

The Fmn-family formin Cappuccino does not contain classical autoihibitory domains but is autoinhibited. The N-terminus inhibits actin nucleation and competes with elongation.
Abstract
Formins are a conserved family of proteins known to enhance actin polymerization. Most formins are regulated by an intramolecular interaction. The Drosophila formin, Cappuccino (Capu), was believed to be an exception. Capu does not contain conserved autoinhibitory domains and can be regulated by a second protein, Spire. We report here that Capu is, in fact, autoinhibited. The N-terminal half of Capu (Capu-NT) potently inhibits nucleation and binding to the barbed end of elongating filaments by the C-terminal half of Capu (Capu-CT). Hydrodynamic analysis indicates that Capu-NT is a dimer, similar to the N-termini of other formins. These data, combined with those from circular dichroism, suggest, however, that it is structurally distinct from previously described formin inhibitory domains. Finally, we find that Capu-NT binds to a site within Capu-CT that overlaps with the Spire-binding site, the Capu-tail. We propose models for the interaction between Spire and Capu in light of the fact that Capu can be regulated by autoinhibition.
INTRODUCTION
Formins are a large family of proteins that regulate growth of the actin cytoskeleton. These proteins help build a variety of structures, including, but not limited to, stress fibers, contractile rings, and filopodia. Formins function as actin nucleators, elongation factors, bundlers, and, in at least one case, as a depolymerizing factor (Chhabra and Higgs, 2006; Goode and Eck, 2007). Formins are defined by their well-conserved formin homology 1 and 2 (FH1 and FH2) domains (Higgs and Peterson, 2005). The FH2 domain forms a donut-shaped homodimer that nucleates new filaments and remains associated with the barbed ends of growing filaments (Pruyne et al., 2002; Higashida et al., 2004; Otomo et al., 2005). Through its association with barbed ends, the FH2 domain can modulate the elongation rate and protect filaments from other barbed end–binding proteins such as capping protein (Goode and Eck, 2007). The proline-rich FH1 domain binds profilin-actin and perhaps some SH3 domain–containing proteins (Ahern-Djamali, 1999; Paul et al., 2008). The FH1 and FH2 domains cooperate to accelerate filament elongation in many cases (Kovar et al., 2006; Neidt et al., 2009).
Actin nucleators must be tightly regulated to control when and where new filaments are created. Actin assembly by formins is commonly inhibited by an intramolecular interaction between the N- and C-terminal halves of the protein. This autoinhibition was first described for diaphanous-related formins (DRFs), specifically mDia1 (Watanabe et al., 1999; Goode and Eck, 2007). Detailed analysis showed that actin nucleation activity is inhibited by a direct interaction between the C-terminal diaphanous autoregulatory domain (DAD) and the N-terminal diaphanous inhibitory domain (DID; Alberts, 2001; Li and Higgs, 2003, 2005). The DID/DAD interaction also controls cellular localization of some formins (Seth et al., 2006; Gorelik et al., 2011). Crystal structures of the DID/DAD complex show that the DAD domain forms a single α-helical peptide that binds in the concave surface formed by the five armadillo repeats of the DID domain (Lammers et al., 2005; Nezami et al., 2006). This interaction is commonly regulated by small GTPases, such as Rho, binding to a GTPase-binding domain (GBD) adjacent to and overlapping with the DID domain, which results in release of the DAD domain (Lammers et al., 2005; Otomo et al., 2005; Rose et al., 2005). Recent studies confirmed these observations with full-length mDia1, showing that it is an autoinhibited dimer that can be at least partially activated by RhoA (Ramalingam et al., 2010; Maiti et al., 2012).
The DID and DAD domains are readily identified in the amino acid sequences of five of the seven metazoan groups of formins (Schönichen and Geyer, 2010), as well as in fungal and amoeba formins (Higgs and Peterson, 2005). Furthermore, interaction between the N- and C-termini has been reported for six of the seven groups (Kobielak et al., 2003; Li and Higgs, 2005; Liu et al., 2008; Schulte et al., 2008; Vaillant et al., 2008; Chhabra et al., 2009). The seventh group, delphilin-family formins, remains untested. In most cases, the DID/DAD interaction inhibits actin nucleation and elongation. There are, however, variations on this theme. For example, in the case of INF2, depolymerization but not polymerization is inhibited (Chhabra et al., 2009). Intriguingly, no functional consequence of the FRL2 DID/DAD interaction has been identified (Vaillant et al., 2008).
How members of the formin (Fmn) group of formins are regulated remains an open question, in part because the N-termini are not well conserved within this group. We and others originally predicted that Cappuccino (Capu), the only Drosophila Fmn-family formin, was not regulated by autoinhibition for two reasons: the canonical DID and DAD domains are absent (Higgs and Peterson, 2005), and an alternative means of regulating Capu's nucleation and elongation activity had been identified, namely the WH2-nucleator Spire (Spir; Quinlan et al., 2007; Vizcarra et al., 2011). Further evidence supporting this idea came from the report that the N- and C-termini of Capu interact in pull-down assays, but addition of the N-terminal half of Capu to its FH2 domain had no effect in pyrene-actin polymerization assays (Rosales-Nieves et al., 2006). However, an autoinhibitory interaction was described for Fmn1, one of two mammalian Fmn isoforms (Kobielak et al., 2003). One explanation for this difference is that Capu and Fmn1 are regulated by distinct mechanisms, an idea that is supported by the poor sequence conservation in the N-termini of Fmn-family formins.
Capu was originally identified in a genetic screen for developmental patterning genes (Manseau and Schüpbach, 1989). Loss of Capu results in premature cytoplasmic streaming during oogenesis, polarity defects in oocytes and embryos, and female sterility (Manseau and Schüpbach, 1989; Theurkauf, 1994; Emmons et al., 1995). Recently it was linked to two actin structures in Drosophila oocytes: an isotropic mesh that traverses the oocyte and a network of filaments extending from the posterior cortex of the oocyte (Dahlgaard et al., 2007; Chang et al., 2011; Tanaka et al., 2011). Knockout experiments show that one of the mammalian isoforms, Fmn2, is essential for an actin mesh in mouse oocytes as well (Azoury et al., 2008; Schuh and Ellenberg, 2008). The Spir/Fmn interaction is conserved in mammals (Quinlan et al., 2007; Pechlivanis et al., 2009) and Spir is essential to both the Drosophila and mammalian oocyte actin structures (Dahlgaard et al., 2007; Chang et al., 2011; Pfender et al., 2011). These similarities made us question whether regulation of Fmn-family formins is, in fact, conserved as well.
Recent in vivo studies provide insight into how Capu is regulated. Dahlgaard et al. (2007) created flies expressing a variant of Capu in which a putative Rho-binding site, residues 1–270, was deleted (green fluorescent protein [GFP]–CapuΔN). Expression of GFP-CapuΔN resulted in a denser ectopic actin mesh in the nurse cells relative to either wild-type or GFP-Capu–expressing flies, suggesting that CapuΔN is more active than full-length Capu. A complimentary observation showed that Capu's N-terminal 100 amino acids exert a dominant-negative effect on Capu's Oskar protein–anchoring activity, possibly by inhibiting formation of long arrays of actin filaments extending from the posterior cortex (Chang et al., 2011). Thus the N-terminus of Capu is important for Capu's activity, and these findings suggested to us that an intramolecular interaction might be necessary. We therefore revisited the question of whether or not Capu is autoinhibited. Here we present evidence that there is a direct and tight interaction between the N- and C-termini of Capu and that this interaction can, in fact, inhibit actin nucleation by the FH2 domain. We mapped the domains and performed hydrodynamic analysis on the N-terminal portion, which we refer to as the Cappuccino inhibitory domain (CID). Of interest, the CID-binding site in the C-terminus of Capu maps to the Capu-tail, a short sequence known to bind Spir (Vizcarra et al., 2011).
RESULTS
Polymerization activity of Capu is autoinhibited
To determine whether Capu is autoinhibited, we purified the N-terminal half of Capu (amino acids [aa] 1–466, Capu-NT; Figure 1, A and B) and tested its ability to inhibit the actin polymerization activity of Capu's C-terminal half (aa 467–1059, Capu-CT; Supplemental Figure S1A) in a pyrene-actin polymerization assay. Capu-NT alone had no effect on actin polymerization (green; Figure 1C). Capu-NT did inhibit Capu-CT's ability to stimulate actin polymerization in a dose-dependent manner, demonstrating that the polymerization activity of Capu can be regulated by autoinhibition (Figure 1C). We plotted the barbed-end concentration at half-maximal polymerization (t1/2) for each concentration of Capu-NT (Figure 1D). These data were fitted with a quadratic binding curve. We chose to analyze our interaction data in terms of subunit concentrations, given that the dimers such as Capu-CT have two binding sites. Thus 20 nM Capu-CT here is equal to 10 nM of the nucleation competent dimer. The inhibition constant (Ki) calculated for inhibition of Capu-CT by Capu-NT is 10 nM, which suggests a tight interaction between the two halves of Capu. Calculation of the Ki based on the nucleation rate gave a similar result (Ki = 8 nM; Supplemental Figure S1, B and C).
