

Abstract
RecQL4, one of the five human RecQ helicases, is crucial for genomic stability and RecQL4 when mutated leads to premature aging phenotypes in humans. Unlike other human RecQ helicases, RecQL4 is found both in the nucleus and the cytoplasm. While the nuclear localization signal (NLS) and the retention domain at the N-terminus are responsible for the nuclear localization of RecQL4, the signal for its cytoplasmic localization is essentially unknown. In this study, two functional nuclear exporting signals (NESs; pNES2 and pNES3) were identified at the C-terminus of RecQL4. Deletion of pNES2 drastically diminished the cytoplasmic localization of RecQL4. Strikingly, addition of ubiquitination tail at the C-terminus of RecQL4 substantially enriched the cytoplasmic fraction of RecQL4 only in the presence of functional pNES2. Immunofluorescence studies revealed that the cytoplasmic RecQL4 was localized in mitochondria. Consistent with its mitochondrial localization, a regulatory role for RecQL4 in the maintenance of mitochondrial DNA (mtDNA) copy number was demonstrated. Elevation of ectopic expression of RecQL4 increased the mtDNA copy number in HEK293 cells while RecQL4 knock down markedly decreased the mtDNA copy number in U2OS cells. Additionally, a substantially increased level of mitochondrial superoxide production, and a markedly decreased repair capacity for oxidative DNA damage were observed in the mitochondria of both RecQL4 deficient human fibroblasts and RecQL4-suppressed cancer cells. These data strongly suggest a regulatory role for RecQL4 in mitochondrial stability and function. Collectively, our study demonstrates that NES-mediated RecQL4 export to the cytoplasm is essential for the maintenance of mitochondrial genome stability.
Keywords: RecQL4, Nuclear exporting signal (NES), Mitochondrial co-localization, ROS (Reactive oxygen species), Oxidative damage repair
1. Introduction
Human RecQ helicases share an extensive homology with E.coli RecQ protein. RecQ protein acts as a suppressor of illegitimate recombination in E. coli and its mutants often exhibit genomic instability due to improper resolution of DNA secondary and tertiary structures arising during diverse DNA metabolic activities (Chu and Hickson, 2009). There are 5 RecQ helicases thus far reported in humans [RecQ1, Werner helicase-(WRN), Bloom helicase (BLM), RecQL4 and RecQL5). Their importance in genomic stability is best illustrated in human, where mutations in three RecQ family members (BLM, WRN and RecQL4) result in severe autosomal recessive cancer prone disorders: Bloom syndrome (BS), Werner syndrome (WS) and Rothmund-Thomson syndrome (RTS) (Kitao et al., 1999b, Monnat, 2010, Singh et al., 2009). Additionally, mutations in RecQL4 also result in RAPADILINO and Baller-Gerold (BGS) syndromes (Dietschy et al., 2007). RecQL4 structurally differs from other human RecQ helicases by having nuclear localization and retention domains at the N-terminus as well as a replication-associated domain homologous to yeast Sld2 which functions in replisome assembly (Sangrithi et al., 2005). RecQL4 was initially shown to have only ATPase activity, but recent studies revealed a DNA unwinding activity on different DNA substrates in vitro (Rossi et al., 2010, Suzuki et al., 2009, Xu and Liu, 2009). RecQL4 participates in diverse DNA repair pathways through interaction with multiple DNA repair proteins (Fan and Luo, 2008, Schurman et al., 2009, Singh et al., 2010) and RecQL4-deficient fibroblasts isolated from RTS patients are extremely sensitive to genotoxic agents (Jin et al., 2008, Werner et al., 2006). Consistent with the pathological features of RTS patients, RecQL4 knockout mice also exhibit growth retardation, early death and other symptoms (Mann et al., 2005) illustrating that the RTS features are primarily due to mutations in RecQL4.
Unlike BLM and WRN, whose proteins are exclusively localized to the nucleus and nucleolus, RecQL4 was found both in the nucleus and the cytoplasm (Hayakawa et al., 2000, Marciniak et al., 1998). Molecular mechanism responsible for RecQL4 nuclear importing has been well studied and the nuclear localization signals (NLSs) have been identified in the amino terminus of RecQL4 (Burks et al., 2007). Acetylation of five lysine residues within one of the NLS domains by p300 histone acetyltransferase significantly decreases the nucleoplasmic localization of RecQL4 (Dietschy et al., 2009). Further, demonstration of co-localization of RecQL4 nuclear foci with Rad51 and poly(ADP-ribose) polymerase-1 (PARP-1), two key proteins involved in DNA repair (Petkovic et al., 2005, Woo et al., 2006) suggests a critical role for RecQL4 in the maintenance of genomic stability. An earlier study demonstrated that RecQL4 is the component of a stable complex consisting of UBR1 and UBR2, two ubiquitin ligases acting on protein degradation (Yin et al., 2004).
Although the nuclear functions of RecQL4 have been fairly well established, the functional significance of RecQL4 in the cytoplasm as well as the molecular signaling responsible for cytoplasmic recruitment of RecQL4 remains an enigma. In this study, we have identified for the first time two potential nuclear export signals (NESs) in RecQL4 by GFP-GFP-NES system and one of the NESs is chiefly responsible for the cytoplasmic localization of RecQL4 as demonstrated by functional deletion analysis. Additionally, our study demonstrates that RecQL4 co-localizes with mitochondria in the cytoplasm and RecQL4 suppression leads to mitochondrial dysfunction involving decreased mtDNA copy number, increased superoxide production, enhanced mitochondrial fragmentation and diminished mtDNA repair capacity after oxidative stress. Collectively, our study demonstrates that cytoplasmic RecQL4 plays a crucial role in mitochondrial genome stability.
2. Materials and Methods
2.1 Plasmid construction
Full-length human RecQL4 cDNA was obtained from Genecopoeia and subcloned into multiple cloning sites of pEGFP-C1 vector (Clontech) at Bgl II/XhoI sites. A series of truncated and mutated versions of RecQL4 were generated from pEGFP-C1-RecQL4 by PCR method. To construct double GFP expression vector system, GFP cDNA was amplified from pEGFP-C1 and re-cloned into pEGFP-C1 vector next to intrinsic GFP. All of the putative nuclear export signals (pNESs) were cloned into the above vector downstream of double GFP (Figure 1A). For retroviral constructs, a pair of oligos containing Kozak and Flag sequences were annealed and ligated into the vector pRetroX-Tight-Pur (Clontech). Wild type, nuclear retention domain-mutated and NESs (nuclear export signals)-mutated RecQL4 cDNAs were cloned into the Flag-tagged retroviral vector. All constructs were verified by sequencing and expected sizes of proteins were confirmed by western blot analysis.
Figure 1.
