

Abstract
Lung adenocarcinoma, the most common type of human non-small cell lung cancer (NSCLC), frequently overexpresses EGFR. However, the mechanisms underlying EGFR overexpression are not completely understood. Recent studies have identified that decreased expression of TIP30 is associated with the metastasis of human NSCLCs, but a causative relationship between TIP30 deficiency and NSCLC development remains unclear. We show here that Tip30 deletion leads to spontaneous development of lung adenomas and adenocarcinomas in mice. Lung tumor development was preceded by aberrant expansion of bronchioalveolar stem/progenitor and alveolar type II cells, as well as increased expression of EGFR and its downstream signaling factors in the lung of Tip30−/− mice. Moreover, TIP30 knockdown in human lung adenocarcinoma cells resulted in prolonged EGFR activity in early endosomes, delayed EGFR degradation, increased EGFR nuclear localization, leading to up-regulated pAKT and pERK1/2 expression. Importantly, in human lung adenocarcinomas, low TIP30 expression correlates with prolonged patient overall and post-progression survival times. Together, these results suggest that TIP30 functions as a tumor suppressor to inhibit EGFR cytoplasmic and nuclear signaling and suppress adenocarcinogenesis in the lung and highlight the potential of therapeutic strategies aiming at inhibiting EGFR signaling for patients with low TIP30 expression lung adenocarcinoma.
Keywords: TIP30, lung adenocarcinoma, EGFR signaling, tumor suppressor
Introduction
Proto-oncogenes and tumor suppressor genes, such as EGFR, k-Ras, c-Myc, p53 and DOK have been implicated in the pathogenesis of NSCLC (1–8). In particular, EGFR overexpression was seen in 50% of lung adenocarcinomas, a subtype of NSCLCs (9, 10), but only 10% of them were found to contain mutations in EGFR exon encoding kinase domain, indicating that other genetic and epigenetic alterations may also activate EGFR signaling, thereby contributing to the pathogenesis of lung adenocarcinomas. Thus, a better understanding of the alterations involved in the initiation and progression of lung cancer can help us not only predict the prognosis but also select the therapeutic intervention with optimal impact on survival and quality of life, and contribute to the development of new effective therapeutic strategies against lung cancer.
Lung adenocarcinomas that develop in mice are similar in their histopathological and molecular features to human lung adenocarcinomas. Therefore, studies on mouse models of lung adenocarcinomas may contribute to our understanding of human lung adenocarcinoma pathogenesis by unraveling the critical oncogenic pathways responsible for the initiation and progression of tumors. Towards this end, many genetically-engineered mouse models including mice expressing EGFR mutants(6, 7) and mice with expression of K-Ras mutants or conditional bitransgenic inducible p53/K-Ras genes have been generated and exhibit spontaneous development of lung adenocarcinomas (1, 3). However, EGFR expression, which is frequently increased in human lung adenocarcinomas (11), is decreased in several mouse models of lung adenocarcinoma including p53/K-Ras models. To date, a tumor suppressor gene-knockout mouse model of lung adenocarcinoma with EGFR overexpression has not been reported yet.
TIP30, also called CC3 or HTATIP2, is a transcriptional cofactor that was initially identified as a putative metastasis suppressor of small cell lung cancer (12, 13). Subsequent studies have revealed that aberrant expression of TIP30 is implicated in a variety of human cancers (14–17). Recently, decreased expression of TIP30 was observed in approximately 37% of human NSCLCs, and was correlated with metastasis (17). Moreover, TIP30 knockdown was demonstrated to enhance survival and metastasis potential of human lung cancer cells in vitro, and promote lung metastasis in xenograft mouse models (17). We previously reported that genetically-engineered Tip30 knockout mice are prone to development of liver tumors and other tissue types of tumors in C57/ B6/129SVJ mixed background and mammary hyperplasia in C57/B6 genetic background (14, 18). Thus, a causative relationship between TIP30 deficiency and NSCLC development still remains unknown despite a strong implication of TIP30 loss in human lung carcinogenesis. Because the Balb/c mouse strain is prone to development of lung and mammary tumors, we examined development of tumors in a cohort of wild type and Tip30 knockout mice in a Balb/c genetic background. Moreover, we investigated the effects of low TIP30 expression on cultured human adenocarcinoma cells and the prognosis of patients with lung adenocarcinomas. This approach demonstrated that TIP30 deletion resulted in spontaneous development of lung adenomas and adenocarcinomas in mice, with tumors closely recapitulating the histological and molecular features of human lung adenocarcinomas with EGFR overexpression. Our results revealed TIP30 as a crucial regulator in suppressing cytoplasmic and nuclear EGFR signaling in the lung, and underlined low TIP30 expression as a potential biological marker to predict overall and post-progression survival times for patients with lung adenocarcinomas at stages I and II.
Results
Tip30−/− mice spontaneously develop lung tumors
In an effort to better understand the roles of Tip30 in tumorigenesis, we generated Tip30+/− and Tip30−/− mice in Balb/c background by backcrossing the Tip30 knockout gene into Balb/c mice for seven generations (14). Balb/c mice bearing Tip30 heterozygous and homozygous deletion were viable and fertile. We monitored tumor development in a cohort of Tip30+/+, Tip30+/− and Tip30−/− Balb/c mice for 78 weeks. Mice were euthanized and subjected to complete necroscopy when they were moribund or reached 78 weeks of age. The survival curves of Tip30+/+ and Tip30−/− mice differed significantly (Supplementary Figure S1, P = 0.013), as 34.4 % of Tip30−/− mice versus 7.7 % of wild type mice died before 78 weeks of age. The exact cause of the spontaneous death of Tip30−/− mice has not been determined except those with tumors. At 78 weeks of age, 43% of Tip30−/− mice of both sexes developed pulmonary tumors and 22% of Tip30−/− female mice developed mammary tumors. In contrast, pulmonary tumors were observed in 8.3% (1/12) of Tip30+/− and 4.2% (1/24) of wild-type mice (Figures 1a and b). Histopathological analysis revealed that Tip30−/− mice exhibited several types of progressive lesions in the lung, such as atypical adenomatous hyperplasias, adenomas and adenocarcinomas (Figures 1c–e); Adenomas and adenocarcinomas developed in approximately 20% and 24% of Tip30−/− mice, respectively. The hyperplasias were frequently observed in the alveoli and alveolar space adjacent to the respiratory bronchioles, but not in the bronchioles as described in K-Ras mouse model of lung adenocarcinoma (4). The hyperplastic lesions consisted mainly of cuboidal pneumocytes that thickened alveolar walls in preserved alveolar spaces. Most of the lung tumors were founded in peripheral portions of the lung. Adenocarcinomas were primarily a mixed subtype with both solid and papillary growth patterns, infiltrative border, cytological pleomorphism and nuclear atypia, which resembled human lung adenocarcinoma (Figure 1e). These results suggest that Tip30 plays an important role in the suppression of lung tumorigenesis.
Figure 1.
