

Abstract
Autophagy is an evolutionarily conserved homeostatic process by which cells deliver cytoplasmic material for degradation into lysosomes. Autophagy may have evolved as a nutrient-providing homeostatic pathway induced upon starvation, but with the acquisition of cargo receptors, autophagy has become an important cellular defence mechanism as well as a generator of antigenic peptides for major histocompatibility complex (MHC) presentation. We propose that autophagy efficiently protects against microbes encountering the cytosolic environment accidentally, for example, upon phagosomal damage, whereas pathogens routinely accessing the host cytosol have evolved to avoid or even benefit from autophagy.
Selective autophagy for innate and adaptive immunity
Autophagy comprises a set of evolutionarily conserved cytoplasmic degradation pathways that deliver cytosolic constituents to lysosomes 1, 2. These include chaperone-mediated autophagy, and micro- and macroautophagy. While chaperone-mediated autophagy and microautophagy import substrates directly into lysosomes, macroautophagy engulfs its substrates into double-membrane vesicles, called autophagosomes. These autophagosomes fuse subsequently with lysosomes or late endosomes for degradation of their cargo. Macroautophagy is the only known mechanism for cells to dispose of cytosolic components too large for proteasomal degradation. Originally thought a nonselective process, autophagosomes are now recognised also to engulf substrates selectively, including damaged organelles and protein aggregates but also bacteria, parasites and virions. Pathogens that invade the cytosolic compartment as part of their regular life cycle must therefore avoid, or at least inhibit, selective autophagy to establish infection. By contrast, pathogens that encounter autophagy sporadically, for example vesicle-dwelling pathogens released into the cytosol from accidentally damaged vesicles, are more likely to be restricted by autophagy in their ability to colonise the cytosol. However, as is true for most evolutionarily conserved defence mechanisms, certain pathogens have evolved the ability to overcome autophagy or even take advantage of this pathway, for example, to gain access to specific cellular compartments. Recently, some of the mechanisms that target pathogens for autophagy have been revealed 3, 4, 5. By targeting cytosolic pathogens for lysosomal destruction, macroautophagy contributes to the ability of cells to present pathogen-derived peptides to T cells. Autophagy is therefore an important contributor to both innate and adaptive immunity.
In this review we will discuss the mechanisms of selective macroautophagy of pathogens and its contribution to innate and adaptive immunity. We will also provide several examples of how certain bacteria and viruses are restricted by macroautophagy, whereas others escape autophagy and sometimes even use this pathway for their propagation. It therefore appears that pathogens not adapted to life in the cytosol are efficiently restricted by macroautophagy, whereas those with a cytosolic lifestyle have evolved to avert autophagy or even subvert it for their benefit.
Molecular mechanisms of autophagosome formation
Before discussing selective macroautophagy of pathogens, we will introduce some aspects of the molecular machinery underlying this process. Thirty-five genes have been identified as essential for macroautophagy in yeast [6], which are called autophagy-related genes or atgs. During autophagosome formation in either yeast or mammals, a membrane is marked with phosphatidylinositol-3-phosphate [PtdIns(3)P; PI3P] by a complex of the type III PI3 kinase Vps34 (vacuolar protein sorting 34), in complex with Vps15, ATG6/BECLIN-1 (Bcl-2-interacting protein 1) and ATG14L. At the PI3P marked site, membrane elongation results in the formation of an isolation membrane (also known as a phagophore), which eventually closes around the substrate into a completely sealed autophagosome (Figure 1 ). Autophagosome formation in all eukaryotes requires two ubiquitin-like systems that act sequentially. The first system comprises the conjugation of the ubiquitin-fold protein ATG12 to ATG5, catalysed by the E1- and E2-like enzymes ATG7 and ATG10. ATG5–ATG12 associates with ATG16L1 into an E3-like enzyme that mediates the covalent attachment of ATG8 (LC3B being the primarily studied mammalian homologue) to phosphatidylethanolamine (PE) in the isolation membrane. Prior to this coupling event, ATG8 requires C-terminal processing by the protease ATG4 and activation by the E1- and E2-like enzymes ATG7 and ATG3, respectively. Lipidated ATG8 facilitates tethering and fusion of vesicular membranes and, therefore, could promote elongation of the isolation membrane 7, 8, 9. In addition, ATG8/LC3 is involved in anchoring substrates to the inside of the emerging autophagosome. Once the autophagsome is completed, it rapidly fuses with lysosomes in a Rab7 (Ras-related in brain)-dependent fashion (half-life of 10–25 min in hepatocytes). This fusion is also facilitated by a PI3 kinase-containing ATG6/BECLIN-1 complex, possibly in conjunction with UVRAG (UV radiation resistance-associated gene) and negatively regulated by Rubicon (RUN domain protein as Beclin 1 interacting and cysteine-rich containing). The de novo formation of autophagosomes enables macroautophagy to engulf structures of variable sizes, for example, protein aggregates, cell organelles or whole pathogens, and accordingly autophagosomes can range from 0.5 to 10 μm in diameter.
Figure 1.
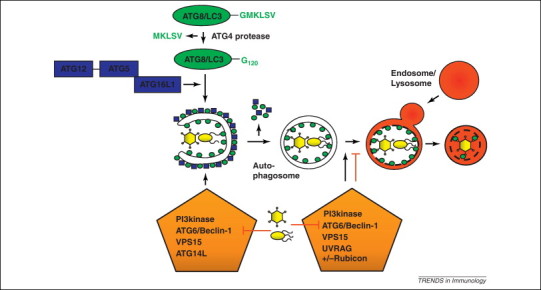
Restriction of pathogens by macroautophagy and their escape. Autophagosomes are formed with the help of two ubiquitin-like systems (ATG8/LC3 and ATG12) and their formation and degradation is guided by phosphatidylinositol-3 (PI3) kinase complexes. The ATG8/LC3 ligase complex ATG5–ATG12–ATG16L1 conjugates ATG8/LC3 to the isolation membrane or phagophore and is then recycled from the outer autophagosomal membrane with ATG8/LC3. Upon fusion with lysosomes, the inner autophagosomal membrane and its content, including pathogens, is degraded. Viruses interfere with this process and either block autophagosome formation or degradation by interacting with ATG6/BECLIN-1, which is contained in autophagosome forming PI3 kinase complexes (VPS34 as PI3 kinase, VPS15, ATG6/BECLIN-1 and ATG14L) and autophagosome degrading (VPS34, VPS15, ATG6/BECLIN-1 and UVRAG) or degradation blocking (VPS34, VPS15, ATG6/BECLIN-1, UVRAG and Rubicon) PI3 kinase complexes.
The membrane source for autophagosome formation is still heavily debated. Isolation membranes have been visualised by electron tomography on the rough endoplasmic reticulum (rER) 10, 11, but membranes of autophagosomes are also found to originate from the Golgi apparatus, the plasma membrane or mitochondria [12]. For selective autophagy, particularly if involving pathogens occurring in unforeseen places, the substrate might determine the location where phagophores must form. The above-described molecular machinery may therefore deploy membranes already existing in close proximity to the pathogen to initiate the formation of the isolation membrane. This flexibility of autophagosome formation, both in size and location, is used by the immune system to engulf invading pathogens for degradation and antigen presentation, as well as by pathogens to generate membrane structures that are useful to them. In addition to the capture of pathogens by canonical autophagy, ATG proteins are also deployed, either individually or in small modules, to fight intracellular pathogens in alternative pathways that do not involve the formation of autophagosomes (discussed later in the review).
Macroautophagy in innate immunity
Pathogen restriction by macroautophagy
Bacteria enter cells either passively by phagocytosis or actively by inducing their uptake into normally nonphagocytic cells, for example, through specialised secretion apparatuses that inject bacterial effector proteins directly into the host cytosol. Although most phagocytosed bacteria are safely killed upon delivery into lysosomes, many intracellular pathogens have evolved the ability to manipulate vesicle maturation, allowing them to either proliferate inside the vesicular compartment or to escape into the cytosol. In an attempt to prevent infection, cells deploy macroautophagy and LC3-associated phagocytosis (LAP; discussed in detail later) to capture bacteria during their inwards journey, followed by delivery into lysosomes for destruction (Figure 2 ). In contrast to LAP, which modifies the limiting membrane of pathogen-containing vesicles, macroautophagy engulfs pathogens with additional autophagosome-derived membranes. The principal targets of antibacterial autophagy are bacteria-containing vesicles, bacteria associated with remnants of damaged vesicles, and bacteria that have already escaped into the cytosol. Once caught safely inside an autophagosome, the autophagosomal double membrane restricts pathogen dispersal and provides an additional barrier against potential attempts of pathogens to manipulate cellular processes. The autophagosomal membranes separate pathogens from cytosolic resources and promote their lysosomal delivery. The ability of macroautophagy to restrict bacteria from colonising the host cytosol has been demonstrated for Streptococcus pyogenes [13] and Salmonella enterica serovar Typhimurium [14].
Figure 2.
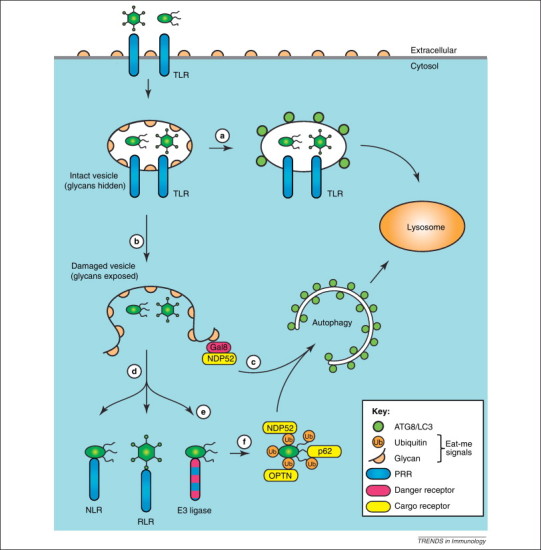
How autophagy and LC3-assisted phagocytosis defend cells against infection. Synergistic lines of defence prevent the entry of pathogens into the cytosol of host cells. (a) LC3-assisted phagocytosis is triggered by Toll-like receptors (TLRs) and potentially other pattern recognition receptors (PRRs) in response to microorganisms that are taken up by phagocytosis or that have actively invaded nonphagocytic cells. LC3-assisted phagocytosis requires a subset of autophagy genes for the labelling of phagosomes with ATG8/LC3, which promotes their lysosomal delivery and the efficient killing of vesicular pathogens. (b) Damage to the limiting membrane of the pathogen-containing vesicle, either accidental or caused by pathogens attempting to escape from the vesicle, exposes the cytosol to glycans previously hidden inside the vesicle. (c) Cytosol-accessible glycans are detected by the danger receptor galectin-8, which, by recruiting the cargo receptor NDP52 (nuclear dot protein 52), triggers autophagy. (d) Pathogens having escaped galectin-8-induced autophagy are met by yet another layer of PRRs in the cytosol. (e) A yet-to-be-identified E3 ubiquitin ligase causes the ubiquitin-coating of invading bacteria. It remains to be established whether this ligase only targets membrane-associated or also free-floating bacteria, whether it is a PRR, and also whether its substrate is of bacterial or host origin. (f) Ubiquitin-coated bacteria are targeted for autophagy by three apparently non-redundant cargo receptors, that is, NDP52, p62, and optineurin.