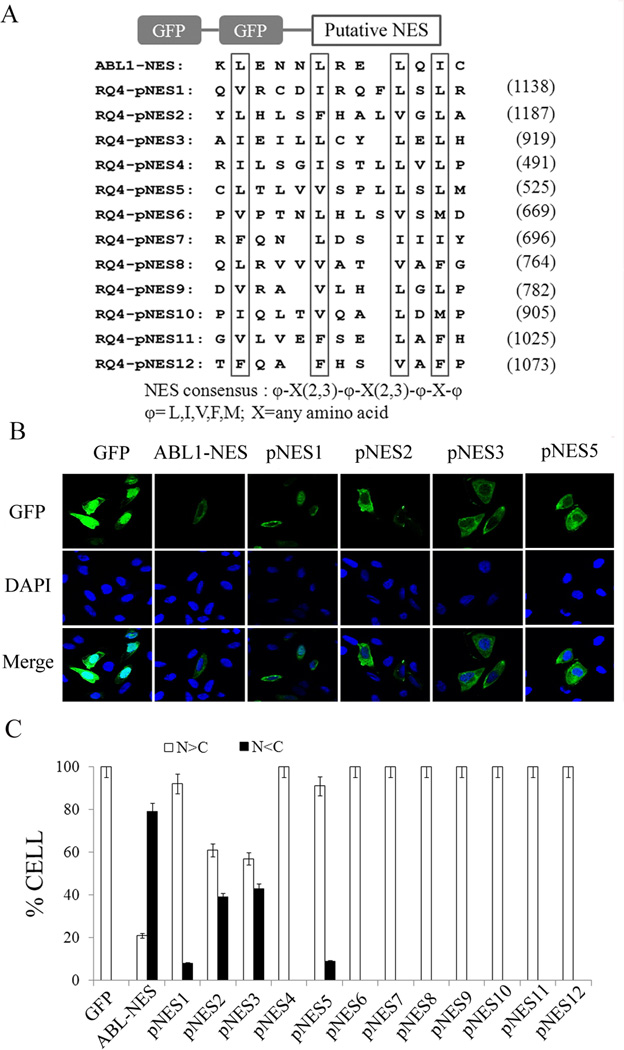
Identification of RecQL4 nuclear export signals by GFP-GFP system. (A) Schematic representation of GFP-GFP-RecQL4_NESs. All of putative NESs in RecQL4 were examined by NES consensus: φ-X(2,3)-φ-X(2,3)-φ-X-φ, φ= L,I,V,F,M; X=any amino acid. The key hydrophobic amino acids were boxed. c-Abl-NES was used as positive control; (B) Subcellular distribution of GFP after transient transfection of GFP-GFP-NES constructs (ABL1-NES, pNES1–3 & pNES5) in U2OS cells; (C) Percentage of cells with more GFP protein in nuclear than in cytoplasm (N>C) or more in cytoplasm than in nuclear (N<C). A total of 200 GFP-positive cells were counted in each of three independent experiments.
For RecQL4 shRNA lentiviral constructs, two 21mer shRNA sequences including shRNA1: GCTCAAGGCCAATCTGAAAGG (366–386, Accession No. NM_004260) and shRNA2: GGGAATCTGTCCTGCAGAAGA (1727–1747) were cloned into pSIF-H1-shRNA vector (SBI Biosystems) and pU2-FH lentiviral vector pU2-FH (Kindly provided by Dr. Yunfeng Feng), respectively. The control scrambled 21mer shRNA sequence is GAAGAGGACACGCCTTAGACT. Lentiviral particles containing shRNA sequence were produced in 293 packaging cells, and used for infecting cells following the recommended protocol by manufacturer.
2.2 Establishment of cell lines with altered RecQL4 expression
Human osteosarcoma cell line U2OS and human embryonic kidney cell line HEK293 were maintained in Dulbecco’s modified Eagle’s medium (DMEM; GIBCO) supplemented with 10% heat-inactivated fetal bovine serum (FBS; Hyclone) at 37°C in humidified 5% CO2 incubator. RecQL4-suppressed U2OS clonal cell lines were established by transduction of cells with lentiviral particles containing either RecQL4 shRNA1 and shRNA2 or scrambled control shRNA and subsequent selection of cells selected with 2 µg/ml puromycin. For inducible system, HEK293 cells were first infected with TetR retroviral particles using a standard procedure. TetR-expressing HEK293 cells were then infected with wild type or NESs-mutated RecQL4 retroviral particles. After selection with puromycin, clonal cells were analyzed by western blot for the inducible expression of either wild type RecQL4 (HEK293Tet-Flag-RQ4) or RecQL4 mutants (HEK293Tet-Flag-RQ4ΔNES2&3) after growth in the presence of 0.5 µg/ml doxycycline.
2.3 Immunofluorescence staining
Cells were grown on 2-well chamber slides for 24 h, and immunofluorescence staining was then performed following the procedures as following. For mitochondrial staining, cells were washed with PBS and then incubated in DMEM supplemented with 500 nM of MitoTracker Red CM-H2XRos (Molecular Probes) for 30 min. For endogenous RecQL4 and PolG staining, cells were fixed in 4% formaldehyde, permeabilized with 0.1% Triton X-100 and blocked with 5% non-fat milk (Santa cruz), and then were incubated with appropriate primary antibodies: rabbit anti-human RecQL4 (SDI, 1;100 dilution) and goat anti-human PolG (Santa cruz, 1:100 dilution) at 4 °C overnight followed by fluorescence-conjugated secondary antibodies: Alexa Fluor 488 Chicken Anti-Goat IgG (H+L) (Invitrogen, 1:100 dilution) and Texas Red Goat Anti-Rabbit IgG antibodies (Vector laboratories, 1:100 dilution) at RT for 2h. After several washes with PBS, the cells were counterstained by DAPI and the fluorescent images were captured using the Leica confocal microscope.
2.4 Genomic DNA isolation and protein isolation
AG18375 and AG18459 human fibroblasts, U2OS-shControl and shRecQL4 cells as well as HEK293Tet-RecQL4 and HEK293Tet-RecQL4ΔNES2&3 cells were cultured in 6-well plates at 1×105 cells per well. For inducible HEK293 cell lines, cells were cultured in the DMEM medium containing 10% serum in the presence or absence of 0.5 µg/ml doxycycline for 48 h. Cells were washed once and then collected in cold PBS. After centrifugation, one half of the cells (0.5×105) were used for genomic DNA isolation (TIANamp Genomic DNA Kit, Tiangen, China). The remaining cells were resuspended in lysis buffer (Cell Signal) for isolating proteins used for western blot approach.
For isolating nucleic and mitochondrial proteins, 1×107 cells were suspended in lysis buffer and pure mitochondrial fraction was isolated by using Qiangen Qproteome mitochondria isolation kit following the manufacture recommended procedures.
2.5 Mitochondrial morphology analyses
RecQL4-suppressed U2OS cells were seeded in 6-well plates at 1×105 cells per well. Scrambled shRNA-transfected U2OS cells were used as control. Cells were treated with 12.5 µM Menadione for 60 min, gently washed three times with PBS, and then maintained in fresh DMEM supplemented with 10%FBS at 37°C in a humidified 5% CO2 incubator for 48 h. Before fixation, cells were incubated in DMEM supplemented with 25 nM DiOC6 (3,3′-dihexyloxacarbocyanine iodide) for 15 min. Mitochondrial morphology was analyzed using Leica Confocal Microscope.
2.6 Real-time PCR and long-range quantitative PCR (QPCR)
The human nuclear encoded 18S rRNA (Nu18S) gene and mitochondrial NADH dehydrogenase 1 (mtND1) were quantified separately by Real-time PCR using SYBR Green detection on a CFX96 Real-Time PCR Detection System Bio-Rad) using the following primers: Nu18S-F, 5’-TAGAGGGACAAGTGGCGTTC; Nu18S-R, 5’-CGCTGAGCCAGTC AGTGT; mtND1_3212F, 5’-CACCCAAGAACAGGGTTTGT; mtND1_3319R, 5’-TGGCCA TGGGTATGTTGTTAA (Petit et al., 2003). PCR was performed at 95°C for 1 min followed by 40 cycles at 95°C for 10 s and 60°C for 30s. All reactions were done in triplicate and three independent experiments were performed. Relative mtDNA in each sample was calculated by CFX Manager.
To facilitate the comparison in long range QPCR result, we have equalized the amount of mtDNA template used for long range QPCR reaction in menodione-untreated and treated cells. First, mtND1 gene was quantified in each DNA sample by Real time RT-PCR using nuclear 18S rRNA as control. The input of mtDNA template was then adjusted based on mtND1 to guarantee equal amount of mtDNA template in each sample was used for the long range QPCR reaction. Nearly full-length (16261bp) mtDNA fragments were amplified using LA Taq (Takara) following manufacturer's protocol. The primers were essentially the same as previously described (Santos et al., 2008): hmt15149F, 5’-TGAGGCCAAATAT CATTCTGAGGGGC and hmt14841R, 5’-TTTCATCATGCGGA GATGTTGGATGG. PCR reactions were performed at 94°C for 1 min followed by 25 cycles at 98°C for 10 s, 60°C for 40 s, 68°C for 16 min and a final elongation for 10 min. The products were separated by electrophoresis on a 0.8% agarose gel stained with ethidium bromide.