FIGURE 1:

Capu-NT inhibits the polymerization activity of Capu-CT. (A) Domain organization of Capu along with diagrams of the constructs used in this study. Previously characterized domains are shown as boxes, including FH1 and FH2 domains (orange and green, respectively) and the Capu-tail (violet). (B) Purified Capu-NT and truncations visualized on a Coomassie-stained SDS–PAGE gel. (C) Pyrene-actin polymerization assays with Capu-CT and Capu-NT. Polymerization of 4 μM actin (5% pyrene labeled) was induced by adding Mg2+ and KCl (see Materials and Methods). Addition of 9–300 nM Capu-NT (shown with increasing shades of blue) to 20 nM Capu-CT (alone in red) before mixing with actin inhibited the polymerization activity of Capu-CT in a dose-dependent manner. Addition of 225 nM Capu-NT to actin (green) does not change polymerization kinetics (black). (D) From the pyrene assay in C, the barbed-end concentration at time until half-maximal polymerization (t1/2) was calculated and plotted for each concentration of Capu-NT. Data were fitted with a quadratic binding equation, yielding an inhibition constant (Ki) of 10 nM.
Mapping the C-terminal binding domain
Most formins have a DAD domain C-terminal to the FH2 domain (Higgs and Peterson, 2005). Capu does not have a DAD domain, but we asked whether Capu-NT binds to the analogous region, where the previously described Capu-tail is located (Vizcarra et al., 2011). Truncation of only 12 residues from the Capu-tail—Capu(467–1047)—dramatically reduced inhibition of actin assembly by Capu-NT as measured in the pyrene assay (Figure 2A). Capu-NT's inhibition activity continues to be reduced as Capu-CT is truncated further, with no activity remaining when residues 1036–1059 are removed. Because C-terminal truncations have a strong effect on Capu-CT's activity, as previously reported (Vizcarra et al., 2011), we also used a competition assay to test for Capu-NT/Capu-tail interaction. We added increasing amounts of purified Capu-tail to a constant concentration of Capu-CT and Capu-NT. Unless otherwise indicated, Capu-tail experiments were performed with the monomeric peptide cleaved from a purified glutathione S-transferase (GST)–Capu-tail construct. Inhibition was relieved by addition of Capu-tail in a dose-dependent manner (Figure 2B). Capu-tail alone did not alter actin polymerization at concentrations as high as 12 μM, confirming that the effect was through interaction with Capu-NT (Supplemental Figure S2A).
FIGURE 2:

Capu-NT binds to the Capu-tail region. (A) Capu-NT does not inhibit C-terminally truncated Capu-CT. The bar graph shows the barbed-end concentrations at t1/2 in the presence of 40 nM Capu-CT or truncations of the construct ending at the residues indicated, alone (black) or with 300 nM Capu-NT added (red). Each bar represents the average of three experiments (error bars, SD). (B) Capu-tail relieves inhibition by Capu-NT. Addition of 0.5–10 μM Capu-tail (shown with increasing shades of blue) to 20 nM Capu-CT plus 100 nM Capu-NT leads to recovery of Capu-CT polymerization activity in a dose-dependent manner. Capu-CT alone (20 nM, red) and with 100 nM Capu-NT (green) are shown. (C) GST–Capu-tail relieves inhibition by Capu-NT. The same experiment as in B but with increasing concentration (0.09–4.5 μM) of GST–Capu-tail. Adding 2.3 μM GST alone to the Capu-CT/Capu-NT mix had a weak effect on polymerization (brown). (D) From the pyrene assays in B and C, the barbed-end concentration at t1/2 was calculated for each concentration of Capu-tail or GST–Capu-tail (concentration of the GST-tagged monomer is indicated) and graphed, yielding an estimated EC50 of ∼3 μM for Capu-tail and ∼1.5 μM for GST–Capu-tail. The dashed red and blue lines are drawn to guide the eye.
By examining the barbed-end concentration at t1/2 versus the Capu-tail concentration, we determined that the half-maximal effective concentration of Capu-tail (EC50) is ∼3 μM (Figure 2D). If we interpret this functional assay as a competition binding assay, then we can estimate the affinity of the Capu-tail for Capu-NT as described in Vinson et al. (1998; see Materials and Methods). An EC50 of 3 μM and a Ki of 10 nM for Capu-NT/Capu-CT predicts a Kd of 333 nM for the Capu-NT/Capu-tail interaction. This calculated affinity is much weaker than the Ki we measured for Capu-NT/Capu-CT. Because Capu-NT is a dimer (see later discussion), we reasoned that dimer (Capu-CT) to dimer (Capu-NT) binding might be tighter than monomer (Capu-tail) to dimer (Capu-NT) binding. To mimic the dimer/dimer interaction, we repeated the pyrene competition experiment with dimeric GST–Capu-tail under the same conditions (Figure 2C). On the basis of the GST crystal structure (Kursula et al., 2005) and the length of the linker region, we expect GST–Capu-tail to be a reasonable spatial approximation for Capu-tail's position on the Capu-CT dimer. GST alone and GST–Capu-tail had no effect on actin polymerization (Supplemental Figure S2A), but GST–Capu-tail did relieve inhibition of Capu-CT by Capu-NT. The EC50 of GST–Capu-tail was ∼1.5 μM (Figure 2D), and based on the Vinson analysis, the Kd was 167 nM, consistent with dimerization of Capu-tail modestly enhancing binding to Capu-NT. Of interest, competition with GST–Capu-tail resulted in complete recovery of Capu-CT's activity (Figure 2C). This was not so for monomeric Capu-tail or monomeric mDia-DAD in analogous experiments (Li and Higgs, 2005; Figure 2, B–D). Taken together, these results show that the Capu-tail is necessary for Capu-NT to regulate Capu-CT's polymerization activity. Although dimerization enhances the activity of Capu-tail, the difference in apparent affinities of Capu-NT for Capu-CT versus Capu-tail or GST-Capu-tail led us to conclude that the actual binding site is likely to extend beyond this short region.
Mapping the N-terminal binding domain
Neither phylogenetic analysis (Higgs and Peterson, 2005) nor a Pfam search (Punta et al., 2012) predicted any domains or motifs in Capu-NT. In fact, the N-terminal halves of Fmn-family formins are poorly conserved. Thus it was difficult to predict what part of Capu-NT would bind to the Capu-tail. Initially we designed and characterized various truncated Capu-NT constructs, based on predicted secondary structure and previous biochemical studies (Rosales-Nieves et al., 2006), in order to identify the Capu inhibitory domain (CID; Figure 3A and Supplemental Figure S3A). Inhibition activities were initially quantified by comparing the t1/2 on a scale according to which Capu-CT alone is 0% and actin alone is 100% inhibited. Capu(1–321) was the shortest construct with inhibition activity similar to Capu-NT (Figure 3A and Supplemental Figure S3A). Two longer constructs had lower activity than Capu(1–321)—Capu(1–350) and Capu(1–402) (Supplemental Figure S3A). Perhaps these truncations fall in the middle of a domain, impairing activity due to its disruption. Capu(1–321) inhibits polymerization activity of Capu-CT in a dose-dependent manner (Figure 3B), with a Ki indistinguishable from that of Capu-NT (9 nM; Figure 3C). Calculation based on the nucleation rate gave a similar inhibition constant (12 nM; Supplemental Figure S4, A and B). Capu-tail also prevents inhibition of Capu-CT by Capu(1–321), with an EC50 of ∼3 μM (Supplemental Figure S4, C and D). In sum, the activity of Capu(1–321) is indistinguishable from that of Capu-NT.
FIGURE 3:

Mapping the CID. (A) Summary of the Capu-NT truncations that have inhibition activity against polymerization by Capu-CT. Four different concentrations (50, 100, 200, and 400 nM) were added to the pyrene assay containing 20 nM Capu-CT. Inhibition activities were quantified by comparing the t1/2 where Capu-CT alone is 0% and actin alone is 100% inhibited. Conditions not tested are indicated by a dashed line (---). (B) Addition of 12.5–800 nM Capu(1–321) (shown with increasing shades of blue) to 10 nM Capu-CT before mixing with 4 μM actin inhibited Capu-CT polymerization activity in a dose-dependent manner. Capu-CT (20 nM) alone is shown in red, and 4 μM actin alone is in black. Capu(1–321) alone does not alter actin polymerization (Supplemental Figure S2B). (C) A Ki of 9 nM for Capu(1–321) was determined as described in Figure 1D. (D) Limited proteolysis of Capu-NT with or without Capu-tail. The proteolysis reaction containing 3 μM Capu-NT and 6 nM trypsin with or without 50 μM Capu-tail was incubated for up to 2 h. Red arrows indicate the band analyzed by N-terminal sequencing and MALDI, and black arrows indicate smaller, relatively stable digest products (see Materials and Methods and Supplemental Figure S3B). Bottom, proteolysis of 50 μM Capu-tail alone. (E) Addition of 0.06–1.2 μM Capu(1–222) (shown with increasing shades of blue) to 20 nM Capu-CT before mixing with 4 μM actin inhibited nucleation by Capu-CT in a dose-dependent manner. Capu-CT (20 nM) alone is in red, and 4 μM actin alone is in black. Adding 1.2 μM Capu(1–222) to actin alone weakly affected spontaneous polymerization of actin (green). (F) Polarization anisotropy of 20 nM Capu-tail–Alexa Flour 488 and increasing concentrations of Capu-NT in blue, Capu(1–321) in green, or Capu(1–222) in red. Capu-NT and Capu(1–321) had indistinguishable equilibrium dissociation constants (Kd = 300 nM). Capu(1–222) binds to Capu-tail–Alexa Fluor 488 less tightly (Kd = 4.8 μM).
We also performed limited proteolysis of Capu-NT. Tryptic digest produced four different products over time (Figure 3D). Adding Capu-tail to this reaction slowed the proteolysis, suggesting that it stabilizes Capu-NT when bound (Figure 3D). Using N-terminal sequencing and matrix assisted laser desorption ionization (MALDI) mass spectrometry, we determined that the largest stable band could be either Capu(1–222) or Capu(1–257) (red arrows; Figure 3D and Supplemental Figure S3B). Capu(1–257) expressed poorly and was relatively insoluble, so we proceeded with only Capu(1–222). The inhibition activity of Capu(1–222) was ∼40% that of Capu-NT at a concentration of 100 nM (Figure 3, A and E). The Ki determined (78 nM) overestimates the inhibitory effect because high concentrations of Capu(1–222) weakly inhibit polymerization of actin alone (Figure 3E and Supplemental Figure S4E).
