

Abstract
New data are revealing a complex landscape of gene regulation shaped by chromatin states that extend into the bodies of transcribed genes and associate with distinct RNA elements such as exons, introns and polyadenylation sites. Exons are characterized by increased levels of nucleosome positioning, DNA methylation and certain histone modifications. As pre-mRNA splicing occurs co-transcriptionally, changes in the transcription elongation rate or epigenetic marks can influence exon splicing. These new discoveries broaden our understanding of the epigenetic code and ascribe a novel role for chromatin in controlling pre-mRNA processing. In this review, we summarize the recently discovered interplay between the modulation of chromatin states and pre-mRNA processing with the particular focus on how these processes communicate with one another to control gene expression.
CONTROL OF PRE-MRNA SPLICING
Eukaryotic cells generate astonishing protein diversity and as a consequence exceedingly complex phenotypes from a finite set of genes. Alternative pre-mRNA splicing and polyadenylation play essential roles in creating this protein diversity by generating multiple RNA variants from a single gene (see 1,2 for recent reviews). In mammals, greater than 90% of multi-exon genes are alternatively spliced producing, on average, at least seven mRNA variants per gene (3,4).
Recognition of the proper splice sites is critical for pre-mRNA splicing and a key point for the regulation of gene expression (see 5,6 for recent reviews). Splice site recognition is governed by cis-regulatory elements typically located in the exons or their surrounding intronic sequences. These sequence elements are recognized by trans-acting factors which act in concert to recruit the spliceosome to the correct splice sites and block nearby pseudo-splice sites. Short introns of 250 nt or less are typically spliced via an intron definition mechanism that involves splicing factor interactions across the intron (7,8). The average size of mammalian introns, which is significantly larger than the 250 nt limit, impairs the direct formation of such bridging complexes (9,10). Instead, the splice sites of exons surrounded by long introns are recognized by an exon definition complex that forms across the exon and is subsequently converted to a cross-intron complex (11). Consequently, both the formation of the exon definition complex and its conversion to the intron definition complex provide points for the control of alternative splicing. Furthermore, pre-mRNA splicing is carried out co-transcriptionally and the rate of elongation by the RNA polymerase II (RNAPII) has a significant effect on the exon recognition process. Reduced rates of transcriptional elongation and RNAPII pausing near exons significantly increase the efficiency of splice site recognition and the inclusion rate of alternative exons (12,13). The co-transcriptional nature of pre-mRNA splicing and its dependence on the elongation rate of RNAPII place the splicing process in a position where it can be influenced by the structure of the chromatin.
CHROMATIN STATES IN THE BODIES OF TRANSCRIBED GENES
An exciting discovery brought forth by recent epigenome and transcriptome studies is that exons of expressed genes are associated with specific chromatin states. Specifically, exons are characterized by increased levels of nucleosome positioning, DNA methylation and certain histone modifications. Establishment of these exon-specific states of the chromatin structure requires nucleosomes and factors involved in chromatin remodeling, histone modification and DNA methylation to be guided to the exonic sequences. Evidence suggests that such guidance is provided both by intrinsic differences in nucleotide composition between introns and exons and by direct interactions between writers of epigenetic marks and the splicing machinery.
One of the striking findings of several recent genome-wide surveys of the chromatin structure was that nucleosomes are preferentially positioned over exons (14–17). Nucleosome positioning has been studied in detail in the context of transcriptional regulation where it is specified by DNA sequence composition, binding of transcription factors and RNAPII to the DNA and the activity of the chromatin remodeling machinery (see 18 for a recent review). Presumably, the same general mechanisms dictate the distribution of nucleosomes in the bodies of genes as well. Evidence suggests that the preference for nucleosome positioning over exons is at least in part controlled by nucleotide sequence composition (Fig. 1). Across eukaryotes, exon sequences tend to have elevated GC content compared with flanking introns (19–21). In mammals, this trend is preserved in exons that are surrounded by long introns and thus rely on exon definition rather than intron definition for their splicing (19). Sequences with moderate GC content are preferred binding sites for nucleosomes, due to their increased flexibility compared with long homopolymeric stretches (22–25). Exons are also depleted of poly-dA:dT stretches of five or more nucleotides compared with introns, creating a sharp transition in the frequency of these homopolymers at the intron/exon border (26,16). The rigid structure of poly-dA:dT DNA destabilizes the nucleosome–DNA interactions and forms nucleosome-free regions in the chromatin (27–30). Taken together, the general characteristics of mammalian spliced exons, i.e. relatively GC-rich sequences of ∼145–170 nt length (9,10) surrounded by AT-rich intronic sequences (Fig. 1A), bear striking similarity to the strong nucleosome-positioning signals termed ‘container sites’ by Valouev et al. (24). These ‘container sites’ consist of ∼150 nt long GC-rich sequence surrounded by AT-rich flanks (Fig. 1B). Nucleosomes bind to the cores of the ‘container sites’ are prevented from sliding out of position by the surrounding low-affinity sequences. In summary, these studies indicate that the nucleotide composition of exons and the surrounding introns favor nucleosome positioning over the exons. It is also possible that direct physical interactions between the splicing and chromatin remodeling machineries can contribute to nucleosome positioning over exons (14–17), although further studies are needed to obtain direct evidence.
Figure 1.
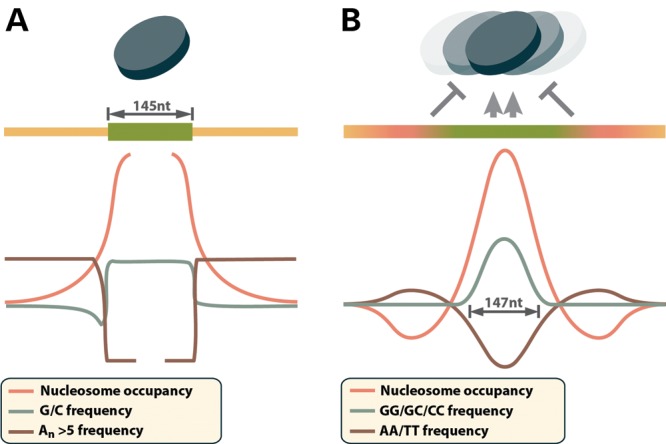
Nucleotide composition of exons and nucleosome ‘container sites’. (A) Exons contain elevated GC content relative to introns (19), which favors nucleosome binding (44), while nearby intron sequences are enriched in nucleosome repellant poly-dA:dT stretches (26). This sequence arrangement mirrors the recently identified strong nucleosome-positioning signals termed ‘container sites’ (B), which consist of relatively G/C-rich nucleosome binding core, surrounded by nucleosome repellant A/T-rich sequence that prevents nucleosomes from sliding out of position (24).
