

Background: AMPK phosphorylates CFTR and inhibits PKA-stimulated CFTR channel gating by unclear mechanisms.
Results: NDPK-A, AMPK, and CFTR exist in a membrane-associated complex. AMPK-CFTR binding and NDPK-A catalytic function are required for CFTR inhibition by AMPK.
Conclusion: NDPK-A plays an integral role in the regulation of CFTR by AMPK.
Significance: Targeting the AMPK-CFTR interaction and NDPK-A function could yield new therapeutic strategies for CF.
Keywords: AMP-activated Kinase (AMPK), CFTR, Chloride Transport, Ion Channels, Oocyte, Patch Clamp Electrophysiology, Phosphorylation, Calu-3 Cells, Nm23, Nucleoside Diphosphate Kinase
Abstract
Cystic fibrosis transmembrane conductance regulator (CFTR) Cl− channel mutations cause cystic fibrosis lung disease. A better understanding of CFTR regulatory mechanisms could suggest new therapeutic strategies. AMP-activated protein kinase (AMPK) binds to and phosphorylates CFTR, attenuating PKA-activated CFTR gating. However, the requirement for AMPK binding to CFTR and the potential role of other proteins in this regulation are unclear. We report that nucleoside diphosphate kinase A (NDPK-A) interacts with both AMPK and CFTR in overlay blots of airway epithelial cell lysates. Binding studies in Xenopus oocytes and transfected HEK-293 cells revealed that a CFTR peptide fragment that binds AMPK (CFTR-1420-57) disrupted the AMPK-CFTR interaction. Introduction of CFTR-1420-57 into human bronchial Calu-3 cells enhanced forskolin-stimulated whole cell conductance in patch clamp measurements. Similarly, injection of CFTR-1420-57 into Xenopus oocytes blocked the inhibition of cAMP-stimulated CFTR conductance by AMPK in two-electrode voltage clamp studies. AMPK also inhibited CFTR conductance with co-expression of WT NDPK-A in two-electrode voltage clamp studies, but co-expression of a catalytically inactive H118F mutant or various Ser-120 NDPK-A mutants prevented this inhibition. In vitro phosphorylation of WT NDPK-A was enhanced by purified active AMPK, but phosphorylation was prevented in H118F and phosphomimic Ser-120 NDPK-A mutants. AMPK does not appear to phosphorylate NDPK-A directly but rather promotes an NDPK-A autophosphorylation event that involves His-118 and Ser-120. Taken together, these results suggest that NDPK-A exists in a functional cellular complex with AMPK and CFTR in airway epithelia, and NDPK-A catalytic function is required for the AMPK-dependent regulation of CFTR.
Introduction
The CFTR4 Cl− channel is localized to the apical membrane of many epithelial tissues including the airway, where the most serious clinical manifestations of cystic fibrosis (CF) occur (1). CF-associated CFTR mutations cause abnormalities in epithelial solute transport and fluid secretion with excessive airway surface liquid reabsorption and inflammation, poor mucociliary clearance, and subsequent life-threatening infections and eventual destruction of lung tissue (2). Excessive CFTR activation in the gut underlies the secretory diarrhea in patients with cholera (3). CFTR activity also plays an important role in the pathogenesis of autosomal dominant polycystic kidney disease (4). CFTR is thus directly involved in the pathophysiology of diseases that cause excess morbidity and mortality in millions of individuals worldwide each year (5). A more thorough understanding of CFTR regulation could yield important clinical benefits, including therapies to boost or inhibit inappropriate levels of CFTR activity.
CFTR is an ATP-binding cassette family transport protein with five major cytoplasmic domains, including two nucleotide-binding domains, a regulatory R domain containing numerous phosphorylation sites, and NH2- and COOH-terminal cytoplasmic tails that interact with various proteins and help regulate the gating and trafficking of the channel (6–8). CFTR activation requires PKA-dependent phosphorylation of the R-domain and ATP binding/hydrolysis at the nucleotide-binding domains (9, 10). Of note, CFTR Cl− channel activity is down-regulated in epithelia in response to metabolic stress, but the specific mechanisms involved are unclear (11, 12). An important link that we have established between metabolic state and ion transport activity is the metabolic sensor AMPK.
AMPK is a highly conserved, ubiquitously distributed Ser/Thr kinase that exists as a heterotrimer with a catalytic α subunit and regulatory β and γ subunits. Each subunit has multiple isoforms with differing tissue distributions (13). AMPK becomes activated in response to metabolic and other cellular stresses and has been shown to phosphorylate a variety of target proteins with pleiotropic effects, including modulation of cellular metabolic pathways, inflammation, cell growth and proliferation, protein synthesis, and gene transcription (14). AMPK-α binds to the COOH-terminal tail of CFTR at residues 1420–1457, phosphorylates CFTR at residue Ser-768 in the R domain, and inhibits PKA-stimulated gating of CFTR by decreasing its open probability (Po) in cells (15–18). AMPK may thus limit CFTR activity in the absence of agonist stimulation. The regulation of CFTR, an ATP-gated channel, by AMPK is consistent with the known homeostatic role of AMPK in maintaining energy stores by inhibiting energy-consuming processes and activating energy-generating processes in the cell under conditions of metabolic stress. However, further molecular details of this regulation, including the importance of AMPK binding to CFTR and the potential involvement of additional proteins, are unknown.
Early studies performed in Xenopus oocytes suggested that both the COOH-terminal half of the AMPK-α1 catalytic subunit, the region of AMPK that interacts with CFTR, and an active kinase domain within AMPK-α1 were required for AMPK-dependent regulation of CFTR (15). However, the role of AMPK binding to CFTR has not been explicitly tested in cells. Additionally, the potential role of other accessory proteins in the AMPK-dependent regulation of CFTR is unclear. Along these lines, the nucleoside diphosphate kinase A (NDPK-A) has been recently reported to interact and co-localize with AMPK in respiratory tract epithelium, and there may be an important functional inter-regulation between these two kinases (19).
Like AMPK, NDPK (also known as Nm23 and NME) is a multifunctional, ubiquitously distributed hexameric histidine kinase with multiple isoforms (including NDPK-A, B, C, and D in humans; group I NDPK isoforms) (20). NDPKs catalyze the transfer of phosphates from nucleoside triphosphates to nucleoside diphosphates and are involved in regulating a variety of fundamental cellular processes, including growth and development, G-protein signaling, metastasis, transcriptional regulation, and DNA repair (20). NDPK catalytic function requires autophosphorylation at the catalytic His-118 residue. The nearby Ser-120 residue may also be a target for phosphorylation, which appears to play an important role in determining NDPK function, because Ser-120 mutations affect motility and the metastatic potential of cancer cells (19, 21, 22).
The studies conducted herein were performed to better define the mechanisms of CFTR inhibition by AMPK. The role of AMPK binding to CFTR was tested by introduction of a blocking peptide corresponding to the tract in CFTR that binds AMPK (CFTR-1420-57) into cells prior to biochemical and electrophysiological studies. The role of NDPK-A in the regulation of CFTR by AMPK was tested through expression of WT and mutant NDPK-A in cells for electrophysiological studies. Finally, the AMPK-dependent regulation of NDPK-A phosphorylation was further clarified.
EXPERIMENTAL PROCEDURES
Chemicals and Reagents
All chemicals and reagents were obtained from Sigma unless otherwise stated.
Overlay Assays
Human bronchial epithelial (16HBE14o−) cells were cultured in medium 199 containing FBS as described (23) until confluent. Proteins separated by SDS-PAGE were transferred to a PVDF membrane (Millipore) by semi-dry electrophoretic transfer, and prestained markers were used to confirm transfer. The blots were probed with primary antibodies to NDPK-A (1:1000), rabbit polyclonal antibody raised to nm23-H1 (Santa Cruz); or AMPK-α (1:5000), rabbit polyclonal antibody, gifts of D. Carling (MRC Hammersmith London) and D. G. Hardie (Dundee); or CFTR (1:1000), monoclonal antibody, R & D Systems (Minneapolis); or CFTR (1:1000), monoclonal antibody, transgene MATG103 (Strasbourg) and HRP-conjugated anti-rabbit secondary antibody (1:2000) followed by SuperSignal chemiluminescence detection (Pierce). NDPK was partially purified from a membrane fraction obtained from ovine tracheal epithelium using a chelating Sepharose fast flow column (20 ml) charged with ferric chloride. The membrane fraction solubilized with 0.25% (v/v) Triton X-100 in buffer A (0.1 m acetic acid/NaOH, pH 5.0, 0.5 m NaCl, 0.4 mm 4-(2-aminoethyl)-benzol-sulfonylfluoride) was centrifuged at 100,000 × g for 30 min at 4 °C. The supernatant fraction was incubated at 4 °C for 1 min with ATP (50 nm) and then applied to a chelating Sepharose fast flow column pre-equilibrated with buffer A. The column was then washed with buffer A, and bound phosphoproteins were selectively eluted with 20 mm sodium biphosphate in buffer A.