Truncation of Capu constructs from the N-terminus generally reduced inhibition activity (Figure 3A and Supplemental Figure S3A). We conclude that the CID domain is situated near the N-terminus of Capu-NT, probably between residues 80 and 222, and its inhibition activity is enhanced by the flanking regions. Residues 1–105 greatly increase the activity of constructs ending at 321 or 466, although residual activity is present in both Capu(105–321) and Capu(105–466) (Figure 3A and Supplemental Figure S3A). Residues beyond 222 may act only by dimerizing the CID, as discussed later. A similar result was observed for mDia1: the affinity for the FH2-DAD construct is doubled by inclusion of the GBD in the DID construct (Nezami et al., 2006), and the inhibitory activity of the N-terminus is enhanced 10-fold in longer constructs (+GBD = mDia1(1–548) vs. –GBD = mDia1(129–548); Li and Higgs, 2005). Likewise, truncation of the dimerization domain C-terminal to the core structural DID domain—mDia1(129–369)—decreased autoinhibitory activity another 10-fold (Li and Higgs, 2005).
We confirmed direct binding between the Capu-tail and three N-terminal Capu constructs—Capu-NT, Capu(1–321), and Capu(1–222)—using fluorescence anisotropy with Alexa Fluor 488–labeled Capu-tail (Figure 3F). Capu-NT and Capu(1–321) bind Capu-tail with the same affinity (Kd = 300 nM). Capu(1–222) binds with lower affinity (Kd = 4.8 μM). The dissociation constants for Capu-NT/Capu-tail and Capu(1–321)/Capu-tail are in excellent agreement with the affinities we calculated from functional competition assays (300 vs. 333 nM). The weaker affinity of Capu(1–222) compared with both Capu-NT and Capu(1–321) is consistent with its weaker inhibitory activity (Figure 3F). These data further support our conclusion that the C-terminal binding site extends beyond the Capu-tail.
Mutational analysis of the interaction between Capu-CT and Capu-NT
Capu-tail also binds to the Spir-kinase noncatalytic C-lobe domain (KIND; Figure 4A; Vizcarra et al., 2011), leading us to ask whether Capu-NT and Spir-KIND bind to the same site within this small domain. To address this question, we asked whether Capu-NT can bind and inhibit Capu-CT with mutations in the Capu-tail domain. We first tested point mutations in residues that have been shown to abolish the Spir-KIND/Capu-tail interaction (L1048, K1049, R1051, M1052, R1055; Vizcarra et al., 2011; Supplemental Figure S5A). Only mutations in R1051 had a notable effect on Capu-NT activity, but the baseline nucleation activity of Capu-CT was also compromised by this change. Thus we tested several more mutations. Based on the cocrystal structure of human Spir1-KIND bound to Fmn2 tail, residues 1048–1055 of the Capu-tail form an α-helix when Spir-KIND is bound (Figure 4A). We tested residues on the opposite face of this helix (L1053 and M1054) from the KIND-binding site, as well as residues N-terminal and C-terminal to the helix (Figure 4 and Supplemental Figure S5). Mutation of K1058, had a stronger effect than others, although it did not abolish the interaction (Figure 4B). When considered on a scale of 100% inhibition equal to Capu-NT inhibition of wild-type Capu-CT and 0% equal to Capu-CT alone (with each respective mutation), Capu-NT was able to inhibit R1051A and K1058A 70–80% as well as wild-type Capu-CT. The activity of Capu-NT was reduced ∼50% when R1051 was mutated to D, which may reflect the effect of charge reversal. We could not carry out the same experiment with double or triple mutants because such changes greatly decreased Capu-CT's polymerization activity, complicating the interpretation of the data (e.g., K1039A/K1058A, Supplemental Figure S5B). In sum, residues critical for Spir-KIND binding do not have the same effect on Capu-NT binding, but the binding sites do overlap. We speculate that Capu-tail may take on a different structure when bound to Capu-NT since mutations likely to disrupt the α-helix are not as deleterious for Capu-NT binding as they are for Spir-KIND (e.g., R1051D or M1052D).
FIGURE 4:

Mutational analysis of CID binding to the Capu-tail. (A) The structure of Spir1-KIND (gray) and Fmn2-tail (green; Vizcarra et al., 2011). Numbering reflects Capu residues, with Fmn2 residues indicated in parentheses. (B) Barbed-end concentrations at t1/2 in the presence of 20 nM Capu-CT or Capu-CT mutants (gray) plus 150 nM Capu-NT (red) or 1 μM Spir-KIND (green). Each bar represents the average of three experiments (error bars, SD). Residues shown in B are indicated in A. Pink are mutations tested in Vizcarra et al. (2011). Blue and black are new mutations. Residues equivalent to R1034 and K1058 were not visible in the crystal structure.
The effect of Capu-NT on nucleation and processive elongation by Capu-CT
Bulk pyrene-actin polymerization assays do not clearly distinguish between the effects that formins have on filament nucleation and elongation. There are several possible molecular mechanisms that could account for the activity of Capu-NT observed in Figure 1C. The Capu-CT/Capu-NT complex could be unable to nucleate new filaments. Alternatively, a Capu-CT/Capu-NT complex could nucleate normally but, when bound to filament barbed ends, slow or block elongation. To better understand the mechanism of inhibition by Capu-NT, we measured the elongation rate of individual actin filaments using total internal reflection fluorescence (TIRF) microscopy in the presence and absence of Capu-CT and Capu-NT.
Formins have been reported to accelerate barbed-end elongation in the presence of profilin (Kovar et al., 2006; Neidt et al., 2009). Capu-CT alone does not have a large effect on the barbed-end elongation rate but accelerates growth approximately fourfold in the presence of the Drosophila profilin, Chickadee (Chic; Figure 5, A and C). We did not originally observe enhanced elongation in bulk assays (Vizcarra et al., 2011). Presumably, the acceleration was masked by the fact that profilin tends to bind unlabeled actin with higher affinity than actin labeled at Cys-374 (Vinson et al., 1998). In TIRF assays, this bias results in dimmer filaments when Capu-CT and profilin are present. We used filament brightness and elongation rate to determine which filaments had Capu-CT bound and to ask how the Capu-CT/Capu-NT complex affects filament elongation. When we incubated Capu-CT with Capu-NT before adding them to profilin/actin monomers, only 2% of the filaments were dim and fast growing (n = 896 filaments), compared with 57% of the filaments in Capu-CT–alone samples (n = 756 filaments; Figure 5, A and B). In addition, slides with both Capu-CT and Capu-NT had fewer filaments than did those with Capu-CT alone. These results indicate that Capu-NT not only inhibits Capu-CT's nucleation activity but also prevents Capu-CT from binding to filament barbed ends.
FIGURE 5:

Capu-NT inhibits nucleation and competes with barbed- end binding by Capu-CT. (A) Direct observation of filament elongation in the presence of Capu-NT and/or Capu-CT by TIRF microscopy. For each condition, TIRF images are shown for 0, 3, 6, and 9 min, and elongation traces from three different slides, for each condition, are plotted to the right. The bright filaments are shown as blue traces and blue arrows, and the dim filaments, which grew approximately fourfold faster than the bright filaments, are shown as red traces and red arrows (open arrow, pointed end; closed arrow, barbed end). The average and SD of the rates, calculated by linear fits, are shown next to each set of traces. Conditions used are as follows: 1 μM actin (16% Oregon green), 5 μM profilin (Chic), 1 nM Capu-CT, and 100 nM Capu-NT. (B) Quantification of number of filaments bound to Capu-CT with and without Capu-NT from slides in A. For each condition, an average percentage bound is given for three different slides. Error bars are SEM for at least three fields of view from a given slide. Taken together, these data show that when Capu-CT is added alone, 57% (n = 756 filaments) of the filaments are dim and fast growing, that is, bound by Capu-CT. When Capu-CT is mixed with an excess of Capu-NT before addition to the actin, only 2% (n = 896 filaments) of filaments are dim and fast growing. (C) Capu(1–321) inhibits binding of Capu-CT to barbed ends when Capu(1–321) is incubated with Capu-CT before mixing with filament seeds. In this assay, 0.25 μM actin seeds were mixed with 0.5 μM actin monomers, 100 nM Capu-CT, 500 nM Capu(1–321), and 0.375 nM mouse capping protein (CP). The order of protein addition is indicated by a number (1 or 2). For both Capu-CT and Capu(1–321), 1 means that proteins were mixed together before they were added to the seeds. (Error bars, SD, n = 3.)
Consistent with the TIRF assay, Capu-CT, in the presence of Capu(1–321), could not protect barbed ends from capping protein (CP) in bulk elongation assays (Figure 5C). Previously we found that Capu-CT alone can protect barbed ends from CP (Vizcarra et al., 2011). When Capu(1–321) and Capu-CT were mixed before adding them to actin, barbed ends were no longer protected (Figure 5C and Supplemental Figure S6A). These results confirm that autoinhibited Capu-CT is unable to bind barbed ends. Spir-KIND can displace Capu-CT from growing barbed ends (Vizcarra et al., 2011). To determine whether Capu(1–321) has the same effect, we added Capu(1–321) or Spir-KIND to the reaction after Capu-CT was already bound to growing filaments (Supplemental Figure S6B). Indeed, the addition of Capu(1–321) or Spir-KIND (in the presence of CP) slowed filament elongation to similar extents, demonstrating that Capu(1–321) can displace Capu-CT from growing barbed ends as well as Spir-KIND.