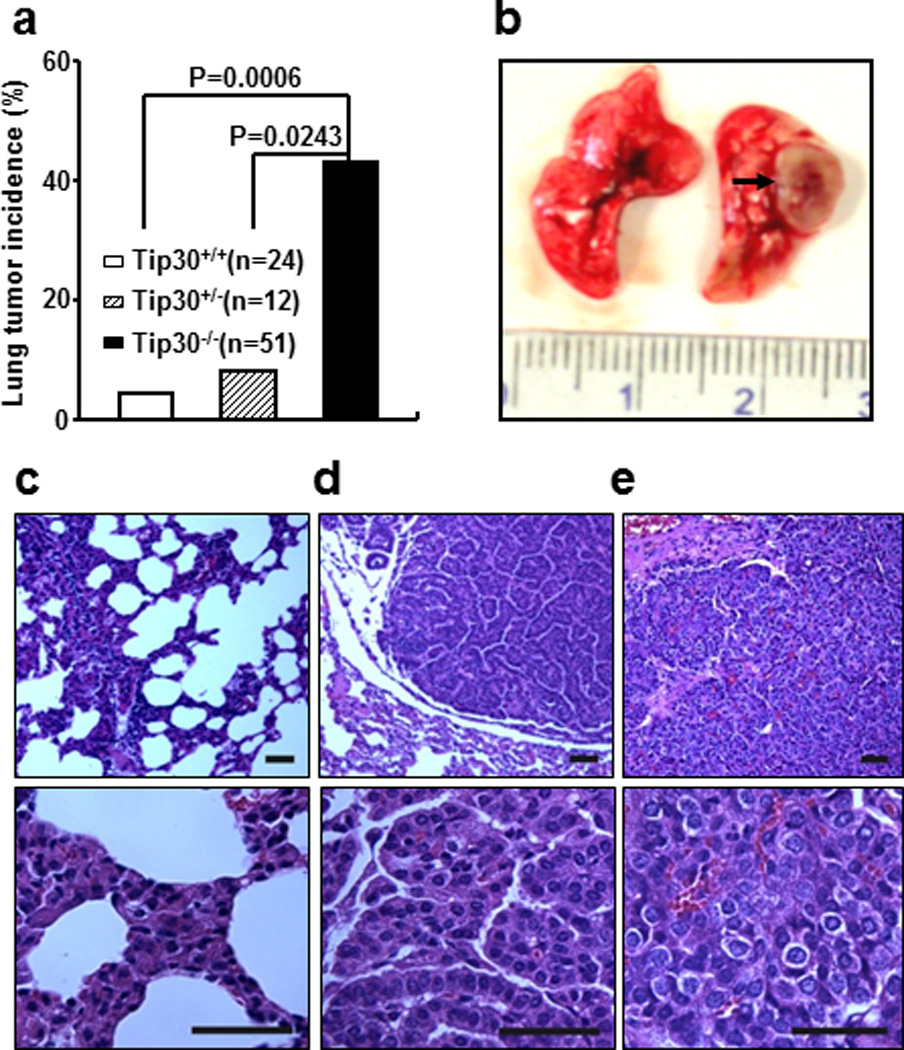
Tip30 deletion resulted in tumor development in mice. (a) Graph shows incidence of lung tumors arising in Tip30+/+, Tip30+/−, and Tip30−/− mice. (b) Representative image of lung adenocarcinoma arising in Tip30−/− mice. The arrow indicates a tumor mass. (c–e) Representative H&E- stained sections of pulmonary hyperplasia (c), pulmonary adenoma (d), pulmonary adenocarcinoma (e) arising in Tip30−/− mice. Scale bar, 20 µm.
Tip30 deletion promotes expansion of AT2 and bronchioalveolar stem/progenitor cells (BASCs)
To investigate the histogenesis of the lung tumors, we first stained lung tumor tissues from Tip30−/− mice with antibodies against surfactant apoprotein C (SP-C, marker for AT2 cell) and Clara cell 10 kDa protein (CC10, marker for Clara cell) to determine the cell type comprising pulmonary hyperplasias and tumors. Similar to the observations in EGFR mutant transgenic mouse models of lung adenocarcinoma, we found that the tumors and hyperplasias were mainly composed of SP-C-positive and CC10-negative cells (Figure 2a). When growth of SP-C positive cells was compared in lungs of Tip30+/+ and Tip30−/− mice prior to development of tumors, we found that the numbers of SP-C positive cells in the alveoli were significantly increased in Tip30−/− lungs (Figures 2b and c). To assess the effect of Tip30 deletion on BASCs (19), we conducted dual immunofluorescent staining for SP-C and CC10 to detect these cells at the bronchoalveolar duct junction (BADJ) in Tip30+/+ and Tip30−/− lung tissues from age-matched mice. A significant increase in the number of dual SP-C and CC10 positive cells was observed in Tip30−/− compared with Tip30+/+ lungs (Figures 2d and e). Moreover, the proliferative cell nuclear antigen (PCNA) was more frequently detected in SP-C positive AT2 cells in Tip30−/− lung alveoli and tumors than Tip30+/+ lung alveoli, whereas the numbers of apoptotic cells was slightly, but not significantly, increased in Tip30−/− lung tissues (Supplementary Figure S2). These results indicate that Tip30 deletion promotes expansion of AT2 and BASCs, supporting BASCs as the possible cell type of origin for lung tumors arising in Tip30−/− mice.
Figure 2.
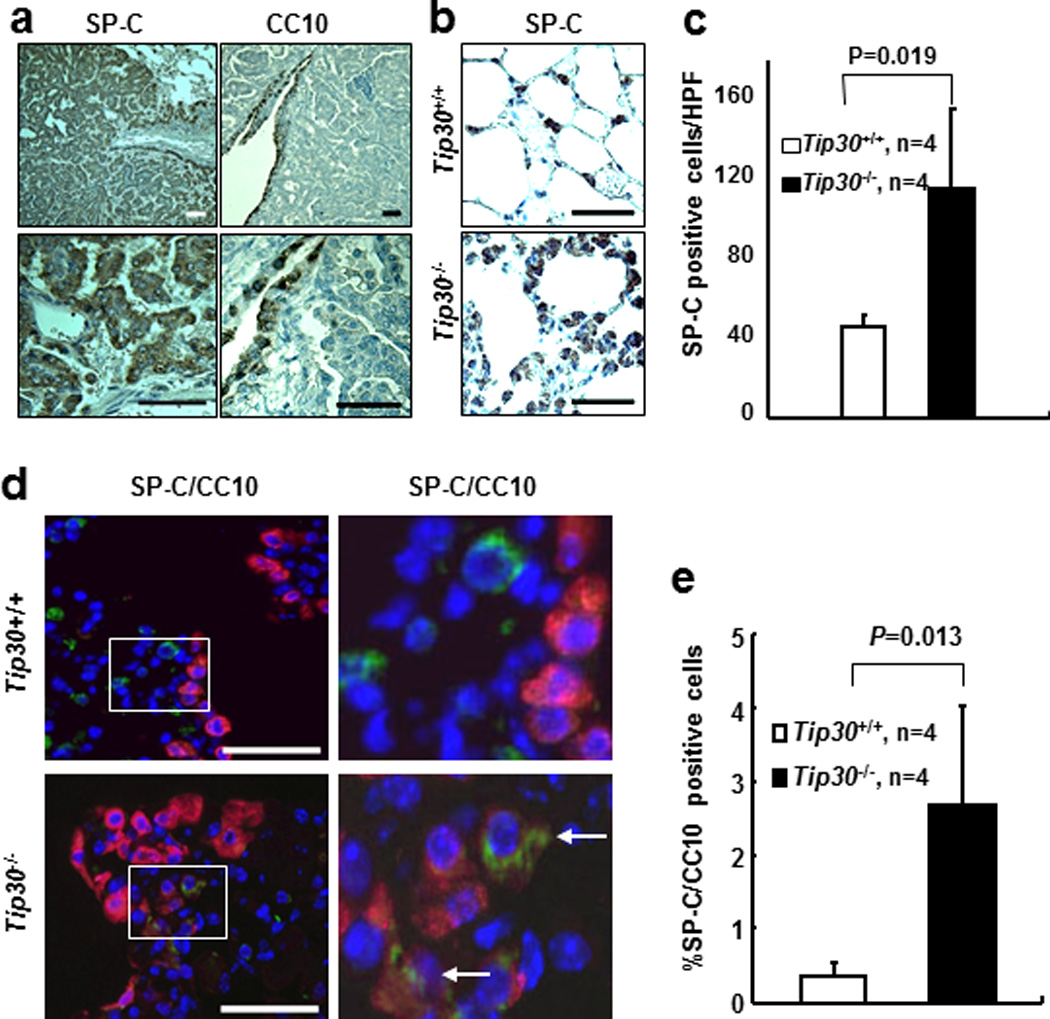
Immunostaining for SP-C, CC10 in lung tissues and tumors. Scales bar, 20µm. (a) Representative Images show SP-C positive cells in lung tumors and CC10 positive cells in the bronchiole. (b) Representative Images show SP-C positive cells in lung alveoli. (c) Quantification of SP-C positive cells in lung alveoli. SP-C positive cells were counted in 5 randomly selected high power fields (HPF) per slide. Graph shows the numbers (mean ± SEM) of SP-C positive cells per HPF. (d) Dual SP-C (green) and CC10 (red) positive cells at bronchoalveolar duct junction (BADJ). Dual SP-C/CC10 positive cells at BADJ are indicated with arrowheads. (e) Graph shows the percentages of dual SP-C/CC10 positive cells relative to CC10-positive cells at BADJ in lung sections.
