted in high potency at A1-receptors. The potency (in some cases extending into the subnanomolar range) and selectivity for A1-receptors (up to 200-fold) suggest that this approach can yield a versatile class of “functionalized congeners” of adenosine receptor antagonists in which distal modifications of the attached moiety (“carrier”) can serve also to improve pharmacodynamic and pharmacokinetic parameters. The water solubility in many of the more potent analogs has been enhanced by two orders of magnitude over that of simple, uncharged 8-phenyl xanthine derivatives. Analogs in which the carrier contains d-tyrosine have potential for development of iodinated radioligands for adenosine receptors. The functionalized congener approach is potentially applicable to other drugs and for development of prodrugs.
Adenosine mediates a wide variety of physiological processes (1–6) including vasodilation, cardiac depression, inhibition of lipolysis, vasoconstriction in the kidney, inhibition of platelet aggregation, inhibition of insulin release and potentiation of glucagon release in the pancreas, inhibition of lymphocyte functions, potentiation of histamine release from mast cells, and inhibition of neurotransmitter release from nerve endings. There are two classes of adenosine receptors, A1 and A2, which in some systems have been shown to be inhibitory and stimulatory, respectively, to adenylate cyclase. The major class of adenosine receptor antagonists consists of the 1,3-dialkylxanthines, e.g., theophylline, 1a. The presence of 1,3-dipropyl and 8-phenyl substituents confers high potency (7) and in some cases selectivity (8), as in 1c. Unfortunately, the low water solubility and high lipophilicity of such xanthines will prevent their widespread use in vivo.
A functionalized “congener” approach to the design of catecholamine analogs has been described recently (9–11). In this approach, isoproterenol analogs having carboxylic acid-functionalized chains were synthesized and attached covalently to various monodisperse carriers, including oligopeptides (12,13), resulting in β-adrenergic agonists certain of which have high potency. In an extension to the approach, we have investigated sites of derivatization of agonists (14) and antagonists (15) for the adenosine receptor. In a series of antagonists derived from 1,3-dipropyl-8-phenylxanthine, the para-position of the phenyl ring appeared to accommodate functional groups for the attachment to carriers. The carboxylic acid congener 2 and the amino congener 3a (see Fig. 1) could be attached to various small molecules by standard amide coupling methods producing analogs with varying binding affinities at adenosine receptors. This report describes the preparation, in vitro biological activity, and stability of a series of amino acid conjugates of 1,3-dipropylxanthines (see Fig. 2). The “functionalized congener” approach, as used here in the development of new adenosine antagonists, would appear applicable to other areas in which such alterations in a drug or other pharmacologically active agent are sought.
Fig. 1.

Analogs of theophylline found to have high potency at the adenosine receptor. Compounds 2 and 3 are functionalized congeners designed for attachment to “carriers.” Structures in bold face enhance potency at A1-adenosine receptors.
Fig. 2.

The functionality on the chain is varied by linking the congeners to amino acids by a two-step synthetic process. The primary pharmacophore remains the same in each case.
Materials and Methods
Compounds 2, 3a, and 3b were prepared as described previously (15). Protected amino acid conjugates 8a through 21a (general formulas 4 and 5) were synthesized by the coupling methods specified in Table 1, following the general procedures previously reported. Active ester derivatives of glutamine, leucine, and phenylalanine were obtained from Sigma Chemical Co. (St. Louis, MO). Derivatives of citrulline and methionine were from Bachem (Torrance, CA), and derivatives of asparagine, glycine, and glycylglycine were from United States Biochemical Corp. (Cleveland, OH). Deblocking of acid-labile protecting groups was carried out for 1 hr at room temperature in anhydrous 30% HBr in acetic acid for Cbz- derivatives and in neat trifluoroacetic acid for Boc- derivatives. After evaporation of the acid solution, the residue obtained was triturated with ether, and the solid product was collected, washed with ether, and dried under vacuum. The purity of the xanthine analogs was monitored by thin layer chromatography in chloroform:methanol:acetic acid (85:10:5 or 50:50:5), and, if necessary, the product was recrystallized from dimethylformamide/ether or methanol/ether. Proton NMR spectroscopy was carried out by Dr. H. Yeh (National Institutes of Health) on a 300-MHz Varian spectrometer.
TABLE 1.