Physical properties of Capu-NT constructs
We characterized Capu-NT's physical properties using various solution-based assays. Circular dichroism (CD) showed that Capu-NT has substantial α-helix and random coil content and low β-sheet content (Figure 6A). Removing residues 322–466 reduced the α-helical content by ∼12%, did not change β-sheet content, and increased the relative amount of random coil by ∼10%. The CD spectra for Capu-NT plus Capu-tail and for Capu-NT alone were similar (Figure 6A). Overall, the CD data suggest that Capu-NT has a large amount of random structure. Proteolysis results with trypsin and proteinase K concur (Figure 3D and unpublished data). Although substructures were detectable within Capu-NT, overall it was rapidly digested, as would be expected for a protein with a substantial amount of random coil.
FIGURE 6:

Capu-NT is a dimer. (A) Capu-NT (black; 2.9 μM), Capu(1–321) (red; 4.5 μM), and Capu-NT plus Capu-tail (blue; 3 μM of each) were analyzed by circular dichroism. Each was scanned three times and averaged for analysis. Inset table shows the calculated percentage secondary structure for each construct. (B) Equilibrium sedimentation showed that Capu-NT is a dimer in solution (monomer is 48.6 kDa). Capu-NT at varying concentrations (0.6, 1.8, and 2.8 μM) was spun to equilibrium at 11,000 rpm (red circles) and 13,000 rpm (green circles). Lines represent the best fit to a single-species model (molecular weight, 83.5 kDa). Residuals are shown below. When fitted to monomer–dimer model, the apparent dimer dissociation constant is 245 nM. (C) SEC-MALS data show that both Capu-NT (black) and Capu(1–321) (green) are dimers, with molecular weights of 102.9 and 64.9 kDa, respectively. Capu(1–222) (red) is a monomer with molecular weight of 22.9 kDa. The differential refractive index and calculated molecular weight of the proteins are plotted vs. retention volume. See Table 1 for a summary.
We used analytical ultracentrifugation and size-exclusion column (SEC) multiangle light scattering (MALS) to measure the sizes and shapes of Capu-NT, Capu(1–321), and Capu(1–222). If we fitted sedimentation equilibrium data for Capu-NT with a single-species model, the molecular mass measured was 83.5 kDa, suggesting that the molecule is a dimer (Figure 6B and Table 1). This result gives a 14% error from the expected mass of a dimer (97.2 kDa), so we also fitted the data with a monomer–dimer equilibrium model. The predicted Kd of 245 nM is consistent with the dominant species being a dimer at 2.4 μM of Capu-NT. In both cases there is small but systematic error in the residuals, which probably reflects the tendency of Capu-NT to aggregate over long periods of time. Capu(1–321) was not stable enough, and Capu(1–222) lacks tryptophan and tyrosine residues necessary to perform equilibrium sedimentation. We therefore obtained independent confirmation that the dominant Capu-NT species is a dimer and measured the molecular mass of Capu(1–321) and Capu(1–222) using SEC-MALS. The molecular mass of Capu-NT as measured with MALS was 102.9 kDa (Figure 6C). SEC-MALS shows that Capu(1–321) has a molecular mass of 64.9 kDa, similar to the predicted molecular weight for a dimer (66.6 kDa; Figure 6C and Table 1). Of interest, the molecular weight of Capu(1–222) is 22.9 kDa, consistent with the monomer weight of 22.3 kDa (Figure 6C and Table 1). This indicates that the dimerization domain of Capu-NT is located between residues 222 and 321. We also measured the sedimentation velocity coefficients of Capu-NT and Capu(1–321) (3.3 and 2.7 S, respectively; Supplemental Figure S7, A and B, and Table 1). Together our analytical ultracentrifugation and SEC-MALS data suggest that Capu-NT and Capu(1–321) are dimers with aspect ratios of ∼17:1, assuming that they are prolate ellipsoids. This ratio is extremely elongated and unlikely. We speculate that the low sedimentation coefficients reflect disordered loops at the surface of Capu-NT, in agreement with our CD data.
TABLE 1:
Size determination of Capu-NT constructs.
| Construct | Mass, calculated (kDa) | Mass, MALDI (kDa) | Mass, equilibrium AUC (kDa)a | Mass, MALS (kDa)a | S20,W collected at 20°C | S20,W collected at 4°C |
|---|---|---|---|---|---|---|
| Capu-NT | 48.6 | 48.7 | 83.5 ± 0.7 (14%)b | 102.9 ± 0.1 (6%) | 3.3 | 3.3 |
| Capu(1–321) | 33.5 | 33.5 | NDc | 64.9 ± 0.1 (2%) | 2.7 | 2.6 |
| Capu(1–222) | 22.3 | 22.3 | ND | 22.88 ± 0.07 (3%) | ND | ND |
AUC, analytical ultracentrifugation; ND, not determined.
aPercentages indicate deviations from the predicted molecular weight.
bMonomer/dimer fit of the same data showed that Capu-NT has a dimerization Kd of 245 nM.
cWe could not determine the mass of Capu(1–321) using equilibrium AUC due to protein instability..
DISCUSSION
We found that the actin assembly activity of the Drosophila formin, Capu, is autoinhibited, despite the absence of canonical DID and DAD domains. This discovery agrees with data suggesting that the N-terminus of Capu plays an important physiological role (Dahlgaard et al., 2007; Chang et al., 2011; Tanaka et al., 2011). In the original test of Capu autoinhibition, no effect of Capu-NT on nucleation by Capu-CT was observed (Rosales-Nieves et al., 2006). In this case Capu(1–415) was used, which may be less potent than Capu(1–466) or Capu(1–321). Consistent with this idea, we find that Capu(1–402) has only minimal inhibitory activity. In addition, a 1:1 mixture of the two constructs was tested. We examined a range of concentrations and see a small effect at this ratio but very potent inhibition when the concentration of Capu-NT is increased. Because autoinhibition was observed for Fmn1 as well, we propose that this mode of regulation is conserved in Fmn-family formins. Whether Fmn2 is autoinhibited remains to be tested.
Comparison of autoinhibition by Capu with DRFs
Capu's autoinhibitory domains are positioned similarly to DID and DAD domains, on either side of the FH1-FH2 domains. Like the N-termini of other formins, Capu-NT is a dimer. However, the structural similarities seem to end there. The classic DAD domain is a well-described motif. DAD domains are contained within the sequence C-terminal to the FH2 domain, but the distance from the FH2 domain varies between formins. The Capu-tail is only ∼30 residues long and highly basic. The Capu-tail is well conserved among Fmn-family formins, but the only similarity to DAD domains is a trivial similarity to the polybasic region at the end of the DAD motif. The classic DID domain consists of armadillo repeats, which are not apparent in the Capu sequence or those of other Fmn-family formins. Capu-NT and the shorter constructs we tested contain a high percentage of loops and random coil, which is very different from the tightly packed DID domain. We performed limited proteolytic digests and made a number of truncations in order to identify a minimal CID domain. Reaching this goal was more difficult than expected based on what we know about DID domains. We deduced that the core CID lies between residues 80 and 321 or even 80 and 222. Residues 1–80 could be contributing directly by binding to Capu-CT or indirectly by influencing the structure of the CID. In vivo data show that expression of Capu(1–100) has negative consequences but only in a sensitized background (Chang et al., 2011). Perhaps the CID is not contained in one stable linear portion of Capu-NT but instead consists of multiple weak-binding sites within Capu-NT that together form the CID. Future studies, including high-resolution structural data, are needed for a clear understanding of the CID, how it interacts with Capu-CT, and how it compares to the DID.
Mechanism of autoinhibition
Ramalingam et al. (2010) and Maita et al. (2012) showed that purified full-length mDia1 is autoinhibited, confirming conclusions from many studies performed on formin subfragments similar to the work presented here. Structural insight into the mechanism of autoinhibition comes from two recent crystal structures of mDia1 N- and C-termini bound to each other (Nezami et al., 2010; Otomo et al., 2010). The crystal structures are complicated tetrameric complexes, which forced the authors to develop multiple models of autoinhibition. In most of the models presented, actin filaments are occluded at the FH2 domain by the DID/DAD interaction. Cryo–electron microscope reconstruction of mDia1 (Maita et al., 2012) is consistent with the “trans model” presented by Nezami et al. (2010) and “Model 1” presented by Otomo et al. (2010). Based on the crystal structure, the autoinhibited complex could bind actin monomers but not in a productive manner. More than two monomers or a growing filament would be sterically blocked. Finally, regions close to the DAD domain have been shown to play a positive role in nucleation, and binding to the DID would prevent this activity (Gould et al., 2011; Heimsath and Higgs, 2012).
How does this relate to Capu autoinhibition? Although Capu does not have a DAD domain, the Capu-tail is required for nucleation in a manner similar to that described for other formins (Gould et al., 2011; Vizcarra et al., 2011; Heimsath and Higgs, 2012). We believe that Capu-NT inhibits Capu's actin assembly activity by binding and effectively sequestering the Capu-tail as proposed for Spir-KIND (Vizcarra et al., 2011); hence the ability of monomeric constructs, which might not occlude filament binding, to inhibit. We note a subtle but perhaps not insignificant difference between Capu-NT and Spir-KIND inhibition. Capu-NT completely inhibits actin assembly, whereas saturating concentrations of Spir-KIND do not. This may reflect the difference in binding sites we detect, and/or it could be due to dimer/dimer binding of Capu-CT and Capu-NT. We favor the former mechanism for two reasons. First, monomeric Capu(1–222) completely inhibits Capu-CT. Second, although GST-Capu-tail is sufficient to compete with Capu-CT for Capu-NT binding, the apparent affinities between Capu-NT and Capu-CT versus either tail construct differ by 10- to 50-fold. We interpret this as evidence that the actual Capu-NT binding site extends into the FH2 domain. This does not exclude a role of Capu-NT as steric inhibitor of polymerization. The combination of mechanisms would ensure that autoinhibition is highly effective.