Since Tip30−/− mice carried a knocked-in β-galactosidase (β-Gal) gene at the Tip30 gene locus under the control of Tip30 promoter, co-immunostaining of SP-C or CC10 with β-Gal was carried out to determine whether Tip30-promoter was active in these tumor cells. Despite the fact that the β-Gal gene was knocked in the genome of all cell types, AT2 cells and SP-C positive hyperplasia cells were found to express detectable β-Gal (Supplementary Figure S3a) whereas the CC10 positive cells were not (Supplementary Figure S3b), indicating that Tip30 promoter was preferentially active in AT2 cells and SP-C positive hyperplasia cells. These results imply that Tip30 deletion may preferentially affect pulmonary AT2 cells and predispose them to malignant transformation.
Down regulation of Tip30 enhances EGFR signaling in mouse lung alveoli and human lung adenocarcinoma cells
We previously reported that down regulation of TIP30 up-regulates EGFR signaling in hepatocytes and mammary epithelial cells (20, 21). To determine whether Tip30 deletion also up-regulates EGFR downstream signaling targets in pulmonary cells, we examined the levels of EGFR, p-Akt, p-Erk1/2 and cyclin D1 in Tip30+/+ and Tip30−/− lung tissues and tumors. EGFR expression was increased in Tip30−/− lung tissues and tumors compared with Tip30+/+ lung tissues (Figure 3a and Supplementary Figure S4). As expected, the numbers of p-Erk1/2, p-Akt or Cyclin D1 positive cells were significantly increased in Tip30−/− lung tissues and lung tumors as compared with Tip30+/+ lung tissues (Figures 3b–e). Western blot analysis of lung tissue protein extracts confirmed that the protein levels of EGFR, p-Akt and p-Erk1/2 were notably elevated in Tip30−/− lung tissues (Supplementary Figure S5). Together, these results suggest that Tip30 deletion enhances EGFR signaling in mouse pulmonary and tumor cells.
Figure 3.
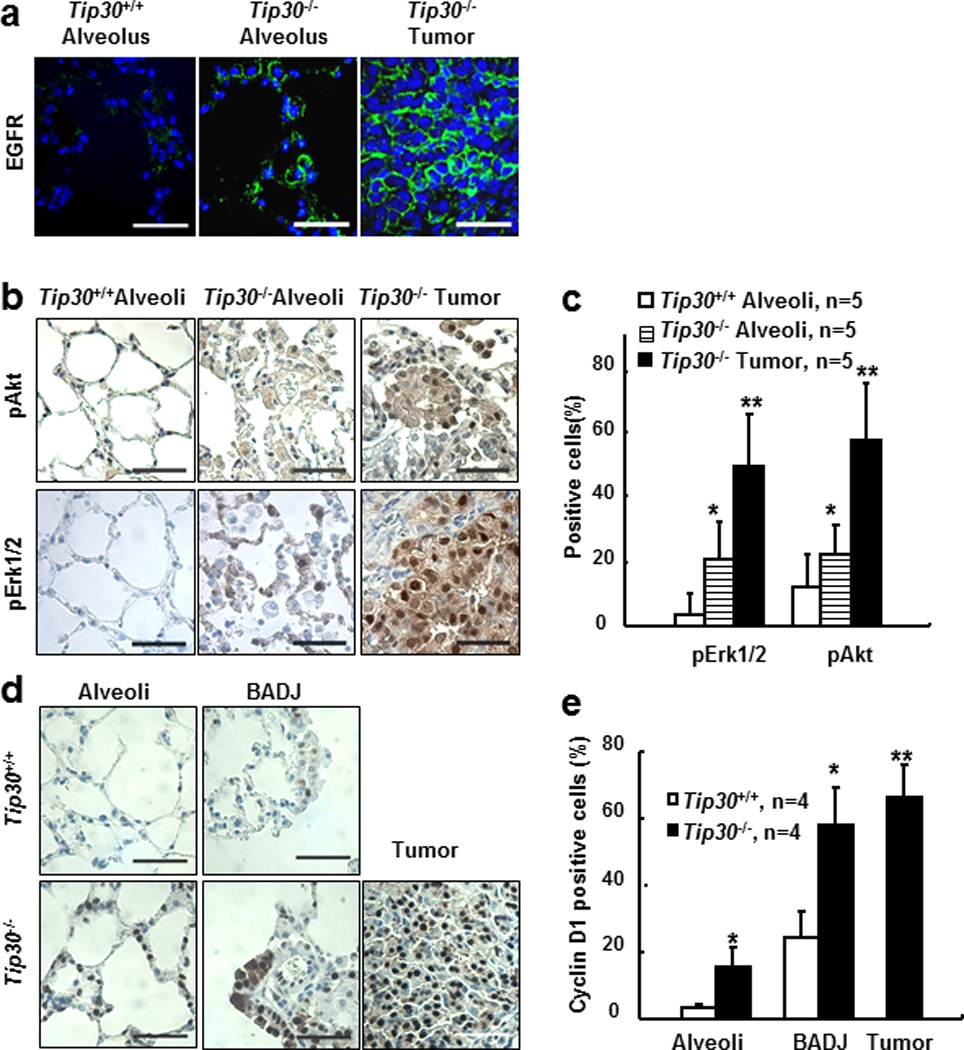
Tip30 deletion enhanced EGFR signaling in mouse lung tissues and tumors. (a–b) Representative images of indicated lung tissues and tumors stained for EGFR (a), pAkt (b), or pErk1/2 (b), scale bars: 20µm. (c) Quantification of pAkt or pErk1/2 positive cells shown in (a) and (b). Graph shows the percentages of pAkt or pErk1/2 positive alveolar cells per field on Tip30+/+, Tip30−/− lung alveoli, and Tip30−/− lung tumors. (d) Representative images of indicated lung tissues and tumors stained for Cyclin D1, scale bars: 20µm. (e) Graph shows the percentages of Cyclin D1 positive alveolar cells per field on the indicated lung alveoli, BADJ, or Tip30−/− mice lung tumors. pAkt, pErk1/2, Cyclin D1-positive and total alveolar cells were counted from five microscopic fields per slide. Error bars denote mean � SEM, # P < 0.05 vs. Tip30+/+ mice. * P < 0.05 vs. Tip30−/− mice alveoli. ** P > 0.05 vs. Tip30−/− mice BADJ. by 2-tailed students t test.
To extend the role of TIP30 in the mouse lung to human lung adenocarcinoma cells, we knocked down TIP30 expression in human lung adenocarcinoma A549 cells using two shRNAs that specifically target TIP30 (Figure 4a) and performed endocytic degradation assays to examine EGF-induced EGFR endocytic degradation and signaling. TIP30 knockdown resulted in delayed EGFR endocytic degradation, prolonged phosphorylation of AKT (Ser473) and ERK1/2 (Thr202/Tyr204) in A549 cells (Figures 4b–g and Supplementary Figure S6). It appeared that p-AKT was dephosphorylated faster than EGFR and ERK1/2. Although we do not know the precise mechanism underlying this phenomenon, it may be due to the autonomic balance of kinase and phosphatase activities for AKT in cells. Moreover, expression of Cyclin D1, which is transcriptionally activated by the nuclear EGFR, was also increased in TIP30 knockdown cells (Supplementary Figure S6b). Consistent with the finding of increased Cyclin D1 expression, confocal microscopy and Western blot analyses revealed that EGFR nuclear localization was significantly increased in TIP30 knockdown cells after EGF induction (Figures 5a–c and Supplementary Figure S7). We then aimed to analyze the mechanism that leads to enhanced EGFR cytoplasmic and nuclear signaling in TIP30 knockdown A549 cells. EGFR internalization analysis revealed that TIP30 knockdown significantly prolonged EGFR in EEA1-positive early endosomes after EGF treatment (Figures 5d and e). Moreover, TIP30 knockdown in immortalized human lung epithelial BEAS-2B cells also results in delayed EGFR degradation. Together, these observations indicate that decreased TIP30 expression blocks endocytic degradation of EGFR, leading to enhanced EGFR cytoplasmic and nuclear signaling pathways, which may offer a biochemical explanation for increased ERK1/2 and AKT activation and Cyclin D1 expression in Tip30−/− lungs and tumors.