2.7 Mitochondrial superoxide production
Both RecQL4-suppressed and scrambled shRNA-transfected U2OS cells were cultured on chamber slides for 48h and then stained with 5 µM MitoSOX Red (Molecular Probes) for 30 min at 37°C. After three gentle washes with pre-warmed PBS, cells were trypsinized and resuspended in BD Falcon tubes (BD Biosciences) at concentration of 1×107 cells/ml, and then analyzed using BD FACSAria (BD Biosciences). For statistical analysis, at least 10,000 events were collected for each of three independent experiments.
3. Results
3.1 Identification of NES in RecQL4
Nuclear localization signals (NLSs) located at N-terminal of RecQL4 protein have been suggested to be responsible for RecQL4 nuclear localization (Burks et al., 2007). To verify the importance of NLS, HEK293 cells were transfected with NLS-deleted RecQL4 construct and sub-cellular distribution of RecQL4 protein was monitored by immunofluorescence staining technique. The result showed that RecQL4 protein with NLS deletion was predominantly cytoplasmic (Suppl. Fig. 1). To identify the potential NESs within RecQL4, the protein sequence was screened for a short leucine-rich NES motif, recognized as the general accepted loose consensus φ-X(2,3)-φ-X(2,3)-φ-X-φ φ=L,I,V,F,M (Kutay and Guttinger, 2005). Twelve putative NESs, ranged from the helicase domain to the carboxyl terminus (C-terminus), were identified. All the putative NESs (named as pNES1–pNES12), together with a well-documented NES as a positive control within the ABL1 gene (Taagepera et al., 1998), were synthesized and inserted into the GFP-GFP plasmid separately (Fig. 1A) as described (See Materials and Methods). The plasmids each with one putative NES (pNES1–pNES12) fused to the C-terminus of double GFP coding sequences were transfected into U2OS cells for determining the subcellular distribution of GFP by fluorescence microscopy. Among the NESs tested, GFP-GFP with pNES2 or pNES3 tail displayed a much higher proportion of cytoplasmic distribution in comparison to other NES signals (Fig. 1B, Suppl. Fig.2). To verify and validate this observation, a total of 200 GFP-positive cells were examined and categorized as N>C (more GFP in nucleus) or N<C (more GFP in cytoplasm) groups based on the GFP intensity in the nucleus and the cytoplasm. Cells transfected with either GFP-pNES2 or GFP-pNES3 alone showed a consistently higher percentage of N<C cells (Fig. 1C). Among the other NESs, cells transfected with either pNES1 or pNES5 also showed more GFP in the cytoplasm (N<C) but the percentage of cells with N<C was much less compared to cells transfected separately with either pNES2 or pNES3. The highest nuclear exporting of RecQL4 was observed in cells transfected with pNES3 followed by pNES2, pNES5 and pNES1.
To provide further evidence, a series of GFP-RecQL4 constructs containing either full length RecQL4 or deletion of each of the three domains (C-, N-terminus or helicase domain) were constructed. Additionally, plasmids containing RecQL4 C-terminal fragments (aa825–1018, 1019–1208) either with pNES2 and/or pNES3 or without pNES2 signal (1187–1198) were also constructed (Fig. 2A). In corroboration with a previous report (Woo et al., 2006), cells transfected with full length GFP-RecQL4 construct displayed a moderate level of GFP in the cytoplasm. However, transfection with GFP-RecQL4_N (1–475) construct with deletion of all the pNESs abolished the cytoplasmic distribution of RecQL4 (Suppl. Fig. 3). As expected, transfection of GFP-RecQL4_M (476–824) plasmid with a functional pNES5 only slightly increased the level of RecQL4 in the cytoplasm. In contrast, the strongest GFP fluorescence in the cytoplasmic region was observed in cells transfected with pNES2-containing plasmids (GFP-RecQL4 aa825–1208) and GFP-RecQL4-C2 (aa1019–1208). Transfectoin of pNES2 deleted plasmid (GFP-RecQL4-C2ΔNES2) results in a reduced cytoplasmic signal to a large extent (Fig. 2B). It should be noted that the weak residual cytoplasmic GFP fluorescence detected in cells transfected with pNES2 deletion plasmid may be probably due to an existing weak functional pNES1. Although GFP-GFP-pNES system identified pNES3 as a potent nuclear export signal (Fig. 1B), it was not confirmed by GFP-RecQL4 deletion analysis (Fig. 2B). Since pNES2 is located at the distal end of the protein relative to pNES3, we assumed that that pNES3 may be masked by protein conformation structure. To verify this assumption, we generated different RecQL4 constructs by either replacing pNES2 with pNES3 or switching the locations of pNES2 & pNES3 (Suppl.Fig.4). The results demonstrate that pNES3 is actually functional when it is placed at the location of pNES2 suggesting that pNES3 motif is normally masked most probably by RecQL4 protein conformation structure. Therefore, we conclude that pNES2 consensus motif with amino acid location of 1187–1198 is the most potent NES that recruits RecQL4 to the cytoplasm.
Figure 2.
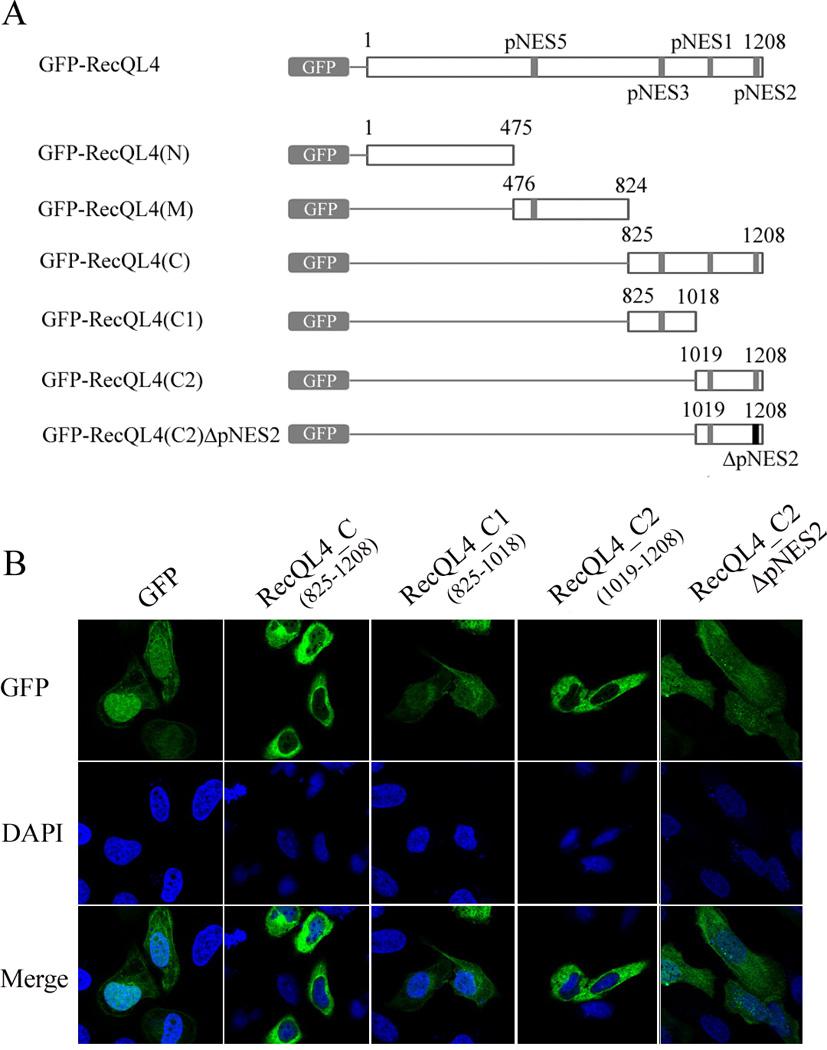
Subcellular distribution of pNES-containing GFP-RecQL4 truncated protein. (A) Schematic representation of GFP-RecQL4 and its deletion constructs. Two regions of potential nuclear export signals were represented by black box. (B) Cytoplasmic distribution of GFP in U2OS cells after transfection with truncated GFP-RecQL4 constructs or pNES2-deleted construct (GFP-RecQL4_C2ΔpNES2). After transfection, the cells were analyzed by confocal microscopy.