Regulation of autoinhibition
How is the intramolecular interaction regulated? At least three of the DRFs are regulated by binding of small GTPases to a region adjacent to and overlapping with the DID. Rosales-Nieves et al. (2006) found a genetic and biochemical interaction between Capu and Rho1, making it a candidate for regulation of Capu autoinhibition. Capu(125–250) binds Rho1 with a preference for GTP- over GDP-Rho1. This fragment of Capu binds to Capu-CT in pull-down assays and overlaps with the constructs we identified in autoinhibition assays. However, our preliminary results indicate that constitutively active Rho1 does not relieve autoinhibition of actin assembly by Capu (unpublished data). Perhaps Rho1's interaction with Capu is similar to Rho1's interaction with DAAM: DAAM is epistatically downstream of Rho1 in a noncanonical Wnt-signaling pathway, but Rho1 does not significantly activate Daam1 (Liu et al., 2008).
Regulation of Capu through autoinhibition and Spir
How does our finding that Capu is autoinhibited fit with data showing that Spir and Capu collaborate and that Spir inhibits Capu? One interpretation is that Capu can function independently of Spir, perhaps when expressed in cells lacking Spir (Supplemental Figure S8A). A clear role for a functional collaboration between Spir and Capu (and their mammalian homologues) has been established in oocytes, but much less is known about either protein in other cells (Dahlgaard et al., 2007; Quinlan et al., 2007; Pfender et al., 2011). Outside of the oocyte, a role for Fmn1 in adherens junctions was described (Kobielak et al., 2003). No role for Spir1 or Spir2 in adherens junctions has been observed, supporting the idea that Fmn-family formins may act independently at times. More work is required to address whether Fmn-family formins are expressed in the absence of Spir, a case in which autoinhibition would be the primary mechanism for regulation.
Autoinhibition may be part of the Spir/Capu regulatory cycle (Supplemental Figure S8B). A speculative model is as follows: 1, 2) Spir plays a positive role, in that when Spir and Capu are bound to each other, Spir nucleates; 2, 3) the new filament is handed off to Capu, and Spir is released; 4a) in time Capu autoinhibition displaces Capu-CT from barbed end and inhibits itself until (1) it binds Spir and is primed to nucleate again. Alternatively, Spir could function as a regulator of Capu elongation (4b), much like Bud14 (Chesarone et al., 2009). The consequence of this mode of ending filament elongation is that Spir and Capu would then be bound to each other and primed to nucleate again upon termination of the previous nucleation/elongation cycle. On the basis of the fact that Spir-KIND and Capu-NT have similar affinities for Capu-CT and that Capu-NT acts in-cis as opposed to in-trans, we expect Capu-NT to outcompete Spir for binding to Capu-CT. Therefore we speculate that external factors, such as small GTPases and/or posttranslational modifications, play an important role in regulating the interaction between Capu and Spir and potentially selecting the paths outlined here.
MATERIALS AND METHODS
DNA constructs
Capu-NT (aa 1–466) and truncations of Capu-NT were generated by PCR amplification from a full-length Capu template (CG3399, Capu-PA). Capu-NT truncations were subcloned into pGEX-6P-2 (GE Healthcare, Piscataway, NJ) between the BamHI and NotI sites. Point mutations were introduced into Capu-CT using QuikChange Site Directed Mutagenesis (Stratagene, Santa Clara, CA).
Protein expression and purification
Acanthamoeba castellani actin, Capu-tail, and Capu-CT constructs were purified according to published protocols (MacLean-Fletcher and Pollard, 1980; Vizcarra et al., 2011). In brief, Capu-tail was expressed as a GST fusion and then cleaved away from the tag (unless otherwise indicated); Capu-CT was expressed as a histidine-tagged construct. We expressed Capu-NT::pGEX-6P-2 in Rosetta (DE3) pLysS-competent cells. Cells were grown in Terrific Broth at 37°C until they reached an optical density at 600 nm of 0.6–0.8, at which point the temperature was lowered to 20°C for 1 h. Then the cells were induced with 0.25 mM isopropyl-β-d-thiogalactoside and harvested after 13–16 h. Cell pellets were flash frozen with liquid nitrogen and stored at −80°C.
Thawed cell pellets were resuspended in lysis buffer (50 mM Tris, pH 7, 150 mM NaCl, 0.2% Triton X-100, 1 mM dithiothreitol [DTT]) supplemented with 1.7 mM phenylmethanesulfonyl fluoride (PMSF) and 1 μg/ml DNaseI. All subsequent steps were carried out at 4°C or on ice. Cells were lysed by two passages through a microfluidizer (Microfluidics, Newton, MA). The lysate was centrifuged at 20,000 × g for 20 min, and the supernatant was nutated with 1.5 ml glutathione–Sepharose 4b beads (GE Healthcare) for 1 h. Capu-NT was cleaved from the bound GST by incubating with PreScission Protease (GE Healthcare) overnight at 4°C. The eluate was concentrated in an Amicon 10-kDa–molecular weight cutoff centrifugal filter unit and then gel filtered using a Superdex 200 10/300 GL column (GE Healthcare; column buffer: 20 mM Tris, pH 8, 100 mM NaCl, 0.5 M l-arginine, 1 mM DTT). Fractions were pooled based on SDS–PAGE analysis, dialyzed into storage buffer (10 mM Tris, pH 7, 1 mM DTT), and then placed in 1:1 glycerol:storage buffer overnight. Protein aliquots were flash frozen in liquid nitrogen and stored at −80°C. The resulting Capu-NT samples were >98% pure (Figure 1B).
While purifying Capu-NT, two problems arose: 1) protein breakdown, resulting in a doublet on gels, and 2) DnaK chaperone contamination. The former was remedied by using the lysis buffer described previously as opposed to the standard phosphate-buffered saline recommended for glutathione resin. Before protease cleavage, an additional wash step using lysis buffer supplemented with 5 mM ATP and 10 mM MgCl2 removed most of the DnaK (Pastorino et al., 2008). Truncations of Capu-NT were purified by the same method.
Concentrations of all proteins are reported in terms of their monomer (subunit) concentrations to simplify analysis of interactions. Thus 20 nM Capu-CT is equal to 10 nM of functional nucleation unit. Similarly, the new extinction coefficients reported here are in terms of monomer concentrations. To determine the extinction coefficient of Capu-NT (204,855 cm−1 M−1), we measured the concentration of a purified sample by amino acid analysis (AAA) at the University of California at Los Angeles Biopolymer Laboratory. The extinction coefficient was calculated using Beer's law (Abs = c*ε*b), where c is the concentration of the protein measured by AAA, Abs is the absorbance at 280 nm measured for the sample analyzed by AAA, and b is path length (1 cm). The extinction coefficient of Capu(50–321) (41,758 cm−1 M−1) was determined similarly. To determine the extinction coefficient of Capu(1–321) (99,582 cm−1 M−1), we quantified bands from five different concentrations of Capu(1–321) on an SDS–PAGE gel stained with SyproRed (Invitrogen). Gels were imaged on a Pharos FX (Bio-Rad, Hercules, CA). The Capu(1–321) concentration was measured using Capu(50–321) as a standard. Because Capu(1–222) lacks tryptophan and tyrosine residues, we determined its concentration using quantitative SyproRed staining with Capu(1–321) as a standard.
Chickadee (Drosophila profilin) was expressed without an affinity tag in the pET17b plasmid in BL21 (DE3) pLysS-competent cells (overnight at 18°C). Cell pellets were resuspended in extraction buffer (10 mM Tris, pH 8, 1 mM EDTA, 1 mM DTT, 1 mM PMSF) and lysed with a microfluidizer. The lysate was centrifuged at 20,000 × g for 20 min, and the supernatant was incubated with DE52 resin for 30 min at 4°C. Both the flowthrough and an extraction buffer wash were collected. Ammonium sulfate was added to 35% saturation with stirring at 4°C for 15 min. The sample was centrifuged at 30,000 × g for 30 min. The supernatant was brought to 61% ammonium sulfate saturation and centrifuged again, and the pellet was dialyzed overnight with 5 mM potassium phosphate, pH 7.5, 1 mM DTT. The sample was filtered through a hydroxyapatite column (Bio-Rad) and then further purified by gel filtration on a HiLoad 26/60 Superdex 75 column (GE Healthcare) in 150 mM NaCl, 10 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES), pH 7, and 0.5 mM Tris(2-carboxyethyl)phosphine (TCEP). The purified protein was flash frozen in 50% glycerol, 50% 10 mM HEPES, pH 7, and 0.5 mM TCEP and stored at −80°C. The concentration of Chic was calculated by quantitative SyproRed staining using Schizosaccharomyces pombe profilin (1.63 OD/mg/ml) as a standard (Lu and Pollard, 2001).
Purified Acanthamoeba castellani actin was labeled with Oregon green 488 iodoacetamide (Life Technologies, Carlsbad, CA) on cysteine 374. Filamentous actin was dialyzed for 3–4 h with 100 mM KCl, 2 mM MgCl2, 25 mM imidazole, pH 7.5, and 0.2 mM ATP to remove reducing reagents. The actin was rocked at 4°C overnight with a 5- to 10-fold molar excess of dye. The reaction was quenched with 10 mM DTT and spun at 195,000 × g. The pellet was resuspended in actin G buffer (2 mM Tris, 0.1 mM CaCl2, 0.2 mM ATP, 0.5 mM TCEP, 0.04% sodium azide) and dialyzed for 3–4 d before gel filtration on a Superdex 200 10/300 GL column. Actin concentration and dye labeling percentage were calculated according to Kovar et al. (2003).