Bacteria-engulfing autophagosomes are created by the core autophagy machinery and have features similar to conventional autophagosomes [15]. Certain differences nevertheless exist, for example the ability to engulf very large cargo such as chains of S. pyogenes. Formation of such giant autophagosomes requires the involvement of Rab7 at early steps of autophagosome formation [16], which contrasts its role in canonical macroautophagy where Rab7 primarily participates in the fusion of autophagosomes with lysosomes [17]. Antibacterial autophagosomes also have the tendency to enwrap their cargo into onion-like membrane layers 16, 18, 19, although the origin and functional importance of these structures remain uncertain.
Selective autophagy of cellular cargo and invading pathogens are related processes and, unsurprisingly, follow similar principles. Selective autophagy relies on the tagging of the prospective cargo with specific ‘eat-me’ signals, which often comprise polyubiquitin chains. Similar to protein aggregates that become ubiquitylated before being engulfed by autophagosomes, bacteria that have damaged their vesicular compartments, and are exposed to the cytosol, can attract a dense polyubiquitin coat [20]. Little is known about the nature of this ubiquitin coat. For example, it remains unknown which bacterial surface proteins and/or host proteins in the bacterial proximity are ubiquitylated. Of particular interest is the identity of the E3 ubiquitin ligases that create the bacterial ubiquitin coat. These ligases likely represent novel pattern recognition or danger receptors. Their identification would enable a rigorous genetic assessment of the importance of the ubiquitin coat for the cell-autonomous antimicrobial defence. The strongest candidate so far is LRSAM1 (leucine-rich, α sterile α motif), a RING (Really Interesting New Gene) domain E3 ubiquitin ligase with leucine-rich repeats required for the autophagy of S. typhimurium [21], but whose role in generating the ubiquitin coat has not been established. Additional pattern recognition receptors with proposed or established affinity for peptidoglycan also contribute to antibacterial autophagy. PGRP-LE (peptidoglycan recognition protein-LE) restricts the growth of Listeria monocytogenes in Drosophila, whereas NOD1 (Nucleotide-Binding Oligomerization Domain) and NOD2 attack Gram-negative and -positive bacteria in human cells, possibly by recruiting ATG16L1 to the site of bacterial entry 22, 23. Mutant NOD2 alleles, found in some patients with Crohn's disease, fail to antagonise the cytosolic growth of several bacteria, including adherent-invasive Escherichia coli (AIEC), a bacterium associated with ileal Crohn's disease 24, 25.
An alternative eat-me signal was recently identified, when the recruitment of galectin-8 to damaged bacteria-containing vesicles was found to restrict the intracellular growth of S. typhimurium by triggering autophagy [5]. Galectins are β-galactoside-binding lectins that accumulate in the cytosol before being secreted in a leader-peptide-independent manner [26]. Although galectins bind carbohydrates of certain pathogens with high specificity [27], S. typhimurium does not display ligands for galectin-8 [5]. The recruitment of galectin-8 to S. typhimurium rather depends on the rupture of Salmonella-containing vesicles (SCVs), which exposes host glycans previously hidden inside the vesicles as ligands for galectin-8. Sterile damage to endosomes or lysosomes also recruits galectin-8 to vesicle remnants, as does damage caused by L. monocytogenes or Shigella flexneri; two bacteria that actively invade the cytosol as part of their life cycle. Galectin-8 is therefore a danger receptor that, by surveying the integrity of the endolysosomal compartment, serves as a versatile sentinel for vesicle-damaging pathogens. The human genome encodes about 12 galectins, of which galectin-1, galectin-3 and galectin-9 are also recruited to damaged vesicles 5, 28, 29. The functional consequences of their recruitment to membrane remnants remain to be identified.
The ubiquitin- and galectin-8-dependent eat-me signals for selective autophagy are detected by adaptor proteins, also known as autophagy or cargo receptors. These receptors deploy LC3-interacting regions (LIRs) to direct cargo to nascent LC3-positive phagophores. LIRs are short β-strands, often preceded by a stretch of acidic amino acids, which, via a W/FxxL/I consensus motif, bind to LC3/GABARAP (GABA receptor-associated protein) family members [30]. P62 (sequestosome 1 or SQSTM1), the best-studied autophagy receptor, selects a variety of ubiquitylated cargos, including protein aggregates, damaged mitochondria, midbody rings and also ubiquitin-coated bacteria for autophagy 18, 30. By contrast, the p62 paralogue NBR1 (neighbour of the BRCA1 gene 1) is not required to restrict the growth of S. typhimurium [18], but may contribute to the autophagy attack against S. flexneri [31]. NDP52 (nuclear dot protein 52) was identified in a search for the ubiquitin-binding adaptor of TBK1 [32], a major immunoregulatory kinase that restricts the intracellular growth of S. typhimurium and S. pyogenes [33]. NDP52 recruits TBK1 (TANK-binding kinase) in complex with SINTBAD (similar to NAP1 TBK1 adaptor) or NAP1 (NAK-associated protein 1) to cytosol-exposed bacteria by detecting either a galectin-8 or a polyubiquitin eat-me signal 5, 32, 34, 35. Binding of NDP52 to ubiquitin and galectin-8 is mediated by its C-terminal zinc finger and an adjacent short peptide, respectively, which distinguishes NDP52 as the first autophagy receptor that responds to two types of eat-me signals [5]. A third cargo receptor, optineurin, that also detects ubiquitin-coated S. typhimurium, binds LC3 with high affinity only if phosphorylated by TBK1 because it lacks the negatively charged residues preceding the LIR motif in other autophagy receptors [3].
Understanding the nonredundant contributions of NDP52, p62 and optineurin to antibacterial autophagy will be an interesting objective for future research. The three receptors clearly differ in their ability to detect unique eat-me signals associated with S. typhimurium, such as galectin-8 and possibly distinct linkage types in the bacterial ubiquitin coat [3]. Differential interactions with eat-me signals are likely to cause the occurrence of NDP52, p62 and optineurin in microdomains around S. typhimurium, populated by either NDP52 or p62 [36], NDP52 and optineurin but not p62 [3], or NDP52 and galectin-8 but not p62 [5]. However, care must be taken when analysing microdomains, because these are dynamic structures as suggested by the recruitment of NDP52 to S. typhimurium via galectin-8 and ubiquitin at early and later time points post-infection, respectively [5].
Another feature distinguishing NDP52, p62 and optineurin from each other might be their ability to recruit selectively different effector molecules for the restriction of bacterial growth. Such evidence has already been obtained for TBK1, which is bound by optineurin and (indirectly) by NDP52 but not by p62 32, 37. A specific function of p62 might be the delivery of ubiquitylated cytosolic proteins to autolysosomes, where they are proteolytically converted into antimicrobial peptides 38, 39. None of the many other p62 binding partners has an established role in antibacterial autophagy. However, for the selective autophagy of ubiquitylated protein aggregates, p62 recruits ALFY (autophagy-linked FYVE protein), a 400-kDa protein that binds ATG12–ATG5–ATG16L1 complexes and PI3P-containing membranes. ALFY may therefore also be required for antibacterial autophagy [40]. The potential recruitment of ATG proteins other than LC3 and its paralogues by the cargo receptors might explain how atg5 −/− or atg7 −/− cells form autophagosome-like structure around S. typhimurium, despite not being able to conjugate LC3 onto membranes 15, 18. These LC3-negative structures are not restricting bacterial growth, possibly because the closure of antibacterial autophagosomes requires ATG8 paralogue-dependent hemifusion activity 7, 41. In contrast to cells deficient in the LC3-conjugation machinery, cells lacking ATG9 or FIP200 (FAK-family interacting protein 200) fail to enclose S. typhimurium in autophagosomal membranes. However, despite the absence of such membranes, atg9 −/− or fip200 −/− cells still accumulate LC3 around the bacteria, probably by conjugating LC3 to the damaged SCV membrane [15]. The current model of antibacterial autophagy, in which NDP52, p62 and optineurin link bacteria to LC3-positive phagophores, is therefore simplistic and further work is required to understand how distinct autophagy proteins are independently recruited to cytosol-exposed bacteria.
Pathogen restriction by LC3-associated phagocytosis and other noncanonical functions of ATG proteins
LC3 conjugated to PE, an invariant feature of canonical macroautophagy, can also occur on single-membrane vesicles. In a process called LC3-associated phagocytosis (LAP), LC3 is recruited to certain phagosomes, for example, those containing bacteria, where it promotes phagosome maturation and fusion with lysosomes to enhance the antimicrobial potential of phagocytosis [42]. LAP requires several proteins that are also involved in canonical autophagy (BECLIN-1, VPS34, ATG5, ATG7) but not others, such as for example FIP200, a component of the ULK1 (UNC-51-like kinase 1) kinase complex necessary for macroautophagy 42, 43. Canonical macroautophagy and LAP are therefore distinct processes, both morphologically and mechanistically, that nevertheless result in the accumulation of LC3 on vesicles. The two pathways may even operate simultaneously and electron microscopy is therefore required to distinguish them with confidence. For example, upon treatment with rapamycin or interferon (IFN)-γ mycobacterial phagosomes acquire LC3 and mature into phagolysosomes, consistent with LAP [44]. However, electron microscopy has revealed the simultaneous presence of additional onion-like multilamellar structures typical for canonical macroautophagy against bacterial targets.