Synthesis of 1,3-dipropytxanthine amino acid conjugates (amino acids of l-configuration unless noted)
| A Derivatives of carboxtylic acid congener (general formula 4) | ||||
| Compound | R1 = | X = | Methoda | Yield |
|
| ||||
| 6a | —H | —C(CH3)3 | A | 56 |
| 6b | —H | —H | E | 78 |
| 7a | —(CH2)4NHCOOCH2ϕ | —C(CH3)3 | A | 85 |
| 7b | —(CH2)4NH2 | —H | D | 100 |
|
| ||||
| B. Derivatives of amino congener (general formula 5) | ||||
|
| ||||
| Compound | R2 = | Y’ = | Method | Yield |
| % | ||||
| 8a | —CH2CONH2 | —COOCH2ϕ | B | 92 |
| 8b | —CH2CONH2 | —H·HBr | D | 76 |
| 9a | —(CH2)3-NHCONH2 | —COOC(CH3)3 | B | 71 |
| 9b | —(CH2)3-NHCONH2 | —H·CF3COOH | E | 68 |
| 10a | —(CH2)2CONH2 | —COOC(CH3)3 | C | 48 |
| 10b | —(CH2)2CONH2 | —H·CF3COOH | E | 80 |
| 11a | —H | —COOCH2ϕ | B | 36 |
| 11b | —H | —H·HBr | D | 93 |
| 12a | —CH2CH(CH3)2 | —COOC(CH3)3 | C | 82 |
| 12b | —CH2CH(CH3)2 | —H·CF3COOH | E | 98 |
| 13a | —(CH2)4NHCOOCH2 | —COOC(CH3)3 | C | 39 |
| 13b | —(CH2)4NH2·HBr | —H·HBr | D | 100 |
| 14a | —(CH2)4NHCOOCH2ϕ(D) | —COOC(CH3)3 | A | 95 |
| 14b | —(CH2)4NH2·HBr(D) | —H·HBr | D | 100 |
| 15a | —(CH2)2SCH3 | —COOC(CH3)3 | C | 47 |
| 15b | —(CH2)2SCH3 | —H·CF3COOH | E | 73 |
| 16a | —CH2ϕ | —COOC(CH3)3 | C | 63 |
| 16b | —CH2ϕ | —H·CF3COOH | E | 100 |
| 17a |
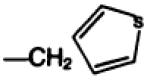
|
—COOC(CH3)3 | C | 71 |
| 17b |
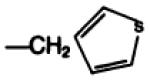
|
—H·CF3COOH | E | 98 |
| 18a |
|
—COOC(CH3)3 | C | 70 |
| 18b |
|
—H·CF3COOH | E | 93 |
| 19a |
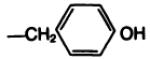
|
—COOCH2ϕ | B | 89 |
| 19b |
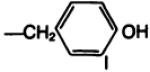
|
—H·HBr | D | 81 |
| 20a |
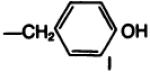
|
—COOC(CH3)3 | C | 41 |
| 20b |
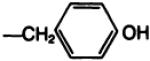
|
—H·CF3COOH | E | 100 |
| 21a |
|
—COOC(CH3)3 | C | 70 |
| 21b |
|
H·CF3COOH | E | 88 |
Methods are as follows: A, carbodiimide coupling (DCC + 1-hydroxybenzotnazole); B, p-nitrophenyl ester coupling to compound 3a: C, N-hydroxysuccinimide ester coupling: D, HBr/HOAc; E, trifluoroacetic acid; F, H2/Pd.
t-Butyloxycarbonyl-d-tyrosine N-hydroxysuccinimide ester (24) was prepared from the Boc-d-tyrosine (Chemical Dynamics, South Plainfield, NJ), N-hydroxysuccinimide, and DCC in DMF in a 95% yield, with m.p. 191–192°, [α]D = 42.3 (c = 1.4, DMF, 23°). C18H22N2O7 analysis, calculated: 57.14% C, 5.86% H, 7.40% N; found: 57.24% C, 6.19% H, 7.74% N.
t-Butyloxycarbonyl-l-3-(2′-thienyl)alanine (25) was prepared from l-3-(2′-thienyl)alanine (Chemical Dynamics) and di-t-butyl-dicarbonate by a standard method (16). The product was isolated as a clear oil in 95% yield.
t-Butyloxycarbonyl-l-3-(2′-thienyl)alanine N-hydroxysuccinimide ester (26) was prepared from compound 25 by the DCC method in an 84% yield, with m.p. 127–130°, [α]D = −32.5° (c = 1.5, DMF, 23°). C16H20N2O6S analysis, calculated: 52.16% C, 5.47% H, 7.60% N; found: 52.56% C, 5.78% H, 7.81% N.
8-(4′-Carboxymethyloxyphenyl)-1,3-dipropylxanthine-2-(N-t-butyloxycarbonyl-3(2′-thienyl)-l-alanylamino)-ethy lamide (17a) was prepared by dissolving compound 26 (32 mg, 87 μmol) in 1 ml of DMF and adding the xanthine amino congener 3b (Ref. 15, 25 mg, 58 μmol). After stirring for 6 hr, 3 ml of dry ether was added forming a precipitate which was filtered and dried in vacuo at 60° to give 28 mg of product, melting with decomposition at 176–180°, [α]D = −2.3° (c = 0.9, DMF, 22°).
8-(4′-Carboxymethyloxyphenyl)-1,3-dipropylxanthine-2-(3-(2′-thienyl)-l-alanylamino)-ethylamide trifluoroacetate (17b) was prepared by treating compound 17a (64 mg) with anhydrous trifluoroacetic acid (2 ml) at room temperature for 1 hr. The solvent was evaporated under a stream of dry argon, and the residue was triturated with ether to give 59 mg of a light tan solid, melting at 185–189°, [α]D = +7.3° (c = 1.1, DMF, 22°).
8-(4′-Carboxymethyloxyphenyl)-1,3-dipropylxanthine-2-(N-benzyloxycarbonyl-diglycylamino)-ethylamide (22a) was prepared by combining 3b (0.20 g, 0.47 mmol), Cbz-glycyl-glycine (0.15 g, 0.56 mmol) and DCC (0.11 g, 0.53 mmol) in 10 ml of DMF. The mixture was agitated for 16 hr and filtered. Dry ether and petroleum ether were added to precipitate the product (0.20 g, 63%), which was recrystallized (DMF/H2O) and melted at 222–225°.