Pyrene-actin polymerization assays
Pyrene-actin assembly assays were carried out essentially as described (Zalevsky et al., 2001). Briefly, 4 μM A. castellani actin (5% pyrene labeled) was incubated for 2 min at 25°C with ME buffer (final concentration, 200 μM ethylene glycol tetraacetic acid [EGTA] and 50 μM MgCl2) to convert Ca-G-actin to Mg-G-actin. Polymerization was initiated by adding KMEH polymerization buffer (final concentration, 10 mM Na-HEPES, pH 7.0, 1 mM EGTA, 50 mM KCl, 1 mM MgCl2) to the Mg-G-actin. Additional components, such as Capu-CT, Capu-tail, and Capu-NT, were combined in the polymerization buffer before addition to Mg-G-actin. Fluorescence was monitored in a spectrofluorometer (Photon Technology, Birmingham, NJ) or a TECAN F200 with λexcitation = 365 nm and λemission = 407 nm.
Barbed-end concentrations at t1/2 were calculated for the bulk pyrene assays using the following equation: [barbed end] = elongation rate/(k+ × [actin monomers] − k−), where k+ = 11.6 μM−1 s−1 and k− = 1.4 s−1 (Pollard, 1986). The elongation rate was determined by the slope of a best-fit line to the raw data at half-maximal polymerization. We assumed that the actin monomer concentration was 2 μM when analyzing the data near t1/2. Calculation of the nucleation rates of Capu-CT in the presence of Capu-NT was done as described previously (Quinlan et al., 2007). In brief, using a custom wavelet analysis–based algorithm, we smoothed the fluorescence intensity data. The data from the first 30 s were plotted versus time, and the slopes of these lines were taken as the rate of barbed-end formation (i.e., nucleation rate).
Competition pyrene polymerization experiments were analyzed using a competition binding model as described in Vinson et al. (1998). To calculate Kd for the Capu-tail/Capu-NT interaction, we used the following equation:
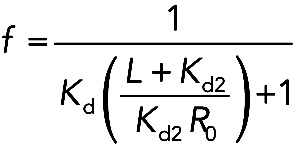 |
where L = [Capu-tail], R0 = [Capu-NT], Kd is the affinity of Capu-CT for Capu-NT, and Kd2 is the affinity of Capu-tail for Capu-NT. At EC50 = 3 μM, f = 0.5 and L = 3 μM. Under these conditions and when Kd is assumed to be 10 nM (Ki of Capu-CT/Capu-NT), Kd2 is 333 nM.
Proteolysis
Capu-NT was dialyzed into 10 mM Tris, 50 mM KCl, and 1 mM DTT and diluted to a final concentration of 3 μM. At time 0 min, 6 nM trypsin was added to the Capu-NT. At time points 1, 2, 5, 10, 15, 30, 60, 90, and 120 min, samples were taken from the reaction and added to 4 mM PMSF to stop proteolysis. Samples were boiled and run on an SDS–PAGE gel for visualization. In the second condition, 50 μM Capu-tail was added to Capu-NT before adding trypsin.
To determine the boundaries of stable bands after tryptic digest, proteolyzed Capu-NT was run on an SDS–PAGE gel and transferred to Immobilon-P membrane (Millipore, Billerica, MA). The bands were visualized by Coomassie staining and cut out from the membrane. The Nevada Proteomics Center (Reno, NV) performed Edman protein sequencing to identify the N-terminus. To determine the molecular weights of the digested bands, both untreated and digested (5 min) Capu-NT were analyzed by MALDI mass spectrometry (Supplemental Figure S3B). Capu-NT (0.5 μl of 3.2 μM) was mixed with 0.5 μl of saturated sinapinic acid and analyzed on a Voyager-DE STR MALDI-TOF Mass Spectrometer (Applied Biosystems, Foster City, CA).
Fluorescence anisotropy
Fluorescence polarization anisotropy of 20 nM Capu-tail–Alexa Fluor 488 (40% labeled), with increasing amounts of Capu-NT, Capu(1–321), or Capu(1–222), was measured with a spectrofluorometer. All assays were carried out at 25°C in 10 mM HEPES, pH 7.0, 1 mM EGTA, 1 mM TCEP, 0.5 mM Thesit, 50 mM KCl, and 1 mM MgCl2. The fluorophore was excited by plane-polarized light at 488 nm, and emission was measured at 520 nm at angles parallel and perpendicular to the angle of incidence. A bandpass filter at 520 ± 5 nm was placed in the emission path. Data were analyzed as previously described (Vizcarra et al., 2011).
TIRF microscopy assays
Coverslips for TIRF elongation assays were prepared essentially as described (Hansen et al., 2010). Briefly, coverslips were washed by sonication with ethanol, 1 M potassium hydroxide, and ethanol, washing with Milli-Q water after each step. The coverslips were then sonicated with isopropanol (15 min), followed by 5% 3-aminopropyltriethoxysilane (Sigma-Aldrich, St. Louis, MO) in isopropanol (30 min). Silanized coverslips were baked at 90°C and stored in 100% ethanol for up to 1 mo. Before PEGylation, coverslips were washed with Milli-Q water. A drop of 30 mg/ml N-hydroxylsuccinimide–functionalized polyethylene glycol (PEG-NHS; 97% methoxy-PEG-NHS and 3% biotin-PEG-NHS; JenKem Technology, Allen, TX) in N,N-dimethyl formamide was placed on half of the silanized coverslips. Another coverslip was placed over each drop to create a sandwich. The PEGylation reaction was carried out by incubating the sandwiches at 75°C for at least 2 h, followed by multiple washes with Milli-Q water. PEGylated coverslips were stored in a sealed container at 4°C for up to 1 mo before use.
Heavy meromyosin (HMM; a gift of the Reisler lab, University of California at Los Angeles) was biotinylated using maleimide-PEG11-biotin (Thermo Scientific, Waltham, MA). HMM (0.5 mg/ml) was incubated with 0.5 mM TCEP for 30 min at room temperature. TCEP was removed using a PD10 column (GE Healthcare), and HMM was eluted in 50 mM sodium phosphate, pH 7, and 100 mM sodium chloride. A 40-fold molar excess of maleimide-PEG11-biotin was incubated with the HMM for 2 h at room temperature, followed by dialysis with 25 mM Tris, pH 8, 30 mM KCl, and 1 mM DTT, and then the same buffer was mixed with equal volume of glycerol. The biotinylated-HMM was flash frozen and stored at −80°C. Streptavidin (VWR, Radnor, PA) was diluted to 30 μM tetramer in Milli-Q water, flash frozen, and stored at −80°C.
Flow cells with volumes of ∼10–15 μl were assembled using double-stick tape. Immediately before imaging, 25 μl of 40 nM streptavidin was applied to the flow cell for 30 s, followed by a wash with 25 μl of 1× TIRF buffer (50 mM KCl, 1 mM MgCl2, 1 mM EGTA, 10 mM HEPES, pH 7, 0.2 mM ATP, 50 mM DTT, 0.2% methylcellulose), 30 s of incubation with 25 μl of 20 nM biotinylated-HMM, and a wash with 25 μl of 1× TIRF buffer. Profilin/actin (final concentration, 1 μM actin, 16% Oregon green labeled, and 5 μM Drosophila profilin) was incubated with ME buffer for 2 min at room temperature. A solution containing 2× TIRF buffer, glucose oxidase (final concentration 0.25 mg/ml), catalase (final concentration 0.05 mg/ml), and any additional proteins (1 nM Capu-CT and/or 100 nM Capu(1–466) final concentrations) was mixed with the Mg-G-actin solution. Filament elongation was visualized on a DMI6000 TIRF microscope (Leica, Wetzlar, Germany) for at least 10 min, capturing images at 10-s intervals. Filament lengths and elongation rates were analyzed with JFilament incorporated in FIJI (Smith et al., 2010).
Bulk elongation assay
Actin (5 μM) was polymerized in 1× KMEH buffer at 25°C for 1 h and aliquoted into 5-μl volumes of 5 μM 1 d before an experiment. To initiate polymerization at the barbed end, we added 20% labeled 0.5 μM Mg-G-actin to the seeds. Filament assembly was tracked for 700 s. Elongation rates were calculated from the slopes between 200 and 700 s. Final concentrations of proteins were 0.25 μM actin seeds, 0.5 μM Mg-G-actin, 100 nM Capu-CT, 500 nM Capu(1–321), and 0.375 nM recombinant mouse capping protein α1β2. Capu-CT was mixed with buffer or Capu(1–321) and incubated for 60 s at 25°C. Subsequently, this mix was added to the seeds and incubated for another 30 s. At this time, Mg-G-actin was added to the ME buffer (see earlier description) and incubated for 2 min at 25°C. After 2 min, seeds, Mg-G-actin, and 1× KMEH buffer containing either buffer blank or CP were combined and analyzed in the fluorometer. To minimize filament shearing, cut pipette tips were used to transfer the polymerization reaction to the cuvette. For the “mid-assay” addition of protein, instead of adding Capu(1–321) or Spir-KIND in the beginning of the assay, we added one or the other after recording elongation for 200 s. Each condition was repeated three times.
Circular dichroism
Proteins were dialyzed into 50 mM potassium phosphate, 1 mM DTT (pH 7). Circular dichroism spectra were measured on a J-715 spectropolarimeter (Jasco, Tokyo, Japan) by averaging three wavelength scans from 195 to 280 nm. The data were analyzed using a previously described method within the Jasco and Sofsec1 software packages (Sreerama and Woody, 1993). The latter compares the input data with a database of spectra from proteins with known secondary structure.