Membrane proteins (100–500 μg) were resuspended in 1 ml of immunoprecipitation (IP) buffer (10 mm Tris-HCl, pH 7.4, containing 2 mm EDTA, 1 mm NaF, 1 mm DTT, 1% (w/v) sodium deoxycholate, 1% v/v Nonidet P-40, 0.3 m aprotonin, 0.2 m PMSF). The mixture was then precleared with protein G-Sepharose beads (30 min at 4 °C) and centrifuged at 4 °C at 350 × g for 5 min, and the supernatant was incubated with primary antibody to CFTR (1 μg) for 1 h at 4 °C. New beads were added, and the mixture was incubated overnight at 4 °C. The mixture was then centrifuged at 350 × g for 5 min. The pelleted beads were washed five times in 1 ml of IP buffer containing 1 m NaCl. The pelleted beads were then washed twice in 10 mm MOPS buffer, pH 7.9, containing 0.05% (v/v) Triton X-100. For the overlay assays, proteins from ovine tracheal epithelial membranes solubilized in 10 mm MOPS buffer, pH 7.9 containing Chaps detergent (0.1% w/v) were then separated by SDS-PAGE and blotted onto PVDF membranes. NDPK-A (100 μg protein) in 1× TBS containing 5% (w/v) nonfat dry milk was then overlaid onto the PVDF and incubated at room temperature for 1 h. Following washes with 1× TBS with 0.1% (v/v) Tween 20 (four times for 15 min each), the blots were probed with antibodies to NDPK-A (1:1000) and AMPK (1:5000).
Co-immunoprecipitation Studies
Co-IP experiments were performed on lysates from Xenopus oocytes injected with cRNAs to express human CFTR and HA-tagged rat AMPK-α1 (5 ng/oocyte of each cRNA) 2 days prior to experimentation. A peptide corresponding to residues 1420–1457 of CFTR (CFTR-1420-57) was synthesized by the University of Pittsburgh Genomics and Proteomics Core Laboratories. This peptide (or vehicle) was microinjected into the oocytes 2–4 h prior to lysis. CFTR was immunoprecipitated (IP'd) from lysates (∼50 oocytes/condition) using the M24-1 anti-CFTR mouse monoclonal antibody (2.5 μg/IP; R & D Systems). Following SDS-PAGE and transfer to a nitrocellulose membrane, immunoblotting was performed using HA.11 (Covance; 1:500) anti-HA and M3A7 (Millipore; 1:700) anti-CFTR mouse monoclonal antibodies. Whole cell lysate samples (5–10% of input) were run alongside co-IP lanes, and bands were quantitated by densitometry to determine the percentage of IP'd and co-IP'd proteins for various conditions, with corrections for differences in exogenous protein expression levels, as described previously (24). Paired t tests were performed to determine the significance of differences between blocking peptide-treated samples and controls.
GST Pulldown Assays in HEK-293 Cells
HEK-293 cells were co-transfected 1 day after plating using Lipofectamine 2000 (Invitrogen) with plasmids to express CFTR and either NH2-terminal GST-tagged AMPK-α1 or GST alone 2 days prior to lysis for in vivo GST pulldown assays, based on previously described methods (15). On the day between transfection and lysis, 100 μm CFTR-1420-57 peptide (or vehicle) was transduced into cells using the BioPORTER® reagent (Genlantis) as per the manufacturer's recommendations. After washes in ice-cold PBS, the cells were lysed with ice-cold lysis buffer (PBS containing 1% Triton X-100, 2 mm EDTA, and complete protease inhibitor mixture (Roche Applied Science)), and total protein concentration was estimated using the Bradford assay. Lysate samples (∼1 mg of total cellular protein lysate/condition in equal volumes) were then incubated with glutathione-Sepharose beads on a rotator overnight at 4 °C to bind GST fusion proteins and any interacting proteins. After washing the beads in lysis buffer, elution at 65 °C for 15 min in sample buffer, SDS-PAGE, and transfer to nitrocellulose membranes, immunoblotting was performed for either CFTR or GST using M3A7 or GST-HRP antibodies, respectively. Whole cell lysate samples (5% of input) were run alongside GST pulldown samples, and the bands were quantitated by densitometry to determine relative binding of AMPK to CFTR, as described above.
Whole Cell Patch Clamp Studies
Calu-3 cells were maintained in 1:1 DMEM/F-12 medium, 10% FBS, and 1% penicillin/streptomycin, and measurements of CFTR whole cell conductance were performed as described (16). Two days prior to patch clamp experiments, the cells were plated onto 25-mm glass coverslips in 35-mm plastic dishes at a concentration of 8–10 × 105 cells/dish. CFTR-1420-57 peptide + GFP (versus GFP alone) were transduced into cells using the BioPORTER® reagent 1 day prior to patch clamp experiments. The cells were identified for patching by their green fluorescence. Whole cell patch clamp recordings were performed with 1 μm forskolin in the bath solution. The bath and pipette solutions used have been described previously (25). Current-time and current-voltage (I-V) plots were generated by stepping from a holding potential of −20 mV to −100 through +100 mV in steps of 20 mV. Comparison of slope conductance values between the two groups was performed at +40 mV, and unpaired t tests were used to test the significance of differences between conditions.
Two-electrode Voltage Clamp Studies
Xenopus oocytes were prepared for two-electrode voltage clamp (TEV) measurements as previously described (15). Briefly, harvested oocytes were injected with cRNAs generated using the mMessage mMachine kit (Ambion) to express wild-type human CFTR (1–2 ng) and NH2-terminal V5 epitope-tagged wild-type or mutant NDPK-A cloned into the dual mammalian and oocyte expression vector pMO (5 ng), and the oocytes were used 2–3 days post-injection for experimentation. Either 32 nl of 40 mm potassium gluconate (control) or potassium 5-aminoimidazole-4-carboxamide ribonucleoside monophosphate (ZMP), a direct cellular AMPK activator (26), was microinjected into oocytes, and the oocytes were used within 6 h for measurements (17). Once a stable base line was achieved, 1 μm forskolin and 0.1 mm 3-isobutyl-1-methylxanthine (IBMX) were bath applied via perfusion manifold to stimulate CFTR conductance. Evoked current was measured using voltage ramps from −20 to +60 mV (15). The degree of AMPK-dependent inhibition of stimulated CFTR conductance was determined in at least 20 oocytes/condition derived from at least three separate frogs. An analysis of variance (ANOVA) factorial model was used to compare TEV data obtained from different batches of oocytes, as described (15). In another set of TEV experiments, CFTR-1420-57 peptide was microinjected into oocytes (final concentration, 20 μm) to disrupt AMPK-CFTR interaction. TEV measurements of peptide-injected oocytes were compared with vehicle-injected oocytes; both groups were co-injected with ZMP to activate AMPK.
In Vitro Phosphorylation Studies
In vitro phosphorylation studies were performed as described (24). HEK-293 cells were transfected with either wild-type NDPK-A or NDPK-A mutants (H118F, S120A, S120D, S120E, S122A, and S122D) and then lysed 1–2 days later for NDPK-A IP using 1.2 μg of anti-V5 antibody (Invitrogen). Highly purified, recombinant active (α1-T172D, β1, γ1) or kinase-deficient (α1-D157A, β1, γ1) rat AMPK holoenzyme (27) or no-kinase control buffer was added to examine direct, AMPK-mediated phosphorylation of IP'd NDPK-A in vitro in the presence of [γ-32P]ATP. In some studies, AMPK-phosphorylated samples were subjected to an established acid treatment protocol to distinguish acid-labile (i.e., His) phosphorylated residues from acid-stable Ser/Thr linkages, as previously described (28). Briefly, IP'd NDPK-A samples on protein A/G beads that had already been phosphorylated in the presence of [γ-32P]ATP were then subjected to acid (or control) washes (± addition of 10 μl of 1 n HCl to the wash buffer to adjust the pH to 1.0) for 30 min prior to elution in Laemmli sample buffer. SDS-PAGE, transfer, immunoblotting, and phosphoscreen imaging of the same nitrocellulose membrane were then performed. For all conditions, the ratio of the phosphorylation signal to the immunoblot signal of the NDPK-A band on the membrane was compared to derive relative phosphorylation levels, as described (24).
RESULTS
We previously found that AMPK activation, either pharmacologically or through overexpression of an activating AMPK mutant subunit, inhibited PKA-mediated CFTR activation in Xenopus oocytes and human bronchial serous (Calu-3) and colonic (T84) epithelial cells (15, 16, 29). More recently, others have reported similar data (18). Specifically, we have shown that this AMPK-dependent inhibition of CFTR activity in Calu-3 cells occurs via an inhibition of CFTR Po in cell-attached patch clamp studies without affecting CFTR surface expression (N), as measured by surface biotinylation assays (16). Taken together, it appears that AMPK acts as a brake on CFTR activity. In current Calu-3 cell studies, however, we have found that the observed Po (∼0.4–0.5) in excised inside-out patch clamp recordings with active PKA catalytic subunit and ATP in the bath (30) was unaffected by addition of purified active AMPK holoenzyme to the bath solution (see supplemental Fig. S1). One interpretation of this unexpected finding is that other cellular co-factors, which potentially could be lost after excision of the patch, may be required to confer the AMPK-dependent inhibition of CFTR in intact Calu-3 cells. Because recent work suggests a potential functional and physical association between NDPK-A and AMPK (19, 31, 32), we tested the hypothesis that NDPK-A is an important co-factor in the inhibition of CFTR by AMPK.