Consistent with autophagy overcoming the maturation block of mycobacterial autophagosomes and leading to enhanced bacterial killing [44], experimental interference with the delivery of LC3 to phagosomes may prevent the killing of phagocytosed microbes, as demonstrated for yeast in atg7 −/− macrophages [42]. A similar strategy is used by Burkholderia pseudomallei, a Gram-negative bacterium causing melioidosis, which suppresses LAP by secreting BopA and BpscN (TTSS effectors) into the macrophage cytosol via its type III secretion apparatuses 45, 46, 47. How BopA and BspnN interfere with LAP remains unknown. Recruitment of LC3 to microbe-containing phagosomes requires Toll-like receptor (TLR) signalling from within the vesicle, rather than from the cell surface, as evidenced by the lack of LC3 conjugation to phagocytosed latex beads if cells are exposed to LPS subsequent to the phagocytic event [42]. Opsonisation can replace TLR signalling, and reactive oxygen species produced by NADPH oxidases appear to be required for the targeting of LC3 to phagosomes [48], which could result from autophagosome fusion with phagosomes or from direct coupling of LC3 to the phagosomal membrane. The generation of diacylglycerol on Salmonella-containing vacuoles also triggers recruitment of LC3, most likely during LAP, although macroautophagy has not been formally excluded [49].
The ability of cells to distinguish phagosomes with ‘innocent’ and ‘infectious’ content may allow the preferential handling of infectious vesicles. In striking contrast to the ability of macrophages to ignore phagocytosed latex beads, macropinosomes and also whole cells in entotic vesicles can be targeted by LC3 [43]. How the engulfment of noninfectious cargo triggers LAP requires further investigations, although cells in entotic vacuoles may expose phosphatidylserine before showing morphological signs of cell death. The phosphatidylserine receptor TIM4 (T cell Ig domain and mucindomain protein 4), which triggers LAP in response to dead cells, may therefore be involved [43]. Experimentally curtailing the degradative potential of phagosomes containing dead cells, for example, by preventing LAP in atg7 −/− macrophages, leads to prolonged macrophage activation and sustained production of proinflammatory cytokines [43]. Therefore, in addition to its antimicrobial importance, LAP also contributes to the anti-inflammatory potential of macrophages.
Another autophagosome-independent defence pathway requiring at least one autophagy gene, atg5, occurs in Toxoplasma gondii-infected macrophages stimulated with IFN-γ, whereupon ATG5-mediated recruitment of the immunity-related p47 GTPase IIGP1 (interferon-inducible GTPase 1) the parasitophorus vacuole is destroyed and the parasite killed [50].
These studies suggest that the autophagy machinery contributes to the defence against pathogens in a variety of ways, either by conventional macroautophagy or by deploying subsets of autophagy genes to accomplish the subcellular trafficking of proteins or the maturation of organelles.
Escape mechanisms of pathogens from autophagy-mediated restriction
The ability of cells to defend themselves against invading pathogens represents an ancient form of immunity preceding the origin of multicellularity. In mammals, in which ironically cell-autonomous immunity is best studied, several lines of defence provide synergistic protection against incoming pathogens. With respect to macroautophagy and LAP, at least three classes of receptors detect invaders and ensure their delivery to the lysosome: (i) extracellular and endosomal pattern recognition receptors such as TLRs, (ii) danger receptors surveying the integrity of the endolysosomal pathway such as galectin-8; and (iii) cytosolic pattern recognition receptors such as NOD-like receptors (NLRs). Although noninvasive bacteria, for example environmental bacteria phagocytosed by lung macrophages, will be safely destroyed, it is obvious how defects in certain defence mechanisms can favour opportunistic pathogens. Pathogens that invade the cytosol as part of their normal life cycle, however, have to slip through all the nets. Much can therefore be learnt from studying how professional cytosol-dwelling pathogens avoid and inhibit cell-autonomous defence.
L. monocytogenes, a Gram-positive food-borne pathogen that causes meningitis in immunocompromised individuals and abortion in pregnant woman, is internalised by many cell types. Using a pore-forming toxin and two lipolytic enzymes, L. monocytogenes escapes from its vacuole into the cytosol with high efficiency. Vacuolar damage by L. monocytogenes is detected by galectin-3, -8 and -9, which may trigger autophagy as bacteria transiently colocalise with LC3 during invasion 5, 28, 29, 51. However, because autophagy merely delays the onset of proliferation, L. monocytogenes appears to escape the autophagic attack actively [52]. Once in the cytosol, L. monocytogenes deploys ActA (actin nucleator A), a cell surface protein and functional analogue of host Wiskott-Aldrich syndrome protein (WASP) proteins, for the recruitment of the actin-nucleating complex Arp2/3 (actin related protein 2/3). Actin-driven mobility, a feature of many cytosol-dwelling bacteria, favours the spreading of bacteria into neighbouring host cells without exposure to extracellular immune effectors. ActA prevents the coating of L. monocytogenes with ubiquitin and thereby interferes with cargo-receptor-mediated selective autophagy, possibly because ActA-recruited host proteins disguise the bacterial surface 20, 53. InlK (internalin-like K), an internalin family member expressed mainly in vivo, similarly conceals the presence of L. monocytogenes from autophagy by coating the bacterial surface with MVP (major vault protein), the major component of large cytosolic ribonucleoprotein complexes called vaults [54]. In contrast to mammals, in which L. monocytogenes colonises the cytosol with the help of ActA and InlK, in Drosophila, L. monocytogenes falls prey to autophagy due to the presence of PGRP-LE, a cytosolic peptidoglycan receptor [22]. How PGRP-LE triggers autophagy and whether it is a LIR-motif-containing autophagy receptor remains unknown. The vertebrate PGRP orthologues, called PGLYRPs, are unlikely to be involved in autophagy because they function extracellularly as peptidoglycan amidases and antimicrobial proteins. Nevertheless, recognition of peptidoglycan fragments still contributes to autophagy induction in mammalian cells because NOD1 and NOD2, the founding members of the NLR family, recruit ATG16L1 to bacterial entry sites [55].
In addition to its cytosolic lifestyle associated with extensive proliferation, L. monocytogenes also occupies a vesicular compartment, termed spacious Listeria-containing phagosomes (SLAPs). Bacteria in SLAPs proliferate at a much lower rate, which may be associated with persistent infections [14]. SLAPs are single membrane vesicles positive for LC3 that are formed in a listeriolysin-O-dependent manner; preferentially if toxin levels are suboptimal for bacterial escape into the cytosol. Although the contribution of autophagy to SLAP formation, possibly via the LAP pathway, is not fully understood, SLAPs clearly represent a stalemate between autophagic attack and bacterial countermeasures since bacterial hyperproliferation occurs in atg5 −/− cells.
S. flexneri is a Gram-negative, enteroinvasive bacterium that can cause severe colonic inflammation. Shigella enforces its uptake into nonphagocytic cells with the help of a type III secretion system, before escaping from its vacuole into the cytosol. NOD1 and ATG16L1 are recruited to the bacterial entry site [55], whereas the membrane remnants of Shigella vacuoles are detected by galectin-3, -8 and -9, similar to the situation in Listeria-infected cells 5, 28, 29. The membrane remnants furthermore codistribute with ubiquitin-, p62- and LC3-positive structures, suggesting that they are targeted by selective autophagy [28]. Once in the cytosol, Shigella deploys the autotransporter IcsA (intracellular spread A) to recruit N-WASP and Arp2/3, which provide the bacterium with actin-dependent cytosolic mobility and the ability to spread from cell to cell. However, IcsA and its biochemical activity are also targets of cell-autonomous defence mechanisms. IcsA is directly recognised by ATG5, which, unless outcompeted by IcsB, restricts bacterial growth by targeting bacteria into autophagosomes [19]. Septins, a family of GTP-binding cytoskeletal components, prevent actin-dependent mobility and cell-to-cell spread by forming cages around Shigella in an IcsA- and actin-polymerisation-dependent manner [56]. Septin-encaged Shigella colocalises with NDP52, p62 and LC3, suggesting they are targeted by selective autophagy. A positive feedback between septin cage formation and autophagy exists, because depletion of SEPT2 or SEPT9 prevents p62 recruitment, whereas depletion of p62 or core autophagy genes (atg5, atg6/beclin-1, atg7) interferes with the entrapment of Shigella in septin cages. The ability of S. flexneri and L. monocytogenes to proliferate in the mammalian cytosol, despite facing a multipronged immune attack, suggests the existence of potent bacterial countermeasures that deserve to be elucidated.
Macroautophagy assisting bacterial and viral replication
Vesicular compartments for bacterial replication and release
Contrary to the mere avoidance of autophagy practiced by L. monocytogenes and S. flexneri, other bacteria take active advantage of the unique opportunities autophagy offers to pathogens able to co-opt the pathway. These possibilities include the generation of vesicular compartments to sustain bacterial replication and to allow unorthodox trafficking.
Rather than being restricted by autophagy, a phylogenetically heterogeneous group of bacteria proliferate in double- or multilamellar LC3+ vesicles, which most likely represent canonical autophagosomes failing to acidify and mature. Examples include Staphylococcus aureus 57, 58, Yersinia pseudotuberculosis [59], Yersinia pestis [60] and Anaplasma phagocytophilum [61]. Except for Y. pestis, which is impervious to the effects of autophagy, these species fail to grow in atg5 −/− cells, whereas autophagy induction by rapamycin enhances their proliferation. How these bacteria co-opt autophagy and how autophagy contributes to bacterial growth remains unknown.
Brucella abortus and Francisella tularensis deploy autophagy not to foster their proliferation, but to travel in an unorthodox manner within and out of their host cells. B. abortus resides in the endosomal pathway for several hours post-infection before acquiring ER markers including membrane-bound ribosomes. Proliferation begins once the ER-derived Brucella-containing vacuole (BCV) is established. Bacterial spreading from cell to cell requires conversion of the replicative BCV into an autophagosome-like structure comprising multiple membranes but lacking LC3 [62]. Consistent with these features, formation of multi-lamellar BCVs is independent of atg4, atg7, atg5 and atg16l1 but requires upstream autophagy genes, namely ulk1, beclin-1 and atg14l. LC3-negative double-membrane structures, formed in an ATG5-independent fashion, also engulf Mycobacterium marinum, an actin-polymerising and therefore professionally cytosol-dwelling species [63]. Conceivably, LC3-negative autophagosomes can be formed if bacteria selectively engage only a subset of autophagy genes and they may represent a physiological pendant to alternative autophagy in atg5 −/− or atg7 −/− cells [64].