Genome-wide surveys reveal organized deposition of various histone modifications across the bodies of transcribed genes (Box 1). In particular, nucleosomes positioned over exons are found to be enriched in specific histone modifications (Box 1) that include H3K36me3 and H3K79me1 (14,15,31,32). Of these modifications, H3K36me3 is strongly associated with exonic sequences and correlates with the level of gene expression and the rate of exon inclusion during alternative splicing (14). The mechanisms by which these marks are established in the gene bodies have been summarized by a number of recent reviews and typically involve the recruitment of the histone modifying factors by the elongating RNAPII (33–36). In particular, the Kmt2a (Set1/MLL/Trx), Kmt3a (Set2/Setd2/HYPB) and Kmt4 (Dot1/Dot1L) methyltransferases responsible for the methylation of histone H3 tails at lysines 4, 36 and 79, respectively, are part of the elongating RNAPII complex. However, the recruitment of these factors by RNAPII alone does not explain the preferential enrichment of the respective marks in exons. An explanation of the observed enrichment of H3K36 and H3K79 methylation over exons is provided by the discovery of a physical association between components of the splicing machinery and histone methyltransferases. Three abundant nuclear proteins involved in pre-mRNA processing and transport, hnRNP-M, hnRNP-U and Aly/Ref1 have been found in complex with Kmt4 (37). Another ubiquitous splicing factor, hnRNP-L, together with Aly/Ref1 is in complex with Kmt3a (38,39). Strikingly, H3K36 trimethylation by Kmt3a in vivo depends to a significant degree on pre-mRNA splicing. Inhibition of splicing by splice site mutagenesis, pharmacological agents or depletion of Sf3b3 (SAP 130), a component of the core spliceosome, decrease Kmt3a recruitment to the chromatin and H3K36me3 levels (38,40,41). In agreement with a mechanism that involves the recruitment of Kmt3a by the spliceosome (Fig. 2), alternative exons in general have lower levels of H3K36me3 than constitutive exons (14,32). However, H3K36me3 deposition is not solely dependent on splicing and the H3K36me3 state over alternative exons may show no or even inverse correlation with the exon inclusion rate (40,42,43). Other histone marks including mono-methylation of H3K27 and H2BK5 follow a pattern of enrichment over exons similar to that of H3K36me3, raising the possibility that their deposition is coordinated by the splicing machinery in a manner similar to H3K36me3 (32,44). In addition to the histone marks generally associated with spliced exons, a subset of exons is marked by H3K9me3, a modification typically associated with heterochromatin (45,46). Further studies are needed to determine the mechanisms guiding the deposition of these marks over exons in actively transcribed genes.
Box 1. Histone modifications enriched in the bodies of transcribed genes. The list is compiled from references (15,31,32,44).
Transcribed region 5′ proximal to the promoter: H2BK5me1, H3K4me1, H3K4me2, H3K9me1, H3K27me1, H3K79me1, H3K79me2, H3K79me3 and H4K20me1.
Exons: H2BK5me1, H3K4me1, H3K4me2, H3K9me1, H3K9me2, H3K9me3*, H3K27me1, H3K27me2, H3K27me3, H3K36me3, H3K79me1, H3R2me1, H3R2me2, H4K20me1, H4K20me3 and H4R3me2.
Introns: H3K36me1**.
*Enrichment is present only over a subset of exons.
**Intron enrichment is observed only in highly expressed genes.
Figure 2.
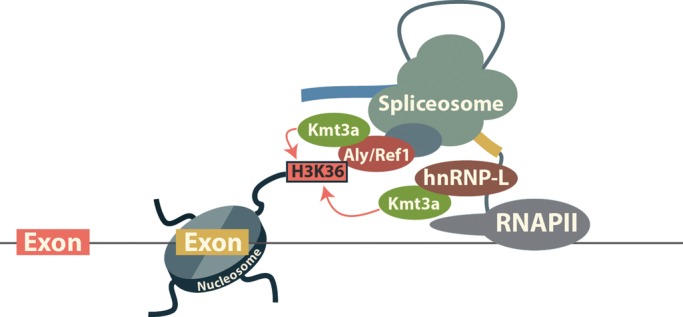
Deposition of H3K36me3 over exons. hnRNP-L directly interacts with RNA elements that place it in close proximity to the exons. Aly/Ref1 is recruited to the pre-mRNA during spliceosome assembly (76). In turn, hnRNP-L and Aly/Ref1 cooperate with RNAPII to bring Kmt3a to the exons where it methylates H3K36.
Methylated cytosine residues in CpG dinucleotides are another epigenetic mark enriched in the exons of transcribed genes (47). The Dnmt3a and Dnmt3b methyltransferases are responsible for the de novo methylation of DNA in mammalian cells and their specificity is determined by interactions with regulatory factors. The two methyltransferases are in a complex that can be recruited to DNA via direct interaction of Dnmt3a with the exon-specific H3K36me3 mark (48).
Transcription termination and polyadenylation sites (PASs) also display distinct nucleosome occupancy patterns. Specifically, genomic regions surrounding PAS are strongly depleted of nucleosomes, while the regions immediately downstream of PAS are enriched in nucleosomes (15). Furthermore, in genes undergoing alternative polyadenylation, the usage of individual PAS is correlated with the nucleosome occupancy downstream of the PAS, suggesting a role of the chromatin structure in the regulation of polyadenylation.
SPLICING IN VIVO IS CARRIED OUT on the chromatin
The physiological substrate for most pre-mRNA processing steps is the nascent transcript emerging from RNAPII. Evidence for the concurrency of transcription and splicing in eukaryotes has long been observed in electron micrographs of Drosophila embryo genes and by reverse transcriptase–polymerase chain reaction on dissected polytene chromosomes (49,50). More recently, the introns of the FN1 and SRC genes were shown to be excised, while the nascent RNA is still attached to the chromatin and only fully spliced transcripts are released into the nucleoplasm (51). The co-transcriptional nature of pre-mRNA splicing was also elegantly illustrated by recent imaging experiments that track the kinetics of transcription and splicing of individual genes in vivo (52). Further evidence for the predominance of co-transcriptional splicing comes from high-throughput RNA sequencing of nascent transcripts, which reveals a pattern of high RNA-Seq signal at the 5′ end of each intron that decreases toward the 3′ end (53,54). This ‘saw-tooth’ pattern is produced by successive rounds of transcription entering the 5′ end of the intron, combined with excision and release of the intron from the nascent transcript shortly after its synthesis is completed (Fig. 3A). It should be noted that a significant proportion of introns are not excised co-transcriptionally and are still present after transcription termination and cleavage of the transcript at the polyadenylation site (53,55). Both imaging and high-throughput RNA sequencing studies show that such fully transcribed but incompletely spliced pre-mRNAs are retained on the chromatin until the introns are excised (52,55).