NDPK-A Association with AMPK and CFTR
Overlay (or far Western) assays were performed to identify potential binding partners for NDPK-A in airway epithelial membranes and examine potential interactions between NDPK-A, AMPK, and CFTR. Our results show that NDPK-A binding to a 63-kDa protein was detected when purified NDPK-A was overlaid on Western blots containing solubilized membrane proteins from ovine tracheal epithelium (Fig. 1A, lane ii). NDPK-A binding to a 63-kDa protein was also observed when the Western blot was overlaid with unpurified or crude protein solution extracted from ovine tracheal epithelial membranes using EDTA (5 mm) (data not shown). Further analysis showed that the 63-kDa protein not only bound NDPK-A but was also immune-reactive with antibody specific to AMPK-α (Fig. 1A, lane iii, aliquot of first lane proteins).
FIGURE 1.
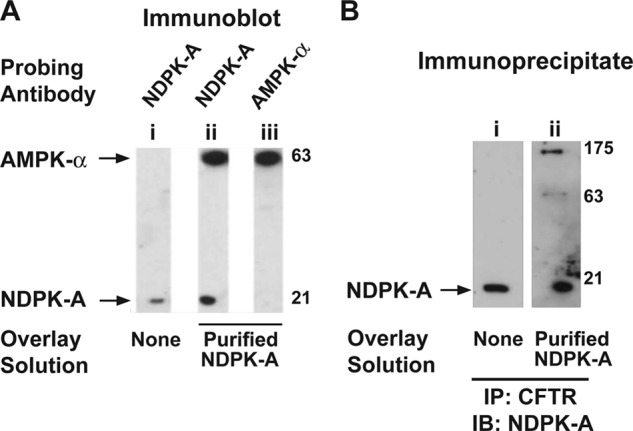
NDPK-A association with AMPK-α and CFTR in overlay assays. A, immunoblots of ovine tracheal epithelium membrane proteins (100 μg) performed in the presence or absence of an overlay solution of purified ovine tracheal epithelium NDPK-A (indicated by labels) and probed with either NDPK-A (lanes i and ii) or AMPK-α (lane iii) antibody. NDPK-A immunoreactivity was detected at ∼20 kDa and additionally at 63 kDa in overlaid blot. AMPK-α immunoreactivity was detected at 63 kDa. B, immunoblots (IB) of immunoprecipitated CFTR from membrane fractions of polarized 16HBE14o− cell lysates performed in the presence or absence of purified NDPK overlay and probed with NDPK-A antibody. NDPK-A immunoreactivity appears at 20 kDa (lanes i and ii) and additionally at 63 and 175 kDa in overlaid blot (lane ii), consistent with its binding to AMPK and CFTR present in the membrane, respectively. The results shown are representative of three replicate experiments.
To further analyze the interaction, CFTR IP'd from human bronchial epithelial (16HBE14o−) cell membrane preparations was electrophoresed and transferred to membranes, which were then overlaid with purified NDPK-A and subsequently immunoblotted with antibody to NDPK-A (Fig. 1B). In addition to showing immunoreactivity at ∼20 kDa (NDPK-A), additional NDPK-A antibody immunoreactivity was detectable at 63 and 175 kDa (Fig. 1B, lane ii). These data suggest that both NDPK-A and AMPK-α co-precipitated with CFTR and that purified NDPK-A protein can interact with denatured AMPK and CFTR in membrane blots.
Interference of CFTR-AMPK Binding by CFTR-1420-57 Peptide
Previous studies led us to propose that the interaction of AMPK with CFTR may help target AMPK and other proteins to CFTR in epithelial cells (15). These interactions may form an efficient CFTR regulatory complex at the apical membrane. To test whether the AMPK-α1 binding tract in the CFTR COOH-terminal tail (CFTR residues 1420–1457) could displace the binding of AMPK to CFTR in cells, we injected Xenopus laevis oocytes with cRNAs to co-express CFTR and HA-tagged AMPK-α1. Two days later we microinjected into the oocytes a purified peptide corresponding to this tract (CFTR-1420-57) or vehicle and then lysed the oocytes 4 h later for co-IP assays. Compared with control-injected oocytes, the amount of AMPK-α1 that co-IP'd with CFTR IP'd from oocytes injected with the CFTR-1420-57 peptide was substantially reduced (by 75 ± 20%; Fig. 2, A, lower blot, right lanes, and B). To confirm this result by independent means, HEK-293 cells were co-transfected to express CFTR and either GST-tagged AMPK-α1 or GST alone. CFTR-1420-57 peptide (or vehicle) was then introduced into cells by protein transduction 1 day prior to lysis, followed by affinity purification on glutathione-Sepharose beads (Fig. 2C, lanes 5–8). Apparent binding of CFTR to the GST-tagged AMPK-α1 was substantially reduced by the addition of the CFTR-1420-57 peptide (Fig. 2C, upper blot, lanes 6 and 8). There was no detectable binding of CFTR to GST alone. Densitometric quantitation from three replicate experiments confirmed that the relative CFTR binding to GST-AMPK-α1 was reduced by 41 ± 6% in the presence of the blocking peptide (Fig. 2D). Taken together, these results indicate that the CFTR-1420-57 peptide effectively disrupts the binding of AMPK to CFTR when introduced into cells.
FIGURE 2.
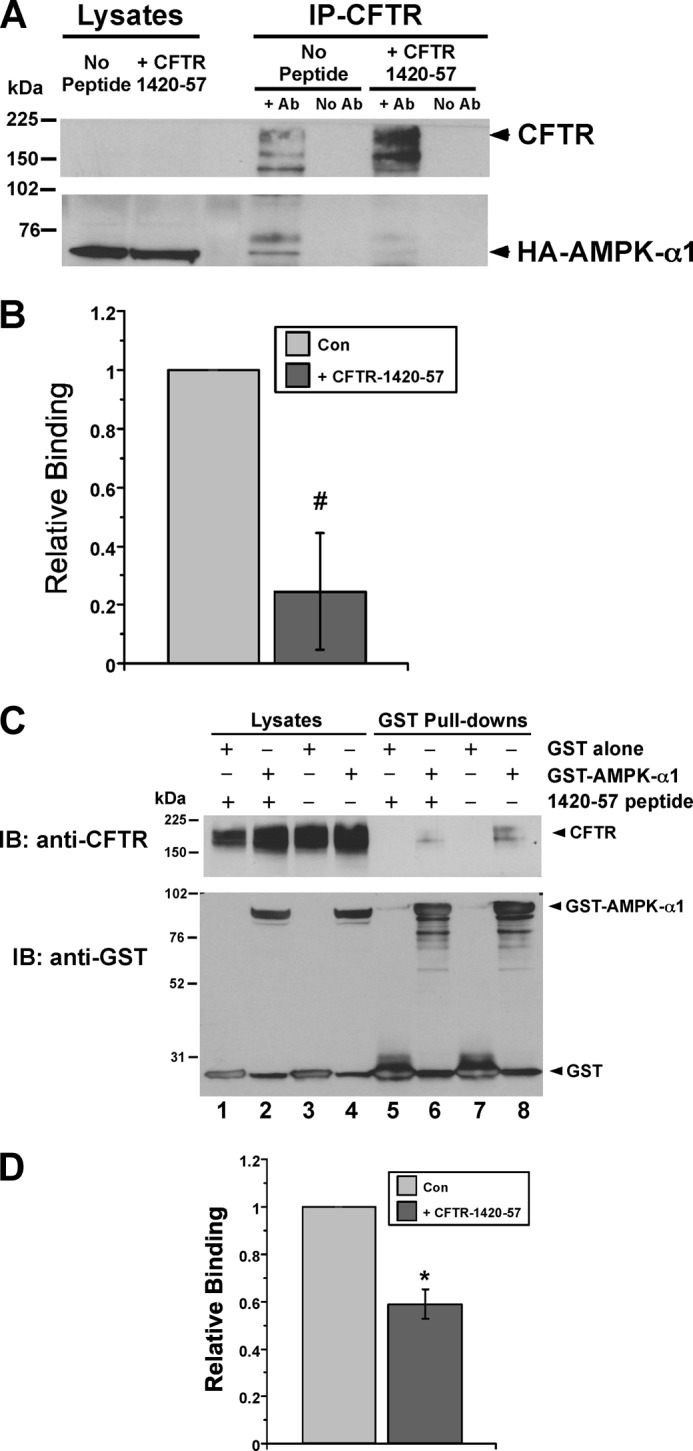
CFTR-1420-57 blocking peptide displaces binding of AMPK-α1 to CFTR in oocytes and HEK-293 cells. A, Xenopus oocytes expressing both CFTR and HA-tagged AMPK-α1 cRNA (injected 2 days prior) were injected with either CFTR-1420-57 peptide or vehicle (no peptide) 4 h prior to lysis. The lysates were used for IP (right) of CFTR using M24-1 antibody (+ Ab lanes) or no antibody as an IP control (No Ab lanes), followed by immunoblotting for CFTR (upper panel) or co-IP'd HA-AMPK-α1 (lower panel). The blots of 5% of whole cell lysates are also shown (left lanes). B, by densitometric analysis, the CFTR-1420-57 peptide reduced the amount of AMPK-α1 co-IP'd with CFTR (relative binding) by 75 ± 20%. #, p = 0.06, paired t test, n = 3 experiments. C, HEK-293 cells were co-transfected with plasmids to express CFTR and either NH2-terminal GST-tagged AMPK-α1 or GST alone 2 days prior to lysis. 100 μm CFTR-1420-57 peptide (or vehicle) was transduced into cells using the BioPORTER reagent 1 day before lysis as described under “Experimental Procedures.” After GST affinity purification of the cell lysates, the proteins were eluted in sample buffer and subjected to SDS-PAGE and immunoblotting for either CFTR (upper) or GST (lower). The amount of CFTR pulled down was substantially reduced following transduction of the blocking CFTR-1420-57 peptide (lane 6, top) relative to vehicle (lane 8, top). D, summary of relative binding (relative amount of CFTR pulled down by GST-AMPK-α1) in the presence of the CFTR-1420-57 peptide versus vehicle. By densitometric analysis, the blocking peptide reduced the relative binding of CFTR to AMPK-α1 by 41 ± 6%. *, p = 0.02, paired t test, n = 3 replicate experiments. Con, control; IB, immunoblot.