F. tularensis, a highly infectious bacterium that was once included in biological warfare programs, replicates in the cytoplasm of mammalian cells, suggesting it avoids autophagy. Following replication, however, F. tularensis is taken up into large double-membrane vacuoles, which measure up to 15 μm in diameter, are positive for LC3 and contain multiple bacteria [65]. Although the replication of F. tularensis is not dependent on autophagy, exposure to the low pH of autolysosomes may trigger the expression of genes required for egress, reinfection or extracellular life. Understanding how F. tularensis switches from a state in which it avoids autophagy to a state in which it induces autophagy could give important insights into the mechanisms of antibacterial autophagy.
Membrane structures for viral replication
Shortly after the discovery of autophagosomes by electron microscopy [66], autophagosome-like vesicles were noticed in poliovirus infected cells [67]. Infection with this picornavirus leads to the accumulation of small double-membrane surrounded vesicles in the perinuclear area of infected cells, on which the virus assembles its replication machinery. These vesicles contain cytosol, the lysosome associated membrane protein 1 (LAMP1) and ATG8/LC3 [68]. Although the two poliovirus proteins 2BC and 3A induce this vesicle accumulation, only 2BC is required to lipidate ATG8/LC3. This extensive membrane reorganisation seems to be required for virus production, because downregulation of macroautophagy via siRNA-mediated silencing of either of the two ubiquitin-like molecules of macroautophagy, ATG12 or ATG8/LC3A and B, results in diminished virus replication [69]. Thus, poliovirus seems to use at least parts of the macroautophagy machinery to assemble membrane structures for its replication.
In addition to poliovirus, many other RNA viruses have been suggested to interfere with membrane trafficking via the molecular machinery of macroautophagy [70]. However, for most of these viruses the relevance of this regulation for their replication is still unresolved and in the case of coronaviruses even controversial 71, 72. Thus, we focus in this section on one other virus, which induces significant membrane remodelling for its replication and uses macroautophagy to do this: the flavivirus hepatitis C virus (HCV). The autophagic vesicles that accumulate in HCV-infected hepatocytes fuse with each other and do not accumulate as a multitude of small autophagosomes as observed in poliovirus-infected cells [73]. Both ATG5 and ATG8/LC3 accumulate at the stabilised membrane structures, which is suggested to result from a virus-induced unfolded protein response (UPR) [74]. This hypothesis is consistent with macroautophagy induction by UPRs in yeast, which is thought to degrade expanded ER to return affected cells to the physiological equilibrium [75]. In addition, the HCV NS3 protein interacts with the immunity-associated GTPase family M (IRGM), which in turn interacts with ATG5, ATG10, ATG8/LC3 and endophilin-1 (SH3GLB1) [76]. RNA silencing of several essential atg products, namely ATG4, ATG5, ATG6/BECLIN-1, ATG7, ATG8/LC3 and ATG12, as well as IRGM, suppresses HCV replication 74, 76, 77. Primarily, translation of HCV RNA, shortly after infection seems to depend on functional macroautophagy machinery [77]. Therefore, HCV also prevents fusion of autophagic membranes with lysosomes to promote its own replication.
In addition to this membrane remodelling for viral replication, another flavivirus, dengue virus, generates energy for its replication via macroautophagy [78]. This catabolic activity leads to the turnover of lipid droplets and triglycerides via autophagosomes, generating free fatty acids, from which ATP can be generated to benefit viral replication. Thus, autophagy does not only generate membrane compartments for viral replication, but also in part the energy necessary to carry out virus production.
Viral release via macroautophagy
In addition to viral replication on autophagic membranes, macroautophagy is involved in the generation of vesicular structures, from which viruses are exported. This was hypothesized initially for poliovirus [69]. Indeed, release of this virus is increased if tethering of these autophagic vesicles to the cytoskeleton is inhibited [79]. In this respect, we will discuss two other viruses of interest to human health, namely HIV and influenza virus. Both block autophagosome maturation in infected cells 80, 81. In addition, HIV downregulates autophagosome formation 82, 83, whereas influenza virus boosts autophagic flux under certain circumstances 84, 85. HIV tat and interleukin (IL)-10, produced by infected cells, have both been implicated in auto- and paracrine inhibition of autophagosome formation [83], whereas HIV nef is involved in blocking autophagosome fusion with lysosomes [80]. Interestingly, it achieves this through interaction with ATG6/BECLIN-1 and IRGM 76, 80. This stabilisation of autophagic vesicles, autophagosomes themselves or multivesicular bodies (MVBs) that receive input from autophagosomes, augments virus production and release of infectious particles 76, 80. RNA silencing of ATG6/BECLIN-1, ATG7 or IRGM inhibits HIV particle release 76, 80. These studies suggest that HIV inhibits autophagosome degradation in order to replicate efficiently in myeloid cells.
Although influenza A virus also causes autophagosome accumulation 76, 81, it remains unclear how this affects virus replication. Although one study has observed inhibition of influenza A virus replication upon RNA silencing of ATG6/BECLIN-1 and ATG8/LC3 [84], two other studies have reported no effect of silencing or deficiency of ATG5 on macroautophagy 76, 81. However, the influenza A virus protein matrix protein 2 (M2) blocks autophagosome fusion with lysosomes and interacts with ATG6/BECLIN-1 [81]. This block of autophagosome maturation causes more cell death in infected cells, which could facilitate influenza A virus infection in vivo. Thus, HIV and influenza A virus both block autophagosome fusion with lysosomes, but one virus seems to benefit from this block for its own replication, whereas the other regulates host cell death via this manipulation.
Irrespective of the underlying mechanism, macroautophagy plays a crucial role for viral replication and protection from viral pathogenesis in vivo for several RNA and DNA viruses. The RNA virus vesicular stomatitis virus (VSV) replicates better in flies in the absence of macroautophagy [86]. In addition, the RNA virus Sindbis virus replicates to similar levels in macroautophagy-deficient neurons, but causes more pathology in ATG5-negative brains [87], suggesting that macroautophagy ensures survival of infected host cells in vivo. Furthermore, the two DNA viruses of the herpesvirus family, herpes simplex virus (HSV) and mouse herpesvirus (MHV) 68 encode inhibitors of macroautophagy, without which their infection in mice is compromised 88, 89. Along these lines, HSV carries with ICP34.5 a viral gene product that binds ATG6/BECLIN-1 and thereby prevents autophagosome formation [88]. Furthermore, MHV blocks ATG3 and ATG6/BECLIN-1 with its viral FLIP [FLICE (Caspase-8)-inhibitory protein] and Bcl-2 (B Cell CLL/Lymphoma) gene products, respectively 89, 90. Interestingly, especially the neurovirulence of both Sindbis virus and HSV is compromised by macroautophagy. For these two viruses, p62/sequestosome and possibly its recruitment via SMURF1 (SMAD-specific E3 ubiquitin protein ligase 1) have been implicated in the recruitment of these pathogens to autophagosomes 4, 87. These studies suggest that macroautophagy can target viral particles and proteins. However, as with bacterial pathogens, successful viruses that cause disease have developed immune escape mechanisms from macroautophagy in their respective hosts. All viruses have a cytosolic stage of their replication cycle, thus, most of them manipulate autophagosome formation or degradation. These escape mechanisms are host species adapted, therefore, the strongest protective effects of macroautophagy can be observed in experimental hosts, for example, in the case of the bird- and mosquito-adapted Sindbis virus in mice.
Regulation of innate cytokine production by macroautophagy
Besides this direct role of macroautophagy in pathogen restriction, macroautophagy has also been shown to influence innate cytokine production, although so far the contribution of this regulation to resistance or susceptibility to pathogens has not been firmly established. Along these lines, macroautophagy can transport viral replication intermediates into endosomes that carry TLRs [91]. TLR-mediated type I IFN production by Sendai virus or VSV-infected plasmacytoid dendritic cells (DCs) depends on macroautophagy. By contrast, parts of the molecular macroautophagy machinery seem to inhibit type I IFN production following cytosolic RNA sensor stimulation 92, 93. Increased reactive oxygen species production or the ATG5–ATG12 complex itself seems to inhibit RIG-I activation. In addition to type I IFN, IL-1 production is also influenced by macroautophagy. Macrophages deficient in this pathway secrete much more of this proinflammatory cytokine upon stimulation [94]. In part, this enhanced IL-1 production results from inflammasome activation by damaged mitochondria, which are no longer degraded by this pathway [95], and decreased inflammasome degradation via autophagosomes [96]. These studies suggest that macroautophagy regulates at least some inflammatory cytokines, which restrict pathogens by innate immune responses and shape adaptive immune responses.
Macroautophagy during adaptive immune responses
Antigen processing via macroautophagy
Both humoral and cell-mediated adaptive immune responses hinge on the efficient induction of helper T cell responses, which play an essential role in antibody affinity maturation and cytotoxic T cell maintenance. Helper T cells recognise antigen fragments presented on MHC class II molecules, which are predominantly generated by lysosomal hydrolysis. Around 20–30% of the MHC class II presented ligands are derived from cytosolic and nuclear antigens [97], suggesting an intracellular antigen processing pathway for MHC class II loading. Among these ligands, self-protein-derived peptides can be found, some of which originate from the mammalian ATG8 homologues LC3 and GABARAP 97, 98. Further evidence that macroautophagy contributes to MHC class II loading of cytosolic and nuclear antigens was provided by the characterisation of the MHC class II derived natural ligand repertoire of starved Epstein–Barr virus (EBV)-transformed B cells (LCLs) [97]. LCLs upregulate macroautophagy after starvation, and under these conditions, MHC class II presentation of cytosolic and nuclear antigens was upregulated by 50%, whereas presentation of membrane-bound protein fragments was not affected. Furthermore, autophagosomes have been found frequently to fuse with late endosomal MHC class II containing compartments (MIICs), in which antigen is loaded onto MHC class II molecules 99, 100 (Figure 3 ). Moreover, targeting of antigens to autophagosomes via fusion to the N terminus of ATG8/LC3 enhances their presentation on MHC class II molecules to CD4+ T cells 85, 99. These studies suggest that autophagosomes deliver intracellular substrates for MHC class II loading in a wide variety of cell types, including DCs, B cells and epithelial cells.