Figure 3.
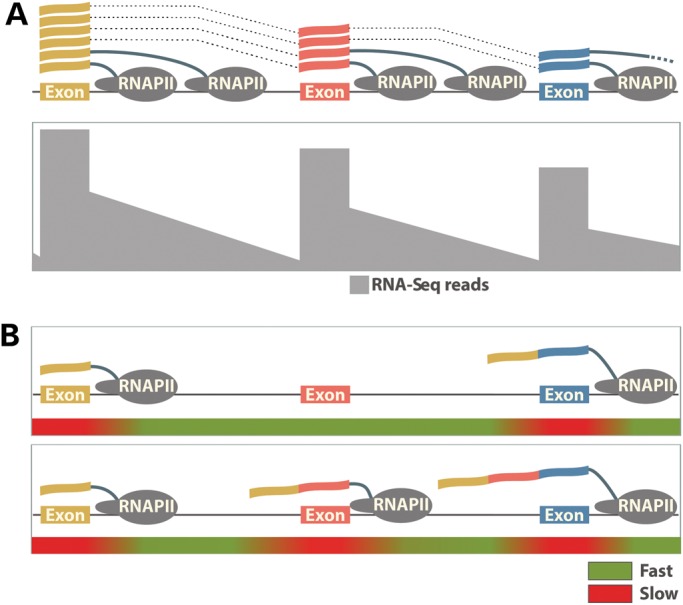
Co-transcriptional splicing and kinetic control of alternative splicing by the elongating RNAPII. (A) The genomic region of a hypothetical gene is illustrated containing three exons. Along the gene body, the frequency of RNA-Seq reads from a cell type where total RNA has been harvested is depicted. A characteristic ‘saw-tooth’ pattern is formed by the frequency of RNA-Seq reads across the genomic region. Exonic reads are most prevalent due to the amount of both nascent and partially processed RNAs containing these sequences. Intronic reads are also present albeit at lower levels representing unprocessed RNA species. The diminished frequency of reads toward the 3′ end of introns arises from rapid co-transcriptional splicing that occurs in this region after RNAPII finishes transcribing each intron. (B) The RNAPII elongation rate controls the exon inclusion. Rapid elongation through an alternative exon defined by weak splicing signals does not allow enough time for exon recognition before the competing strong splicing signals of the downstream exon are presented (top). As a result, the alternative exon is skipped. Slowing down RNAPII within and downstream of the alternative exon provides time for the splicing machinery to recognize and splice the weak exon in the mature transcript (bottom).
The close association between chromatin and splicing leads to a significant cross-talk in which pre-mRNA splicing influences the chromatin state, while the chromatin state facilitates pre-mRNA splicing and controls the rate of alternative exon inclusion. Two models explaining how pre-mRNA splicing is controlled by the chromatin state have emerged from recent studies. In the first, termed the ‘kinetic’ model, the chromatin state controls exon inclusion by modulating the rate of transcription elongation (Fig. 3B). In the second, the ‘recruitment’ model, histone modifications associated with the exons in cooperation with the C-terminal domain of RNAPII function as a ‘landing pad’ to load the spliceosome components necessary for splice site recognition onto the emerging nascent transcript (Fig. 4). The two models are not mutually exclusive and both are supported by ample evidence; however, the relative contribution of each mechanism remains unresolved.
Figure 4.
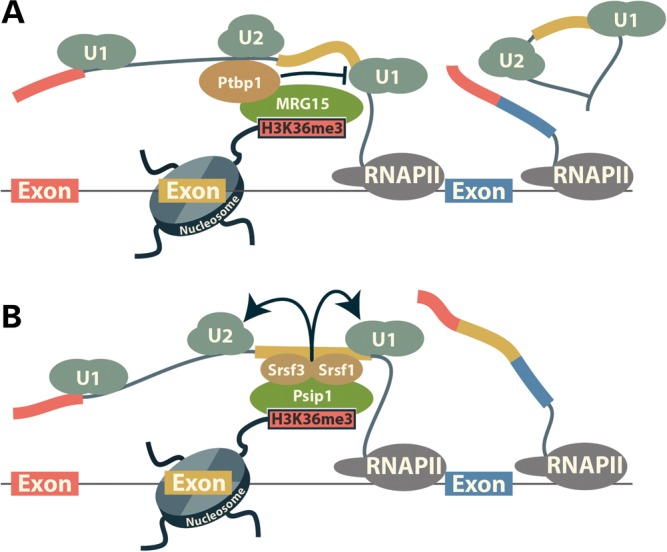
Recruitment model for the control of splicing by H3K36me3. The H3K36me3 mark is read by MRG15 (A) or Psip1 (B) protein. In turn, MRG15 and Psip1 recruit Ptbp1 and SR protein family members (Srsf1 and Srsf3), respectively. Ptbp1 represses splicing when bound to the alternative exons or the upstream of them by preventing the conversion of exon definition to an intron definition complex (77). Srsf1 and Srsf3 bind to cis-elements in the exons and promote exon recognition and splicing by recruiting the U1 and U2 snRNPs (6).
KINETIC CONTROL OF ALTERNATIVE SPLICING BY THE CHROMATIN STATE
Evidence that the efficiency of exon recognition is dependent on the rate of transcription was first provided by experiments showing that the inclusion of alternative exons is favored in cells expressing a slow mutant of RNAPII or by transcription factors reducing the RNAPII elongation rate (12,13,54). Slowing down transcription allows more time for the spliceosome to recognize exons with poor splicing signals before competing downstream exons are presented to the splicing machinery (Fig. 3B). Consistent with this model, Pandya-Jones and Black (51) show that the excision of introns surrounding certain alternative exons is significantly slower when compared with neighboring introns that bridge constitutive exons. Elevated levels of histone acetylation which facilitate elongation through the nucleosome result in increased skipping of such alternative exons (56–58). Similarly, in yeast RNAPII pausing in the terminal exon is required for high-efficiency co-transcriptional splicing of intron containing genes (59).