8-(4′-Carboxymethyloxyphenyl)-1,3-dipropylxanthine-2-(N-t-butyloxycarbonyl-b-tyrosyl-d-(ε-benzyloxycarbonyl)-lysyl-amino)-ethylamide (23a) was prepared by aminolysis of compound 24 (30 mg, 80 μmol) with the trifluoroacetate salt of d-(ε-benzyloxycarbonyl)lysyl conjugate of 3b (58 mg, 72 μmol, prepared by the method for synthesis of 17b) and in 2 ml of DMF containing 9 μl of N-ethylmorpholine. After 1 day the solution was treated with 0.1 M sodium phosphate, pH 6, buffer resulting in 60 mg of product, 86% yield, as a white precipitate, homogeneous by thin layer chromatography.
8-(4′-Carboxymethyloxyphenyl)-1,3-dipropylxanthine-2-(d-tyrosyl-d-lysylamino)-ethylamide dihydrobromide (23b) was prepared from compound 23a (26 mg, 27 μmol) by deprotection in 30% HBr/acetic acid (2 ml) at room temperature for 1 hr. Most of the solvent was evaporated in vacuo, and the residue was triturated with dry ether. Recrystallization from methanol/ether and drying under high vacuum gave 24 mg of a white solid (100% yield) melting at 220° with decomposition beginning at 207°.
Biochemical assays
Inhibition of binding of 1 nM [3H]N6-cyclohexyladenosine to A1-adenosine receptors in rat cerebral cortical membranes was assayed as described previously (17). Inhibition of binding by a range of concentrations of each xanthine was assessed in triplicate for at least two separate experiments. Inhibition of 2-chloroadenosine-stimulated accumulation of [3H]cyclic AMP in [3H]adenine-labeled guinea pig cerebral cortical slices was assayed essentially as described previously (8, 17). In the present experiments 10 μg/ml of adenosine deaminase was present in incubations with slices to prevent effects of endogenous adenosine, and 30 μM 4-(3-cyclopentyloxy-4-methoxy-phenyl)-2-pyrrolidone (rolipram, ZK 62711) (18) was present to inhibit phosphodiesterases. Under these conditions 2-chloroadenosine elicited a maximal 10–20-fold increase in levels of radioactive cyclic AMP in guinea pig cortical slices with an EC50 of about 8 μM (8). Inhibition of the response to 15 μM 2-chloroadenosine by a range of concentrations of each xanthine was assessed in triplicate in at least two separate experiments.
Results
In our earlier work (15), several analogs of 1,3-dipropylxanthine (for example 3a, Fig. 1) bearing distal amino groups on the chain, separated from the phenyl ring by as many as 13 bond lengths, had affinity constants for the A1-receptor subtype in the nanomolar range. The synthesis of a series of amino acid conjugates seemed a convenient strategy to attach a wide range of substituents including polar groups for water solubility, to amino functionalized congeners. In each case the amino acid “carrier” was linked through an amide bond to a functionalized xanthine congener. The “carrier” is defined as an attached moiety that is separate from the primary pharmacophore. The “carrier” may have an influence on binding and/or may lend desired chemical, physical, or pharmacokinetic characteristics to the new analog.
Fig. 2 shows the general structures of the protected (X = alkyl, Y = urethane blocking group) and deprotected (X, Y = H) amino acid conjugates derived from the functionalized congeners of Fig. 1. Amino acids protected as t-butyl esters were coupled to the carboxylic acid functionalized congener, 2, using a carbodiimide in DMF with 1-hydroxybenzotriazole (19) as a catalyst. The xanthine amino congener, 3a, of 1,3-dipropylxanthine derived from ethylene diamine was coupled to various urethane protected amino acids using N-hydroxysuccinimide or p-nitrophenyl active ester intermediates. Since conditions required in this coupling procedure are quite mild, certain amino acids were introduced without side chain protection (e.g., tyrosine and asparagine). During the reaction in DMF, the amino congener substrate dissolved gradually as the acylation proceeded, thus limiting exposure to excess base and therefore decreasing the potential for racemization of the amino acid. The t-butyl ester protecting group (X) of 4 and the urethane protecting groups (Y) of 5 were subsequently cleaved in acid, as specified, without serious side reactions on the xanthine portion of the molecule. Details of each synthesis are included in Table 1. Assignment of proton NMR resonances for each analog in d6-dimethylsulfoxide was carried out by chemical shift analysis and by analogy with other xanthines (15) and amino acid derivatives (20). Because of the possibility of succinimide formation in the synthesis of the asparagine congener, compound 8a was also studied by californium plasma desorption mass spectrometry (21). A positively charged parent ion of the expected molecular weight + 1 (m/z = 543) was observed, thus eliminating the possibility that a succinimide rearrangement had occurred.