Figure 4.
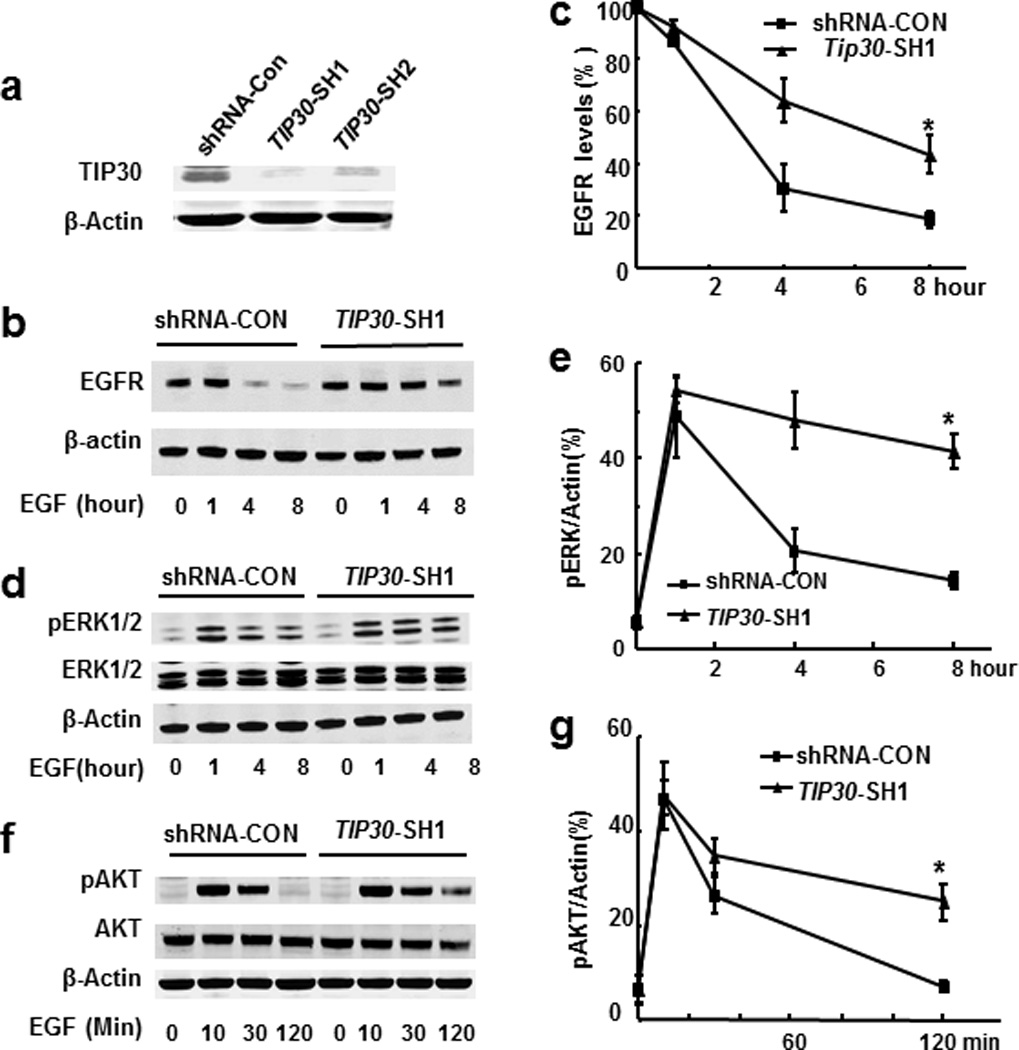
Effects of TIP30 deficiency on EGFR degradation and signaling in human A549 cells. (a) Western blot analysis of A549 cells with shRNA-control or TIP30 knockdown. Whole cell lysates were made from cells expressing a scramble shRNA-CON, TIP30-SH1 or TIP30-SH2 that specifically target TIP30. (b–g) Western blot analysis of EGFR (b, c), pERK1/2 (d, e) and pAKT (f, g) in extracts of shRNA-CON and TIP30-SH1 knockdown A549 cells. Cells were treated with EGF, then incubated in DMEM including cycloheximide and collected at various time points after EGF treatment and subjected to Western blot analysis with indicated antibodies. The amount of EGFR in each lane was quantified and normalized to the amount of β-actin. The values are expressed as percent of EGFR at time zero (c) pAKT or pERK1/2 levels were expressed as the percentages of β-actin. Results are representative of three independent experiments. * P < 0.05 versus shRNA control.
Figure 5.
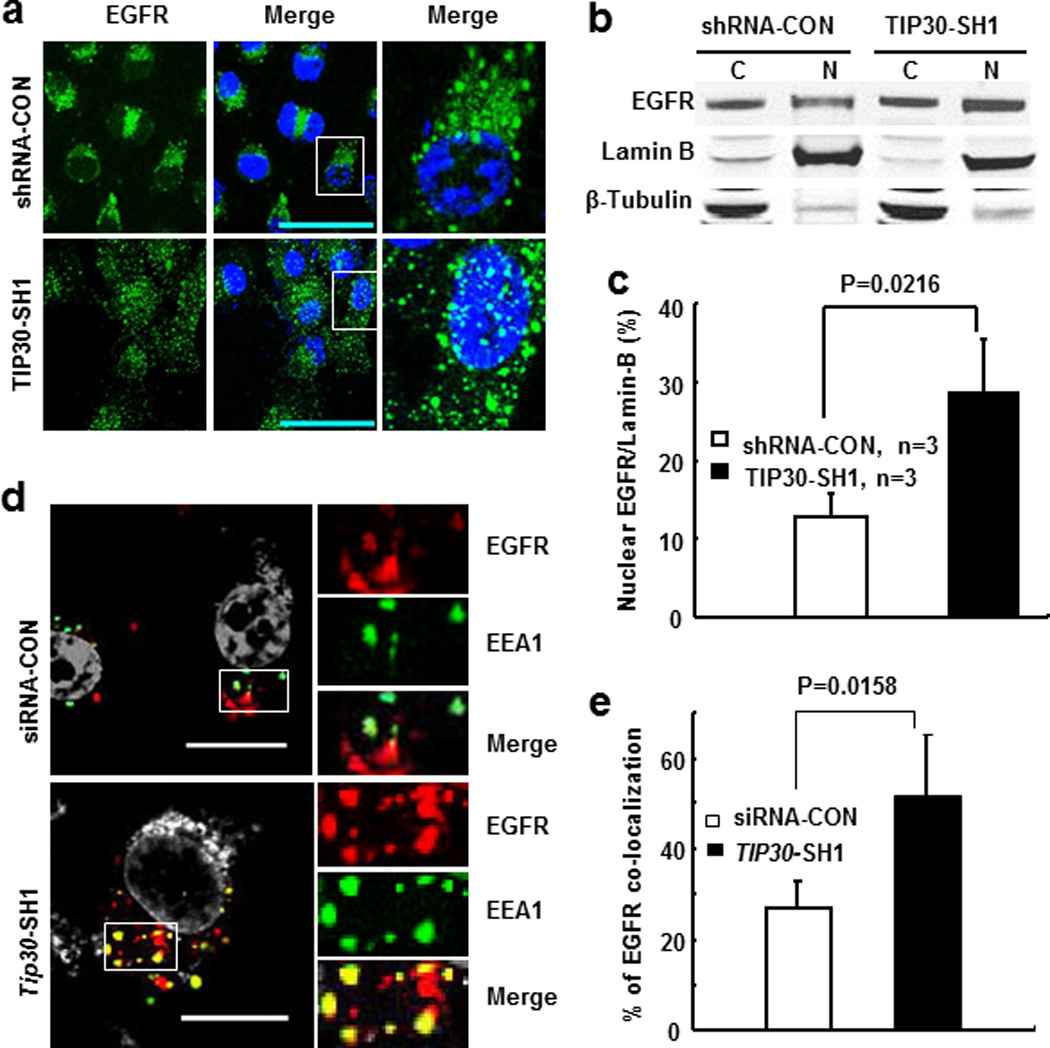
Effect of TIP30 knockdown on EGFR trafficking and nuclear localization inducing by EGF in A549 cells. (a) Representative images of EGFR (green) in human A549 cells. Cells were treated with 5 ng/ml EGF and 10 nM leptomycin B in DMEM for 2 hours were then stained for EGFR. Nuclei were stained by DAPI (blue). (b) Western blot analysis of EGFR nuclear localization. A549 cells were collected after EGF treatment for 3 hours. (c) Quantification of nuclear EGFR levels detected in Western blot analysis. Values are nuclear EGFR levels relative to Lamin-B levels. Data are represented as the means of three independent experiments. (d) Representative images show the localization of EGFR (red) in EEA-1 positive early endosomes (green) after 2 hours of EGF internalization. Yellow is due to overlap between red and green. Boxed areas are magnified. (e) Quantification of EGFR and EEA1 colocalization. * P < 0.05 versus control. Shown are representative of three experiments. Scale bars, 10 µm.