3.2 RecQL4 protein is co-localized with mitochondria and present in mitochondrial protein fraction
Observation of DNA replication and repair functions of nuclear RecQL4 (Liu, 2010, Ouyang et al., 2008) prompted us to verify whether or not the cytoplasmic RecQL4 plays similar roles in mitochondria. First, we wished to determine whether or not the cytoplasmic RecQL4 is localized in mitochondria by co-localization analysis of endogenous RecQL4 and mitochondria in U2OS cells by an indirect immunofluorescence-based approach. As shown in Figure 3A, co-localization between endogenous RecQL4 and mitochondria in U2OS cell was clearly observed. Quantitatively analysis in 10 randomly chosen cells by CoLocalize Express showed the Mander’s overlap coefficient value of 1, suggestive of 100% colocalization between endogenous RecQL4 and mitochondria. Since PolG is localized to mitochondria, we wished to determine whether or not the cytoplasmic RecQL4 co-localizes with DNA polymerase gamma (PolG) in U2OS cells (Suppl. Fig 5). Quantitative analysis performed in more than 10 randomly selected cells using CoLocalize Express software showed the Pearson’s correlation coefficient value of 0.9117. Since PolG is a mitochondria-specific protein, our finding indicates that both RecQL4 and PolG proteins are localized in mitochondria.
Figure 3.

(A)Colocalization of endogenous RecQL4 with mitochondria in U2OS by an indirect immunofluorescence approach. The cells were stained by 500 nM of Mirotracker Red CM-H2Xros for 30 mins, fixed by 4% formaldehyde for 10min followed by 0.1% Triton X-100 for 5min, and then incubated with first RecQL4 antibody(SDI, 1:100 dilution) overnight at 4°C followed by fluorescein goat anti-Rabbit IgG secondary Antibody (Vector laboratories) at RT for 2 h. The staining result was visualized under the Leica Confocal microscope; (B) Level of RecQL4 protein in both nucleic and mitochondrial fractions of U2OS cells with and without RecQL4 suppression as well as in HEK293-Tet/On cells before and after RecQL4 induction determined by western blot approach. Presence of RecQL4 protein in mitochondrial fraction of both cell types was observed, RecQL4 antibody was purchased from Cell Signal (Catalog: #2814).
To verify the co-localization results, western blot analysis was performed to detect the presence of RecQL4 protein in the mitochondrial fraction isolated from the whole cell lysates. Consistent with our co-localization result, RecQL4 protein was clearly detected in the mitochondrial fraction as well as in the nuclear fraction, and RecQL4 silencing significantly decreased their levels in both fractions of U2OS cells (Fig. 3B). Likewise, treatment of HEK293-Tet/On cells with 0.5 µg/ml of doxycycline for 48 h substantially increased the RecQL4 level both in the nuclear and mitochondrial fractions. In corroboration with the immunofluorescence studies, mitochondrial fraction exhibited a much lower level of RecQL4 protein relative to nuclear fraction. Lack of detectable level of lamin B indicated that the mitochondrial fraction was largely free of nuclear contamination (Fig. 3B).
To further verify the importance of NES in cytoplasmic recruitment of RecQL4, subcellular distribution of Tet-induced Flag-tagged RecQL4 protein was analyzed by immunoflurorescence staining and western blot techniques in HEK293 cells. As shown in Figure 4A, cytoplasmic distribution was observed in HEK293 cells when wild type RecQL4 protein was induced, whereas deletion of NES2&3 motifs led to exclusively nuclear distribution of RecQL4. In support, western blot analysis demonstrated that RecQL4 protein was present both in cytosolic and mitochondrial fractions of Dox-treated HEK293 cells, whereas when nuclear exporting signals NES2&3 were deleted, cytoplasmic RecQL4 was undetectable (Fig. 4B).
Figure 4.

(A) Subcellular distribution of Flag-RecQL4 in Dox-treated HEK293-tet/on cells detected by immunofluroscence staining analysis; (B) Level of RecQL4 protein in nucleic, cytosolic and mitochondrial fractions of HEK293-Tet/On cells before and after deletion of NES2&3 motifs.
3.3 RecQL4 modulates mtDNA copy number
Co-localization of RecQL4 and mitochondria led us to investigate whether or not RecQL4 has the potential role in maintaining mitochondrial DNA copy number. For this purpose, the relative mtDNA copy number was analyzed in HEK293 cells after inducible expression of either wild type or pNES2&3-deleted Flag-RecQL4 constructs. As shown in Figure 5A, an increased level of wild type RecQL4 expression induced by doxycycline treatment in HEK293 cells leads to a substantial elevation of mtDNA copy number relative to mock treated cells (Fig. 5A). However, over-expression of mutated Flag-RecQL4 with pNES2&3 deletions at the C-terminus did not exert any effect on mtDNA copy number owing to the abolition of RecQL4 recruitment to the cytoplasm. These findings suggest that the potential NESs identified in this study are crucial for RecQL4 localization in the cytoplasm, where it plays an important role in regulating mtDNA copy number. To further verify and explore the dependency of mtDNA copy number on RecQL4 expression, mtDNA copy number was determined in U2OS cells with a high endogenous level of RecQL4 after RecQL4 suppression. For this purpose, two stable clonal cell lines, C2 from shRNA1 (shRecQL4-C2) and C4 from shRNA2 (shRecQL4-C4) with highly reduced RecQL4 expression were established. Both of these cell lines with stable RecQL4 knockdown showed a significantly less mtDNA copy numbers as compared to scrambled shRNA transfected cells (Fig. 5B). These findings indicate that the cytoplasmic localization of RecQL4 plays a critical role in modulating mtDNA copy number.
Figure 5.