Recent studies have described diverse mechanisms by which chromatin states control the RNAPII elongation rate in the vicinity of alternative exons. Preferential nucleosome positioning over exons provides one way to assist both constitutive and alternative splicing by modulating the elongation rate of RNAPII. Such positioned nucleosomes form a barrier to transcription at the exons that provides time for the splicing machinery to assemble on the emerging transcript. H3K36me3 further contributes to slowing down the RNAPII by recruiting HDAC1 which removes histone acetylation and maintains a repressive chromatin state (60). Another mechanism for kinetic control of splicing involves H3K9me3 marks that are deposited specifically over a subset of alternative exons (45). These H3K9me3 marks recruit the heterochromatin protein Cbx3 (HP1γ), which in turn reduces the RNAPII elongation rate. In addition to controlling alternative splicing, the same Cbx3-dependent mechanism is also critical for constitutive splicing of a large number of transcripts, whose genes are marked by H3K9me3 (46). The control of the elongation rate and splicing is a new role for H3K9me3, which has been studied primarily in relation to establishing heterochromatin by the recruitment of HP1 family members (see 61 for a recent review). This new role of H3K9me3 is specifically dependent on Cbx3 as the knockdown of the other members of the HP1 family, Cbx5 and Cbx1 (HP1α and HP1β), has no effect on splicing. Finally, chromatin states modulate RNAPII elongation and pre-mRNA splicing by controlling binding of the insulator protein CTCF to the bodies of genes. CTCF reduces the transcription elongation rate thereby increasing the inclusion rate of alternative exons when the CTCF-binding sites are located in downstream introns (62). The effect of CTCF on splicing is dependent on the methylation status of its binding sites as CTCF cannot bind to methylated DNA. Another DNA-binding protein, Vezf1, is also capable of modulating alternative splicing using the same kinetic control mechanism as CTCF (63). However, it is still unclear if the binding of Vezf1 to DNA is controlled by epigenetic marks.
While the kinetic competition between transcription and splicing emerges as an important control point for exon inclusion, it only affects a subset of exons. Most introns, including many surrounding alternative exons are excised shortly after being transcribed and thus are not affected significantly by altered elongation rates (51,53).
RECRUITMENT OF SPLICING FACTORS AND SPLICEOSOME COMPONENTS BY EPIGENETIC MARKS
In addition to modulating RNAPII elongation rates, chromatin states can affect pre-mRNA splicing by directly recruiting splicing factors to the nascent pre-mRNA. Early work showed the recruitment of the U2 snRNP to the H3K4me3 mark by a direct interaction between the H3K4me3-binding protein Chd1 and the U2 snRNP component Sf3a1 (64). Chd1 knockdown impairs the removal of intron 3 of the IRF3 gene, providing evidence that the recruitment of the U2 snRNP by H3K4me3 is required for splicing in vivo. The H3K4me3 mark is deposited primarily over the promoter and 5′-transcribed regions of genes; thus, it will mostly affect the splicing of the first intron. In the case of IRF3, due to the relatively short 5′ introns, the peak of H3K4me3 extends to intron 3, explaining the effect on intron 3 excision by Chd1 knockdown. The role of Chd1 in splicing is a significant expansion of its function as a chromatin remodeling factor involved in transcription elongation and maintenance of open chromatin (35,65).
Unlike H3K4me3, the H3K36me3 mark is associated with exons in the gene bodies and emerges as a major recruitment center for spliceosome components (Fig. 4). The MRG15 (Morf4l1) and Psip1 proteins that ‘read’ the H3K36me3 mark by binding to it have been implicated in controlling pre-mRNA splicing by recruiting splicing factors to the chromatin. MRG15 recognizes H3K36me3 via its chromodomain and controls the splicing of a number of exons by bringing the Ptbp1 splicing factor to the nascent transcripts (66,67) (Fig. 4A). Psip1, a protein previously implicated in the control of pre-mRNA splicing, was shown to interact directly with H3K36me3 (68). In immunoprecipitation experiments, Psip1 associates with a large number of factors involved in pre-mRNA splicing that include SR and hnRNP proteins, DEAD-box helicases and core spliceosome components (68). While most of these interactions are unlikely to be direct, Psip1 does bind two members of the SR protein family, Srsf1 (SF2/ASF) and Srsf3 (SRp20), and is required for the recruitment of Srsf1 to chromatin (Fig. 4B). Deletion of Psip1 alters the rate of inclusion of several exons. However, the number of exons affected by the loss of Psip1 is relatively small, considering its association with major splicing factors like SR family members and snRNP components (68). Thus, rather than being critical for splicing Psip1 may function as a facilitator that brings splicing factors to nascent transcripts.
An intriguing consequence of the involvement of H3K36me3 in pre-mRNA splicing is that it completes a regulatory circuit, where pre-mRNA splicing stimulates H3K36me3 over the exons, which in turn promotes exon recognition by recruiting pre-mRNA splicing factors. Such self-perpetuating loops are a hallmark of epigenetic inheritance that is required for maintaining phenotypes across consecutive cell division. Here, the loop centered on the H3K36me3 mark likely functions to increase splicing efficiency and reinforce successful splice site choice made in previous rounds of transcription. It still needs to be investigated if other epigenetic marks associated with exons fulfill functions in splicing similar to those of H3K4me3 and H3K36me3. In particular, while the H3K9me3 histone mark is known to recruit splicing factors of the hnRNP family (69), it is unclear if this recruitment occurs on transcribed genes or on the heterochromatin and whether its function is to control pre-mRNA splicing.
FUTURE PERSPECTIVES
Discovery of the ubiquitous cross-talk between the cellular machineries involved in transcription and RNA processing has greatly expanded our understanding of eukaryotic gene expression. Recent studies reveal a prominent role of chromatin structure and epigenetic marks in coordinating these processes and specifying exon utilization. These new data are fundamentally advancing our understanding of the ‘splicing code’, i.e. mechanistic models that explain exon splicing patterns based on genomic and RNA sequence features. Historically, studies of splicing regulation focus on cis-regulatory RNA elements and their interactions with trans-acting splicing factors (5,6). Computational models built on RNA sequence features have been developed to identify constitutive and alternative exons and to predict tissue-specific alternative splicing (70–73). A recent landmark study by Barash et al. (70) developed a ‘splicing code’ with ∼200 RNA features to predict tissue-specific exons in the mouse genome. Such predictive models constructed from RNA sequence features only partially account for the splicing patterns of exons, suggesting that there is a significant involvement of factors that act independently of the RNA sequence to control pre-mRNA splicing. The newly established connection between chromatin and splicing, therefore, provides a critical missing layer in our understanding of splicing regulation. Future models of the ‘splicing code’ that incorporate chromatin states and epigenetic features may substantially improve our ability to predict splicing patterns in normal and diseased states. It should also be noted that the relative contributions of chromatin states and canonical splicing regulators may vary between exons and cell states. For example, the recruitment of Ptbp1 to H3K36me3 by MRG15 has a measurable contribution to the repression of the FGFR2 exon IIIb in mesenchymal cells (66). Nonetheless, the dramatic difference in the splicing levels of this exon between epithelial and mesenchymal cells is predominantly determined by the canonical splicing regulators ESRP1 and ESRP2, epithelial-specific splicing factors that maintain a genome-wide epithelial alternative splicing program (74).