Figure 3.
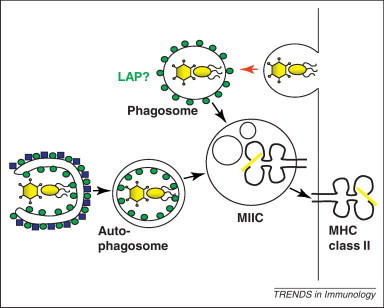
Antigen for major histocompatibility complex (MHC) class II presentation can be provided by phagocytosis or macroautophagy. Cytosolic pathogens are delivered to MHC class II containing compartments (MIICs) via macroautophagy for antigen loading. Extracellular pathogens reach this MHC class II loading compartment via endocytosis, which might be in some instances be facilitated by ATG8/LC3 coupled to the phagosome.
In addition to self-proteins, this antigen processing pathway has been suggested to deliver pathogen-derived antigens for MHC class II loading. Two of these have been investigated in more detail in several studies. The first is the nuclear antigen 1 of EBV (EBNA1). This protein ensures replication of the viral genome in the nucleus prior to host cell division and anchors EBV DNA to mitotic chromosomes during mitosis to ensure viral maintenance in replicating cells. It carries a glycine–alanine repeat domain that protects it from proteasomal degradation 101, 102, and accumulates upon lysosomal inhibition [103]. In addition, it is presented to CD4+ T cells after intracellular processing [104]. This processing is mediated by macroautophagy, because EBNA1 accumulates in autophagosomes after lysosomal inhibition, and RNA silencing of macroautophagy decreases CD4+ T cell recognition of EBV-infected B cells 103, 105. This intracellular antigen processing of EBNA1 for MHC class II presentation via macroautophagy is enhanced, when the nuclear import of EBNA1 is compromised [105]. These studies suggest that a cytosolic pool of EBNA1, either prior to import into the nucleus or liberated after dissociation of the nuclear envelope during mitosis gains access to intracellular MHC class II antigen processing by macroautophagy. A second antigen for macroautophagic processing towards MHC class II presentation to CD4+ T cells is the bacterial transposon-derived neomycin phosphotransferase II (NeoR) 85, 106. MHC class II presentation of this antigen by transfected cells to a specific CD4+ T cell clone is sensitive to macroautophagy inhibition [106], and antigens fused to NeoR are more efficiently presented on MHC class II molecules [85]. In contrast to EBNA1, however, nuclear localisation of NeoR after insertion of a nuclear import sequence does not compromise macroautophagy-dependent antigen processing, and even slightly improves MHC class II presentation [107]. Thus, it is not nuclear localisation in general, but maybe subcompartmentalisation in this organelle that determines accessibility of antigens to macroautophagic antigen processing for MHC class II presentation.
In addition to intracellular antigen processing, macroautophagy might also play a role in extracellular antigen processing for MHC class II presentation. This suggestion is based on a study in mice with macroautophagy deficiency after atg5 knockout in CD11c-positive cells, (mostly DCs) [108]. In addition to diminished CD4+ T cell responses after HSV infection in these mice, addition of soluble ovalbumin (OVA) or OVA-coated splenocytes resulted in decreased MHC class II, but unaffected MHC class I presentation to specific T cell receptor (TCR) transgenic T cells. Thus, the authors suggested that, in addition to intracellular antigen processing for MHC class II presentation after HSV or recombinant Listeria infection, macroautophagy also contributes to extracellular antigen processing for CD4+ T cell stimulation. This correlates with diminished maturation of phagosomes, possibly resulting from slower fusion with lysosomes. This phenotype would fit with three studies showing a role for parts of the macroautophagy machinery in phagosome fusion with lysosomes (LAP), suggesting ATG8/LC3-dependent facilitation of this fusion event 42, 43, 109. Alternatively, autophagosome cargo could render phagosomes more processive by delivering hydrolases to these vesicles through fusion. Along these lines, it has recently been shown that citrullination, a post-translational modification detected on some autoantigens and facilitating their immune recognition, for example, during rheumatoid arthritis, depends on macroautophagy [110]. More specifically, the peptidylarginine deiminase (PAD) responsible for citrullination of endocytosed proteins was only able to reach phagosomes via autophagosomes. Therefore, fusion of phagosomes with lysosomes and composition of phagosomal content could be modified by macroautophagy to result in more efficient extracellular antigen processing onto MHC class II molecules.
Lymphocyte education via macroautophagy
Another physiological setting, in which antigen processing via macroautophagy might be particularly important is T cell education in the thymus. In order to shape a useful and self-tolerant T cell repertoire, developing T cells are educated to recognize self-MHC molecules in the thymic cortex in a process called positive selection. In addition, self-reactive T cell specificities are eliminated in the thymic medulla via negative selection [111]. Thymic epithelial cells (TECs) interact with the developing T cells in these two processes and express both MHC class I and II molecules for this purpose. In order to display peptides not just derived from thymic proteins during these processes, TECs also express at low levels peripheral-organ-specific proteins with the help of the transcription factor autoimmune regulator (AIRE) [112]. However, for processing of these proteins towards MHC class II presentation, it is suggested that TECs require intracellular pathways, because they are poorly endocytic [113]. Along these lines, a role for macroautophagy in both of these processes was then defined [114]. During positive selection of TCR transgenic T cells in atg5-deficient thymuses, some CD4+ T cell specificities were efficiently selected, whereas positive selection of others was compromised, probably due to insufficient MHC class II presentation of the positively selecting peptide ligands. By contrast, positive CD8+ T cell selection was unaffected. Furthermore, negative selection of a wild-type precursor repertoire through atg5-negative thymic transplants in thymus-deficient nude mice resulted in autoreactive T cells, causing autoimmune colitis and lymphocyte infiltration in other organs, including Harderian glands, uterus, liver and lung. In agreement with these findings, autophagosomes were found frequently to fuse with MIICs in TECs [115]. These findings suggest that intracellular proteins are presented on MHC class II molecules of TECs after macroautophagy, and that the T cell repertoire is altered in the absence of this process. Of particular interest is the fact that such an altered T cell repertoire contains specificities that mediate autoimmune colitis, reminiscent in its manifestation of Crohn's disease. Indeed mutations have been found associated with the familial form of this autoimmune disease of the digestive tract in two proteins linked to macroautophagy, namely ATG16L1 and IRGM 116, 117, 118, 119. Furthermore, the Crohn's disease associated ATG16L1 variants have been shown to compromise MHC class II restricted antigen presentation of DCs matured with NOD2 ligands [24], and NOD2 is another risk locus for this disease 120, 121. Therefore, it is tempting to speculate that also in humans compromised macroautophagy in the thymus might allow the development of an autoimmune T cell repertoire that could contribute to Crohn's disease.
Lymphocyte survival via macroautophagy
In addition to this antigen processing function of macroautophagy that shapes lymphocyte repertoires, this pathway plays also a role during lymphocyte development and responses. Haematopoietic progenitor cells (HPCs) rely on this process for their maintenance 122, 123. In its absence, myeloproliferation develops, seemingly because both T and B cell lineage development is compromised. By contrast, plasmacytoid and conventional DC development is unaffected by loss of macroautophagy 91, 108. Both T cell development and proliferation during immune responses are compromised in the absence of macroautophagy [124]. Several mechanisms are responsible for these T cell defects in the absence of macroautophagy. For one, mature T cells need to decrease their mitochondria content during transition from the thymus to the periphery. Macroautophagy deficiency prevents this due to loss of mitochondria degradation. This leads to increased levels of reactive oxygen species, which compromises the survival of T cells without macroautophagy [125]. In addition, macroautophagy-deficient T cells cannot efficiently mobilize Ca2+ upon TCR stimulation, resulting in impaired activation [126]. Therefore, macroautophagy is required for T cell development and function. A less severe, but also detectable dependency of B cell development and maintenance on macroautophagy has been identified. B cell precursor survival in the bone marrow is affected by loss of macroautophagy, particularly at the pro- to pre-B cell transition 127, 128. Mature B cells, in contrast, are mostly unaffected by macroautophagy loss for their survival. Only the innate B-1a subset is compromised in its maintenance by macroautophagy deficiency [128]. Therefore, lymphocytes, but not leukocytes in general, need macroautophagy for their development and survival. Selective lymphocyte lineages are more affected than others by macroautophagy deficiency, and the role of macroautophagy in cell organelle homeostasis, primarily mitochondria and ER turnover, is required during lymphocyte development and their immune responses.
Concluding remarks
Autophagy, particularly macroautophagy, has emerged as an important cellular pathway for pathogen restriction and replication, as well as antigen processing. Recent studies show how pathogens that escape endosomes are imported into autophagosomes and how pathogens adapted to life in the cytosol have evolved escape mechanisms from macroautophagic degradation. In its coevolution with pathogens, the adaptive immune system of higher eukaryotes has also learned to utilise macroautophagy. Autophagy provides peptides for presentation on MHC molecules from self-antigens and pathogens during thymic selection and during peripheral immune responses, respectively. Therefore, as with classical phagocytosis, macroautophagy has evolved from a nutrient-providing pathway to fulfil additional effector mechanisms of innate, cell-intrinsic and adaptive immunity in higher eukaryotes.
The role of the molecular macroautophagy machinery during infection and immune responses may extend even further. At least some ATG proteins are involved in phagocytosis by modulating phagosome maturation and in exocytosis of signal-peptide-lacking substrates 129, 130, 131, possibly even whole virions and exosomes 132, 133. This alternative use of macroautophagy modules needs to be better understood in order to harness pathogen restriction by macroautophagy therapeutically without boosting pathogen replication.
Furthermore, macroautophagy plays a role in MHC class II ligand generation for T cell education and intracellular antigen processing for CD4+ T cell recognition. In addition, this pathway might also influence the compartmentalisation of antigen to vesicles or protein aggregates, which allow processing towards MHC class I presentation for CD8+ T cell responses 134, 135, 136. The nature of these MHC class I antigen compartments and how macroautophagy regulates them needs to be more clearly defined in order to use them for efficient induction of immune responses after vaccination.
The importance of canonical macroautophagy in pathogen infection and immune responses is now well established. Alternative deployment of parts of the macroautophagy machinery seems to have an impact on an even wider spectrum of cell biological processes for the benefit or restriction of invading microbes. These later areas, in particular, constitute a fruitful field of investigation for the near future.