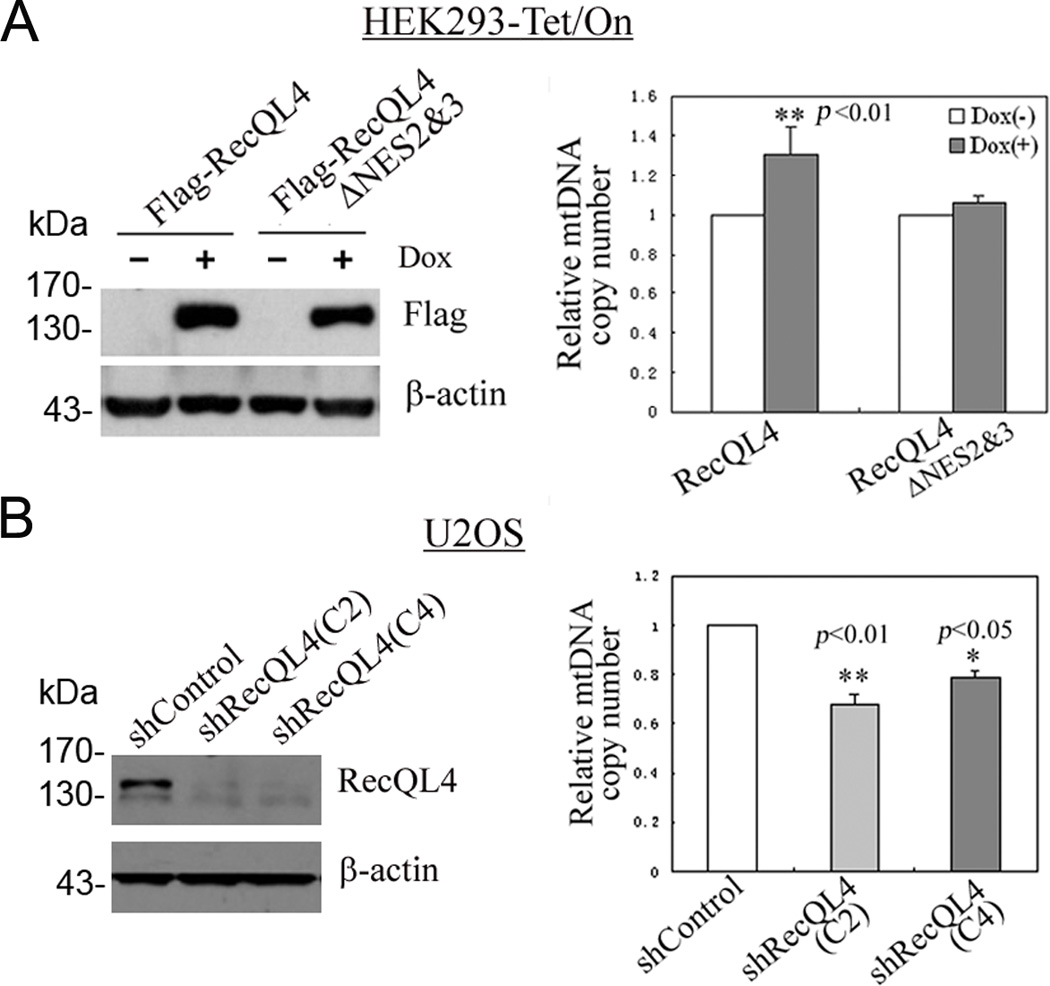
(A) An increased mtDNA copy number in HEK293 cells after an induced expression of wild type RecQL4, not RecQL4 ΔNES2&3 in the presence of 0.5µg/ml of doxycycline for 48 h; (B) Analysis of mtDNA copy number in U2OS cells after shRNA-mediated RecQL4 suppression. The mtDNA copy number was quantified by real-time quantitative PCR by comparing the ratio of mtDNA and nDNA. *, p<0.05; **, p<0.01.
3.4 RecQL4 silencing elevates oxidative stress and causes morphological changes in mitochondria
We next determined whether or not RecQL4 is essential for mitochondrial integrity, mitochondrial oxidative stress was assessed in RecQL4 suppressed U2OS cells using MitoSOX™ Red (Payne et al., 2007). Both control and RecQL4 shRNA transfected cells were stained with MitoSOX™ Red for 30 min and subsequently analyzed by flow cytometry. As shown in Fig. 6A, the curve formed by MitoSOX™ Red-positive cells after RecQL4 suppression shifted markedly to the right of shControl cells, indicating an elevated level of oxidative stress in U2OS cells after RecQL4 silencing.
Figure 6.
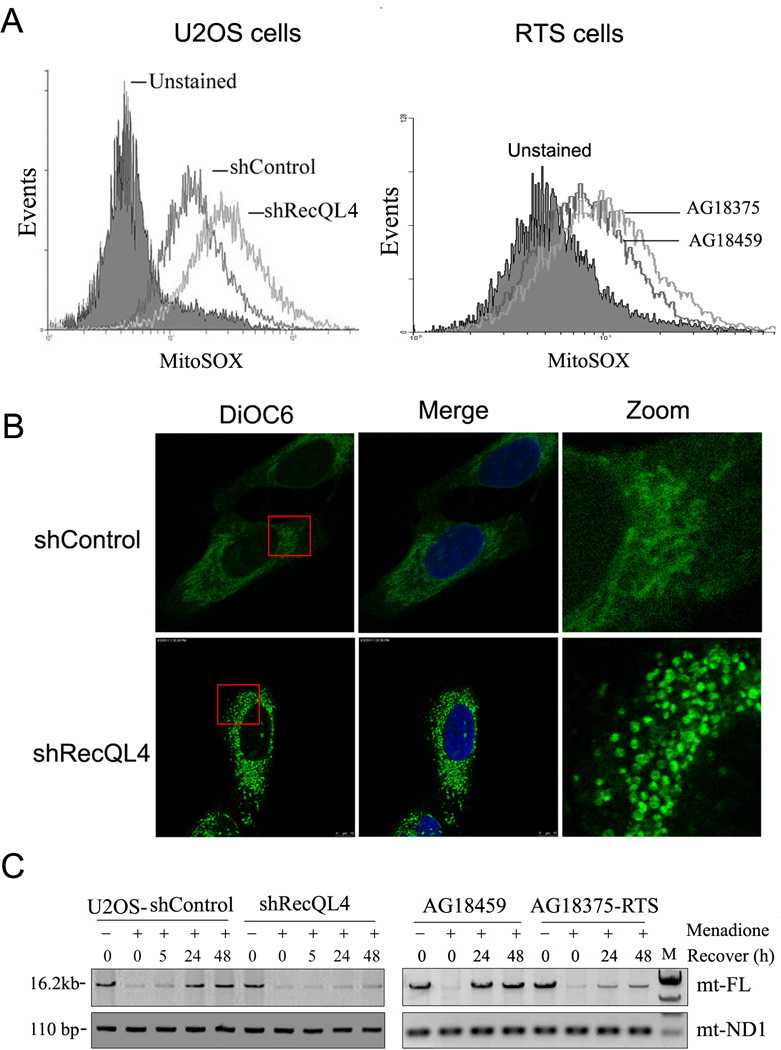
(A) Mitochondrial ROS level in scrambled shRNA-transfected (shControl) and RecQL4-suppressed U2OS cells (shRecQL4) as well as in human fibroblasts derived from unaffected heterozygous mother (AG18459) and affected homozygous son (AG18375). Cells were stained with MitoSOX™ Red for 30 min and then analyzed by flow cytometry; (B) Alteration of mitochondrial morphology in shControl and RecQL4-suppressed U2OS cells 48 h post menadione treatment. Cells were stained with 25 nM DiOC6 for 15 min, fixed and analyzed by confocal microscopy analysis; (C) Long range QPCR result of full length mitochondrial DNA (mt-FL) in shControl and shRecQL4 U2OS cells at 0, 5, 24, 48 h post menadione treatment, and in AG18459 and AG18375-RTS cells at 0, 24, 48 h post menadione treatment. Equal amount of mtDNA template DNA in each sample normalized by mtND1 gene was used for long range QPCR reaction.
We next determined whether or not the human fibroblasts inherently deficient in RecQL4 also exhibit increased ROS production. For this purpose, human fibroblasts derived from unaffected heterozygous mother (AG18459) and affected homozygous son (AG18375) were utilized. Consistent with our observation in U2OS cells, RecQL4 homozygous deficient fibroblasts (AG18375) displayed an increased mitochondrial ROS production (Fig. 6A).
3.5 RecQL4 regulates the efficiency of mitochondrial DNA repair
A potential role for RecQL4 in regulating mitochondrial repair efficiency was further evidenced in cells challenged with menadione, a potent chemical agent that can induce oxidative stress in both cytosol and mitochondrial matrix. We first examined the morphological changes in mitochondria in RecQL4-silenced and proficient U2OS cells with and without menadione treatment. As shown in Suppl. Figure 6, mitochondria in RecQL4-suppressed cells stained with DiOC6 tends to be aggregate together when compared with control shRNA-transfected U2OS cells. Moreover, after treatment with 12.5 µM of menadione for 60 min, mitochondrial fragmentation was observed in both control shRNA and RecQL4 shRNA transfected U2OS cells at 24h post menadione treatment. However, at 48 h post treatment, the morphological change observed in menadione-treated control cells was grossly recovered to untreated cells, whereas RecQL4 shRNA transfected cells failed to recover and still showed severe mitochondrial fragmentation (Fig. 6B).