Transduction of CFTR-1420-57 Peptide Enhances Forskolin-stimulated CFTR Whole Cell Conductance in Calu-3 Cells
We previously reported that AMPK inhibits PKA-stimulated CFTR activity in Calu-3 cells via effects on CFTR Po (16). To investigate whether the disruption of endogenous AMPK-α1 binding to CFTR modulates CFTR whole cell currents in Calu-3 cells, we performed whole cell patch clamp studies of forskolin-stimulated CFTR conductance in Calu-3 cells with or without introduction of the CFTR-1420-57 peptide. Representative I-V plots are shown (Fig. 3A) in the absence or presence of the CFTR-1420-57 peptide (upper and lower panels, respectively). Cells expressing the CFTR-1420-57 blocking peptide had significantly enhanced whole cell conductances in the presence of PKA stimulation by ∼60% (Fig. 3B).
FIGURE 3.
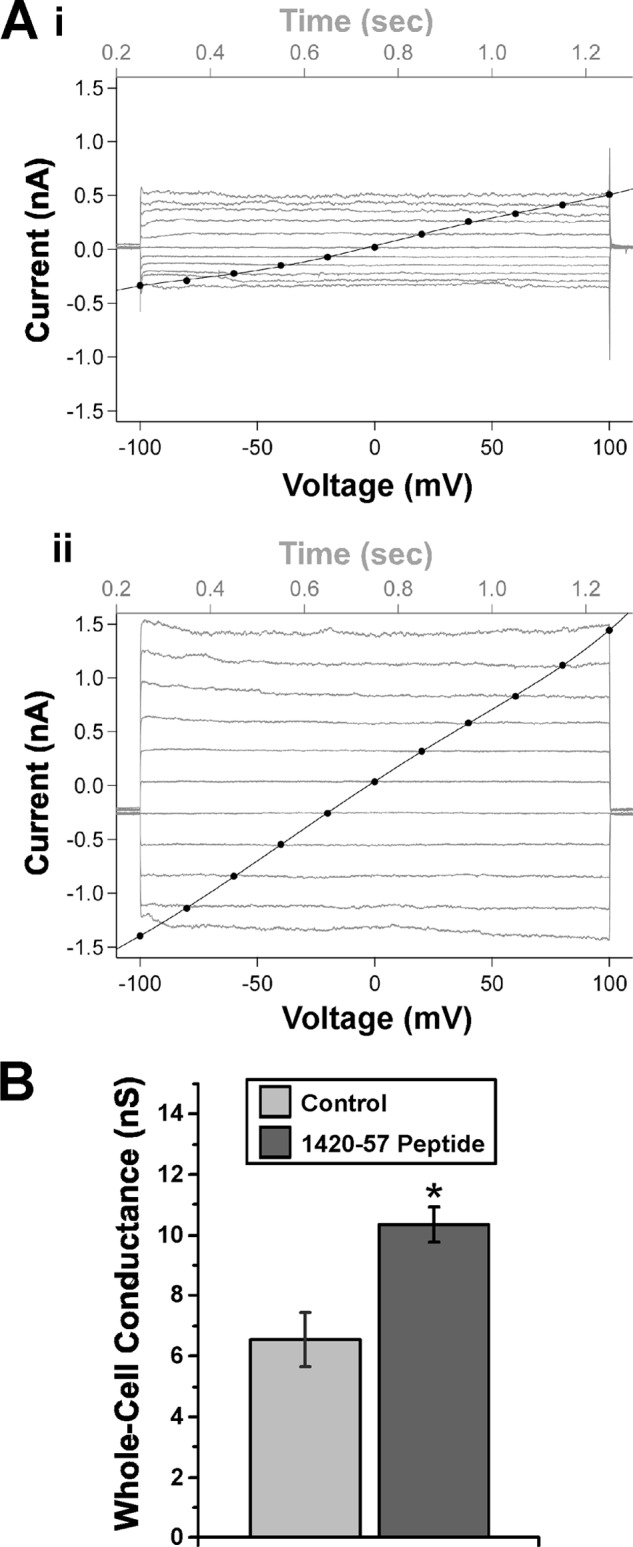
CFTR-1420-57 blocking peptide transduced into Calu-3 cells enhances CFTR whole cell conductance in the presence of forskolin. A, representative current-time sweeps (gray) and current-voltage (I-V) plots (black) are shown from whole cell patch clamp recordings performed in the absence (panel i) or presence (panel ii) of CFTR-1420–57 blocking peptide. B, summary of mean (± S.E.) slope conductances following mock transduction or transduction with blocking peptide 1 day prior to measurements. *, p < 0.02, unpaired t test, n = 4–5 patches/condition.
AMPK Regulation of CFTR with Co-expression of Wild-type versus H118F Mutant NDPK-A in Oocytes
Various NDPK isoforms are ubiquitously expressed in mammalian species and in Xenopus laevis frogs (33). We have previously shown through TEV measurements that microinjection of the AMPK activator ZMP into CFTR-expressing Xenopus oocytes inhibits cAMP-activated CFTR conductance (17). This AMPK-dependent inhibition of peak CFTR conductance following forskolin/IBMX stimulation was preserved in Xenopus oocytes co-expressing CFTR and WT NDPK-A (Fig. 4, A and B). Additional TEV measurements were performed in Xenopus oocytes expressing NDPK-A with a Phe mutation at the site where His phosphorylation is required for NDPK-A phosphotransfer, which is a prerequisite for its catalytic function as a phosphotransferase (NDPK-A-H118F) (19). The AMPK-dependent inhibition of cAMP-stimulated CFTR conductance was abrogated in oocytes expressing the catalytically inactive NDPK-A-H118F mutant (Fig. 4, C and D). Together, these results suggest that expression of this mutant has a dominant-negative effect on endogenous NDPK activity in the oocytes and that NDPK-A phosphotransferase function is required for the AMPK-dependent inhibition of CFTR.
FIGURE 4.

AMPK fails to inhibit CFTR in oocytes expressing catalytically inactive NDPK-A-H118F mutant. TEV recordings were performed 2 days after cRNA injection of CFTR and either wild-type NDPK-A (NDPK-A WT) or the catalytically inactive NDPK-A-H118F mutant into Xenopus oocytes. The oocytes were injected 2–4 h prior to recording with either potassium gluconate (Control) or the AMPK activator K-ZMP (ZMP). A and C, the mean (± S.E.) whole cell conductances are shown over time following addition of the cAMP agonists forskolin (1 μm) and IBMX (0.1 mm) at time 0 to activate CFTR in oocytes expressing either NDPK-A WT (A) or the NDPK-A-H118F mutant (C). B, ZMP injection inhibited the peak CFTR conductance in oocytes co-expressing CFTR and NDPK-A WT relative to control-injected oocytes. *, p < 0.001, ANOVA; data normalized to peak control-injected current. D, ZMP failed to inhibit CFTR conductance with co-expression of the catalytically inactive NDPK-A-H118F mutant (p > 0.10, ANOVA). Starting conductances were lower than peak conductances for all conditions. #, p < 0.001, ANOVA; n = 15–30 oocytes/condition from four to seven separate frogs for all conditions shown.