Reduced TIP30 expression correlates with better overall survival (OS) and post progression survival (PPS) of patients with stage I or II lung adenocarcinomas
To test the clinical relevance of these findings to human lung adenocarcinomas, we first attempted to determine whether TIP30 expression levels in lung adenocarcinomas could predict the prognosis of patients with this type of cancer using a previously published dataset of human lung adenocarcinoma tissue microarrays (22). This dataset contains gene expression profiles from 292 patients with lung adenocarcinomas at stage I or II and at least 60 months of follow-up if survival. Patients were divided into 2 groups: patients with high TIP30 mRNA levels (above or equal the median levels) and patients with low TIP30 mRNA levels (below the median levels). Kaplan-Meier analysis revealed that patients with low TIP30 expression showed significantly better outcome in OS than those with high TIP30 expression (Figure 6a, log rank test, P = 0.0270); This correlation was even more obvious when the OS times for the highest and lowest quartiles were analyzed (Supplementary Figure S8a, p=0.0209). Importantly, although no statistically significant difference in progression-free survival was identified (Supplementary Figure S8b), there was a significant difference of PPS between patients with high and low TIP30 expression levels (Figure 6b, long rank test, P = 0.0093). Patients with low TIP30 expression had a median time of 16.3 months from recurrence to death, whereas patients with high TIP30 expression had only a median time of 7.0 months from recurrence to death. These results suggest that decreased TIP30 expression is associated with better OS and PPS in lung adenocarcinoma patients at stage I and II.
Figure 6.
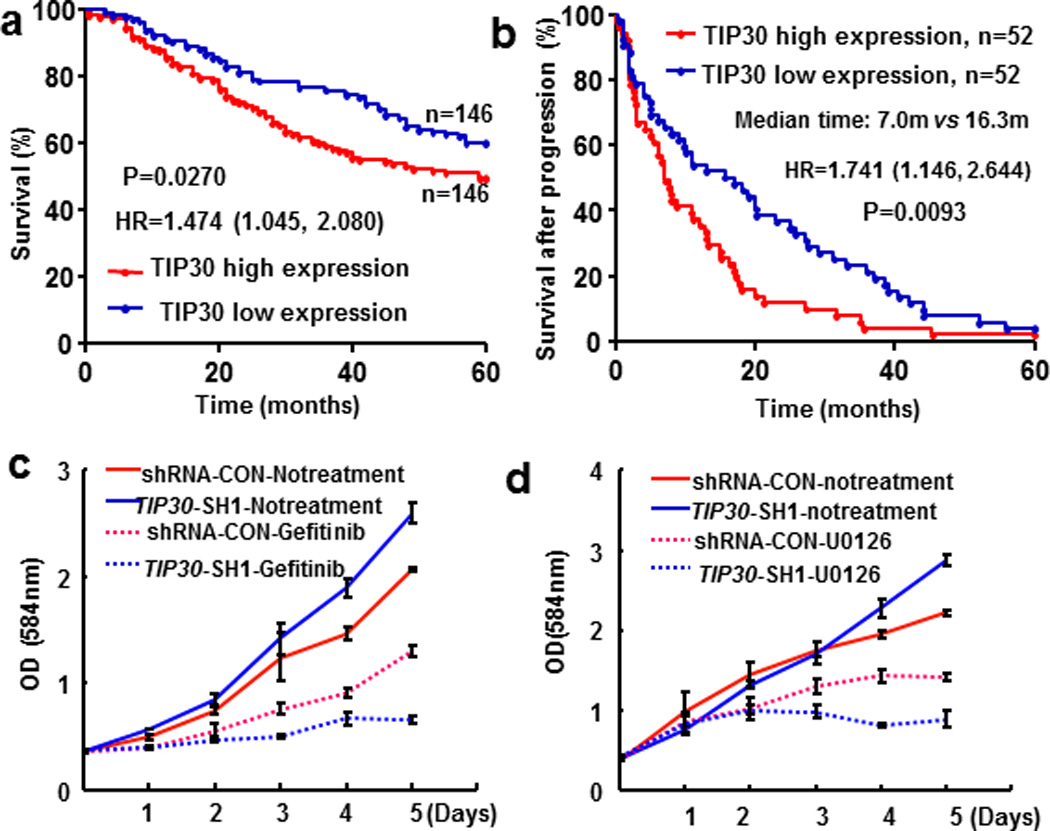
Effects of decreased TIP30 expression on human lung adenocarcinoma. (a–b) Correlation of TIP30 mRNA expression with OS and PPS of lung adenocarcinoma patients. Kaplan-Meier analysis was used to assess survival of stage I/II lung cancer patients from the NCI’s caArray database. (c) Growth curves of shRNA-CON and TIP30-SH1 cells treated with or without gefitinib. Cells were exposed to 10 µM gefitinib for indicating times. (d) Growth curves of shRNA-CON and Tip30-SH1 cells treated with or without U0126. Cells were exposed to 500nM U0126 for indicating times. Data were mean ± SEM of three independent experiments.
Because the associations of decreased TIP30 expression with better OS and PPS for these patients could be due to their better responses to the adjuvant therapies after surgery, we evaluated the inhibitory effects of EGFR and ERK inhibitors, gefitinib and U0126 on growth of shRNA control and TIP30 knockdown A549 and H322 cells. We observed that TIP30 knockdown significantly increased the growth inhibitory effects of EGFR and ERK inhibitors, gefitinib and U0126 on A549 cells as well as gefitinib on H322 cells (Figures 6c and d, Supplementary Figures S9 and 10). This result implies that EGFR signaling is essential for the proliferation of lung tumor cells with decreased TIP30 expression, so that EGFR targeting therapy might be effective for treating TIP30-deficient lung adenocarcinoma.
Discussion
Inactivation of tumor suppressor gene(s) in cells can trigger the onset of tumorigenesis. Despite emerging evidence that has linked decreased expression of TIP30 to the progression and metastasis of human NSCLC (17), whether TIP30 loss causes the initiation of NSCLC had not been determined before. In this study, we demonstrate for the first time that Balb/c mice lacking Tip30 are prone to spontaneous development of lung adenomas and adenocaricnomas. Our current data, coupled with previous findings on the role of TIP30 in the pathogenesis of lung cancers, strongly suggest that TIP30 functions as a tumor suppressor in the lung. To our knowledge, this is the first mouse model of lung adenocarcinoma with enhanced EGFR signaling that was created by conventionally knocking out a tumor suppressor gene. Pulmonary adenocarcinomas arising in Tip30−/− mouse model appeared reminiscent of human mixed papillar and solid subtype of adenocarcinoma. Thus, this model is suitable for studies on the mechanism of development of lung adenocarcinomas with enhanced EGFR signaling.