A long range QPCR approach (Santos et al., 2002) for amplification of 16.2 kb mtDNA was subsequently used to assess the repair efficiency of mtDNA. A major advantage of this method is that any damaged and unrepaired bases will terminate the polymerase elongation during PCR amplification thereby reducing the efficiency of DNA amplification. The mtND1 PCR product (110 bp) was used as a control to normalize the input of the mtDNA template amount in each sample. As shown in Fig. 6C, both control and RecQL4 shRNA-transfected U2OS cells showed a markedly decreased level of mtDNA PCR product at early time-points after menadione treatment. In contrast to control cells, which showed the restoration of full-length mtDNA PCR product by 24 h after treatment, RecQL4 silenced cells displayed a very low level of PCR product even at 48 h post treatment. In consistent with the findings in U2OS, RecQL4 homozygous deficient fibroblasts (AG18375) also displayed a much delayed mtDNA repair post menadione treatment as compared to RecQL4 heterozygous (AG18459) cells (Fig. 6C). These findings clearly illustrate that RecQL4 is essential for the efficient repair of oxidative DNA damage in mitochondria. In contrast to increased ROS production and mtDNA repair deficiency, no significant differences were observed in the copy number of mtDNA between heterozygous and RecQL4 homozygously deficient fibroblasts probably due to the dosage effect as the heterozygous status of RecQL4 gene in AG18459 cells.
3.6 An increased recruitment of RecQL4 protein in mitochondria under oxidative stress condition
We then tested whether there is any change in the mitochondrial distribution of RecQL4 protein under oxidative stress condition. U2OS cells were treated with 25µM menadione for 60 mins in serum-free DMEM, washed with PBS and then cultured in full growth medium. Cell lysates were collected at 30mins post menadione treatment and applied to western blot analysis (Fig. 7A). The result demonstrated that mitochondrial RecQL4 level was markedly increased after menadione treatment with 5.4-fold higher than that in untreated control cells, suggesting that recruitment of RecQL4 protein in mitochondria is largely enhanced when mtDNA was damaged by oxidative stress inducing agent.
Figure 7.
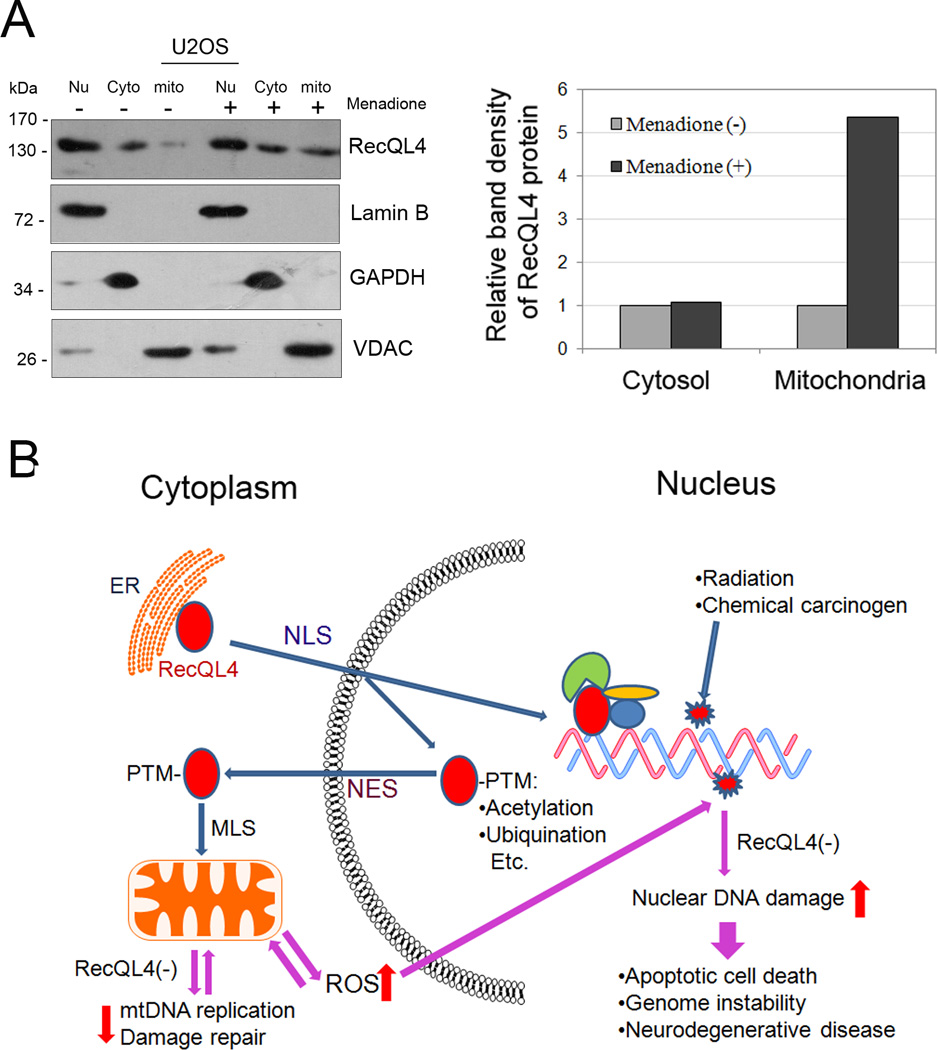
(A)Sub-cellular distribution of RecQL4 protein before and after menadione treatment. Mitochondrial proportion of RecQL4 protein was quantified using the ImageJ software (http://rsbweb.nih.gov/ij/) and normalized to mitochondrial loading control (VDAC); (B)Dynamic regulation of RecQL4 nuclear/cytoplasmic shuttling. NLS: nuclear localization signal; NES: nuclear export signal; MLS: mitochondrial localization signal; PTM: post translational modification.
4. Discussion
Available data suggest that RecQL4 participates in essential nuclear functions involving DNA replication and repair (Liu, 2010, Ouyang et al., 2008). However, a fair degree of controversy exists in the published literature regarding the sub-cellular distribution of RecQL4 in the nucleus and the cytoplasm (Petkovic et al., 2005, Yin et al., 2004, Kitao et al., 1999a). In immortalized and malignantly transformed human cells, RecQL4 localization was observed both in the cytoplasm and the nucleus although the relative abundance of RecQL4 reported in both of the sub-cellular compartments showed some variations (Woo et al., 2006, Yin et al., 2004). Thus, it appears that the sub-cellular distribution of RecQL4 is dependent on cell type and probably cell cycle phases (Petkovic et al., 2005). It is likely that some of the conflicting results obtained on the sub-cellular distribution of RecQL4 stem from antibody specificity issues. In this study, we opted to use a direct approach to identify its locale more precisely. A GFP-RecQL4 expression construct was generated and then transfected into U2OS cells. Consistent with the previous report (Woo et al., 2006), we found that nuclear is the main sub-cellular site for RecQL4 localization, whereas only a very small percentage of RecQL4 protein is localized to cytoplasm. The same approach was also utilized to determine whether or not RecQL4 has potential NES domains, which could explain the nuclear export of RecQL4 to the cytoplasm.