The xanthine derivatives were screened for A1-adenosine receptor affinity on the basis of competitive inhibition of [3H] cyclohexyladenosine binding in rat cerebral cortex membranes (17). The IC50 values are listed in Table 2. The previously noted pattern of high potency observed for analogs having a free amino group was evident in amino acid conjugates of the general formula 5. In the present series the amino group is located between 8 and 14 bond lengths from the phenyl ring, and the receptor binding affinity was in the low nanomolar range. As shown graphically in Fig. 3, the potency of each of the free amino analogs was greater than that of the corresponding ammo-protected intermediates. Representative binding curves for several deprotected amino acid conjugates, including an enantiomeric pair, are shown in Fig. 4. As a control experiment, the amino acid and spacer chain portion of the most potent free-amino derivative, consisting of d-lysyl-NH-(CH2)2-NHCOCH3, was synthesized separately and found to be totally inactive at a concentration several hundred times that of the IC50 for compound 14b. A Cbz- protected derivative, 11a, was the most potent among the protected amino acid conjugates.
TABLE 2.
A comparison of potencies of the xanthine derivatives at adenosine receptor subtypes.
| Compound | IC50 at A1a | IC50 A2a | IC50(A2)/IC50(A1) | ||
|---|---|---|---|---|---|
| nM | nM | ||||
| 1a | 24,000, | 32,000 | 36,000, | 48,000 | 1.5 |
| 1b | 1,000 | 1,800 | 6,600 | 9,600 | 5.8 |
| 1c | 4.8 | 6.8 | 130 | 170 | 26 |
| 2 | 112 | 120 | 75 | 130 | 0.88 |
| 3a | 1.8 | 3.0 | 114 | 180 | 61 |
| 3b | 44 | 52 | 180 | 192 | 3.9 |
| 6a | 22 | 26 | 147 | 153 | 6.3 |
| 6b | 134 | 158 | 144 | 174 | 1.1 |
| 7a | 32 | 36 | 540 | 720 | 19 |
| 7b | 37 | 39 | 180 | 198 | 5.0 |
| 8a | 12.8 | 13.6 | 178 | 182 | 9.1 |
| 8b | 8.1 | 8.7 | 335 | 385 | 29 |
| 9a | 103 | 105 | 250 | 250 | 2.4 |
| 9b | 5.7 | 5.9 | 220 | 220 | 38 |
| 10a | 38 | 42 | 220 | 260 | 6.0 |
| 10b | 35 | 41 | 360 | 420 | 10 |
| 11a | 10.4 | 11.6 | 138 | 222 | 16 |
| 11b | 4.1 | 4.3 | 120 | 180 | 36 |
| 12a | 57 | 59 | 510 | 570 | 9.3 |
| 12b | [4.1 | 4.3]b | 680 | 880 | [190] |
| 13a | 19.6 | 22.0 | 480 | 510 | 24 |
| 13b | 2.04 ±0.14 (4) | 108 | 150 | 63 | |
| 14a | 24 | 28 | 360 | 360 | 14 |
| 14b | 1.74 ±0.18 (4) | 138 | 180 | 91 | |
| 15a | 40 | 48 | 330 | 450 | 8.9 |
| 15b | 2.7 | 3.3 | 310 | 410 | 120 |
| 16a | 78 | 106 | 625 | 635 | 6.8 |
| 16b | [4.5 | 5.9]b | 282 | 306 | [57] |
| 17a | 34 ± 3.3 (4) | 465 | 615 | 16 | |
| 17b | [2.6 ±0.2]b (4) | 550 | 710 | [240] | |
| 18a | 112 | 124 | 1,260 | 11 | |
| 18b | 11.0 | 13.4 | 270 | 310 | 24 |
| 19a | 38 | 42 | 500 | 520 | 13 |
| 19b | [4.0 | 4.4]b | 230 | 310 | 64 |
| 20a | 230 | 234 | 1,500 | 1,800 | 7.1 |
| 20b | 10.4 | 12.0 | 900 | 1,200 | 94 |
| 21a | 92 ± 4.0 (4) | 470 | 610 | 5.9 | |
| 21b | 9.0 ± 0.2 (4) | 210 | 330 | 30 | |
| 22a | 5.5 | 7.0 | 180 | 200 | 15 |
| 22b | 6.7 | 7.5 | 130 | 180 | 22 |
| 23a | 103 | 109 | 1,550 | 2,050 | 17 |
| 23b | 4.2 | 5.4 | 240 | 50 | |
IC50 values are for two separate determinations or are means ± SE with the number of determinations in parentheses. All determinations in a single experiment are in triplicate. In other reports (8, 14, 15) Ki values for xaithines have been calculated from the IC50 values using the Cheng-Prusoff equation, a KB value for binding of [3H]N6-cyclohexyladenosine to A1 receptors in rat cerebral cortical membranes of 1.0 nM (17), and an EC50 value for activation of cyclic AMP-generating systems by 2-chloroadenosine in guinea pig cerebral cortical slices of 8 μM (8) have also been reported.
Values in brackets are uncertain, since the conjugates undergo partial proteolytic cleavage during A1 assay (see Results).
Fig. 3.

A comparison of apparent A1-adenosine receptor affinities of blocked (○) and free amino (●) conjugates of 1,3-dipropylxanthines. The amino acid carrier and amine-protecting groups are listed on the abscissa. All amino acids are of the l-configuration unless noted. Except for compounds 3a and 3b, compound b of each pair corresponds to the free amino form. Cit, citrulline; Tha, 3-(2′-thienyl)alanine.