Our study provides new insight into the molecular mechanism by which TIP30 deletion promotes pulmonary tumorigenesis. Previous studies have established that ectopically increased or decreased expression of TIP30 in immortalized fibroblasts and various human cancer cells significantly influences multiple cellular processes including cell proliferation, apoptosis, DNA damage repair, metabolic adaptation and metastasis (15, 17, 23, 24). TIP30-mediated regulations can be explained by two actions: TIP30 may mediate these processes in the nucleus through direct effects on transcription of genes (25, 26), and indirectly influence gene expression in the cytoplasm through altering signal transduction pathways or inhibiting importin β-mediated nuclear import (27, 28). These views are supported by the observations that TIP30 directly regulates transcription of c-Myc and osteopontin genes (26, 28). Recently, we reported that TIP30 forms a protein complex to regulate EGFR signaling through controlling EGFR endocytic degradation in hepatocytes (21), and that Tip30 loss accelerates mammary tumor development by enhancing EGFR signaling in MMTV-Neu mouse model (20). Moreover, TIP30 inhibition could impair the localizations of Rab5a and V-ATPases in endosomes, leading to the trapping of EGF-EGFR complexes in endocytic vesicles and enhanced EGFR signaling (20). In agreement with these findings, the current study demonstrates that TIP30 knockdown in lung adenocarcinoma cells also leads to sustained EGFR in early endosomes, delayed EGFR endocytic degradation and enhanced EGFR signaling. Importantly, our results indicate that TIP30 is a critical factor in the regulation of EGFR nuclear localization. We speculate that inhibition of TIP30 may block EGFR trafficking to the lysosome and increase EGFR nuclear import (27), thereby increasing EGFR nuclear localization and expression of its two direct targets, CyclinD1 and PCNA. Since nuclear EGFR has been documented to play an important role in the pathogenesis of human cancers (29, 30), these data suggest that Tip30 deletion-induced lung tumorigenesis is driven, at least in part, by increasing both EGFR cytoplasmic and nuclear signaling pathways.
Understanding the cellular origin of lung adenocarcinoma is critical to the development of new therapeutic strategies for human lung cancers. Although whether BASCs are lung stem cells remains debated, a growing body of evidences has suggested BASCs as the origin of lung adenocarcinoma. In support of this view, BASCs were progressively expanded in several mouse models of lung adenocarcinomas (4, 19, 31–33). Similarly, Tip30−/− mice also exhibited expansion and proliferation of BASCs as well as differentiated AT2 cells in the lung, and developed SP-C positive alveolar cell tumors, most in the distal axis of the lung. However, in contrast to K-Ras mouse models that develop both airway and alveolar hyperplasia as well as tumors containing SP-C and CC10 positive cells in the lung (4), Tip30−/− mice only developed pulmonary alveolar hyperplasia and SP-C positive tumors. Moreover, Tip30 deletion preferentially influenced on the proliferation of SP-C positive alveolar cells, as Tip30 promoter was predominantly activated in SP-C positive AT2 cells. While these findings suggest TIP30 as a growth regulator of BASC and AT2 cells, they raise the intriguing possibility that Tip30 deletion may exploit a dysregulated TIP30-mediated pathway to promote differentiation of BASCs to AT2 cells and predispose AT2 cells to malignant transformation. The precise molecular mechanism by which Tip30 deletion promotes BASCs to AT2 differentiation remains to be determined. Nevertheless, our data has not demonstrated BASCs as the cells of origin of tumors arising in Tip30−/− mice in the discussion.
Our findings may have important implication for the evaluation of prognosis and selection of therapeutic strategies for human lung adenocarcinomas, as patients with low TIP30 expression lung adenocarcinomas exhibited better outcomes in OS and PPS (Figures 6a and b). Clearly, these findings need to be validated in larger prospective studies and identify underlying mechanisms of the outcomes. Although we do not know why low TIP30 expression correlates with better outcomes in OS and PPS, we speculate that these should be due to their better responses to anticancer therapies. To date, it is well known that NSCLC patients with mutant EGFR confer greater sensitivity to EGFR inhibitors than those with wild type EGFR (34–36). However, approximately 1–20% of patients without detectable EGFR mutations in clinical trials showed a radiographic response to EGFR inhibitors (37–39). Moreover, patients with either high EGFR gene copy number or protein levels showed higher response to EGFR inhibitors than patients with negative or low EGFR expression (9, 40). These observations suggest that a subset of lung adenocarcinomas with enhanced EGFR signaling caused by other genetic alternations may also respond to EGFR inhibitors. It is plausible that this subgroup of patients would derive the most benefit from treatments targeting EGFR signaling in the neoadjuvant, adjuvant, and metastatic settings. Nevertheless, we do not exclude the possibility that patients with low TIP30 expression lung adenocarcinomas might also respond better to adjuvant chemotherapies or radiotherapies (Supplementary Figures S8c and d). Thus, the finding of the better OS and PPS for the patients with low TIP30 expression lung adenocarcinomas is not disagreement with TIP30 as a lung tumor suppressor, but rather is consistent with the mechanism whereby TIP30 suppresses lung tumorigenesis partly through the negative regulation of EGFR signaling. Regardless of the mechanisms, the results reported here imply that decreased TIP30 expression may define a distinct molecular subset of lung adenocarcinomas that are sensitive to EGFR inhibitors, thereby helping guide the administration of specific types of EGFR-targeted therapy. Therefore, our findings warrant further investigation into the exploitation of TIP30 expression as an additional marker for predicting the response to EGFR inhibitors in the treatment of lung adenocarcinomas.
Materials and methods
Animals
Tip30−/− mice in Balb/c background were generated by backcrossing the Tip30 knockout gene into Balb/c mice for seven generations as described previously (14). Mice were housed in the Animal Facility at Michigan State University. All animal experiments were carried out in accordance with a protocol approved by Michigan State University IACUC Committee.
Histopathology
The lungs of mice were inflated with PBS. Fresh lung tissues used for Western blot assay were frozen with liquid nitrogen after lavage with PBS and stored at −80°C. Lung and other tissues for H&E staining were fixed with 4% paraformaldehyde solution, and then paraffin-embedded. Histopathology of lung sections was reviewed according to the classification and criteria recommended by the mouse models of human cancers consortium by A. L and H. X. and confirmed by Dr. Ying Qin, Pathologist who read slides blind to experimental detail (41).
Cell culture and shRNAs
Human lung cancer cell lines A549, NCI-H322 and human immortalized lung cell line BEAS-2B were obtained from American Type Culture Collection (ATCC) and cultured in DMEM (Invitrogen) supplemented with10% FBS and 1% penicillin/streptomycin at 37° in 5% CO2 incubator. Lentiviral plasmids producing shRNAs against TIP30 were purchased from Sigma-Aldrich as described previously (20, 21).
Antibodies and chemicals
Rabbit anti-SP-C, anti-CC10, and anti-cyclin D1 were from Millipore (Temecula,CA). Mouse anti-CC10 and anti-PCNA were from Santa Cruz Biotechnology (Santa Cruz, CA). U0126, rabbit anti-EGFR, anti-pAkt(473), anti-Akt, anti-pErk1/2, anti-Erk1/2, and mouse anti-EEA1 were from Cell Signaling Technology (Danvers, MA). Antigen-purified rabbit anti-TIP30 was produced by our laboratory (13). Mouse anti-β-Actin (AC-15) was from Sigma.DAPI, Biotin conjugated goat anti-rabbit, anti-mouse second antibodies, and ABC kits for immunohistochemistry were from Vector Laboratories (Burlingame, CA). Mouse anti-β-Gal was from Promega (Madison, WI). Fluorescence-labeled secondary antibodies, donkey anti-mouse Alexa-594, chicken anti-rabbit Alexa-594, goat anti-rabbit Alexa-488, donkey anti-goat Alexa-488, were from Invitrogen (Eugene, Oregon). Donkey anti-rabbit IRDye800CW and donkey anti-mouse IRDye680 were from LI-COR Biosicences (Lincoln, Nebraska). Gefitinib was from AstraZeneca.
Immunohistochemistry and immunofluorescence
Immunohistochemistry was performed according to the protocol supplied by Cell Signaling Technology. Anti-SP-C, CC10, pAkt, p-ERK1/2, or cyclin D1 antibodies were used as described previously(20, 21). Immunofluorescent analysis of EGFR, SP-C, CC10, PCNA, and β-Gal were performed according to the protocol supplied by Cell Signaling Technology. Nuclei were counterstained with DAPI. 0.3% Sudan black B was used to reduce the autofluorescence of the tissue.