It is fairly well established that N-terminal nuclear localization signals (NLSs) are responsible for RecQL4 nuclear localization (Burks et al., 2007). In an effort to search signaling motifs that mediate RecQL4 nuclear exporting, NES predictor program was utilized to analyze the leucine-rich motif, a consensus signal mediating c-Abl protein nuclear-exporting (Taagepera et al., 1998), in the RecQL4 coding region. Using GFP-GFP-NES constructs, we have identified two NESs: pNES2 and pNES3 that are located at the C-terminus of RecQL4 are sufficient for GFP nuclear exporting. Although two potential NESs (pNES2 and pNES3) were identified by GFP-GFP-NES system, only the cells transfected with NES2-containing RecQL4 plasmid showed the strongest cytoplasmic GFP fluorescence. This finding was further confirmed by transfection of cells with pNES2-deleted GFP-RecQL4 construct that almost abolished the cytoplasmic export of RecQL4, suggesting that this signaling domain is chiefly responsible for the cytoplasmic localization of RecQL4. Taken together, our data attribute an essential role to pNES2 in mediating the nuclear export of RecQL4 to the cytoplasm.
After establishing the molecular basis for the nuclear export of RecQL4, we wished to determine the functional significance of RecQL4 in the cytoplasm. Since mitochondria are the most abundant organelle in the cytoplasm, we first analyzed whether or not the cytoplasmic RecQL4 is present in mitochondria. Our immunofluorescence staining results obtained on U2OS tumorigenic cells, together with the western blot result observed in mitochondrial fractions of U2OS and inducible HEK293 cells, clearly illustrated that cytoplasmic RecQL4 is present in and co-localizes with mitochondria. Further, two research articles that appeared very recently convincingly demonstrated the localization of a fraction of RecQL4 in mitochondria (Croteau et al., 2012, De et al., 2012). RecQL4 has been shown to play a critical role in the replication of nuclear DNA through its interaction with proteins involved in DNA replisome assembly (Sangrithi et al., 2005, Korhonen et al., 2004). Further, depletion of RecQL4 has been shown to diminish the replication potential of mammalian cells (Thangavel et al., 2010). However, it is not clear whether or not the cytoplasmic RecQL4 exerts a similar role in mtDNA replication. It is well established that mtDNA replication is performed by a machinery including Twinkle DNA helicase, a single-stranded DNA-binding protein and mitochondrial DNA polymerase gamma (PolG) (Korhonen et al., 2004). Our immunofluroscence staining data clearly showed the co-localization of RecQL4 and PolG in mitochondria (Suppl. Fig. 5). Since PolG is specifically found in mitochondria, observation of RecQL4 co-localization with PolG unequivocally confirms the mitochondrial localization of RecQL4. Although the data from Dr. Croteau did not provide any compelling evidence for the modulatory role of RecQL4 in PolG-mediated mtDNA synthesis (Croteau et al., 2012), RecQL4 has been implicated in mtDNA replication by a cellular assay (De et al., 2012). This finding is further substantiated by our data showing the correlation between mtDNA copy number and RecQL4 expression in both RecQL4-overexpressed (HEK293) and silenced (U2OS) cell lines suggesting that RecQL4 regulates mtDNA copy number but the precise mode of regulation awaits further studies.
Although precise mechanism(s) responsible for RecQL4 nuclear-cytoplasmic shuttling is largely unclear, post translational modifications such as acetylation or ubiquitination may play an important role during this process. In support, a relatively recent report demonstrated that p300-mediated acetylation at lysine residues of NLS motif led to a significant shift of a proportion of RecQL4 protein from the nuclear to the cytoplasm (Dietschy et al., 2009). In addition, protein mono-ubiquitination, an important post-translational modification, has been shown to be involved in diverse processes such as nuclear/cytoplasmic shuttling of proteins (Hoeller et al., 2006). It has been shown that addition of monoubiquitin tag to p53 or MDM2-mediated p53 ubiquitination promotes p53 nuclear export presumably by exposing and activating a nuclear export signal(NES) (Nie et al., 2007). To provide a clue specifying whether or not ubiquination modification mediates RecQL4 nuclear export, an ubiquitin tag was added to C-terminus of RecQL4 protein. Western blot analysis demonstrated that both cytosolic and mitochondrial proportions of RecQL4 protein are substantially increased after addition of mono-ubiquitin tail (Suppl. Fig. 7). It has been shown previously that RecQL4 protein forms a stable complex with UBR1 and UBR2 (Yin et al., 2004). Thus, it is worthy of pursuing to determine whether or not ubiquitination modification promotes RecQL4 nuclear exporting. It should be noted that in addition to post translational modification, NES is also required for RecQL4 nuclear exporting since NES2&3 deletions lead to a total absence of RecQL4 cytoplasmic localization even though a monoubiquitin tail was added to the C-terminus of RecQL4 (Suppl. Fig. 7).
The dynamic regulation of RecQL4 nuclear/cytoplasmic shuttling was summarized in Figure 7B. C-terminal NES of RecQL4 mediates its recruitment to the cytoplasm. Post-translational modifications such as acetylation or ubiquitination of RecQL4 appear to be essential for its cytoplasmic recruitment probably through masking of nuclear localization sequence (NLS) at the N-terminal domain (NT) of RecQL4 or exposing/activating the NES at the CT. Once in the cytoplasm, the MLS motif may target RecQL4 to mitochondria (De et al., 2012) where it serves to protect mitochondrial genome integrity through regulation of replication and repair activities of mtDNA. Any defects in the dynamic regulation of nuclear and cytoplasmic distribution of RecQL4 may drastically affect the replication and repair fidelities in both nuclear and mitochondrial DNA genomes. These perturbations in DNA replication and repair may lead to either apoptotic death or various genomic instability features manifested in diverse human diseases such as neurodegeneration and cancer.
Mitochondria are intracellular organelles responsible for ATP production by enzymes of the electron transport chain located in the inner mitochondrial membrane, therefore, are the primary source of ROS in the cells (Sage et al., 2010). Because of its close proximity to this membrane as well as limited DNA repair capacities (de Souza-Pinto et al., 2009), oxidative stress is constitutively elevated in mitochondria. Aberrant alterations in mitochondrial functions are observed in a wide range of human pathological conditions including neurodegenerative diseases, aging and cancer (Chatterjee et al., 2006, Chinnery et al., 2002). There are several recent papers dealt with oxidative stress in RTS cells (Werner et al., 2006, Woo et al., 2006, Schurman et al., 2009). Among them, the most notable one is that of Werner et al. who examined the effect of oxidative damage/stress induced by hydrogen peroxide on DNA synthesis, cell growth, cell cycle distribution and viability in normal and RTS fibroblasts. As compared to normal cells, RTS cells showed irreversible growth arrest, reduced DNA synthesis and highly reduced S-phase cells. Collectively, these findings do indicate that RecQL4 is essential for dealing with oxidative stress. In support, we have convincingly demonstrated that the loss of RecQL4 leads to mitochondrial dysfunction evidenced by increased ROS and diminished mtDNA repair efficiency. In particular, an increased recruitment of RecQL4 protein in mitochondria was observed in U2OS cells when the cells were exposed to oxidative stress inducing agent-menadione. It is highly likely that some of the pathological features observed in RTS patients may be due to mitochondrial dysfunction triggered by RecQL4 deficiency. It is worth noting that most of the RTS mutations have been mapped to the RecQL4 helicase domain (Larizza et al., 2006). In AG18375 RTS cells used in this study, RecQL4 gene contains a single base deletion in exon 9 and both full length and predicted truncated form of RecQL4 protein were undetectable by western blot analysis (Suppl. Fig. 8). Thus, mutations occurred in RTS may lead to either a truncated form of protein with compromised gene function or a total loss of gene product. Moreover, both conditions will negatively affect the regulatory function of RecQL4 on mitochondria. As a result, incapability of repairing of damaged mtDNA will further elevates the ROS production in mitochondria (Cook and Higuchi, 2011). It has been documented that ROS-mediated nuclear DNA damage plays a role in initiation of carcinogenesis as well as in malignant transformation (Ziech et al., 2011). Therefore, it is highly likely that the mitochondrial dysfunction triggered by RecQL4 deficiency may contribute to the pathogenesis of RTS through ROS triggered nuclear DNA damage.