Acknowledgements
Research in the laboratory of C.M. is supported by grants from the National Cancer Institute (R01CA108609), the Sassella Foundation (10/02), Cancer Research Switzerland (KFS-02652-08-2010), the Association for International Cancer Research (11-0516), the Vontobel Foundation, the Baugarten Foundation, Novartis and the Swiss National Science Foundation (310030_126995). Work in the laboratory of F.R. is supported by the Medical Research Council (U105170648) and The National Association for Colitis and Crohn's Disease (M/11/3).
Contributor Information
Felix Randow, Email: randow@mrc-lmb.cam.ac.uk.
Christian Münz, Email: christian.muenz@uzh.ch.
References
- 1.Mizushima N. The role of atg proteins in autophagosome formation. Annu. Rev. Cell Dev. Biol. 2011;27:107–132. doi: 10.1146/annurev-cellbio-092910-154005. [DOI] [PubMed] [Google Scholar]
- 2.Mizushima N., Komatsu M. Autophagy: renovation of cells and tissues. Cell. 2011;147:728–741. doi: 10.1016/j.cell.2011.10.026. [DOI] [PubMed] [Google Scholar]
- 3.Wild P. Phosphorylation of the autophagy receptor optineurin restricts Salmonella growth. Science. 2011;333:228–233. doi: 10.1126/science.1205405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Orvedahl A. Image-based genome-wide siRNA screen identifies selective autophagy factors. Nature. 2011;480:113–117. doi: 10.1038/nature10546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Thurston T.L. Galectin 8 targets damaged vesicles for autophagy to defend cells against bacterial invasion. Nature. 2012;482:414–418. doi: 10.1038/nature10744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Klionsky D.J. A comprehensive glossary of autophagy-related molecules and processes (2nd edition) Autophagy. 2011;7:1273–1294. doi: 10.4161/auto.7.11.17661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nakatogawa H. Atg8, a ubiquitin-like protein required for autophagosome formation, mediates membrane tethering and hemifusion. Cell. 2007;130:165–178. doi: 10.1016/j.cell.2007.05.021. [DOI] [PubMed] [Google Scholar]
- 8.Weidberg H. LC3 and GATE-16/GABARAP subfamilies are both essential yet act differently in autophagosome biogenesis. EMBO J. 2010;29:1792–1802. doi: 10.1038/emboj.2010.74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Weidberg H. LC3 and GATE-16 N termini mediate membrane fusion processes required for autophagosome biogenesis. Dev. Cell. 2011;20:444–454. doi: 10.1016/j.devcel.2011.02.006. [DOI] [PubMed] [Google Scholar]
- 10.Hayashi-Nishino M. A subdomain of the endoplasmic reticulum forms a cradle for autophagosome formation. Nat. Cell Biol. 2009;11:1433–1437. doi: 10.1038/ncb1991. [DOI] [PubMed] [Google Scholar]
- 11.Yla-Anttila P. 3D tomography reveals connections between the phagophore and endoplasmic reticulum. Autophagy. 2009;5:1180–1185. doi: 10.4161/auto.5.8.10274. [DOI] [PubMed] [Google Scholar]
- 12.Tooze S.A., Yoshimori T. The origin of the autophagosomal membrane. Nat. Cell Biol. 2010;12:831–835. doi: 10.1038/ncb0910-831. [DOI] [PubMed] [Google Scholar]
- 13.Nakagawa I. Autophagy defends cells against invading group A Streptococcus. Science. 2004;306:1037–1040. doi: 10.1126/science.1103966. [DOI] [PubMed] [Google Scholar]
- 14.Birmingham C.L. Listeriolysin O allows Listeria monocytogenes replication in macrophage vacuoles. Nature. 2008;451:350–354. doi: 10.1038/nature06479. [DOI] [PubMed] [Google Scholar]
- 15.Kageyama S. The LC3 recruitment mechanism is separate from Atg9L1-dependent membrane formation in the autophagic response against Salmonella. Mol. Biol. Cell. 2011;22:2290–2300. doi: 10.1091/mbc.E10-11-0893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yamaguchi H. An initial step of GAS-containing autophagosome-like vacuoles formation requires Rab7. PLoS Pathog. 2009;5:e1000670. doi: 10.1371/journal.ppat.1000670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kimura S. Dissection of the autophagosome maturation process by a novel reporter protein, tandem fluorescent-tagged LC3. Autophagy. 2007;3:452–460. doi: 10.4161/auto.4451. [DOI] [PubMed] [Google Scholar]
- 18.Zheng Y.T. The adaptor protein p62/SQSTM1 targets invading bacteria to the autophagy pathway. J. Immunol. 2009;183:5909–5916. doi: 10.4049/jimmunol.0900441. [DOI] [PubMed] [Google Scholar]
- 19.Ogawa M. Escape of intracellular Shigella from autophagy. Science. 2005;307:727–731. doi: 10.1126/science.1106036. [DOI] [PubMed] [Google Scholar]
- 20.Perrin A.J. Recognition of bacteria in the cytosol of mammalian cells by the ubiquitin system. Curr. Biol. 2004;14:806–811. doi: 10.1016/j.cub.2004.04.033. [DOI] [PubMed] [Google Scholar]
- 21.Ng A.C. Human leucine-rich repeat proteins: a genome-wide bioinformatic categorization and functional analysis in innate immunity. Proc. Natl. Acad. Sci. U.S.A. 2011;108(Suppl. 1):4631–4638. doi: 10.1073/pnas.1000093107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yano T. Autophagic control of listeria through intracellular innate immune recognition in drosophila. Nat. Immunol. 2008;9:908–916. doi: 10.1038/ni.1634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Travassos L.H. Nod proteins link bacterial sensing and autophagy. Autophagy. 2010;6:409–411. doi: 10.4161/auto.6.3.11305. [DOI] [PubMed] [Google Scholar]
- 24.Cooney R. NOD2 stimulation induces autophagy in dendritic cells influencing bacterial handling and antigen presentation. Nat. Med. 2010;16:90–97. doi: 10.1038/nm.2069. [DOI] [PubMed] [Google Scholar]
- 25.Lapaquette P. Crohn's disease-associated adherent-invasive E. coli are selectively favoured by impaired autophagy to replicate intracellularly. Cell. Microbiol. 2010;12:99–113. doi: 10.1111/j.1462-5822.2009.01381.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rabinovich G.A., Toscano M.A. Turning ‘sweet’ on immunity: galectin-glycan interactions in immune tolerance and inflammation. Nat. Rev. Immunol. 2009;9:338–352. doi: 10.1038/nri2536. [DOI] [PubMed] [Google Scholar]
- 27.Vasta G.R. Roles of galectins in infection. Nat. Rev. Microbiol. 2009;7:424–438. doi: 10.1038/nrmicro2146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dupont N. Shigella phagocytic vacuolar membrane remnants participate in the cellular response to pathogen invasion and are regulated by autophagy. Cell Host Microbe. 2009;6:137–149. doi: 10.1016/j.chom.2009.07.005. [DOI] [PubMed] [Google Scholar]
- 29.Paz I. Galectin-3, a marker for vacuole lysis by invasive pathogens. Cell. Microbiol. 2010;12:530–544. doi: 10.1111/j.1462-5822.2009.01415.x. [DOI] [PubMed] [Google Scholar]
- 30.Johansen T., Lamark T. Selective autophagy mediated by autophagic adapter proteins. Autophagy. 2011;7:279–296. doi: 10.4161/auto.7.3.14487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mostowy S. p62 and NDP52 proteins target intracytosolic Shigella and Listeria to different autophagy pathways. J. Biol. Chem. 2011;286:26987–26995. doi: 10.1074/jbc.M111.223610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Thurston T.L. The TBK1 adaptor and autophagy receptor NDP52 restricts the proliferation of ubiquitin-coated bacteria. Nat. Immunol. 2009;10:1215–1221. doi: 10.1038/ni.1800. [DOI] [PubMed] [Google Scholar]
- 33.Radtke A.L. TBK1 protects vacuolar integrity during intracellular bacterial infection. PLoS Pathog. 2007;3:e29. doi: 10.1371/journal.ppat.0030029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Fujita F. Identification of NAP1, a regulatory subunit of IkappaB kinase-related kinases that potentiates NF-kappaB signaling. Mol. Cell. Biol. 2003;23:7780–7793. doi: 10.1128/MCB.23.21.7780-7793.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ryzhakov G., Randow F. SINTBAD, a novel component of innate antiviral immunity, shares a TBK1-binding domain with NAP1 and TANK. EMBO J. 2007;26:3180–3190. doi: 10.1038/sj.emboj.7601743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cemma M. The ubiquitin-binding adaptor proteins p62/SQSTM1 and NDP52 are recruited independently to bacteria-associated microdomains to target Salmonella to the autophagy pathway. Autophagy. 2011;7:341–345. doi: 10.4161/auto.7.3.14046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Morton S. Enhanced binding of TBK1 by an optineurin mutant that causes a familial form of primary open angle glaucoma. FEBS Lett. 2008;582:997–1002. doi: 10.1016/j.febslet.2008.02.047. [DOI] [PubMed] [Google Scholar]
- 38.Alonso S. Lysosomal killing of Mycobacterium mediated by ubiquitin-derived peptides is enhanced by autophagy. Proc. Natl. Acad. Sci. U.S.A. 2007;104:6031–6036. doi: 10.1073/pnas.0700036104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ponpuak M. Delivery of cytosolic components by autophagic adaptor protein p62 endows autophagosomes with unique antimicrobial properties. Immunity. 2010;32:329–341. doi: 10.1016/j.immuni.2010.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Filimonenko M. The selective macroautophagic degradation of aggregated proteins requires the PI3P-binding protein Alfy. Mol. Cell. 2010;38:265–279. doi: 10.1016/j.molcel.2010.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Noda T. The late stages of autophagy: how does the end begin? Cell Death Differ. 2009;16:984–990. doi: 10.1038/cdd.2009.54. [DOI] [PubMed] [Google Scholar]
- 42.Sanjuan M.A. Toll-like receptor signalling in macrophages links the autophagy pathway to phagocytosis. Nature. 2007;450:1253–1257. doi: 10.1038/nature06421. [DOI] [PubMed] [Google Scholar]
- 43.Martinez J. Microtubule-associated protein 1 light chain 3 alpha (LC3)-associated phagocytosis is required for the efficient clearance of dead cells. Proc. Natl. Acad. Sci. U.S.A. 2011;108:17396–17401. doi: 10.1073/pnas.1113421108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gutierrez M.G. Autophagy is a defense mechanism inhibiting BCG and Mycobacterium tuberculosis survival in infected macrophages. Cell. 2004;119:753–766. doi: 10.1016/j.cell.2004.11.038. [DOI] [PubMed] [Google Scholar]