An important question is whether and how individual epigenetic marks selectively regulate the splicing of exon targets. A mechanism for such selective regulation becomes evident from studies on the H3K36me3 mark. This mechanism involves both cooperative and competitive interactions between epigenetic mark readers, splicing factors recruited by these readers and cis-RNA elements recognized by the recruited splicing factors. H3K36me3 can repress the inclusion of a subset of exons by recruiting Ptbp1 via its interaction with MRG15 (66). At the same time, the splicing of other exons is facilitated by the recruitment of Srsf1 and Srsf3 to H3K36me3 by a different reader, Psip1 (68). In another example, DNA methylation and CTCF affect exon 5 of CD45, but not the alternatively spliced exons 4 and 6 (62). It must be noted that the enrichment of specific chromatin states and epigenetic marks in exons reflects the aggregated effects across the entire genome and does not apply equally to all exons. Furthermore, epigenetic modifications specific to a subset of exons may not be evident in the aggregate genomic data. Such is the case of H3K9me3 which is deposited over the exons of certain transcribed genes or over specific exons within genes that otherwise lack this mark (45,46). It is also possible that combinatorial deposition of several epigenetic modifications produces a series of unique scaffolds for splicing factor recruitment. In the future, large-scale studies that concurrently measure epigenome and transcriptome profiles across divergent cell types or cellular states may allow us to establish a robust association between chromatin states and splicing profiles at the individual exon level.
Finally, epigenetic regulation of gene expression is a major mechanism involved in organism development, cellular response to external stimuli or environmental perturbation and human pathology (75). While changes in epigenetic marks and their readers have a major impact on gene transcription, it is reasonable to expect that they will also impact splicing of select exons, which contribute to the cellular phenotypes or disease pathogenesis. For example, epigenetic perturbation is an integral part of oncogenesis. Future work should determine the extent to which aberrant epigenetic regulation underlies widespread dysregulation of alternative splicing in cancer cells.
FUNDING
Y.X. is supported by National Institutes of Health (R01GM088342) and a junior faculty grant from the Edward Mallinckrodt Jr Foundation. S.J.B. is supported by National Institutes of Health postdoctoral fellow training grant (T32HL007638).
ACKNOWLEDGEMENTS
We are grateful to Drs Lisa Salati, Russ Carstens, Yang Shi and Alexey Ivanov for critical reading of the review.
Conflict of Interest statement. None declared.
REFERENCES
- 1.Nilsen T.W., Graveley B.R. Expansion of the eukaryotic proteome by alternative splicing. Nature. 2010;463:457–463. doi: 10.1038/nature08909. doi:10.1038/nature08909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Di Giammartino D.C., Nishida K., Manley J.L. Mechanisms and consequences of alternative polyadenylation. Mol. Cell. 2011;43:853–866. doi: 10.1016/j.molcel.2011.08.017. doi:10.1016/j.molcel.2011.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wang E.T., Sandberg R., Luo S., Khrebtukova I., Zhang L., Mayr C., Kingsmore S.F., Schroth G.P., BURGE C.B. Alternative isoform regulation in human tissue transcriptomes. Nature. 2008;456:470–476. doi: 10.1038/nature07509. doi:10.1038/nature07509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pan Q., Shai O., Lee L.J., Frey B.J., Blencowe B.J. Deep surveying of alternative splicing complexity in the human transcriptome by high-throughput sequencing. Nat. Genet. 2008;40:1413–1415. doi: 10.1038/ng.259. doi:10.1038/ng.259. [DOI] [PubMed] [Google Scholar]
- 5.Hertel K.J. Combinatorial control of exon recognition. J. Biol. Chem. 2008;283:1211–1215. doi: 10.1074/jbc.R700035200. doi:10.1074/jbc.R700035200. [DOI] [PubMed] [Google Scholar]
- 6.Wang Z., Burge C.B. Splicing regulation: from a parts list of regulatory elements to an integrated splicing code. RNA. 2008;14:802–813. doi: 10.1261/rna.876308. doi:10.1261/rna.876308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fox-Walsh K.L., Dou Y., Lam B.J., Hung S., Baldi P.F., Hertel K.J. The architecture of pre-mRNAs affects mechanisms of splice-site pairing. Proc. Natl Acad. Sci. USA. 2005;102:16176–16181. doi: 10.1073/pnas.0508489102. doi:10.1073/pnas.0508489102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sterner D.A., Carlo T., Berget S.M. Architectural limits on split genes. Proc. Natl Acad. Sci. USA. 1996;93:15081. doi: 10.1073/pnas.93.26.15081. doi:10.1073/pnas.93.26.15081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Koralewski T.E., Krutovsky K.V. Evolution of exon-intron structure and alternative splicing. PLoS One. 2011;6:e18055. doi: 10.1371/journal.pone.0018055. doi:10.1371/journal.pone.0018055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lander E.S., Linton L.M., Birren B., Nusbaum C., Zody M.C., Baldwin J., Devon K., Dewar K., Doyle M., FitzHugh W., et al. Initial sequencing and analysis of the human genome. Nature. 2001;409: 860–921. doi: 10.1038/35057062. doi:10.1038/35057062. [DOI] [PubMed] [Google Scholar]
- 11.Schneider M., Will C.L., Anokhina M., Tazi J., Urlaub H., Lührmann R. Exon definition complexes contain the Tri-snRNP and can be directly converted into B-like precatalytic splicing complexes. Mol. Cell. 2010;38:223–235. doi: 10.1016/j.molcel.2010.02.027. [DOI] [PubMed] [Google Scholar]
- 12.Kadener S., Fededa J.P., Rosbash M., Kornblihtt A.R. Regulation of alternative splicing by a transcriptional enhancer through RNA Pol II elongation. Proc. Natl Acad. Sci. USA. 2002;99:8185–8190. doi: 10.1073/pnas.122246099. doi:10.1073/pnas.122246099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.de la Mata M., Alonso C.R., Kadener S., Fededa J.P., Blaustein M., Pelisch F., Cramer P., Bentley D., Kornblihtt A.R. A slow RNA polymerase II affects alternative splicing in vivo. Mol. Cell. 2003;12:525–532. doi: 10.1016/j.molcel.2003.08.001. doi:10.1016/j.molcel.2003.08.001. [DOI] [PubMed] [Google Scholar]