EGFR internalization and nuclear localization assay
EGFR internalization assay was carried out as described previously (21). Briefly, after pretreatment with 100 ng/ml EGF and 20 µg/ml cycloheximide on ice for 1 hour, A549 cells were incubated at 37 °C in DMEM containing 20 µg/ml cycloheximide for various times. EGFR nuclear localization assay was carried out as described (42). A549 cells were treated with 5 ng/ml EGF, 10 nM leptomycin B and 20 µg/ml cycloheximide in DMEM at 37°C for 2 hours.
Western blot
Lung issues prepared from 10-month-old mice and human lung adenocarcinoma A549 cells (ATCC, Rockville, MD) were lysed in RIPA buffer containing 1 mM DTT and protease inhibitors for the total protein extracts. Cytoplasmic and nuclear proteins were extracted following the manufacturer’s protocol (Nuclear Extraction Kit, Chemicon). For the detection of EGFR, p-ERK1/2, ERK1/2, p-Akt, Akt, β-Actin, in lung tissue and A549 cell lysates, aliquots containing 70 µg of protein for each sample were separated by electrophoresis on NuPAGE 4–12% Bis-Tris Gels (Invitrogen) and then subjected to Western blot analysis. The protein levels were quantified using Odyssey 2.1 software.
TUNEL assay
Apoptotic cells in mouse lungs were visualized using a TUNEL assay kit (Roche, Basel, Switzerland) according to the manufacture’s instruction. Numbers of TUNEL-positive cells in the lung were counted from five randomly selected high-power fields per sample.
Cell growth assay
shRNA-control, TIP30-SH1 or TIP30-SH2 A549 cells were seeded and cultured in 96-well plates (3000 cells per well). Cell viability was measured every day in triplicate for 5 days using cell count kit-8 (CCK-8, Dojindo Molecular technologies, Rockville, Maryland). Cells were incubated with 10 µl CCK-8 for 1 h at 37 °C, and absorbance was measured at 584 nm using a FLUOstar OPTIMA spectrophotometer.
Survival analysis
Microarray dataset were downloaded from the NCI’s caArray database involving 442 patients with human lung adenocarcinomas and clinical annotation (22). Overall survival of patients with lung adenocarcinomas at stage I or II was defined as the time interval from surgery to death or to the date of 5-year follow-up contact. Post progression survival was defined as the time interval from the first progression to death of patients with lung adenocarcinomas at stage I or II. The patients without documented progression date were excluded.
Statistics
The differences between groups with parametric distributions were analyzed using Student’s 2-tailed t test. Survival curves were compared using a Log-Rank (mantel-cox) test. Tumor incidences were compared using Chi-square test. Data represent mean ± SEM. P values of 0.05 or less were considered statistically significant.
Supplementary Material
Acknowledgments
We are grateful to Jill Pecha for backcrossing Tip30 knockout gene into Balb/c mice, Ying Qin and Zuguo Li for histological examination of preneoplastic lesions and tumors, Eran Andrechek and Inez Yuwanita for helping patient survival analysis, and Sandra Z. Haslam for critical reading of the manuscript. S.G and F.C equally contributed to this work. This work was supported by grants RO1 DK066110-01 and W81XWH-08-1-0377 (to H.X.) from NIDDK, NIH and DOD. A.L. is partly supported by a fellowship from Nanfang Hospital, China.
Abbreviations
- TIP30
30 kDa HIV-1 Tat interacting protein
- EGFR
epidermal growth factor receptor
Footnotes
Conflict of interest
The authors declare no competing financial interests.
References
- 1.Jackson EL, Olive KP, Tuveson DA, Bronson R, Crowley D, Brown M, et al. The differential effects of mutant p53 alleles on advanced murine lung cancer. Cancer Res. 2005;65:10280–10288. doi: 10.1158/0008-5472.CAN-05-2193. [DOI] [PubMed] [Google Scholar]
- 2.Tchou-Wong KM, Jiang Y, Yee H, LaRosa J, Lee TC, Pellicer A, et al. Lung-specific expression of dominant-negative mutant p53 in transgenic mice increases spontaneous and benzo(a)pyrene-induced lung cancer. Am J Respir Cell Mol Biol. 2002;27:186–193. doi: 10.1165/ajrcmb.27.2.4799. [DOI] [PubMed] [Google Scholar]
- 3.Fisher GH, Wellen SL, Klimstra D, Lenczowski JM, Tichelaar JW, Lizak MJ, et al. Induction and apoptotic regression of lung adenocarcinomas by regulation of a K-Ras transgene in the presence and absence of tumor suppressor genes. Genes Dev. 2001;15:3249–3262. doi: 10.1101/gad.947701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jackson EL, Willis N, Mercer K, Bronson RT, Crowley D, Montoya R, et al. Analysis of lung tumor initiation and progression using conditional expression of oncogenic K-ras. Genes Dev. 2001;15:3243–3248. doi: 10.1101/gad.943001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ehrhardt A, Bartels T, Geick A, Klocke R, Paul D, Halter R. Development of pulmonary bronchiolo-alveolar adenocarcinomas in transgenic mice overexpressing murine c-myc and epidermal growth factor in alveolar type II pneumocytes. Br J Cancer. 2001;84:813–818. doi: 10.1054/bjoc.2000.1676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ji H, Li D, Chen L, Shimamura T, Kobayashi S, McNamara K, et al. The impact of human EGFR kinase domain mutations on lung tumorigenesis and in vivo sensitivity to EGFR-targeted therapies. Cancer Cell. 2006;9:485–495. doi: 10.1016/j.ccr.2006.04.022. [DOI] [PubMed] [Google Scholar]
- 7.Politi K, Zakowski MF, Fan PD, Schonfeld EA, Pao W, Varmus HE. Lung adenocarcinomas induced in mice by mutant EGF receptors found in human lung cancers respond to a tyrosine kinase inhibitor or to down-regulation of the receptors. Genes Dev. 2006;20:1496–1510. doi: 10.1101/gad.1417406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Berger AH, Niki M, Morotti A, Taylor BS, Socci ND, Viale A, et al. Identification of DOK genes as lung tumor suppressors. Nat Genet. 2010;42:216–223. doi: 10.1038/ng.527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tsao MS, Sakurada A, Cutz JC, Zhu CQ, Kamel-Reid S, Squire J, et al. Erlotinib in lung cancer - molecular and clinical predictors of outcome. N Engl J Med. 2005;353:133–144. doi: 10.1056/NEJMoa050736. [DOI] [PubMed] [Google Scholar]
- 10.Rusch V, Baselga J, Cordon-Cardo C, Orazem J, Zaman M, Hoda S, et al. Differential expression of the epidermal growth factor receptor and its ligands in primary non-small cell lung cancers and adjacent benign lung. Cancer Res. 1993;53:2379–2385. [PubMed] [Google Scholar]
- 11.Gazdar AF, Minna JD. Deregulated EGFR signaling during lung cancer progression: mutations, amplicons, and autocrine loops. Cancer Prev Res (Phila) 2008;1:156–160. doi: 10.1158/1940-6207.CAPR-08-0080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Shtivelman E. A link between metastasis and resistance to apoptosis of variant small cell lung carcinoma. Oncogene. 1997;14:2167–2173. doi: 10.1038/sj.onc.1201059. [DOI] [PubMed] [Google Scholar]
- 13.Xiao H, Tao Y, Greenblatt J, Roeder RG. A cofactor, TIP30, specifically enhances HIV-1 Tat-activated transcription. Proc Natl Acad Sci U S A. 1998;95:2146–2151. doi: 10.1073/pnas.95.5.2146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ito M, Jiang C, Krumm K, Zhang X, Pecha J, Zhao J, et al. TIP30 deficiency increases susceptibility to tumorigenesis. Cancer Res. 2003;63:8763–8767. [PubMed] [Google Scholar]