Analytical ultracentrifugation
Sedimentation velocity analytical ultracentrifugation was performed on Capu-NT and Capu(1–321) essentially as described (Chen et al., 2012). Both proteins were dialyzed into 10 mM Tris, 150 mM NaCl, and 0.5 mM TCEP (pH 7.5) and diluted to a final concentration of 2.4 μM Capu-NT or 5.0 μM Capu(1–321). Proteins were spun at 50,000 rpm at 4°C for 3 h in an Optima XL-A Analytical Ultracentrifugation system (Beckman Coulter, Brea, CA). Data were acquired every 3 min for 50 scans. Data were analyzed with Beckman Origin-based software, version 3.01. Capu(1–321) was unstable over the course of an experiment at 20°C. This breakdown is apparent in the analysis (Supplemental Figure S6A), where we see a clear peak at 2.7S and then trailing shoulders from that peak. Data acquired at 4°C were corrected for the viscosity change.
Sedimentation equilibrium analytical ultracentrifugation was performed at two speeds (11,000 and 13,000 rpm) with three different concentrations of Capu-NT (0.6, 1.8, and 2.8 μM). Scans collected between 24 and 28 h after speed was attained were analyzed. The same buffer conditions, centrifuge, and analysis software were used as in the velocity sedimentation experiments.
Size-exclusion column with multiangle light scattering
To analyze Capu(1–222), Capu(1–321), and Capu-NT with SEC-MALS, the proteins were dialyzed into 10 mM Tris, 150 mM NaCl, and 0.5 mM TCEP overnight and spin concentrated using an Amicon 10-kDa–molecular weight cutoff centrifugal filter unit. An analytical size-exclusion column (5 μm, 300 Å, 7.8 × 300 mm; Wyatt Technology, Goleta, CA) was equilibrated with phosphate-buffered saline (140 mM NaCl, 2.7 mM KCl, 10 mM Na2HPO4, 1.8 mM KH2PO4). In each case a 100-μl sample was loaded: 43.5 μM Capu(1–222), 18.3 μM Capu(1–321), or 5.8 μM Capu-NT. The smaller the size of the protein, the more protein was needed to detect light scattering signals. During elution, light scattering was measured with a miniDAWN TREOS and the refractive index (n) was measured with an Optilab T-rEX system (Wyatt Technology). The data were analyzed by ASTRA 6 software to obtain average molecular weights (Table 1). The dn/dc (where c is concentration) for the calculation was set to 0.185 ml/g, a typical value for proteins.
Supplementary Material
Acknowledgments
We thank Quinlan lab and Reisler lab members for invaluable discussion and feedback. We thank Elizabeth Roth and Andrey Shur for help purifying the Capu-tail mutants, Dan McNamara for help in using the MALS instrument, and R. Loo and J. Loo for help in analyzing the proteolyzed Capu-NT by MALDI. Last but not least, we thank O. Akin for his feedback and for providing software for pyrene assay analysis. This work was supported in part by grants from the National Institutes of Health (1R01GM096133-01), the Burroughs-Welcome Fund (Career Award in the Biomedical Sciences), and March of Dimes Foundation Grant 5-FY10-81 (M.E.Q.) and National Institutes of Health National Research Service Award Postdoctoral Fellowship F32GM087857 (C.L.V.).
Abbreviations used:
- CID
Cappuccino inhibitory domain
- CP
capping protein
- DAD
diaphanous autoregulatory domain
- DID
diaphanous inhibitory domain
- Fmn
formin
- GBD
GTPase-binding domain
- KIND
kinase noncatalytic C-lobe domain
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E12-04-0288) on August 8, 2012.
REFERENCES
- Ahern-Djamali SM. Identification of profilin and src homology 3 domains as binding partners for Drosophila Enabled. Proc Natl Acad Sci USA. 1999;96:4977–4982. doi: 10.1073/pnas.96.9.4977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alberts AS. Identification of a carboxyl-terminal diaphanous-related formin homology protein autoregulatory domain. J Biol Chem. 2001;276:2824–2830. doi: 10.1074/jbc.M006205200. [DOI] [PubMed] [Google Scholar]
- Azoury J, Lee KW, Georget V, Rassinier P, Leader B, Verlhac M-H. Spindle positioning in mouse oocytes relies on a dynamic meshwork of actin filaments. Curr Biol. 2008;18:1514–1519. doi: 10.1016/j.cub.2008.08.044. [DOI] [PubMed] [Google Scholar]
- Chang C-W, Nashchekin D, Wheatley L, Irion U, Dahlgaard K, Montague TG, Hall J, Johnston DS. Anterior-posterior axis specification in Drosophila oocytes: identification of novel bicoid and oskar mRNA localization factors. Genetics. 2011;188:883–896. doi: 10.1534/genetics.111.129312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen CK, Sawaya MR, Phillips ML, Reisler E, Quinlan ME. Multiple forms of Spire-actin complexes and their functional consequences. J Biol Chem. 2012;287:10684–10692. doi: 10.1074/jbc.M111.317792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chesarone M, Gould CJ, Moseley JB, Goode BL. Displacement of formins from growing barbed ends by bud14 is critical for actin cable architecture and function. Dev Cell. 2009;16:292–302. doi: 10.1016/j.devcel.2008.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chhabra ES, Higgs HN. INF2 Is a WASP homology 2 motif-containing formin that severs actin filaments and accelerates both polymerization and depolymerization. J Biol Chem. 2006;281:26754–26767. doi: 10.1074/jbc.M604666200. [DOI] [PubMed] [Google Scholar]
- Chhabra ES, Ramabhadran V, Gerber SA, Higgs HN. INF2 is an endoplasmic reticulum-associated formin protein. J Cell Sci. 2009;122:1430–1440. doi: 10.1242/jcs.040691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dahlgaard K, Raposo AASF, Niccoli T, Johnston DS. Capu and Spire assemble a cytoplasmic actin mesh that maintains microtubule organization in the Drosophila oocyte. Dev Cell. 2007;13:539–553. doi: 10.1016/j.devcel.2007.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emmons S, Phan H, Calley J, Chen W, James B, Manseau L. Cappuccino, a Drosophila maternal effect gene required for polarity of the egg and embryo, is related to the vertebrate limb deformity locus. Genes Dev. 1995;9:2482–2494. doi: 10.1101/gad.9.20.2482. [DOI] [PubMed] [Google Scholar]
- Goode BL, Eck MJ. Mechanism and function of formins in the control of actin assembly. Annu Rev Biochem. 2007;76:593–627. doi: 10.1146/annurev.biochem.75.103004.142647. [DOI] [PubMed] [Google Scholar]
- Gorelik R, Yang C, Kameswaran V, Dominguez R, Svitkina T. Mechanisms of plasma membrane targeting of formin mDia2 through its amino terminal domains. Mol Biol Cell. 2011;22:189–201. doi: 10.1091/mbc.E10-03-0256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gould CJ, Maiti S, Michelot A, Graziano BR, Blanchoin L, Goode BL. The formin DAD domain plays dual roles in autoinhibition and actin nucleation. Curr Biol. 2011;21:384–390. doi: 10.1016/j.cub.2011.01.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen SD, Mullins RD. VASP is a processive actin polymerase that requires monomeric actin for barbed end association. J Cell Biol. 2010;191:571–584. doi: 10.1083/jcb.201003014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heimsath EG Jr, Higgs HN. The C terminus of formin FMNL3 accelerates actin polymerization and contains a WH2 domain-like sequence that binds both monomers and filament barbed ends. J Biol Chem. 2012;287:3087–3098. doi: 10.1074/jbc.M111.312207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higashida C, Miyoshi T, Fujita A, Oceguera-Yanez F, Monypenny J, Andou Y, Narumiya S, Watanabe N. Actin polymerization-driven molecular movement of mDia1 in living cells. Science. 2004;303:2007–2010. doi: 10.1126/science.1093923. [DOI] [PubMed] [Google Scholar]
- Higgs HN, Peterson KJ. Phylogenetic analysis of the formin homology 2 domain. Mol Biol Cell. 2005;16:1–13. doi: 10.1091/mbc.E04-07-0565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobielak A, Pasolli HA, Fuchs E. Mammalian formin-1 participates in adherens junctions and polymerization of linear actin cables. Nat Cell Biol. 2003;6:21–30. doi: 10.1038/ncb1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kovar DR, Harris ES, Mahaffy R, Higgs HN, Pollard TD. Control of the assembly of ATP- and ADP-actin by formins and profilin. Cell. 2006;124:423–435. doi: 10.1016/j.cell.2005.11.038. [DOI] [PubMed] [Google Scholar]
- Kovar DR, Kuhn JR, Tichy AL, Pollard TD. The fission yeast cytokinesis formin Cdc12p is a barbed end actin filament capping protein gated by profilin. J Cell Biol. 2003;161:875–887. doi: 10.1083/jcb.200211078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kursula I, Heape AM, Kursula P. Crystal structure of non-fused glutathione S-transferase from Schistosoma japonicum in complex with glutathione. Protein Pept Lett. 2005;12:709–712. doi: 10.2174/0929866054696154. [DOI] [PubMed] [Google Scholar]