Disruption of CFTR Regulation by AMPK with the CFTR-1420-57 Peptide
To test whether AMPK binding to CFTR and NDPK-A catalytic function are both necessary components of the AMPK-dependent regulation of CFTR within the same experiment, TEV studies were performed on oocytes co-expressing CFTR and either WT or the catalytically inactive H118F mutant form of NDPK-A. All oocytes were then microinjected with the AMPK activator ZMP, with or without co-injection of the CFTR-1420-57 peptide 2–4 h prior to TEV measurements (Fig. 5). In oocytes expressing WT NDPK-A, addition of the CFTR-1420-57 blocking peptide significantly increased CFTR conductance relative to controls by ∼60% (Fig. 5, A and B). In contrast, oocytes expressing NDPK-A-H118F showed no significant increase in conductance with addition of the blocking peptide in both raw trace and normalized data (Fig. 5, C and D). In summary, these data indicate that under conditions when AMPK is activated (ZMP-injected oocytes), blockade of AMPK binding to CFTR by the CFTR-1420-57 peptide prevented CFTR inhibition by AMPK, which reinforces the importance of AMPK binding to CFTR for this inhibitory regulation. Moreover, the CFTR-1420-57 peptide failed to further enhance peak CFTR conductance when the catalytically inactive form of NDPK-A (H118F) was expressed, a condition that also blocked the AMPK-dependent inhibition of CFTR (Fig. 4). Taken together, the lack of significant further enhancement of CFTR conductance by the CFTR-1420-57 peptide in this NDPK dysfunctional setting (where CFTR inhibition by AMPK is already prevented) suggests that the CFTR-1420-57 peptide is acting, as expected, by preventing the AMPK-dependent regulation of CFTR rather than via a distinct, AMPK-independent mechanism.
FIGURE 5.

Importance of both AMPK binding to CFTR and NDPK-A catalytic function for the inhibition of CFTR by AMPK in oocytes. TEV experiments were performed 2 days after microinjection of Xenopus oocytes with cRNAs to express CFTR and either NDPK-A WT or NDPK-A-H118F mutant. The oocytes were microinjected with either ZMP alone or ZMP + CFTR-1420-57 peptide 4 h prior to TEV recordings. Forskolin and IBMX were infused at time 0 to activate CFTR conductance. A, summary of mean (± S.E.) changes in whole cell conductance over time are shown in oocytes expressing NDPK-A WT. B, mean (± S.E.) conductances are shown relative to the peak conductance for the ZMP alone condition. The addition of the CFTR-1420-57 blocking peptide induced a significant increase in peak conductance relative to control. *, p = 0.002 (ANOVA). C, summary of mean (± S.E.) changes in whole cell conductance over time is shown in oocytes expressing NDPK-A-H118F. D, normalized data for CFTR and NDPKA-H118F condition show no significant change in either peak or starting conductances by the CFTR-1420-57 peptide. n = 23–25 oocytes from five separate frogs for all conditions shown).
AMPK Enhances NDPK-A Autophosphorylation in Vitro
Previous studies (15, 19) and our current data (Fig. 1) suggest that CFTR, NDPK-A, and AMPK-α1 can exist together in a tripartite signaling complex in airway epithelial cells. A potential role for AMPK in the phosphorylation and function of NDPK-A has not been well established. NDPK-A harboring an H118F point mutation is catalytically inactive in cells (34), and AMPK enhances NDPK-A phosphorylation (19, 35), but the specific residue(s) involved are unclear. A recent study suggests that Ser-120 is a target for AMPK-dependent phosphorylation, but direct AMPK phosphorylation at that residue was not demonstrated (35). To examine direct AMPK-mediated phosphorylation of NDPK-A in vitro and test which residues may be involved, we performed [γ-32P]ATP labeling of immunoprecipitated NDPK-A in the presence and absence of purified active AMPK holoenzyme (Fig. 6). In the absence of AMPK, NDPK-A phosphorylation was apparent in the WT, S120A, S122A, and S122D mutant constructs, but minimal phosphate labeling occurred in the H118F, S120D, and S120E mutants (Fig. 6B). The addition of AMPK enhanced phosphorylation of the WT and S120A and S122 mutant constructs, but not the H118F, S120D, or S120E mutants, as summarized in Fig. 6B. The simplest interpretation of these data is that AMPK does not directly phosphorylate NDPK-A at Ser-120 or Ser-122 (or at any other site) but rather enhances NDPK-A autophosphorylation at His-118. Moreover, although this autophosphorylation event is unaffected by a nearby S120A substitution, it is strongly inhibited by a negatively charged residue at position 120, which may mimic phosphorylation at that site or retard His-118 phosphorylation. Ser-120 has been previously suggested to be phosphorylated (e.g., by casein kinase 2 (34)) and plays an important regulatory role in NDPK-A function (21, 36). Together, these results are consistent with a recently described “trans-model” wherein the His-118 residue of NDPK-A is transiently autophosphorylated by intermolecular phosphate transfer between adjacent NDPK hexamers, an event that is required for NDPK-A catalytic function, but then the high energy phosphate group on His-118 subsequently migrates to the nearby (lower energy) Ser-120 site (22).
FIGURE 6.

AMPK enhances NDPK-A autophosphorylation at His-118 in vitro. A, WT or indicated NDPK-A mutants were immunoprecipitated and subjected to in vitro phosphorylation by [γ-32P]ATP labeling in the presence (+) or absence (−) of purified active AMPK holoenzyme AMPK-α1-T172D, -β1, -γ1. Immunoblotting (lower panel) and phosphoscreen imaging (upper panel) were then performed on the same nitrocellulose membrane, as described under “Experimental Procedures.” B, summary of mean (± S.E.) NDPK-A phosphorylation signal normalized to expression levels and reported relative to WT NDPK-A phosphorylation in the presence of AMPK. *, p < 0.02, paired t test relative to same NDPK-A construct in presence of AMPK; #, p < 0.001 relative to WT NDPK-A in presence of AMPK, unpaired t-tests; data pooled from three to eleven experiments for each condition. These results suggest that AMPK does not directly phosphorylate NDPK-A at Ser-120 or Ser-122 (or any other site) but rather enhances an NDPK-A autophosphorylation event at His-118. C, in vitro phosphorylation assays were performed using active AMPK holoenzyme (AMPK-α1-T172D, -β1, -γ1), kinase-dead holoenzyme (AMPK-α1-D157A, -β1, -γ1), or no kinase, and mean (± S.E.) phosphorylation values relative to + active AMPK condition are shown. *, p < 0.001, unpaired t test relative to active AMPK; n = 8. D, acid (or control) washes (± addition of 10 μl of 1 n HCl to wash buffer to adjust the pH to 1.0 for 30 min) of the IP'd NDPK-A on protein A/G beads were performed following in vitro phosphorylation to detect whether phosphorylation had occurred at an acid-labile (i.e., His) residue in NDPK-A. *, p < 0.05; #, p < 0.05, relative to + AMPK condition of same NDPK-A species, unpaired t-tests, n = 3–4 experiments for each condition.
To test whether AMPK catalytic activity is required for the enhancement of NDPK-A autophosphorylation, we compared in vitro phosphate labeling of NDPK-A in the presence of catalytically active AMPK (AMPK-α1-T172D, β1, γ1), catalytically inactive AMPK (AMPK-α1-D157A, β1, γ1) or no AMPK (Fig. 6C). Only active AMPK enhanced the autophosphorylation of NDPK-A, suggesting that AMPK catalytic function is required for this effect. Inactive AMPK failed to enhance NDPK-A autophosphorylation relative to no kinase control.
Finally, to confirm that this phosphorylation event occurs at an acid-labile (i.e., His) residue rather than an acid-stable (e.g., Ser or Thr) residue, we used an established protocol to compare the phosphate signal of [32P]phosphate-labeled NDPK-A with or without subsequent acid treatment (28, 37). The prior phosphorylation signal of either WT or S120A mutant NDPK-A was largely abolished (by ∼90%) by a 30-min acid (pH 1.0) exposure, consistent with predominant NDPK-A phosphorylation occurring at the His-118 site (Fig. 6D).
CFTR Inhibition by AMPK Is Blocked by Co-expression of NDPK-A Ser-120 Mutants
The NDPK-A catalytic residue His-118 is of clear functional importance in the inhibition of CFTR by AMPK (Figs. 4 and 5). In addition, it appears that AMPK does not directly phosphorylate NDPK-A in vitro but rather enhances NDPK-A autophosphorylation at His-118. A negatively charged residue at Ser-120 inhibited this autophosphorylation event (Fig. 6). The Ser-120 site has been previously identified as a putative site of phosphorylation by another regulator of CFTR, casein kinase 2 (38), and plays an important role in NDPK-A function (21, 36). More recently, it has been shown that both the His-118 and Ser-120 residues contribute to autophosphorylation and NTP-synthesizing processes of NDPK-A (22). Hence, we tested the effects of Ser-120 mutations on CFTR whole cell conductance in Xenopus oocytes (Fig. 7). Under conditions in which CFTR and NDPK-A wild type are expressed, ZMP injection to activate AMPK significantly reduced whole cell CFTR conductance (stimulated by forskolin/IBMX). However, this modulation was lost when mutated NDPK-A was expressed (S120A and S120D). No significant differences in starting conductances were observed prior to forskolin/IBMX stimulation. We conclude that both the His-118 and Ser-120 residues are involved in the AMPK-dependent modulation of CFTR. Specifically, mutations of either His-118 or Ser-120 disrupt the AMPK-dependent inhibition of CFTR, suggesting that autophosphorylation of NDPK-A at His-118, and potentially the ability to transfer the phosphate group (for example, to Ser-120 or to some other protein), may play an important role in this regulation.