Fig. 4.
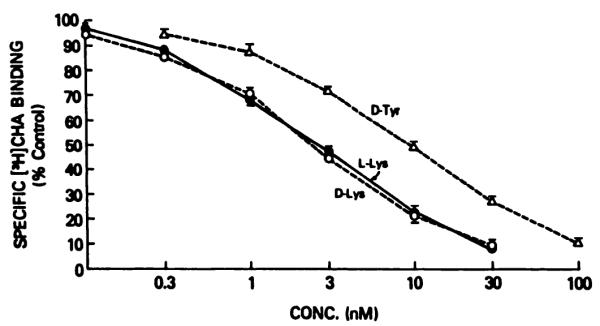
Inhibition of specific binding of [3H]N6-cyclohexyladenosine to A1-adenosine receptors of rat cerebral cortical membranes by amino acid conjugates 13b (l-Lys, ●), 14b (d-Lys, ○), and 21b (d-Tyr, Δ).
The possibility of hydrolytic cleavage of the attached amino acid from the pharmacophore during the 2-hr incubation with membranes was investigated. After incubation with xanthine conjugates at initial concentrations of 0.3 and 1.0 μM, the membrane suspensions were centrifuged, and the supernatant was examined by HPLC. Most of the xanthine derivatives, i.e., those containing urethane-blocked amino acids, unbranched aliphatic amino acids (e.g., 9b, 10b, 11b, and 13b), and d-amino acids, were unaltered. In contrast, certain of the free amino conjugates of aromatic and branched amino acids of the l-configuration were partially degraded during the 2-hr incubation. Thus, at a concentration of 300 nM, 19b, the conjugate of l-tyrosine, was hydrolyzed to compound 3a to the extent of 25%, and even at concentrations as low as 30 nM, some cleavage was detectable. Compounds 12b, 16b, and 17b were cleaved to a comparable extent. The conjugates of free amino conjugates of l-tryptophan and l-3-iodotyrosine were much less susceptible to hydrolysis (less than 5%). Addition of 0.1 mM bacitracin to the incubation medium reduced the hydrolysis of 19b to 12%.
Biological activity at the A2-adenosine receptor (Table 2) was measured by inhibition of 2-chloroadenosine-stimulated accumulation of cyclic AMP in guinea pig brain slices (17). Xanthine derivatives bearing a free carboxylate group, e.g., compounds 2 and 6b, are among the least potent at the A1-receptors, whereas many of the free amino conjugates show a high degree of selectivity for A1-receptors. Among the most selective for the A1-receptor are conjugates of l-citrulline, 9b; l-lysine, 13b; d-lysine, 14b; and l-methionine, 15b. Certain of the present analogs have proven selective in blocking A1-receptor-mediated cardiac depression in vivo compared to A2-receptor-mediated vasodilation (22).
Many of the highly potent A1-antagonists also had greatly enhanced water solubility (Fig. 5). Conjugate 9b, derived from citrulline and the amino congener, was 3 times more soluble than the amino congener itself (pH 7.2, 0.1 M sodium phosphate). The neutral, polar side chain of citrulline has been noted for improving water solubility in oligopeptides (12). The lysyl conjugates, having an additional ammonium group on the carrier, displayed an aqueous solubility of 0.34 mM, 100-fold greater than that of 1,3-dipropyl-8-p-hydroxyphenylxanthine. Octanol/water partition coefficients demonstrate further the improved polarity characteristics of the amino acid conjugates (Table 3).
Fig. 5.

Water solubility of potent xanthine analogs enhanced by charged or uncharged polar groups in the “carrier.”
TABLE 3.
Partition coefficients for selected xanthine analogs
| Compound | Log (conc, in octanol/conc, in 0.01 M Na phosphate, pH 7.2) |
|---|---|
| 6b | 0.40 |
| 7b | 0.29 |
| 8b | 2.1 |
| 9b | 0.81 |
| 11b | 1.40 |
| 12b | 1.9 |
| 14b | −0.06 |
| 17b | 2.0 |
| 18a | 2.5 |
| 18b | 2.1 |
| 19b | 2.0 |
| 22b | 1.00 |
| 23b | −0.19 |
The favorable water solubility made possible effective HPLC procedures for analytical and semipreparative purposes using a C-18 bonded silica column (Altex Ultrasphere ODS, 4.6 × 25 mm) with 60% methanol in 0.1 M triethylammonium acetate buffer (pH 5.2). At 1.0 ml/min, flow rate retention times for selected xanthines were as follows: compounds 2, 7.3 min; 3a, 21.0 min; 9b, 18.7 min; 10b, 22.2 min.
The amino acid “carrier” may also be viewed as a means of introducing a prosthetic group for radiolabeling. As a model for the radioiodination reaction, the monoiodinated derivative, 20b, of the l-tyrosyl conjugate 19b was prepared by two convergent routes; either from Boc-protected 3-iodotyrosine, resulting in compounds 20a and subsequently 20b, or by electrophilic iodination of 19b. The potency at A1-adenosine receptors of the free amino conjugate was comparable before and after iodination. The ability to separate these xanthines by HPLC suggests that HPLC may be appropriate for purification of the radioiodinated analogs. For example, treatment of 19b with one equivalent of chloramine T (0.1 m in dioxane) and sodium iodide converted most of the 19b present (room temperature, 7.8 min. 60% MeOH/0.05 sodium phosphate, pH 6, column as above, 1.0-ml/min flow rate) to a peak with retention time of 11.4 min (identical to that of the monoiodinated 20b) and a minor peak (room temperature, 14.3 min) for the 3,5-diiodo species. With an excess of iodinating reagent the products gradually decomposed, possibly by oxidative cleavage, as has been described for tyrosyl amides (23).