- 15.Zhao J, Ni H, Ma Y, Dong L, Dai J, Zhao F, et al. TIP30/CC3 expression in breast carcinoma: relation to metastasis, clinicopathologic parameters, and P53 expression. Human Pathology. 2007;38:293–298. doi: 10.1016/j.humpath.2006.08.005. [DOI] [PubMed] [Google Scholar]
- 16.Chen X, Cao X, Dong W, Luo S, Suo Z, Jin Y. Expression of TIP30 tumor suppressor gene is down-regulated in human colorectal carcinoma. Dig Dis Sci. 2006;55:2219–2226. doi: 10.1007/s10620-009-0992-0. [DOI] [PubMed] [Google Scholar]
- 17.Tong X, Li K, Luo Z, Lu B, Liu X, Wang T, et al. Decreased TIP30 Expression Promotes Tumor Metastasis in Lung Cancer. The American Journal of Pathology. 2009;174:1931–1939. doi: 10.2353/ajpath.2009.080846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pecha J, Ankrapp D, Jiang C, Tang W, Hoshino I, Bruck K, et al. Deletion of Tip30 leads to rapid immortalization of murine mammary epithelial cells and ductal hyperplasia in the mammary gland. Oncogene. 2007;26:7423–7431. doi: 10.1038/sj.onc.1210548. [DOI] [PubMed] [Google Scholar]
- 19.Kim CF, Jackson EL, Woolfenden AE, Lawrence S, Babar I, Vogel S, et al. Identification of bronchioalveolar stem cells in normal lung and lung cancer. Cell. 2005;121:823–835. doi: 10.1016/j.cell.2005.03.032. [DOI] [PubMed] [Google Scholar]
- 20.Zhang C, Mori M, Gao S, Li A, Hoshino I, Aupperlee MD, et al. Tip30 deletion in MMTV-Neu mice leads to enhanced EGFR signaling and development of estrogen receptor-positive and progesterone receptor-negative mammary tumors. Cancer Res. 2010;70:10224–10233. doi: 10.1158/0008-5472.CAN-10-3057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhang C, Li A, Zhang X, Xiao H. A novel TIP30 protein complex regulates EGF receptor signaling and endocytic degradation. J Biol Chem. 2011;286:9373–9381. doi: 10.1074/jbc.M110.207720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Shedden K, Taylor JM, Enkemann SA, Tsao MS, Yeatman TJ, Gerald WL, et al. Gene expression-based survival prediction in lung adenocarcinoma: a multi-site, blinded validation study. Nat Med. 2008;14:822–827. doi: 10.1038/nm.1790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chen V, Shtivelman E. CC3/TIP30 regulates metabolic adaptation of tumor cells to glucose limitation. Cell Cycle. 2010;9:4941–4953. doi: 10.4161/cc.9.24.14230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fong S, King F, Shtivelman E. CC3/TIP30 affects DNA damage repair. BMC Cell Biol. 2010;11:23. doi: 10.1186/1471-2121-11-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Xiao H, Palhan V, Yang Y, Roeder RG. TIP30 has an intrinsic kinase activity required for up-regulation of a subset of apoptotic genes. EMBO J. 2000;19:956–963. doi: 10.1093/emboj/19.5.956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jiang C, Ito M, Piening V, Bruck K, Roeder RG, Xiao H. TIP30 interacts with an estrogen receptor alpha-interacting coactivator CIA and regulates c-myc transcription. J Biol Chem. 2004;279:27781–27789. doi: 10.1074/jbc.M401809200. [DOI] [PubMed] [Google Scholar]
- 27.King FW, Shtivelman E. Inhibition of nuclear import by the proapoptotic protein CC3. Mol Cell Biol. 2004;24:7091–7101. doi: 10.1128/MCB.24.16.7091-7101.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhao J, Lu B, Xu H, Tong X, Wu G, Zhang X, et al. Thirty-kilodalton Tat-interacting protein suppresses tumor metastasis by inhibition of osteopontin transcription in human hepatocellular carcinoma. Hepatology. 2008;48:265–275. doi: 10.1002/hep.22280. [DOI] [PubMed] [Google Scholar]
- 29.Lin S-Y, Makino K, Xia W, Matin A, Wen Y, Kwong KY, et al. Nuclear localization of EGF receptor and its potential new role as a transcription factor. Nat Cell Biol. 2001;3:802–808. doi: 10.1038/ncb0901-802. [DOI] [PubMed] [Google Scholar]
- 30.Wang SC, Nakajima Y, Yu YL, Xia W, Chen CT, Yang CC, et al. Tyrosine phosphorylation controls PCNA function through protein stability. Nat Cell Biol. 2006;8:1359–1368. doi: 10.1038/ncb1501. [DOI] [PubMed] [Google Scholar]
- 31.Besson A, Hwang HC, Cicero S, Donovan SL, Gurian-West M, Johnson D, et al. Discovery of an oncogenic activity in p27Kip1 that causes stem cell expansion and a multiple tumor phenotype. Genes Dev. 2007;21:1731–1746. doi: 10.1101/gad.1556607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Morimoto M, Kopan R. rtTA toxicity limits the usefulness of the SP-C-rtTA transgenic mouse. Dev Biol. 2009;325:171–178. doi: 10.1016/j.ydbio.2008.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yanagi S, Kishimoto H, Kawahara K, Sasaki T, Sasaki M, Nishio M, et al. Pten controls lung morphogenesis, bronchioalveolar stem cells, and onset of lung adenocarcinomas in mice. J Clin Invest. 2007;117:2929–2940. doi: 10.1172/JCI31854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lynch TJ, Bell DW, Sordella R, Gurubhagavatula S, Okimoto RA, Brannigan BW, et al. Activating mutations in the epidermal growth factor receptor underlying responsiveness of non-small-cell lung cancer to gefitinib. N Engl J Med. 2004;350:2129–2139. doi: 10.1056/NEJMoa040938. [DOI] [PubMed] [Google Scholar]
- 35.Paez JG, Janne PA, Lee JC, Tracy S, Greulich H, Gabriel S, et al. EGFR mutations in lung cancer: correlation with clinical response to gefitinib therapy. Science. 2004;304:1497–1500. doi: 10.1126/science.1099314. [DOI] [PubMed] [Google Scholar]
- 36.Sun S, Schiller JH, Spinola M, Minna JD. New molecularly targeted therapies for lung cancer. J Clin Invest. 2007;117:2740–2750. doi: 10.1172/JCI31809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pao W, Chmielecki J. Rational, biologically based treatment of EGFR-mutant non-small-cell lung cancer. Nat Rev Cancer. 2010;10:760–774. doi: 10.1038/nrc2947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mok TS, Wu YL, Thongprasert S, Yang CH, Chu DT, Saijo N, et al. Gefitinib or carboplatin-paclitaxel in pulmonary adenocarcinoma. N Engl J Med. 2009;361:947–957. doi: 10.1056/NEJMoa0810699. [DOI] [PubMed] [Google Scholar]
- 39.Yang CH, Yu CJ, Shih JY, Chang YC, Hu FC, Tsai MC, et al. Specific EGFR mutations predict treatment outcome of stage IIIB/IV patients with chemotherapy-naive non-small-cell lung cancer receiving first-line gefitinib monotherapy. J Clin Oncol. 2008;26:2745–2753. doi: 10.1200/JCO.2007.15.6695. [DOI] [PubMed] [Google Scholar]
- 40.Cappuzzo F, Hirsch FR, Rossi E, Bartolini S, Ceresoli GL, Bemis L, et al. Epidermal growth factor receptor gene and protein and gefitinib sensitivity in non-small-cell lung cancer. J Natl Cancer Inst. 2005;97:643–655. doi: 10.1093/jnci/dji112. [DOI] [PubMed] [Google Scholar]
- 41.Nikitin AY, Alcaraz A, Anver MR, Bronson RT, Cardiff RD, Dixon D, et al. Classification of proliferative pulmonary lesions of the mouse: recommendations of the mouse models of human cancers consortium. Cancer Res. 2004;64:2307–2316. doi: 10.1158/0008-5472.can-03-3376. [DOI] [PubMed] [Google Scholar]
- 42.Bitler BG, Goverdhan A, Schroeder JA. MUC1 regulates nuclear localization and function of the epidermal growth factor receptor. J Cell Sci. 2010;123:1716–1723. doi: 10.1242/jcs.062661. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.