FIGURE 7.
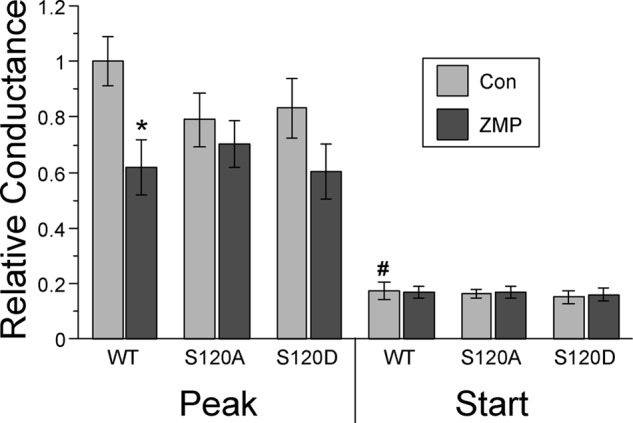
NDPK-A S120 mutants disrupt the AMPK-dependent inhibition of CFTR in oocytes. TEV recordings were performed 2 days after cRNA injection of CFTR and NDPK-A WT or S120A and S120D mutants. 2–4 h prior to recording, oocytes were injected with either potassium gluconate (Con) or the AMPK activator K-ZMP (ZMP). Starting and peak conductances were measured before and after the addition of forskolin (1 μm) and IBMX (0.1 mm). A significant decrease in peak conductance was observed following ZMP treatment with co-expression of WT NDPK-A (*, p < 0.01 relative to Con, ANOVA), whereas this inhibition was lost with co-expression of NDPK-A-S120A and -S120D mutants (p > 0.10 between control and ZMP conditions, ANOVA). Starting conductances for all NDPK-A species were significantly lower than corresponding peak conductances. #, p < 0.001; n = 12 oocytes for each condition from three separate frogs.
DISCUSSION
We and others have shown that the ubiquitous metabolic sensor AMPK regulates CFTR and other transport proteins, which may couple their activities to cellular metabolic status (39, 40). Our previous studies demonstrated that AMPK binds to the COOH-terminal tail of CFTR, phosphorylates CFTR, and inhibits PKA-stimulated CFTR gating (Po) in epithelial cells (15–18).
Here we have further explored the molecular and cellular mechanisms underpinning this regulation with respect to binding of AMPK to CFTR within a multiprotein regulatory complex and demonstrate the importance of NDPK-A function in this regulation. As previously suggested (19), NDPK-A appears to exist along with AMPK and CFTR in an apical membrane multiprotein complex in airway epithelial cells. Purified NDPK-A bound to gel-denatured AMPK-α1 and CFTR derived from airway epithelial cell membrane fractions (Fig. 1), consistent with a putative role for NDPK-A in the regulation of CFTR by AMPK. Introduction of the CFTR-1420-57 blocking peptide into cells interfered with the CFTR-AMPK-α1 interaction (Fig. 2), reduced the tonic inhibition induced by AMPK on CFTR in Calu-3 whole cell patch clamp recordings (Fig. 3), and disrupted the AMPK-dependent inhibition of CFTR in oocytes (Fig. 5). Together, these results confirm the requirement of AMPK binding to CFTR in its capacity to inhibit the degree of CFTR activity.
Overexpression of wild-type NDPK-A in oocytes (Fig. 4) had no qualitative effect on the previously described AMPK-dependent inhibition of cAMP-stimulated CFTR conductance in the absence of exogenous NDPK expression (17). This finding is consistent with the notion that there is endogenous NDPK in the oocytes (33), which allows AMPK to regulate CFTR there in the absence of exogenous NDPK co-expression. Expression of the catalytically inactive H118F NDPK-A, on the other hand, abrogated the inhibition of CFTR by AMPK in oocytes (Fig. 4, C and D). This finding suggests that the overexpression of mutant NDPK-A has a dominant-negative effect on endogenous oocyte NDPK, presumably by getting incorporated into endogenous NDPK hexamers and thereby interfering with NDPK catalytic function in the oocytes. This interference presumably blocks the AMPK-dependent inhibition of CFTR, suggesting that NDPK-A plays a key role in this regulation. Under conditions of AMPK activation, the lack of additive response on the enhancement of CFTR conductance in oocytes expressing both NDPK-A-H118F and the blocking CFTR-1420-57 peptide (Fig. 5) is consistent with the idea that these two factors act within the same pathway for CFTR regulation (i.e., they are both important components of the AMPK regulatory mechanism). This idea fits well with a recent report that the homologue of NDPK-A found in the amoeba Dictyostelium is required to transmit the energy conservation signal induced by activated AMPK (32). Together, this recently published study and our current results suggest that AMPK and NDPK lie within the same functional cellular pathways and that NDPK may play an important role in transducing cellular AMPK signals.
Our in vitro phosphorylation studies confirm that AMPK enhances the phosphorylation of NDPK-A, as recently reported (19, 35). AMPK catalytic activity appears to be required for this phosphorylation enhancement, which is also acid-labile and almost entirely blocked in the NDPK-A-H118F mutant or with negatively charged, acidic residue substitutions at the nearby Ser-120 residue (Fig. 6). Of note, there was no decrement in phosphorylation in vitro with the NDPK-A-S120A mutant relative to WT NDPK-A. Taken together, these results suggest that AMPK does not directly phosphorylate NDPK-A at Ser-120 (or at any other residue) but instead enhances NDPK-A autophosphorylation predominantly at the catalytic His-118 residue. We speculate that negatively charged residue substitutions at Ser-120 inhibit this NDPK-A autophosphorylation at His-118 via local electrostatic charge effects. Additionally, mutation of the Ser-120 site disrupts AMPK-dependent inhibition of CFTR in oocytes (Fig. 7), suggesting that phosphate transfer from His-118 to Ser-120, as recently proposed (22), could be a key component in the regulatory mechanism. Alternatively, neutral or charged substitutions at this site may interfere with NDPK-A catalytic function by disrupting the local conformation at or near the catalytic site of the kinase. Regardless of the mechanism, these data support the idea that NDPK-A catalytic function is required for the AMPK-dependent inhibition of CFTR.
At first glance, our results may appear to be inconsistent with a recent study reporting that the NDPK-A Ser-120 residue is a target for AMPK phosphorylation in vivo (35). However, direct AMPK-mediated phosphorylation at that residue was not tested in vitro. Rather, Onyenwoke et al. (35) showed that NDPK-A phosphorylation was reduced in brain AMPK-α1/2 double knock-out mice by two-dimensional differential in-gel electrophoresis analyses, and they showed AMPK-dependent modulation of Ser-120 phosphorylation in vivo by mass spectrometry.
AMPK and NDPK regulate adjacent enzymes in fatty acid metabolism. AMPK controls acetyl CoA carboxylase (13), a gatekeeper of fatty acid balance, and NDPK controls the cytosolic enzyme ATP-citrate lyase (41), which cleaves the acetyl CoA from citrate recently exported from the mitochondrion. Thus, our finding that both proteins interact with an ion channel (and regulate one another) creates a new dimension to the links between ion transport processes that consume a large proportion of cellular energy and the cellular energy charge. That both AMPK and NDPK-A function to retard CFTR function is consistent with such a linking role and is of particular interest given the reported relationship between in vitro Cl− concentration and the intensity of phosphorylation of NDPK in airway membranes (42).
The notion that an additional accessory protein, such as NDPK-A, is required for the regulation of CFTR by AMPK is attractive, because the AMPK-mediated phosphorylation of CFTR at Ser-768 in the R domain is clearly not sufficient to account for the inhibition by AMPK of CFTR (17). Specifically, PKA can also phosphorylate this residue, but PKA has the opposite (stimulatory) effect on CFTR (43). There are at least two potential mechanisms, not necessarily mutually exclusive, to explain the necessary role of NDPK-A in the regulation of CFTR by AMPK. The first involves a previously proposed “substrate channeling” model whereby nucleotides (e.g., ATP) are directly supplied (enzymic mouth-to-mouth coupling) to AMPK via its close association with NDPK-A, which catalyzes the interconversion of cellular nucleotide pools (e.g., GTP + ADP → ATP + GDP) (31). This process could provide the catalytic AMPK-α1 subunit with a local supply of ATP that could be used to phosphorylate and inhibit the CFTR Cl− channel even under conditions when bulk cellular pools of ATP may be reduced, as with metabolic depletion. A second potential mechanism for the involvement of NDPK-A in the AMPK-dependent regulation of CFTR is that NDPK-A acts downstream of AMPK between AMPK and CFTR. In support of this model, our current data and previous work suggest that AMPK reciprocally modulates NDPK-A function. NDPK-A may, in turn, modulate signaling mediators that regulate CFTR activity. For example, Gs proteins are known signaling mediators modulated by NDPK-B (44), and CFTR activity at the plasma membrane has been shown to be modulated by G-proteins (45). Moreover, CFTR trafficking/localization is regulated by Rab-GTPases (46), some of which are linked to NDPK function (47). Further studies are warranted to explore the mechanism(s) by which NDPK-A contributes to the regulation of CFTR by AMPK.