The achievement of high potency in monoamino acid derivatives prompted extension of the chain to oligopeptides. The amino congener 3a was coupled to Cbz-glycyl-glycine p-nitrophenyl ester resulting in a dipeptide conjugate, 22a, which had high potency at A1-receptors relative to other amino-protected conjugates. Upon deprotection the resulting diglycyl conjugate 22b had an IC50 value of 7.1 nM, which was comparable to other amino-functionalized analogs, logs.
The dipeptide conjugate 23a containing the protected peptide carrier Boc-d-Tyr-d-(ε-Cbz)Lys- was prepared. Cleavage of the urethane groups gave the d-Tyr-d-Lys conjugate 23b shown in Fig. 6. In contrast to compound 22a, which was synthesized by the coupling of an activated dipeptide fragment to the amino congener 3a, the protected dipeptide conjugate 23a was synthesized by the stepwise addition of urethane-protected amino acid residues.
Fig. 6.

Design features of a dipeptide conjugate (23b) of 1,3-dipropylxanthine. The IC50 at the A1-receptor is 4.8 nM.
Discussion
Recent work in our laboratory (14, 15) and in others (9–13) indicates that the attachment of drugs to “carrier” molecules is a useful approach in the design of new, potent analogs. We call this the “functionalized congener” approach after the nomenclature of Goodman and co-workers (9–13). The “carrier” molecule may be considerably larger than the parent drug. Thus, there is practically no maximum size limitation for a fully potent drug analog, as demonstrated with congeners attached to biotin-avidin complexes which bind to the adenosine receptor (24). Unlike the prodrug approach or the immobilization of drugs for slow release, the “functionalized congener” approach is directed toward analogs for which no metabolic cleavage step is necessary for activation. The attachment of the drug to a “carrier” such as a peptide may result in the improved affinity at an extracellular receptor site.
Although the “functionalized congener” approach is designed to identify analogs which might act in vivo as the intact conjugates, the strategy is also amenable to the development of prodrugs. Several conjugates in this series, e.g., 19a, were observed to be cleaved enzymatically during a 2-hr incubation with brain membranes to a more active species. Further study of peptidases which may act specifically upon various conjugates in vivo should indicate which analogs may be developed as prodrugs using this strategy. With a wide range of polarities (Table 3), certain analogs may be more readily absorbed in the gut subsequently to be hydrolyzed to a more active form by peptidases.
The functionalized congener approach is potentially applicable to any drug. A drug analog is synthesized with the regiospecific inclusion of a chain terminating in a chemically reactive group such as a carboxylic acid or an amine. As illustrated here, these functional groups are suited for highly efficient coupling to a wide variety of substances to serve as “carriers.” In using this approach, simple derivatives are prepared first, and then-activities provide a guide to whether biological activity will be affected using that site of attachment and what structures will be tolerated in a further elongated chain. This strategy involving distal modifications then is used to optimize in the same derivatives both potency and other desirable properties such as water solubility, with the added potential for incorporating radiolabels, photoaffinity labels, fluorescent moieties, and chemically reactive or antigenic groups.
We have used the functionalized congener approach to assemble systematically a structure-activity relationship for xanthine analogs having amino acids attached through spacer chains. This study has resulted in a large number of unusually potent adenosine receptor antagonists. Structural changes on the attached carrier result in significant changes in receptor affinity and biological activity.
This relatively high affinity of xanthines with a free amino group on the elongated chain parallels previous observations with simpler amino-functionalized analogs of both antagonists (15) and agonists (14) of the adenosine receptor. It appears likely that the amino group itself, perhaps as the charged ammonium form interacting with an anionic site of the receptor or another membrane component such as a phospholipid, is responsible for high potency and selectivity for the A1-receptor (compare 3a with 3b, 3b with 11b). Steric factors of the carrier, however, influence activity (compare conjugates of Tyr and iodo-Tyr).
The conjugates of amino acids of the d-configuration, 14 and 21, were prepared to reduce potential enzymatic cleavage of the amino acid residue in vivo. Since, in general, optical isomerism at a position close to a pharmacophore is likely to have a major effect on receptor affinity, it was important to determine whether the same magnitude of effect would be seen for a distal chiral center. In the case of the lysyl conjugate, l-and d-enantiomers (Fig. 4) were equipotent at the A1-receptor. The small apparent difference in observed EC50 between enantiomers of the tyrosyl conjugate may be due to the partial cleavage of the l-isomer (see above). In A2-activity, no significant difference between enantiomers of either lysyl or tyrosyl conjugates was observed.