- Lammers M, Rose R, Scrima A, Wittinghofer A. The regulation of mDia1 by autoinhibition and its release by Rho*GTP. EMBO J. 2005;24:4176–4187. doi: 10.1038/sj.emboj.7600879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li F, Higgs HN. The mouse formin mDia1 is a potent actin nucleation factor regulated by autoinhibition. Curr Biol. 2003;13:1335–1340. doi: 10.1016/s0960-9822(03)00540-2. [DOI] [PubMed] [Google Scholar]
- Li F, Higgs HN. Dissecting requirements for auto-inhibition of actin nucleation by the formin, mDia1. J Biol Chem. 2005;280:6986–6992. doi: 10.1074/jbc.M411605200. [DOI] [PubMed] [Google Scholar]
- Liu W, Sato A, Khadka D, Bharti R, Diaz H, Runnels LW, Habas R. Mechanism of activation of the formin protein Daam1. Proc Natl Acad Sci USA. 2008;105:210–215. doi: 10.1073/pnas.0707277105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu J, Pollard TD. Profilin binding to poly-L-proline and actin monomers along with ability to catalyze actin nucleotide exchange is required for viability of fission yeast. Mol Biol Cell. 2001;12:1161–1175. doi: 10.1091/mbc.12.4.1161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacLean-Fletcher S, Pollard TD. Identification of a factor in conventional muscle actin preparations which inhibits actin filament self-association. Biochem Biophys Res Commun. 1980;96:18–27. doi: 10.1016/0006-291x(80)91175-4. [DOI] [PubMed] [Google Scholar]
- Maiti S, Michelot A, Gould C, Blanchoin L, Sokolova O, Goode BL. Structure and activity of full-length formin mDia1. Cytoskeleton (Hoboken) 2012;69:393–405. doi: 10.1002/cm.21033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manseau LJ, Schüpbach T. Cappuccino and spire: two unique maternal-effect loci required for both the anteroposterior and dorsoventral patterns of the Drosophila embryo. Genes Dev. 1989;3:1437–1452. doi: 10.1101/gad.3.9.1437. [DOI] [PubMed] [Google Scholar]
- Neidt EM, Scott BJ, Kovar DR. Formin differentially utilizes profilin isoforms to rapidly assemble actin filaments. J Biol Chem. 2009;284:673–684. doi: 10.1074/jbc.M804201200. [DOI] [PubMed] [Google Scholar]
- Nezami A, Poy F, Toms A, Zheng W, Eck MJ. Crystal structure of a complex between amino and carboxy terminal fragments of mDia1: insights into autoinhibition of diaphanous-related formins. PLoS One. 2010;5:e12992. doi: 10.1371/journal.pone.0012992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nezami AG, Poy F, Eck MJ. Structure of the autoinhibitory switch in formin mDia1. Structure. 2006;14:257–263. doi: 10.1016/j.str.2005.12.003. [DOI] [PubMed] [Google Scholar]
- Otomo T, Tomchick DR, Otomo C, Machius M, Rosen MK. Crystal structure of the formin mDia1 in autoinhibited conformation. PLoS One. 2010;5:e12896. doi: 10.1371/journal.pone.0012896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otomo T, Tomchick DR, Otomo C, Panchal SC, Machius M, Rosen MK. Structural basis of actin filament nucleation and processive capping by a formin homology 2 domain. Nature. 2005;433:488–494. doi: 10.1038/nature03251. [DOI] [PubMed] [Google Scholar]
- Pastorino B, Rolland D, Peyrefitte CN, Wurtz N, Almeras L, Bessaud M, Tolou HJ. Improvement of the purification of Saint Louis encephalitis virus NS2B-NS3 recombinant protease expressed in Escherichia coli. J Chromatogr B Analyt Technol Biomed Life Sci. 2008;868:58–63. doi: 10.1016/j.jchromb.2008.04.035. [DOI] [PubMed] [Google Scholar]
- Paul AS, Paul A, Pollard TD, Pollard T. The role of the FH1 domain and profilin in formin-mediated actin-filament elongation and nucleation. Curr Biol. 2008;18:9–19. doi: 10.1016/j.cub.2007.11.062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pechlivanis M, Samol A, Kerkhoff E. Identification of a short Spir interaction sequence at the C-terminal end of formin subgroup proteins. J Biol Chem. 2009;284:25324–25333. doi: 10.1074/jbc.M109.030320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfender S, Kuznetsov V, Pleiser S, Kerkhoff E, Schuh M. Spire-type actin nucleators cooperate with formin-2 to drive asymmetric oocyte division. Curr Biol. 2011;21:955–960. doi: 10.1016/j.cub.2011.04.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pollard TD. Rate constants for the reactions of ATP- and ADP-actin with the ends of actin filaments. J Cell Biol. 1986;103:2747–2754. doi: 10.1083/jcb.103.6.2747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pruyne D, Evangelista M, Yang C, Bi E, Zigmond S, Bretscher A, Boone C. Role of formins in actin assembly: nucleation and barbed-end association. Science. 2002;297:612–615. doi: 10.1126/science.1072309. [DOI] [PubMed] [Google Scholar]
- Punta M, et al. The Pfam protein families database. Nucleic Acids Res. 2012;40:D290–D301. doi: 10.1093/nar/gkr1065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinlan ME, Hilgert S, Bedrossian A, Mullins RD, Kerkhoff E. Regulatory interactions between two actin nucleators, Spire and Cappuccino. J Cell Biol. 2007;179:117–128. doi: 10.1083/jcb.200706196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramalingam N, Zhao H, Breitsprecher D, Lappalainen P, Faix J, Schleicher M. Phospholipids regulate localization and activity of mDia1 formin. Eur J Cell Biol. 2010;89:723–732. doi: 10.1016/j.ejcb.2010.06.001. [DOI] [PubMed] [Google Scholar]
- Rosales-Nieves AE, Johndrow JE, Keller LC, Magie CR, Pinto-Santini DM, Parkhurst SM. Coordination of microtubule and microfilament dynamics by Drosophila Rho1, Spire and Cappuccino. Nat Cell Biol. 2006;8:367–376. doi: 10.1038/ncb1385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rose R, Weyand M, Lammers M, Ishizaki T, Ahmadian MR, Wittinghofer A. Structural and mechanistic insights into the interaction between Rho and mammalian Dia. Nature. 2005;435:513–518. doi: 10.1038/nature03604. [DOI] [PubMed] [Google Scholar]
- Schönichen A, Geyer M. Fifteen formins for an actin filament: A molecular view on the regulation of human formins. Biochim Biophys Acta. 2010;1803:152–163. doi: 10.1016/j.bbamcr.2010.01.014. [DOI] [PubMed] [Google Scholar]
- Schuh M, Ellenberg J. A new model for asymmetric spindle positioning in mouse oocytes. Curr Biol. 2008;18:1986–1992. doi: 10.1016/j.cub.2008.11.022. [DOI] [PubMed] [Google Scholar]
- Schulte A, Stolp B, Schönichen AS, Pylypenko O, Rak A, Fackler OT, Geyer M. The human formin FHOD1 contains a bipartite structure of FH3 and GTPase-binding domains required for activation. Cell. 2008;16:1313–1323. doi: 10.1016/j.str.2008.06.008. [DOI] [PubMed] [Google Scholar]
- Seth A, Otomo C, Rosen MK. Autoinhibition regulates cellular localization and actin assembly activity of the diaphanous-related formins FRLalpha and mDia1. J Cell Biol. 2006;174:701–713. doi: 10.1083/jcb.200605006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith MB, Li H, Shen T, Huang X, Yusuf E, Vavylonis D. Segmentation and tracking of cytoskeletal filaments using open active contours. Cytoskeleton. 2010;67:693–705. doi: 10.1002/cm.20481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sreerama N, Woody RW. A self-consistent method for the analysis of protein secondary structure from circular dichroism. Anal Biochem. 1993;209:32–44. doi: 10.1006/abio.1993.1079. [DOI] [PubMed] [Google Scholar]
- Tanaka T, Kato Y, Matsuda K, Hanyu-Nakamura K, Nakamura A. Drosophila Mon2 couples Oskar-induced endocytosis with actin remodeling for cortical anchorage of the germ plasm. Development. 2011;138:2523–2532. doi: 10.1242/dev.062208. [DOI] [PubMed] [Google Scholar]
- Theurkauf W. Premature microtubule-dependent cytoplasmic streaming in cappuccino and spire mutant oocytes. Science. 1994;265:2093–2096. doi: 10.1126/science.8091233. [DOI] [PubMed] [Google Scholar]
- Vaillant DC, Copeland SJ, Davis C, Thurston SF, Abdennur N, Copeland JW. Interaction of the N- and C-terminal autoregulatory domains of FRL2 does not inhibit FRL2 activity. J Biol Chem. 2008;283:33750–33762. doi: 10.1074/jbc.M803156200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vinson VK, Cruz EMDL, Higgs HN, Pollard TD. Interactions of Acanthamoeba profilin with actin and nucleotides bound to actin. Biochemistry. 1998;37:10871–10880. doi: 10.1021/bi980093l. [DOI] [PubMed] [Google Scholar]
- Vizcarra CL, Kreutz B, Rodal AA, Toms AV, Lu J, Zheng W, Quinlan ME, Eck MJ. Structure and function of the interacting domains of Spire and Fmn-family formins. Proc Natl Acad Sci USA. 2011;108:11884–11889. doi: 10.1073/pnas.1105703108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe N, Kato T, Fujita A, Ishizaki T, Narumiya S. Cooperation between mDia1 and ROCK in Rho-induced actin reorganization. Nat Cell Biol. 1999;1:136–143. doi: 10.1038/11056. [DOI] [PubMed] [Google Scholar]
- Zalevsky J, Grigorova I, Mullins RD. Activation of the Arp2/3 complex by the Listeria acta protein. Acta binds two actin monomers and three subunits of the Arp2/3 complex. J Biol Chem. 2001;276:3468–3475. doi: 10.1074/jbc.M006407200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.