- 14.Hon G., Wang W., Ren B. Discovery and annotation of functional chromatin signatures in the human genome. PLoS Comput. Biol. 2009;5:e1000566. doi: 10.1371/journal.pcbi.1000566. doi:10.1371/journal.pcbi.1000566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Spies N., Nielsen C.B., Padgett R.A., Burge C.B. Biased chromatin signatures around polyadenylation sites and exons. Mol. Cell. 2009;36:245–254. doi: 10.1016/j.molcel.2009.10.008. doi:10.1016/j.molcel.2009.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Schwartz S., Meshorer E., Ast G. Chromatin organization marks exon-intron structure. Nat. Struct. Mol. Biol. 2009;16:990–995. doi: 10.1038/nsmb.1659. doi:10.1038/nsmb.1659. [DOI] [PubMed] [Google Scholar]
- 17.Tilgner H., Nikolaou C., Althammer S., Sammeth M., Beato M., Valcárcel J., Guigó R. Nucleosome positioning as a determinant of exon recognition. Nat. Struct. Mol. Biol. 2009;16:996–1001. doi: 10.1038/nsmb.1658. doi:10.1038/nsmb.1658. [DOI] [PubMed] [Google Scholar]
- 18.Iyer V.R. Nucleosome positioning: bringing order to the eukaryotic genome. Trends Cell Biol. 2012;22:250–256. doi: 10.1016/j.tcb.2012.02.004. doi:10.1016/j.tcb.2012.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Amit M., Donyo M., Hollander D., Goren A., Kim E., Gelfman S., Lev-Maor G., Burstein D., Schwartz S., Postolsky B., et al. Differential GC content between exons and introns establishes distinct strategies of splice-site recognition. Cell Rep. 2012;1:543–556. doi: 10.1016/j.celrep.2012.03.013. doi:10.1016/j.celrep.2012.03.013. [DOI] [PubMed] [Google Scholar]
- 20.Zhu L., Zhang Y., Zhang W., Yang S., Chen J.-Q., Tian D. Patterns of exon-intron architecture variation of genes in eukaryotic genomes. BMC Genomics. 2009;10:47. doi: 10.1186/1471-2164-10-47. doi:10.1186/1471-2164-10-47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhang M. Statistical features of human exons and their flanking regions. Hum. Mol. Genet. 1998;7:919–932. doi: 10.1093/hmg/7.5.919. doi:10.1093/hmg/7.5.919. [DOI] [PubMed] [Google Scholar]
- 22.Tillo D., Hughes T.R. G+C content dominates intrinsic nucleosome occupancy. BMC Bioinformatics. 2009;10:442. doi: 10.1186/1471-2105-10-442. doi:10.1186/1471-2105-10-442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kaplan N., Moore I.K., Fondufe-Mittendorf Y., Gossett A.J., Tillo D., Field Y., LeProust E.M., Hughes T.R., Lieb J.D., Widom J., et al. The DNA-encoded nucleosome organization of a eukaryotic genome. Nature. 2008;458:362–366. doi: 10.1038/nature07667. doi:10.1038/nature07667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Valouev A., Johnson S.M., Boyd S.D., Smith C.L., Fire A.Z., Sidow A. Determinants of nucleosome organization in primary human cells. Nature. 2011;474:516–520. doi: 10.1038/nature10002. doi:10.1038/nature10002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Geggier S., Vologodskii A. Sequence dependence of DNA bending rigidity. Proc. Natl Acad. Sci. USA. 2010;107:15421–15426. doi: 10.1073/pnas.1004809107. doi:10.1073/pnas.1004809107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cohanim A.B., Haran T.E. The coexistence of the nucleosome positioning code with the genetic code on eukaryotic genomes. Nucleic Acids Res. 2009;37:6466–6476. doi: 10.1093/nar/gkp689. doi:10.1093/nar/gkp689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Suter B., Schnappauf G., Thoma F. Poly(dA·dT) sequences exist as rigid DNA structures in nucleosome-free yeast promoters in vivo. Nucleic Acids Res. 2000;28:4083. doi: 10.1093/nar/28.21.4083. doi:10.1093/nar/28.21.4083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bao Y., White C.L., Luger K. Nucleosome core particles containing a poly(dA·dT) sequence element exhibit a locally distorted DNA structure. J. Mol. Biol. 2006;361:617–624. doi: 10.1016/j.jmb.2006.06.051. doi:10.1016/j.jmb.2006.06.051. [DOI] [PubMed] [Google Scholar]
- 29.Iyer V., Struhl K. Poly(dA:dT), a ubiquitous promoter element that stimulates transcription via its intrinsic DNA structure. EMBO J. 1995;14:2570. doi: 10.1002/j.1460-2075.1995.tb07255.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Field Y., Kaplan N., Fondufe-Mittendorf Y., Moore I.K., Sharon E., Lubling Y., Widom J., Segal E. Distinct modes of regulation by chromatin encoded through nucleosome positioning signals. PLoS Comput. Biol. 2008;4:e1000216. doi: 10.1371/journal.pcbi.1000216. doi:10.1371/journal.pcbi.1000216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ernst J., Kellis M. Discovery and characterization of chromatin states for systematic annotation of the human genome. Nat. Biotechnol. 2010;28:817–825. doi: 10.1038/nbt.1662. doi:10.1038/nbt.1662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dhami P., Saffrey P., Bruce A.W., Dillon S.C., Chiang K., Bonhoure N., Koch C.M., Bye J., James K., Foad N.S., et al. Complex exon-intron marking by histone modifications is not determined solely by nucleosome distribution. PLoS One. 2010;5:e12339. doi: 10.1371/journal.pone.0012339. doi:10.1371/journal.pone.0012339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Shilatifard A. The COMPASS family of histone H3K4 methylases: mechanisms of regulation in development and disease pathogenesis. Annu. Rev. Biochem. 2012;81:65–95. doi: 10.1146/annurev-biochem-051710-134100. doi:10.1146/annurev-biochem-051710-134100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Greer E.L., Shi Y. Histone methylation: a dynamic mark in health, disease and inheritance. Nat. Rev. Genet. 2012;13:343–357. doi: 10.1038/nrg3173. doi:10.1038/nrg3173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Selth L.A., Sigurdsson S., Svejstrup J.Q. Transcript elongation by RNA polymerase II. Annu. Rev. Biochem. 2010;79:271–293. doi: 10.1146/annurev.biochem.78.062807.091425. doi:10.1146/annurev.biochem.78.062807.091425. [DOI] [PubMed] [Google Scholar]