The attachment of free amino acids to the chain not only favors high potency in these xanthine conjugates but has led to improved solubility characteristics due to the presence of the amino group, which is predominantly charged at physiological pH. It has been observed that the 8-phenyl xanthine analogs noted for high potency, such as 8-phenyltheophylline (7,8), are too hydrophobic to be absorbed well into circulation after intraperitoneal injection. This would not be a limitation in the present series, which combines nanomolar potency with greatly increased water solubility (EC50 values at A1-receptors and maximum aqueous solubility differ by a factor of approximately 10,000). The attached carrier in general may have a substantial effect on the overall solubility of the analog even in organic solvents. For example, compound 13a, which has two bulky hydrophobic groups on a lysine residue, is unusually soluble in ethyl acetate in contrast to smaller analogs, e.g., the ethyl ester of compound 2. The asparagine congener 8a, designed to take advantage of the polar side chain, surprisingly showed both low potency and a high octanol/water partition value.
The wide range of incorporated amino acid side chains that lead to high potency suggests considerable versatility in this approach for constructing receptor probes and labels. The conjugates of tyrosine, 19b, 21b, and 23b; tryptophan, 18b; and the unusual amino acid thienylalanine, 17b, are expected to undergo iodination by virtue of electron-rich aromatic rings. These conjugates illustrate a general approach to radiolabeled drug analogs. If a functionalized chain is incorporated in the structure at a position not essential for receptor binding and is systematically extended with a view of maximizing potency, it is then possible to attach prosthetic groups for radiolabeling (24). It is conceivable that prosthetic groups designed to accept specifically other radioisotopes, such as metal chelators, may also be linked to functionalized congeners as receptor probes. Alternately, the peptide carrier may be coupled to a nonradioactive marker, such as colloidal gold or the biotin-avidin complex (25), intended for visualization of receptors.
The fact that high potency was observed for simple amino acid and dipeptide conjugates suggests that monodisperse oligopeptides may be suitable covalent carriers for the xanthines as adenosine receptor antagonists. The choice of tyrosyl and lysyl side chains of 23b demonstrates the versatility of the congener approach in that this analog features a multiply charged group for increased water solubility and a phenol to permit facile radioiodination. The effective permanent charge on the dipeptide is intended to prevent cellular penetration, thereby limiting effects to external receptors and precluding effects on cytosolic enzymes such as phosphodiesterase (26). There is reason to expect that even longer oligopeptides may serve as drug “carriers” for high affinity xanthine analogs.
Linkage of a functionalized drug congener to amino acids or peptides as carriers has several advantages in analog design. The variety of side chains available allows great flexibility in the charge, steric characteristics, hydrophobicity, and functionality of the carrier. A systematic approach as in the present study should lead to the identification of specific carriers that favorably alter the physicochemical or pharmacological properties of a given drug.
ABBREVIATIONS
- Cbz
carbobenzyloxy
- Boc
t-butyloxycarbonyl
- DCC
dicycbhexylcarbodiimide
- DMF
dimethylformamide
- HPLC
high pressure liquid chromatography
References
- 1.Daly JW. Adenosine receptors: target for new drugs. J. Med. Chem. 1982;25:197–207. doi: 10.1021/jm00345a001. [DOI] [PubMed] [Google Scholar]
- 2.Daly JW. Adenosine receptors. Adv. Cycl Nucleotide Protein Phosphorylation Res. 1985;19:29–49. [PubMed] [Google Scholar]
- 3.Fredholm BB. On the mechanism of action of theophylline and caffeine. Acta Med. Scand. 1985;217:149–153. doi: 10.1111/j.0954-6820.1985.tb01650.x. [DOI] [PubMed] [Google Scholar]
- 4.Olsson RA. Structure of the coronary artery adenosine receptor. Trends Pharmacol. Sci. 1984;5:113–116. [Google Scholar]
- 5.Dunnwiddie TV, Fredholm BB. Adenosine receptor mediating inhibitory electrophysical responses in rat hippocampus are different from receptors mediating cyclic AMP accumulation. Naunyn-Schmiedebergs Arch. Pharmacol. 1984;326:294–301. doi: 10.1007/BF00501433. [DOI] [PubMed] [Google Scholar]
- 6.Churchill PC, Churchill MC. A1 and A2 adenosine receptor activation inhibits and stimulates renin secretion of rat renal cortical slices. J. Pharmacol Exp. Ther. 1985;233:589–594. [PubMed] [Google Scholar]