Of note, AMPK, a kinase thought to be AMP-activated, is now known to be additionally activatable by ADP, which prevents AMPK dephosphorylation (48). These recent findings are of particular interest given that ADP is a substrate for NDPK that could be either fed or removed from the vicinity of AMPK. Thus, it is conceivable that NDPK-A also plays an important role in the AMPK-dependent regulation of other transport proteins and in the pleiotropic effects of AMPK on a variety of cellular pathways and functions.
A better understanding of NDPK-A function and regulation is of broad interest to understanding human health and disease. Previous work has shown that phosphorylation of WT NDPK decreases the metastatic potential of cancer cells, whereas S120G or S120A mutants did not (21, 36). Another study implicated Ser-120 as a target for casein kinase 2 phosphorylation, which inhibits the autophosphorylation of His-118 (34). Phosphorylation of the His-118 residue has also been shown to be defective in the pancreatic islets of a diabetic rat model (49).
Relative to CF therapeutics, the strategy of blocking the CFTR-AMPK interaction in vivo (e.g., through the use of our CFTR-1420-57 blocking peptide or a small molecule compound found to disrupt the AMPK-CFTR interaction) could be beneficial in ameliorating CF lung disease by enhancing CFTR function. CF patients have diminished CFTR function and increased (but mislocalized) AMPK levels (50). Therefore, in CF patients with residual CFTR function in the airway, it may be attractive to enhance AMPK activation by treatment with an AMPK activator such as metformin to inhibit excessive airway inflammation and epithelial Na+ channel-mediated fluid reabsorption (51) while selectively preventing the AMPK-dependent inhibition of CFTR with such a compound.
Supplementary Material
Acknowledgments
We thank Drs. D. G. Hardie and D. Carling for gifts of antibodies and the University of Pittsburgh Genomics and Proteomics Core Laboratories for synthesizing the CFTR-1420-57 peptide.
This work was supported, in whole or in part, by National Institutes of Health Grants P30 DK079307 ( to the Pittsburgh Kidney Research Center), R01 DK075048 (to K. R. H.), and T32 HL007563 (to J. D. K.). This work was also supported by Cystic Fibrosis Foundation Grants KING10F0 (to J. D. K.) and HALLOW06P0 (to K. R. H.).

This article contains supplemental text and Fig. S1.
- CFTR
- cystic fibrosis transmembrane conductance regulator
- AMPK
- AMP-activated protein kinase
- ANOVA
- analysis of variance
- CF
- cystic fibrosis
- CFTR-1420-57
- CFTR, blocking peptide corresponding to residues 1420–1457
- IBMX
- 3-isobutyl-1-methylxanthine
- IP
- immunoprecipitation
- IP'd
- immunoprecipitated
- NDPK
- nucleoside diphosphate kinase
- TEV
- two-electrode voltage clamp
- ZMP
- potassium 5-aminoimidazole-4-carboxamide ribonucleoside monophosphate.
REFERENCES
- 1. Welsh M. J., Smith A. E. (1995) Cystic fibrosis. Sci. Am. 273, 52–59 [DOI] [PubMed] [Google Scholar]
- 2. Rowe S. M., Miller S., Sorscher E. J. (2005) Cystic fibrosis. N. Engl. J. Med. 352, 1992–2001 [DOI] [PubMed] [Google Scholar]
- 3. Thiagarajah J. R., Verkman A. S. (2003) CFTR pharmacology and its role in intestinal fluid secretion. Curr. Opin. Pharmacol. 3, 594–599 [DOI] [PubMed] [Google Scholar]
- 4. Davidow C. J., Maser R. L., Rome L. A., Calvet J. P., Grantham J. J. (1996) The cystic fibrosis transmembrane conductance regulator mediates transepithelial fluid secretion by human autosomal dominant polycystic kidney disease epithelium in vitro. Kidney Int. 50, 208–218 [DOI] [PubMed] [Google Scholar]
- 5. Guerrant R. L., Hughes J. M., Lima N. L., Crane J. (1990) Diarrhea in developed and developing countries. Magnitude, special settings, and etiologies. Rev. Infect. Dis. 12, (suppl.) 41–50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Riordan J. R., Rommens J. M., Kerem B., Alon N., Rozmahel R., Grzelczak Z., Zielenski J., Lok S., Plavsic N., Chou J. L. (1989) Identification of the cystic fibrosis gene. Cloning and characterization of complementary DNA. Science 245, 1066–1073 [DOI] [PubMed] [Google Scholar]
- 7. Li C., Naren A. P. (2005) Macromolecular complexes of cystic fibrosis transmembrane conductance regulator and its interacting partners. Pharmacol. Ther. 108, 208–223 [DOI] [PubMed] [Google Scholar]
- 8. Guggino W. B., Stanton B. A. (2006) New insights into cystic fibrosis. Molecular switches that regulate CFTR. Nat. Rev. Mol. Cell Biol. 7, 426–436 [DOI] [PubMed] [Google Scholar]
- 9. Cheng S. H., Rich D. P., Marshall J., Gregory R. J., Welsh M. J., Smith A. E. (1991) Phosphorylation of the R domain by cAMP-dependent protein kinase regulates the CFTR chloride channel. Cell 66, 1027–1036 [DOI] [PubMed] [Google Scholar]
- 10. Carson M. R., Travis S. M., Welsh M. J. (1995) The two nucleotide-binding domains of cystic fibrosis transmembrane conductance regulator (CFTR) have distinct functions in controlling channel activity. J. Biol. Chem. 270, 1711–1717 [DOI] [PubMed] [Google Scholar]
- 11. Quinton P. M., Reddy M. M. (1992) Control of CFTR chloride conductance by ATP levels through non-hydrolytic binding. Nature 360, 79–81 [DOI] [PubMed] [Google Scholar]
- 12. Bell C. L., Quinton P. M. (1993) Regulation of CFTR Cl− conductance in secretion by cellular energy levels. Am. J. Physiol. 264, C925–C931 [DOI] [PubMed] [Google Scholar]
- 13. Hardie D. G., Carling D., Carlson M. (1998) The AMP-activated/SNF1 protein kinase subfamily. Metabolic sensors of the eukaryotic cell? Annu. Rev. Biochem. 67, 821–855 [DOI] [PubMed] [Google Scholar]
- 14. Hardie D. G., Ross F. A., Hawley S. A. (2012) AMPK. A nutrient and energy sensor that maintains energy homeostasis. Nat. Rev. Mol. Cell Biol. 13, 251–262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Hallows K. R., Raghuram V., Kemp B. E., Witters L. A., Foskett J. K. (2000) Inhibition of cystic fibrosis transmembrane conductance regulator by novel interaction with the metabolic sensor AMP-activated protein kinase. J. Clin. Invest. 105, 1711–1721 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Hallows K. R., McCane J. E., Kemp B. E., Witters L. A., Foskett J. K. (2003) Regulation of channel gating by AMP-activated protein kinase modulates cystic fibrosis transmembrane conductance regulator activity in lung submucosal cells. J. Biol. Chem. 278, 998–1004 [DOI] [PubMed] [Google Scholar]
- 17. King J. D., Jr., Fitch A. C., Lee J. K., McCane J. E., Mak D. O., Foskett J. K., Hallows K. R. (2009) AMP-activated protein kinase phosphorylation of the R domain inhibits PKA stimulation of CFTR. Am. J. Physiol. Cell Physiol. 297, C94–C101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kongsuphol P., Cassidy D., Hieke B., Treharne K. J., Schreiber R., Mehta A., Kunzelmann K. (2009) Mechanistic insight into control of CFTR by AMPK. J. Biol. Chem. 284, 5645–5653 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Treharne K. J., Best O. G., Mehta A. (2009) The phosphorylation status of membrane-bound nucleoside diphosphate kinase in epithelia and the role of AMP. Mol. Cell Biochem. 329, 107–114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Boissan M., Dabernat S., Peuchant E., Schlattner U., Lascu I., Lacombe M. L. (2009) The mammalian Nm23/NDPK family. From metastasis control to cilia movement. Mol. Cell Biochem. 329, 51–62 [DOI] [PubMed] [Google Scholar]
- 21. MacDonald N. J., Freije J. M., Stracke M. L., Manrow R. E., Steeg P. S. (1996) Site-directed mutagenesis of nm23-H1. Mutation of proline 96 or serine 120 abrogates its motility inhibitory activity upon transfection into human breast carcinoma cells. J. Biol. Chem. 271, 25107–25116 [DOI] [PubMed] [Google Scholar]
- 22. Dar H. H., Chakraborti P. K. (2010) Intermolecular phosphotransfer is crucial for efficient catalytic activity of nucleoside diphosphate kinase. Biochem. J. 430, 539–549 [DOI] [PubMed] [Google Scholar]