- 36.Weake V.M., Workman J.L. Histone ubiquitination: triggering gene activity. Mol. Cell. 2008;29:653–663. doi: 10.1016/j.molcel.2008.02.014. doi:10.1016/j.molcel.2008.02.014. [DOI] [PubMed] [Google Scholar]
- 37.Park G., Gong Z., Chen J., Kim J.-E. Characterization of the DOT1L network: implications of diverse roles for DOT1L. Protein J. 2010;29:213–223. doi: 10.1007/s10930-010-9242-8. doi:10.1007/s10930-010-9242-8. [DOI] [PubMed] [Google Scholar]
- 38.Yuan W., Xie J., Long C., Erdjument-Bromage H., Ding X., Zeng Y., Tempst P., Chen S., Zhu B., Reinberg D. Heterogeneous nuclear ribonucleoprotein L is a subunit of human KMT3a/Set2 complex required for H3 Lys-36 trimethylation activity in vivo. J. Biol. Chem. 2009;284:15701–15707. doi: 10.1074/jbc.M808431200. doi:10.1074/jbc.M808431200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yoh S.M., Lucas J.S., Jones K.A. The Iws1:Spt6:CTD complex controls cotranscriptional mRNA biosynthesis and HYPB/Setd2-mediated histone H3K36 methylation. Genes Dev. 2008;22:3422–3434. doi: 10.1101/gad.1720008. doi:10.1101/gad.1720008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.de Almeida S.F., Grosso A.R., Koch F., Fenouil R., Carvalho S., Andrade J., Levezinho H., Gut M., Eick D., Gut I., et al. Splicing enhances recruitment of methyltransferase HYPB/Setd2 and methylation of histone H3 Lys36. Nat. Struct. Mol. Biol. 2011;18:977–983. doi: 10.1038/nsmb.2123. doi:10.1038/nsmb.2123. [DOI] [PubMed] [Google Scholar]
- 41.Kim S., Kim H., Fong N., Erickson B., Bentley D.L. Pre-mRNA splicing is a determinant of histone H3K36 methylation. Proc. Natl Acad. Sci. USA. 2011;108:13564–13569. doi: 10.1073/pnas.1109475108. doi:10.1073/pnas.1109475108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Schor I.E., Rascovan N., Pelisch F., Alló M., Kornblihtt A.R. Neuronal cell depolarization induces intragenic chromatin modifications affecting NCAM alternative splicing. Proc. Natl Acad. Sci. USA. 2009;106:4325–4330. doi: 10.1073/pnas.0810666106. doi:10.1073/pnas.0810666106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Huff J.T., Plocik A.M., Guthrie C., Yamamoto K.R. Reciprocal intronic and exonic histone modification regions in humans. Nat. Struct. Mol. Biol. 2010;17:1495–1499. doi: 10.1038/nsmb.1924. doi:10.1038/nsmb.1924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Andersson R., Enroth S., Rada-Iglesias A., Wadelius C., Komorowski J. Nucleosomes are well positioned in exons and carry characteristic histone modifications. Genome Res. 2009;19:1732–1741. doi: 10.1101/gr.092353.109. doi:10.1101/gr.092353.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Saint-André V., Batsché E., Rachez C., Muchardt C. Histone H3 lysine 9 trimethylation and HP1γ favor inclusion of alternative exons. Nat. Struct. Mol. Biol. 2011;18:337–344. doi: 10.1038/nsmb.1995. doi:10.1038/nsmb.1995. [DOI] [PubMed] [Google Scholar]
- 46.Smallwood A., Hon G.C., Jin F., Henry R.E., Espinosa J.M., Ren B. CBX3 regulates efficient RNA processing genome-wide. Genome Res. 2012 doi: 10.1101/gr.124818.111. doi:10.1101/gr.124818.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Choi J.K. Contrasting chromatin organization of CpG islands and exons in the human genome. Genome Biol. 2010;11:R70. doi: 10.1186/gb-2010-11-7-r70. doi:10.1186/gb-2010-11-7-r70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Dhayalan A., Rajavelu A., Rathert P., Tamas R., Jurkowska R.Z., Ragozin S., Jeltsch A. The Dnmt3a PWWP domain reads histone 3 lysine 36 trimethylation and guides DNA methylation. J. Biol. Chem. 2010;285:26114–26120. doi: 10.1074/jbc.M109.089433. doi:10.1074/jbc.M109.089433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Beyer A.L., Osheim Y.N. Splice site selection, rate of splicing, and alternative splicing on nascent transcripts. Genes Dev. 1988;2:754–765. doi: 10.1101/gad.2.6.754. doi:10.1101/gad.2.6.754. [DOI] [PubMed] [Google Scholar]
- 50.Wetterberg I., Baurén G., Wieslander L. The intranuclear site of excision of each intron in Balbiani ring 3 pre-mRNA is influenced by the time remaining to transcription termination and different excision efficiencies for the various introns. RNA. 1996;2:641. [PMC free article] [PubMed] [Google Scholar]
- 51.Pandya-Jones A., Black D.L. Co-transcriptional splicing of constitutive and alternative exons. RNA. 2009;15:1896–1908. doi: 10.1261/rna.1714509. doi:10.1261/rna.1714509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Brody Y., Neufeld N., Bieberstein N., Causse S.Z., Böhnlein E.-M., Neugebauer K.M., Darzacq X., Shav-Tal Y. The in vivo kinetics of RNA polymerase II elongation during co-transcriptional splicing. PLoS Biol. 2011;9:e1000573. doi: 10.1371/journal.pbio.1000573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ameur A., Zaghlool A., Halvardson J., Wetterbom A., Gyllensten U., Cavelier L., Feuk L. Total RNA sequencing reveals nascent transcription and widespread co-transcriptional splicing in the human brain. Nat. Struct. Mol. Biol. 2011;18:1435–1440. doi: 10.1038/nsmb.2143. doi:10.1038/nsmb.2143. [DOI] [PubMed] [Google Scholar]
- 54.Khodor Y.L., Rodriguez J., Abruzzi K.C., Tang C.-H.A., Marr M.T., Rosbash M. Nascent-seq indicates widespread cotranscriptional pre-mRNA splicing in Drosophila. Genes Dev. 2011;25:2502–2512. doi: 10.1101/gad.178962.111. doi:10.1101/gad.178962.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Bhatt D.M., Pandya-Jones A., Tong A.-J., Barozzi I, Lissner M.M., Natoli G., Black D.L., Smale S.T. Transcript dynamics of proinflammatory genes revealed by sequence analysis of subcellular RNA fractions. Cell. 2012;150:279–290. doi: 10.1016/j.cell.2012.05.043. doi:10.1101/gad.178962.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Protacio R.U., Li G., Lowary P.T., Widom J. Effects of histone tail domains on the rate of transcriptional elongation through a nucleosome. Mol. Cell. Biol. 2000;20:8866–8878. doi: 10.1128/mcb.20.23.8866-8878.2000. doi:10.1128/MCB.20.23.8866-8878.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Schor I.E., Alló M., Kornblihtt A.R. Intragenic chromatin modifications: a new layer in alternative splicing regulation. Epigenetics. 2010;5:174–179. doi: 10.4161/epi.5.3.11316. doi:10.4161/epi.5.3.11316. [DOI] [PubMed] [Google Scholar]