- 7.Bruns RF, Daly JW, Snyder SH. Adenosine receptor binding: structure-activity analysis generates extremely potent xanthine antagonists. Proc. Natl Acad. Sci. USA. 1983;80:2077–2080. doi: 10.1073/pnas.80.7.2077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Daly JW, Padgett W, Shamim MT, Butts-Lamb P, Waters J. 1,3-Dialkyl-8-p-sulfophenylxanthines: potent water-soluble antagonists for Aland A1- and A2-adenosine receptors. J. Med. Chem. 1985;28:487–492. doi: 10.1021/jm00382a018. [DOI] [PubMed] [Google Scholar]
- 9.Jacobson KA, Marr-Leisy D, Rosenkranz RP, Verlander MS, Melmon KL, Goodman M. Conjugates of catecholamines. I. N-Alkyl functionalized carboxylic acid congeners and amides related to isoproterenol. J. Med. Chem. 1983;26:492–499. doi: 10.1021/jm00358a007. [DOI] [PubMed] [Google Scholar]
- 10.Rosenkranz RP, Hoffman BB, Jacobson KA, Verlander MS, Klevans L, O’Donnell M, Goodman M, Melmon KL. Conjugates of catecholamines. II. In vitro and in vivo pharmacological activity of N-alkyl functionalized carboxylic acid congeners and amides related to isoproterenol. Mol. Pharmacol. 1983;24:429–435. [PubMed] [Google Scholar]
- 11.Verlander MS, Jacobson KA, Rosenkranz RP, Melmon KL, Goodman M. Some novel approaches to the design and synthesis of peptidecatecholamine conjugates. Biopolymers. 1983;22:531–545. doi: 10.1002/bip.360220166. [DOI] [PubMed] [Google Scholar]
- 12.Jacobson KA, Rosenkranz RP, Verlander MS, Melmon KL, Goodman M. Conjugates of catecholamines. III. Synthesis and characterization of monodisperse oligopeptide conjugates related to isoproterenol. Int. J. Pept. Protein Res. 1983;22:284–304. doi: 10.1111/j.1399-3011.1983.tb02095.x. [DOI] [PubMed] [Google Scholar]
- 13.Rosenkranz RP, Jacobson KA, Verlander MS, Klevans L, O’Donnel M, Goodman M, Melmon KL. Conjugates of catecholamines. IV. In vivo pharmacological activity of monodisperse oligopeptide conjugates. J. Pharmacol Exp. Ther. 1983;227:267–273. [PubMed] [Google Scholar]
- 14.Jacobson KA, Kirk KL, Padgett WL, Daly JW. Functionalized congeners of adenosine: preparation of analogs with high affinity for A1-adenosine receptors. J. Med. Chem. 1985;28:1341–1346. doi: 10.1021/jm00147a039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jacobson KA, Kirk KL, Padgett WL, Daly JW. Functionalized congeners of 1,3-dialkylxanthines: preparation of analogs with high affinity for adenosine receptors. J. Med. Chem. 1985;28:1334–1340. doi: 10.1021/jm00147a038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Moroder L, Hallett A, Wünsch E, Keller O, Wersin G. Di-tertbutyldicarbonate, a useful tert-butylating reagent. Hoppe-Seyler’s Z. Physiol Chem. 1976;357:1651–1653. [PubMed] [Google Scholar]
- 17.Daly JW, Butts-Lamb P, Padgett W. Subclasses of adenosine receptors in the central nervous system: interaction with caffeine and related methylxanthines. Cell. Mol. Neurobiol. 1983;3:69–80. doi: 10.1007/BF00734999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Schwabe U, Miyake M, Ohga Y, Daly JW. 4-(3-Cyclopentyloxy-4-methoxyphenyl)-2-pyrrolidone (ZK 62711): a potent inhibitor of adenosine cyclic 3′,5′-monophosphate phosphodiesterases in homogenates and tissue slices from rat brain. Mol. Pharmacol. 1976;12:900–910. [PubMed] [Google Scholar]
- 19.Kdnig W, Geiger R. Eine neue Methode zur Synthese von Peptiden: Aktivierung der Carboxylgruppe mit Dicyclohexylcarbodiimide unter Zusatz von 1-Hydroxy-benzotriazolen. Chem. Ber. 1970;103:788–798. doi: 10.1002/cber.19701030319. [DOI] [PubMed] [Google Scholar]
- 20.Bundi A, Grathwohl C, Hochmann J, Keller RM, Wagner G, Wüthrich K. Proton NMR of the protected tetrapeptides TFA-Gly-Gly-L-X-L-Ala-OCH3 where X stands for one of the 20 common amino acids. J. Magn. Reson. 1975;18:191–198. [Google Scholar]
- 21.MacFarlane RD. Californium-252 plasma desorption mass spectrometry. Anal. Chem. 1983;55:1247A–1264A. doi: 10.1021/ac00285a026. [DOI] [PubMed] [Google Scholar]
- 22.Jacobson KA, Kirk KL, Daly JW, Jonzon B, Li Y-O, Fredholm BB. A novel 8-phenyl-substituted xanthine derivative is a selective antagonist at adenosine A1-receptors in vivo. Acta Physiol. Scand. 1985;125:341–342. doi: 10.1111/j.1748-1716.1985.tb07725.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Junek H, Kirk KL, Cohen LA. The oxidative cleavage of tyrosylpeptide bonds during iodination. Biochemistry. 1969;8:1844–1848. doi: 10.1021/bi00833a010. [DOI] [PubMed] [Google Scholar]
- 24.Seevers RH, Counsell RE. Radioiodination techniques for small organic molecules. Chem. Rev. 1982;82:575–690. [Google Scholar]
- 25.Jacobson KA, Kirk KL, Padgett W, Daly JW. Probing the adenosine receptor with adenosine and xanthine biotin conjugates. FEBS Lett. 1985;184:30–35. doi: 10.1016/0014-5793(85)80646-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gustafsson LE. Adenosine antagonism and related effects of theophylline derivatives in guinea pig ileum longitudinal muscle. Acta Physiol Scand. 1984;122:191–198. doi: 10.1111/j.1748-1716.1984.tb07498.x. [DOI] [PubMed] [Google Scholar]