- 23. Cozens A. L., Yezzi M. J., Kunzelmann K., Ohrui T., Chin L., Eng K., Finkbeiner W. E., Widdicombe J. H., Gruenert D. C. (1994) CFTR expression and chloride secretion in polarized immortal human bronchial epithelial cells. Am. J. Respir. Cell Mol. Biol. 10, 38–47 [DOI] [PubMed] [Google Scholar]
- 24. Carattino M. D., Edinger R. S., Grieser H. J., Wise R., Neumann D., Schlattner U., Johnson J. P., Kleyman T. R., Hallows K. R. (2005) Epithelial sodium channel inhibition by AMP-activated protein kinase in oocytes and polarized renal epithelial cells. J. Biol. Chem. 280, 17608–17616 [DOI] [PubMed] [Google Scholar]
- 25. Huang P., Trotter K., Boucher R. C., Milgram S. L., Stutts M. J. (2000) PKA holoenzyme is functionally coupled to CFTR by AKAPs. Am. J. Physiol. Cell Physiol. 278, C417–C422 [DOI] [PubMed] [Google Scholar]
- 26. Corton J. M., Gillespie J. G., Hawley S. A., Hardie D. G. (1995) 5-aminoimidazole-4-carboxamide ribonucleoside. A specific method for activating AMP-activated protein kinase in intact cells? Eur. J. Biochem. 229, 558–565 [DOI] [PubMed] [Google Scholar]
- 27. Neumann D., Woods A., Carling D., Wallimann T., Schlattner U. (2003) Mammalian AMP-activated protein kinase. Functional, heterotrimeric complexes by co-expression of subunits in Escherichia coli. Protein Expr. Purif. 30, 230–237 [DOI] [PubMed] [Google Scholar]
- 28. Muimo R., Hornickova Z., Riemen C. E., Gerke V., Matthews H., Mehta A. (2000) Histidine phosphorylation of annexin I in airway epithelia. J. Biol. Chem. 275, 36632–36636 [DOI] [PubMed] [Google Scholar]
- 29. Hallows K. R., Kobinger G. P., Wilson J. M., Witters L. A., Foskett J. K. (2003) Physiological modulation of CFTR activity by AMP-activated protein kinase in polarized T84 cells. Am. J. Physiol. Cell Physiol. 284, C1297–C1308 [DOI] [PubMed] [Google Scholar]
- 30. Raghuram V., Mak D. O., Foskett J. K. (2001) Regulation of cystic fibrosis transmembrane conductance regulator single-channel gating by bivalent PDZ-domain-mediated interaction. Proc. Natl. Acad. Sci. U.S.A. 98, 1300–1305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Muimo R., Crawford R. M., Mehta A. (2006) Nucleoside diphosphate kinase A as a controller of AMP-kinase in airway epithelia. J. Bioenerg. Biomembr. 38, 181–187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Annesley S. J., Bago R., Mehta A., Fisher P. R. (2011) A genetic interaction between NDPK and AMPK in Dictyostelium discoideum that affects motility, growth and development. Naunyn-Schmiedeberg's Arch. Pharmacol. 384, 341–349 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Bilitou A., Watson J., Gartner A., Ohnuma S. (2009) The NM23 family in development. Mol. Cell Biochem. 329, 17–33 [DOI] [PubMed] [Google Scholar]
- 34. Biondi R. M., Engel M., Sauane M., Welter C., Issinger O. G., Jiménez de Asúa L., Passeron S. (1996) Inhibition of nucleoside diphosphate kinase activity by in vitro phosphorylation by protein kinase CK2. Differential phosphorylation of NDP kinases in HeLa cells in culture. FEBS Lett. 399, 183–187 [DOI] [PubMed] [Google Scholar]
- 35. Onyenwoke R. U., Forsberg L. J., Liu L., Williams T., Alzate O., Brenman J. E. (2012) AMPK directly inhibits NDPK through a phosphoserine switch to maintain cellular homeostasis. Mol. Biol. Cell 23, 381–389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Freije J. M., Blay P., MacDonald N. J., Manrow R. E., Steeg P. S. (1997) Site-directed mutation of Nm23-H1. Mutations lacking motility suppressive capacity upon transfection are deficient in histidine-dependent protein phosphotransferase pathways in vitro. J. Biol. Chem. 272, 5525–5532 [DOI] [PubMed] [Google Scholar]
- 37. Biondi R. M., Walz K., Issinger O. G., Engel M., Passeron S. (1996) Discrimination between acid and alkali-labile phosphorylated residues on Immobilon. Phosphorylation studies of nucleoside diphosphate kinase. Anal. Biochem. 242, 165–171 [DOI] [PubMed] [Google Scholar]
- 38. Treharne K. J., Xu Z., Chen J. H., Best O. G., Cassidy D. M., Gruenert D. C., Hegyi P., Gray M. A., Sheppard D. N., Kunzelmann K., Mehta A. (2009) Inhibition of protein kinase CK2 closes the CFTR Cl channel, but has no effect on the cystic fibrosis mutant ΔF508-CFTR. Cell Physiol. Biochem. 24, 347–360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Hallows K. R. (2005) Emerging role of AMP-activated protein kinase in coupling membrane transport to cellular metabolism. Curr. Opin. Nephrol. Hypertens. 14, 464–471 [DOI] [PubMed] [Google Scholar]
- 40. Pastor-Soler N. M., Hallows K. R. (2012) AMP-activated protein kinase regulation of kidney tubular transport. Curr. Opin. Nephrol. Hypertens. 21, 523–533 [DOI] [PubMed] [Google Scholar]
- 41. Wagner P. D., Vu N. D. (1995) Phosphorylation of ATP-citrate lyase by nucleoside diphosphate kinase. J. Biol. Chem. 270, 21758–21764 [DOI] [PubMed] [Google Scholar]
- 42. Treharne K. J., Marshall L. J., Mehta A. (1994) A novel chloride-dependent GTP-utilizing protein kinase in plasma membranes from human respiratory epithelium. Am. J. Physiol. 267, L592–L601 [DOI] [PubMed] [Google Scholar]
- 43. Berger H. A., Anderson M. P., Gregory R. J., Thompson S., Howard P. W., Maurer R. A., Mulligan R., Smith A. E., Welsh M. J. (1991) Identification and regulation of the cystic fibrosis transmembrane conductance regulator-generated chloride channel. J. Clin. Invest. 88, 1422–1431 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Hippe H. J., Wolf N. M., Abu-Taha I., Mehringer R., Just S., Lutz S., Niroomand F., Postel E. H., Katus H. A., Rottbauer W., Wieland T. (2009) The interaction of nucleoside diphosphate kinase B with Gβγ dimers controls heterotrimeric G protein function. Proc. Natl. Acad. Sci. U.S.A. 106, 16269–16274 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Reddy M. M., Quinton P. M. (2001) cAMP-independent phosphorylation activation of CFTR by G proteins in native human sweat duct. Am. J. Physiol. Cell Physiol. 280, C604–C613 [DOI] [PubMed] [Google Scholar]
- 46. Silvis M. R., Bertrand C. A., Ameen N., Golin-Bisello F., Butterworth M. B., Frizzell R. A., Bradbury N. A. (2009) Rab11b regulates the apical recycling of the cystic fibrosis transmembrane conductance regulator in polarized intestinal epithelial cells. Mol. Biol. Cell 20, 2337–2350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Mizrahi A., Molshanski-Mor S., Weinbaum C., Zheng Y., Hirshberg M., Pick E. (2005) Activation of the phagocyte NADPH oxidase by Rac guanine nucleotide exchange factors in conjunction with ATP and nucleoside diphosphate kinase. J. Biol. Chem. 280, 3802–3811 [DOI] [PubMed] [Google Scholar]
- 48. Xiao B., Sanders M. J., Underwood E., Heath R., Mayer F. V., Carmena D., Jing C., Walker P. A., Eccleston J. F., Haire L. F., Saiu P., Howell S. A., Aasland R., Martin S. R., Carling D., Gamblin S. J. (2011) Structure of mammalian AMPK and its regulation by ADP. Nature 472, 230–233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Kowluru A. (2003) Defective protein histidine phosphorylation in islets from the Goto-Kakizaki diabetic rat. Am. J. Physiol. Endocrinol. Metab. 285, E498–E503 [DOI] [PubMed] [Google Scholar]
- 50. Hallows K. R., Fitch A. C., Richardson C. A., Reynolds P. R., Clancy J. P., Dagher P. C., Witters L. A., Kolls J. K., Pilewski J. M. (2006) Up-regulation of AMP-activated kinase by dysfunctional cystic fibrosis transmembrane conductance regulator in cystic fibrosis airway epithelial cells mitigates excessive inflammation. J. Biol. Chem. 281, 4231–4241 [DOI] [PubMed] [Google Scholar]
- 51. Myerburg M. M., King J. D., Jr., Oyster N. M., Fitch A. C., Magill A., Baty C. J., Watkins S. C., Kolls J. K., Pilewski J. M., Hallows K. R. (2010) AMPK agonists ameliorate sodium and fluid transport and inflammation in cystic fibrosis airway epithelial cells. Am. J. Respir. Cell Mol. Biol. 42, 676–684 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.