- 58.Hnilicová J., Hozeifi S., Dušková E., Icha J., Tománková T., Staněk D. Histone deacetylase activity modulates alternative splicing. PLoS One. 2011;6:e16727. doi: 10.1371/journal.pone.0016727. doi:10.1371/journal.pone.0016727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Carrillo Oesterreich F., Preibisch S., Neugebauer K.M. Global analysis of nascent RNA reveals transcriptional pausing in terminal exons. Mol. Cell. 2010;40:571–581. doi: 10.1016/j.molcel.2010.11.004. doi:10.1016/j.molcel.2010.11.004. [DOI] [PubMed] [Google Scholar]
- 60.Jelinic P., Pellegrino J., David G. A novel mammalian complex containing Sin3B mitigates histone acetylation and RNA polymerase II progression within transcribed loci. Mol. Cell. Biol. 2011;31:54–62. doi: 10.1128/MCB.00840-10. doi:10.1128/MCB.00840-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kwon S.H., Workman J.L. The changing faces of HP1: from heterochromatin formation and gene silencing to euchromatic gene expression. BioEssays. 2011;33:280–289. doi: 10.1002/bies.201000138. doi:10.1002/bies.201000138. [DOI] [PubMed] [Google Scholar]
- 62.Shukla S., Kavak E., Gregory M., Imashimizu M., Shutinoski B., Kashlev M., Oberdoerffer P., Sandberg R., Oberdoerffer S. CTCF-promoted RNA polymerase II pausing links DNA methylation to splicing. Nature. 2011;479:74–79. doi: 10.1038/nature10442. doi:10.1038/nature10442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Gowher H., Brick K., Camerini-Otero R.D., Felsenfeld G. Vezf1 protein binding sites genome-wide are associated with pausing of elongating RNA polymerase II. Proc. Natl Acad. Sci. 2012;109:2370–2375. doi: 10.1073/pnas.1121538109. doi:10.1073/pnas.1121538109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Sims R.J., III, Millhouse S., Chen C.-F., Lewis B.A., Erdjument-Bromage H., Tempst P., Manley J.L., Reinberg D. Recognition of trimethylated histone H3 lysine 4 facilitates the recruitment of transcription postinitiation factors and pre-mRNA splicing. Mol. Cell. 2007;28:665–676. doi: 10.1016/j.molcel.2007.11.010. doi:10.1016/j.molcel.2007.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Gaspar-Maia A., Alajem A., Polesso F., Sridharan R., Mason M.J., Heidersbach A., Ramalho-Santos J., McManus M.T., Plath K., Meshorer E., et al. Chd1 regulates open chromatin and pluripotency of embryonic stem cells. Nature. 2009;460:863–868. doi: 10.1038/nature08212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Luco R.F., Pan Q., Tominaga K., Blencowe B.J., Pereira-Smith O.M., Misteli T. Regulation of alternative splicing by histone modifications. Science. 2010;327:996–1000. doi: 10.1126/science.1184208. doi:10.1126/science.1184208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Zhang P., Du J., Sun B., Dong X., Xu G., Zhou J., Huang Q., Liu Q., Hao Q., Ding J. Structure of human MRG15 chromo domain and its binding to Lys36-methylated histone H3. Nucleic Acids Res. 2006;34:6621–6628. doi: 10.1093/nar/gkl989. doi:10.1093/nar/gkl989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Pradeepa M.M., Sutherland H.G., Ule J., Grimes G.R., Bickmore W.A. Psip1/Ledgf p52 binds methylated histone H3K36 and splicing factors and contributes to the regulation of alternative splicing. PLoS Genet. 2012;8 doi: 10.1371/journal.pgen.1002717. e1002717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Vermeulen M., Eberl H.C., Matarese F., Marks H., Denissov S., Butter F., Lee K.K., Olsen J.V., Hyman A.A., Stunnenberg H.G., et al. Quantitative interaction proteomics and genome-wide profiling of epigenetic histone marks and their readers. Cell. 2010;142:967–980. doi: 10.1016/j.cell.2010.08.020. doi:10.1016/j.cell.2010.08.020. [DOI] [PubMed] [Google Scholar]
- 70.Barash Y., Calarco J.A., Gao W., Pan Q., Wang X., Shai O., Blencowe B.J., Frey B.J. Deciphering the splicing code. Nature. 2010;465:53–59. doi: 10.1038/nature09000. doi:10.1038/nature09000. [DOI] [PubMed] [Google Scholar]
- 71.Leparc G.G., Mitra R.D. Non-EST-based prediction of novel alternatively spliced cassette exons with cell signaling function in Caenorhabditis elegans and human. Nucleic Acids Res. 2007;35:3192–3202. doi: 10.1093/nar/gkm187. doi:10.1093/nar/gkm187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Sorek R., Shemesh R., Cohen Y., Basechess O., Ast G., Shamir R. A non-EST-based method for exon-skipping prediction. Genome Res. 2004;14:1617–1623. doi: 10.1101/gr.2572604. doi:10.1101/gr.2572604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Yeo G.W., Van Nostrand E., Holste D., Poggio T., Burge C.B. Identification and analysis of alternative splicing events conserved in human and mouse. Proc. Natl Acad. Sci. USA. 2005;102:2850–2855. doi: 10.1073/pnas.0409742102. doi:10.1073/pnas.0409742102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Warzecha C.C., Sato T.K., Nabet B., Hogenesch J.B., Carstens R.P. ESRP1 and ESRP2 are epithelial cell-type-specific regulators of FGFR2 splicing. Mol. Cell. 2009;33:591–601. doi: 10.1016/j.molcel.2009.01.025. doi:10.1016/j.molmed.2009.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Handel A.E., Ebers G.C., Ramagopalan S.V. Epigenetics: molecular mechanisms and implications for disease. Trends Mol. Med. 2010;16:7–16. doi: 10.1016/j.molmed.2009.11.003. doi:10.1016/j.molmed.2009.11.003. [DOI] [PubMed] [Google Scholar]
- 76.Zhou Z., Luo M., Straesser K., Katahira J., Hurt E., Reed R. The protein Aly links pre-messenger-RNA splicing to nuclear export in metazoans. Nature. 2000;407:401–405. doi: 10.1038/35030160. doi:10.1038/35030160. [DOI] [PubMed] [Google Scholar]
- 77.Sharma S., Kohlstaedt L.A., Damianov A., Rio D.C., Black D.L. Polypyrimidine tract binding protein controls the transition from exon definition to an intron defined spliceosome. Nat. Struct. Mol. Biol. 2008;15:183–191. doi: 10.1038/nsmb.1375. doi:10.1038/nsmb.1375. [DOI] [PMC free article] [PubMed] [Google Scholar]