- 45.Gong L. The Burkholderia pseudomallei type III secretion system and BopA are required for evasion of LC3-associated phagocytosis. PLoS ONE. 2011;6:e17852. doi: 10.1371/journal.pone.0017852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Cullinane M. Stimulation of autophagy suppresses the intracellular survival of Burkholderia pseudomallei in mammalian cell lines. Autophagy. 2008;4:744–753. doi: 10.4161/auto.6246. [DOI] [PubMed] [Google Scholar]
- 47.D’Cruze T. Role for the Burkholderia pseudomallei type three secretion system cluster 1 bpscN gene in virulence. Infect. Immun. 2011;79:3659–3664. doi: 10.1128/IAI.01351-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Huang J. Activation of antibacterial autophagy by NADPH oxidases. Proc. Natl. Acad. Sci. U.S.A. 2009;106:6226–6231. doi: 10.1073/pnas.0811045106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Shahnazari S. A diacylglycerol-dependent signaling pathway contributes to regulation of antibacterial autophagy. Cell Host Microbe. 2010;8:137–146. doi: 10.1016/j.chom.2010.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zhao Z. Autophagosome-independent essential function for the autophagy protein Atg5 in cellular immunity to intracellular pathogens. Cell Host Microbe. 2008;4:458–469. doi: 10.1016/j.chom.2008.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Birmingham C.L. Listeria monocytogenes evades killing by autophagy during colonization of host cells. Autophagy. 2007;3:442–451. doi: 10.4161/auto.4450. [DOI] [PubMed] [Google Scholar]
- 52.Py B.F. Autophagy limits Listeria monocytogenes intracellular growth in the early phase of primary infection. Autophagy. 2007;3:117–125. doi: 10.4161/auto.3618. [DOI] [PubMed] [Google Scholar]
- 53.Yoshikawa Y. Listeria monocytogenes ActA-mediated escape from autophagic recognition. Nat. Cell Biol. 2009;11:1233–1240. doi: 10.1038/ncb1967. [DOI] [PubMed] [Google Scholar]
- 54.Dortet L. Recruitment of the major vault protein by InlK: a Listeria monocytogenes strategy to avoid autophagy. PLoS Pathog. 2011;7:e1002168. doi: 10.1371/journal.ppat.1002168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Travassos L.H. Nod1 and Nod2 direct autophagy by recruiting ATG16L1 to the plasma membrane at the site of bacterial entry. Nat. Immunol. 2010;11:55–62. doi: 10.1038/ni.1823. [DOI] [PubMed] [Google Scholar]
- 56.Mostowy S. Entrapment of intracytosolic bacteria by septin cage-like structures. Cell Host Microbe. 2010;8:433–444. doi: 10.1016/j.chom.2010.10.009. [DOI] [PubMed] [Google Scholar]
- 57.Mestre M.B. Alpha-hemolysin is required for the activation of the autophagic pathway in Staphylococcus aureus-infected cells. Autophagy. 2010;6:110–125. doi: 10.4161/auto.6.1.10698. [DOI] [PubMed] [Google Scholar]
- 58.Schnaith A. Staphylococcus aureus subvert autophagy for induction of caspase-independent host cell death. J. Biol. Chem. 2007;282:2695–2706. doi: 10.1074/jbc.M609784200. [DOI] [PubMed] [Google Scholar]
- 59.Moreau K. Autophagosomes can support Yersinia pseudotuberculosis replication in macrophages. Cell. Microbiol. 2010;12:1108–1123. doi: 10.1111/j.1462-5822.2010.01456.x. [DOI] [PubMed] [Google Scholar]
- 60.Pujol C. Yersinia pestis can reside in autophagosomes and avoid xenophagy in murine macrophages by preventing vacuole acidification. Infect. Immun. 2009;77:2251–2261. doi: 10.1128/IAI.00068-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Niu H. Subversion of cellular autophagy by Anaplasma phagocytophilum. Cell. Microbiol. 2008;10:593–605. doi: 10.1111/j.1462-5822.2007.01068.x. [DOI] [PubMed] [Google Scholar]
- 62.Starr T. Selective subversion of autophagy complexes facilitates completion of the Brucella intracellular cycle. Cell Host Microbe. 2012;11:33–45. doi: 10.1016/j.chom.2011.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Collins C.A. Atg5-independent sequestration of ubiquitinated mycobacteria. PLoS Pathog. 2009;5:e1000430. doi: 10.1371/journal.ppat.1000430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Nishida Y. Discovery of Atg5/Atg7-independent alternative macroautophagy. Nature. 2009;461:654–658. doi: 10.1038/nature08455. [DOI] [PubMed] [Google Scholar]
- 65.Checroun C. Autophagy-mediated reentry of Francisella tularensis into the endocytic compartment after cytoplasmic replication. Proc. Natl. Acad. Sci. U.S.A. 2006;103:14578–14583. doi: 10.1073/pnas.0601838103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Ashford T.P., Porter K.R. Cytoplasmic components in hepatic cell lysosomes. J. Cell Biol. 1962;12:198–202. doi: 10.1083/jcb.12.1.198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Dales S. Electron microscopic study of the formation of poliovirus. Virology. 1965;26:379–389. doi: 10.1016/0042-6822(65)90001-2. [DOI] [PubMed] [Google Scholar]
- 68.Taylor M.P., Kirkegaard K. Modification of cellular autophagy protein LC3 by poliovirus. J. Virol. 2007;81:12543–12553. doi: 10.1128/JVI.00755-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Jackson W.T. Subversion of cellular autophagosomal machinery by RNA viruses. PLoS Biol. 2005;3:e156. doi: 10.1371/journal.pbio.0030156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Lin L.T. Viral interactions with macroautophagy: a double-edged sword. Virology. 2010;402:1–10. doi: 10.1016/j.virol.2010.03.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Prentice E. Coronavirus replication complex formation utilizes components of cellular autophagy. J. Biol. Chem. 2004;279:10136–10141. doi: 10.1074/jbc.M306124200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Zhao Z. Coronavirus replication does not require the autophagy gene ATG5. Autophagy. 2007;3:581–585. doi: 10.4161/auto.4782. [DOI] [PubMed] [Google Scholar]
- 73.Ait-Goughoulte M. Hepatitis C virus genotype 1a growth and induction of autophagy. J. Virol. 2008;82:2241–2249. doi: 10.1128/JVI.02093-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Sir D. Induction of incomplete autophagic response by hepatitis C virus via the unfolded protein response. Hepatology. 2008;48:1054–1061. doi: 10.1002/hep.22464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Bernales S. Autophagy counterbalances endoplasmic reticulum expansion during the unfolded protein response. PLoS Biol. 2006;4:e423. doi: 10.1371/journal.pbio.0040423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Gregoire I.P. IRGM is a common target of RNA viruses that subvert the autophagy network. PLoS Pathog. 2011;7:e1002422. doi: 10.1371/journal.ppat.1002422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Dreux M. The autophagy machinery is required to initiate hepatitis C virus replication. Proc. Natl. Acad. Sci. U.S.A. 2009;106:14046–14051. doi: 10.1073/pnas.0907344106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Heaton N.S., Randall G. Dengue virus-induced autophagy regulates lipid metabolism. Cell Host Microbe. 2010;8:422–432. doi: 10.1016/j.chom.2010.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Taylor M.P. Role of microtubules in extracellular release of poliovirus. J. Virol. 2009;83:6599–6609. doi: 10.1128/JVI.01819-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Kyei G.B. Autophagy pathway intersects with HIV-1 biosynthesis and regulates viral yields in macrophages. J. Cell Biol. 2009;186:255–268. doi: 10.1083/jcb.200903070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Gannage M. Matrix protein 2 of influenza A virus blocks autophagosome fusion with lysosomes. Cell Host Microbe. 2009;6:367–380. doi: 10.1016/j.chom.2009.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Blanchet F.P. Human immunodeficiency virus-1 inhibition of immunoamphisomes in dendritic cells impairs early innate and adaptive immune responses. Immunity. 2010;32:654–669. doi: 10.1016/j.immuni.2010.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Van Grol J. HIV-1 inhibits autophagy in bystander macrophage/monocytic cells through Src-Akt and STAT3. PLoS ONE. 2010;5:e11733. doi: 10.1371/journal.pone.0011733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Zhou Z. Autophagy is involved in influenza A virus replication. Autophagy. 2009;5:321–328. doi: 10.4161/auto.5.3.7406. [DOI] [PubMed] [Google Scholar]
- 85.Comber J.D. Functional macroautophagy induction by influenza A virus without a contribution to MHC-class II restricted presentation. J. Virol. 2011;482:414–418. doi: 10.1128/JVI.02122-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Shelly S. Autophagy is an essential component of Drosophila immunity against vesicular stomatitis virus. Immunity. 2009;30:588–598. doi: 10.1016/j.immuni.2009.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Orvedahl A. Autophagy protects against Sindbis virus infection of the central nervous system. Cell Host Microbe. 2010;7:115–127. doi: 10.1016/j.chom.2010.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Orvedahl A. HSV-1 ICP34.5 confers neurovirulence by targeting the Beclin 1 autophagy protein. Cell Host Microbe. 2007;1:23–35. doi: 10.1016/j.chom.2006.12.001. [DOI] [PubMed] [Google Scholar]
- 89.E X. Viral Bcl-2-mediated evasion of autophagy aids chronic infection of gammaherpesvirus 68. PLoS Pathog. 2009;5:e1000609. doi: 10.1371/journal.ppat.1000609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Lee J.S. FLIP-mediated autophagy regulation in cell death control. Nat. Cell Biol. 2009;11:1355–1362. doi: 10.1038/ncb1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Lee H.K. Autophagy-dependent viral recognition by plasmacytoid dendritic cells. Science. 2007;315:1398–1401. doi: 10.1126/science.1136880. [DOI] [PubMed] [Google Scholar]
- 92.Tal M.C. Absence of autophagy results in reactive oxygen species-dependent amplification of RLR signaling. Proc. Natl. Acad. Sci. U.S.A. 2009;106:2770–2775. doi: 10.1073/pnas.0807694106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Jounai N. The Atg5 Atg12 conjugate associates with innate antiviral immune responses. Proc. Natl. Acad. Sci. U.S.A. 2007;104:14050–14055. doi: 10.1073/pnas.0704014104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Saitoh T. Loss of the autophagy protein Atg16L1 enhances endotoxin-induced IL-1beta production. Nature. 2008;456:264–268. doi: 10.1038/nature07383. [DOI] [PubMed] [Google Scholar]
- 95.Zhou R. A role for mitochondria in NLRP3 inflammasome activation. Nature. 2011;469:221–225. doi: 10.1038/nature09663. [DOI] [PubMed] [Google Scholar]
- 96.Shi C.S. Activation of autophagy by inflammatory signals limits IL-1beta production by targeting ubiquitinated inflammasomes for destruction. Nat. Immunol. 2012;13:255–263. doi: 10.1038/ni.2215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Dengjel J. Autophagy promotes MHC class II presentation of peptides from intracellular source proteins. Proc. Natl. Acad. Sci. U.S.A. 2005;102:7922–7927. doi: 10.1073/pnas.0501190102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Suri A. First signature of islet {beta}-cell-derived naturally processed peptides selected by diabetogenic class II MHC molecules. J. Immunol. 2008;180:3849–3856. doi: 10.4049/jimmunol.180.6.3849. [DOI] [PubMed] [Google Scholar]
- 99.Schmid D. MHC class II antigen loading compartments continuously receive input from autophagosomes. Immunity. 2007;26:79–92. doi: 10.1016/j.immuni.2006.10.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.van den Boorn J.G. Skin-depigmenting agent monobenzone induces potent T-cell autoimmunity toward pigmented cells by tyrosinase haptenation and melanosome autophagy. J. Invest. Dermatol. 2011;131:1240–1251. doi: 10.1038/jid.2011.16. [DOI] [PubMed] [Google Scholar]
- 101.Levitskaya J. Inhibition of antigen processing by the internal repeat region of the Epstein–Barr virus nuclear antigen-1. Nature. 1995;375:685–688. doi: 10.1038/375685a0. [DOI] [PubMed] [Google Scholar]
- 102.Levitskaya J. Inhibition of ubiquitin/proteasome-dependent protein degradation by the Gly-Ala repeat domain of the Epstein–Barr virus nuclear antigen 1. Proc. Natl. Acad. Sci. U.S.A. 1997;94:12616–12621. doi: 10.1073/pnas.94.23.12616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Paludan C. Endogenous MHC class II processing of a viral nuclear antigen after autophagy. Science. 2005;307:593–596. doi: 10.1126/science.1104904. [DOI] [PubMed] [Google Scholar]
- 104.Münz C. Human CD4+ T lymphocytes consistently respond to the latent Epstein–Barr virus nuclear antigen EBNA1. J. Exp. Med. 2000;191:1649–1660. doi: 10.1084/jem.191.10.1649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Leung C.S. Nuclear location of an endogenously expressed antigen, EBNA1, restricts access to macroautophagy and the range of CD4 epitope display. Proc. Natl. Acad. Sci. U.S.A. 2010;107:2165–2170. doi: 10.1073/pnas.0909448107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Nimmerjahn F. Major histocompatibility complex class II-restricted presentation of a cytosolic antigen by autophagy. Eur. J. Immunol. 2003;33:1250–1259. doi: 10.1002/eji.200323730. [DOI] [PubMed] [Google Scholar]
- 107.Riedel A. Endogenous presentation of a nuclear antigen on MHC class II by autophagy in the absence of CRM1-mediated nuclear export. Eur. J. Immunol. 2008;38:2090–2095. doi: 10.1002/eji.200737900. [DOI] [PubMed] [Google Scholar]
- 108.Lee H.K. In vivo requirement for Atg5 in antigen presentation by dendritic cells. Immunity. 2010;32:227–239. doi: 10.1016/j.immuni.2009.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Florey O. Autophagy machinery mediates macroendocytic processing and entotic cell death by targeting single membranes. Nat. Cell Biol. 2011;13:1335–1343. doi: 10.1038/ncb2363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Ireland J.M., Unanue E.R. Autophagy in antigen-presenting cells results in presentation of citrullinated peptides to CD4 T cells. J. Exp. Med. 2011;208:2625–2632. doi: 10.1084/jem.20110640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Kyewski B., Klein L. A central role for central tolerance. Annu. Rev. Immunol. 2006;24:571–606. doi: 10.1146/annurev.immunol.23.021704.115601. [DOI] [PubMed] [Google Scholar]
- 112.Anderson M.S. Projection of an immunological self shadow within the thymus by the aire protein. Science. 2002;298:1395–1401. doi: 10.1126/science.1075958. [DOI] [PubMed] [Google Scholar]
- 113.Klein L., Kyewski B. Self-antigen presentation by thymic stromal cells: a subtle division of labor. Curr. Opin. Immunol. 2000;12:179–186. doi: 10.1016/s0952-7915(99)00069-2. [DOI] [PubMed] [Google Scholar]
- 114.Nedjic J. Autophagy in thymic epithelium shapes the T-cell repertoire and is essential for tolerance. Nature. 2008;455:396–400. doi: 10.1038/nature07208. [DOI] [PubMed] [Google Scholar]
- 115.Kasai M. Autophagic compartments gain access to the MHC class II compartments in thymic epithelium. J. Immunol. 2009;183:7278–7285. doi: 10.4049/jimmunol.0804087. [DOI] [PubMed] [Google Scholar]
- 116.Hampe J. A genome-wide association scan of nonsynonymous SNPs identifies a susceptibility variant for Crohn disease in ATG16L1. Nat. Genet. 2007;39:207–211. doi: 10.1038/ng1954. [DOI] [PubMed] [Google Scholar]
- 117.Rioux J.D. Genome-wide association study identifies new susceptibility loci for Crohn disease and implicates autophagy in disease pathogenesis. Nat. Genet. 2007;39:596–604. doi: 10.1038/ng2032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Parkes M. Sequence variants in the autophagy gene IRGM and multiple other replicating loci contribute to Crohn's disease susceptibility. Nat. Genet. 2007;39:830–832. doi: 10.1038/ng2061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.McCarroll S.A. Deletion polymorphism upstream of IRGM associated with altered IRGM expression and Crohn's disease. Nat. Genet. 2008;40:1107–1112. doi: 10.1038/ng.215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Hugot J.P. Association of NOD2 leucine-rich repeat variants with susceptibility to Crohn's disease. Nature. 2001;411:599–603. doi: 10.1038/35079107. [DOI] [PubMed] [Google Scholar]
- 121.Ogura Y. A frameshift mutation in NOD2 associated with susceptibility to Crohn's disease. Nature. 2001;411:603–606. doi: 10.1038/35079114. [DOI] [PubMed] [Google Scholar]
- 122.Mortensen M. The autophagy protein Atg7 is essential for hematopoietic stem cell maintenance. J. Exp. Med. 2011;208:455–467. doi: 10.1084/jem.20101145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Salemi S. Autophagy is required for self-renewal and differentiation of adult human stem cells. Cell Res. 2012;22:432–435. doi: 10.1038/cr.2011.200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Pua H.H. A critical role for the autophagy gene Atg5 in T cell survival and proliferation. J. Exp. Med. 2007;204:25–31. doi: 10.1084/jem.20061303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Pua H.H. Autophagy is essential for mitochondrial clearance in mature T lymphocytes. J. Immunol. 2009;182:4046–4055. doi: 10.4049/jimmunol.0801143. [DOI] [PubMed] [Google Scholar]
- 126.Jia W. Autophagy regulates endoplasmic reticulum homeostasis and calcium mobilization in T lymphocytes. J. Immunol. 2011;186:1564–1574. doi: 10.4049/jimmunol.1001822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Miller B.C. The autophagy gene ATG5 plays an essential role in B lymphocyte development. Autophagy. 2008;4:309–314. doi: 10.4161/auto.5474. [DOI] [PubMed] [Google Scholar]
- 128.Arsov I. A role for autophagic protein Beclin 1 early in lymphocyte development. J. Immunol. 2011;186:2201–2209. doi: 10.4049/jimmunol.1002223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Manjithaya R. Unconventional secretion of Pichia pastoris Acb1 is dependent on GRASP protein, peroxisomal functions, and autophagosome formation. J. Cell Biol. 2010;188:537–546. doi: 10.1083/jcb.200911149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Duran J.M. Unconventional secretion of Acb1 is mediated by autophagosomes. J. Cell Biol. 2010;188:527–536. doi: 10.1083/jcb.200911154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Dupont N. Autophagy-based unconventional secretory pathway for extracellular delivery of IL-1beta. EMBO J. 2011;30:4701–4711. doi: 10.1038/emboj.2011.398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Uhl M. Autophagy within the antigen donor cell facilitates efficient antigen cross-priming of virus-specific CD8+ T cells. Cell Death Differ. 2009;16:991–1005. doi: 10.1038/cdd.2009.8. [DOI] [PubMed] [Google Scholar]
- 133.Li Y. Efficient cross-presentation depends on autophagy in tumor cells. Cancer Res. 2008;68:6889–6895. doi: 10.1158/0008-5472.CAN-08-0161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.English L. Autophagy enhances the presentation of endogenous viral antigens on MHC class I molecules during HSV-1 infection. Nat. Immunol. 2009;10:480–487. doi: 10.1038/ni.1720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Wenger T. Autophagy inhibition promotes defective neosynthesized proteins storage in ALIS, and induces redirection toward proteasome processing and MHCI-restricted presentation. Autophagy. 2012;8:350–363. doi: 10.4161/auto.18806. [DOI] [PubMed] [Google Scholar]
- 136.Luca A.D. CD4+ T cell vaccination overcomes defective cross-presentation of fungal antigens in a mouse model of chronic granulomatous disease. J. Clin. Invest. 2012;122:1816–1831. doi: 10.1172/JCI60862. [DOI] [PMC free article] [PubMed] [Google Scholar]