Supplementary Material
Supplemental Figure 1. Subcellular distribution of RecQL4 with deletion of NLS motif by immunoflurescence staining analysis. Flag-tagged RecQL4_NLS(−) vector was transfected in HEK293 cells. After staining with MitoTracker Red, cells were fixed and examined by immunoflurescence staining using anti-Flag antibody followed by DyLight 488 horse anti-mouse IgG antibody. Cells were counterstained by DAPI and the fluorescent images were captured using the Leica Confocal microscope.
Supplemental Figure 2. Subcellular distribution of GFP after transient transfection of GFP-GFP-NES constructs including pNES4 & 6–12 in U2OS cells. No strong GFP fluorescence in the cytoplasm was observed after transfection with the above pNES-containing constructs.
Supplemental Figure 3. Cytoplasmic distribution of GFP in U2OS cells after transfection with either full length or truncated GFP-RecQL4 constructs without any potential pNES: RecQL4_N(1–475) or containing a weak pNES5: RecQL4_M(476–824).
Supplemental Figure 4. RecQL4 nuclear exporting mediated by pNES3. An significantly increased cytoplasmic proportion of RecQL4 was observed when putting pNES3 to the location of pNES2 (GFP-RecQL4_C-pNES2→3) or switching the locations of pNES2 and pNES3.
Supplemental Figure 5. Mitochondrial localization of endogenous RecQL4 and PolG in U2OS cells. Immunofluorescence staining was performed by incubating the cells with primary antibodies: goat anti-human PolG (Santa Cruz, catalog: SC-5931, 1:100 dilution) and rabbit anti-human RecQL4 (SDI, Catalog: 2547, 1:100 dilution)] at 4 °C overnight followed by fluorescence-conjugated secondary antibodies: Alexa Fluor 488 Chicken Anti-Goat IgG (H+L) (Invitrogen, Catalog: A-21467, 1:100 dilution) and Texas Red Goat Anti-Rabbit IgG antibodies (Vector laboratories, 1:100 dilution) at RT for 2h. After several washes with PBS, the cells were counterstained by DAPI and the fluorescent images were captured using the Leica confocal microscope. After quantitative analysis in 10 randomly selected cells by CoLocalize Express, the Pearson’s correlation coefficient value is 0.9117.
Supplemental Figure 6. Mitochondrial morphological change in U2OS cells after RecQL4 suppression. Cells were stained with 25 nM DiOC6 for 15 min, fixed and analyzed by confocal microscopy analysis. Mitochondria in RecQL4-suppressed cells stained with DiOC6 tend to be aggregate together when compared with control shRNA-transfected U2OS cells.
Supplemental Figure 7. Addition of mono-ubiquitin tail to C-terminus of RecQL4 protein promotes its nuclear exporting. U2OS cells were transfected with Flag-RecQL4, Flag-RecQL4+Ub or Flag-RecQL4ΔNES2&3+Ub expression vector. Subcellular distribution of individual protein was examined by western blot analysis. The membrane was reprobed with control antibodies: anti-lamin B (nulear marker), GAPDH (cytosolic marker) and VDAC (mitochondrial marker). The result showed that mitochondrial proportion is free of nucleoic and cytosolic contaminations.
Supplemental Figure 8. Western blot analysis of RecQL4 protein level in AG18375 RTS cells relative to AG18459 heterozygous cells.
Highlights.
-
►
11 Amino acid residues (1187–1198) at the C-terminal domain of RecQL4 contains the most potent NES that mediate the export of RecQL4 to the cytoplasm
-
►
Cytoplasmic RecQL4 protein is found localized in mitochondria.
-
►
RecQL4 has a modulatory role in the maintenance of mtDNA copy number
-
►
RecQL4 suppression leads to elevated oxidative stress and morphological changes in mitochondria
-
►
RecQL4 regulates the efficiency of oxidative DNA damage repair in mitochondria
Acknowledgements
The authors thank Dr. Yunfeng Feng of the Longmore lab (Washington University) for providing pU2-FH lentiviral vector. This research was supported by National Natural Science Foundation of China (grant number 30971602) and National Cancer Institute, National Institutes of Health (CA127120).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Burks LM, Yin J, Plon SE. Nuclear import and retention domains in the amino terminus of RECQL4. Gene. 2007;391:26–38. doi: 10.1016/j.gene.2006.11.019. [DOI] [PubMed] [Google Scholar]
- Chatterjee A, Mambo E, Sidransky D. Mitochondrial DNA mutations in human cancer. Oncogene. 2006;25:4663–4674. doi: 10.1038/sj.onc.1209604. [DOI] [PubMed] [Google Scholar]
- Chinnery PF, Samuels DC, Elson J, Turnbull DM. Accumulation of mitochondrial DNA mutations in ageing, cancer, and mitochondrial disease: is there a common mechanism? Lancet. 2002;360:1323–1325. doi: 10.1016/S0140-6736(02)11310-9. [DOI] [PubMed] [Google Scholar]
- Chu WK, Hickson ID. RecQ helicases: multifunctional genome caretakers. Nat Rev Cancer. 2009;9:644–654. doi: 10.1038/nrc2682. [DOI] [PubMed] [Google Scholar]
- Cook CC, Higuchi M. The awakening of an advanced malignant cancer: An insult to the mitochondrial genome. Biochimica et biophysica acta. 2011 doi: 10.1016/j.bbagen.2011.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Croteau DL, Rossi ML, Canugovi C, Tian J, Sykora P, Ramamoorthy M, Wang Z, Singh DK, Akbari M, Kasiviswanathan R, Copeland WC, Bohr VA. RECQL4 localizes to mitochondria and preserves mitochondrial DNA integrity. Aging Cell. 2012 doi: 10.1111/j.1474-9726.2012.00803.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De S, Kumari J, Mudgal R, Modi P, Gupta S, Futami K, Goto H, Lindor NM, Furuichi Y, Mohanty D, Sengupta S. RECQL4 is essential for the transport of p53 to mitochondria in normal human cells in the absence of exogenous stress. Journal of cell science. 2012 doi: 10.1242/jcs.101501. [DOI] [PubMed] [Google Scholar]
- de Souza-Pinto NC, Mason PA, Hashiguchi K, Weissman L, Tian J, Guay D, Lebel M, Stevnsner TV, Rasmussen LJ, Bohr VA. Novel DNA mismatch-repair activity involving YB-1 in human mitochondria. DNA Repair (Amst) 2009;8:704–719. doi: 10.1016/j.dnarep.2009.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dietschy T, Shevelev I, Pena-Diaz J, Huhn D, Kuenzle S, Mak R, Miah MF, Hess D, Fey M, Hottiger MO, Janscak P, Stagljar I. p300-mediated acetylation of the Rothmund-Thomson-syndrome gene product RECQL4 regulates its subcellular localization. J Cell Sci. 2009;122:1258–1267. doi: 10.1242/jcs.037747. [DOI] [PubMed] [Google Scholar]
- Dietschy T, Shevelev I, Stagljar I. The molecular role of the Rothmund-Thomson-, RAPADILINO- and Baller-Gerold-gene product, RECQL4: recent progress. Cell Mol Life Sci. 2007;64:796–802. doi: 10.1007/s00018-007-6468-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan W, Luo J. RecQ4 facilitates UV light-induced DNA damage repair through interaction with nucleotide excision repair factor xeroderma pigmentosum group A (XPA) J Biol Chem. 2008;283:29037–29044. doi: 10.1074/jbc.M801928200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayakawa S, Kaneko H, Fukao T, Kasahara K, Matsumoto T, Furuichi Y, Kondo N. Characterization of the nuclear localization signal in the DNA helicase responsible for Bloom syndrome. Int J Mol Med. 2000;5:477–484. doi: 10.3892/ijmm.5.5.477. [DOI] [PubMed] [Google Scholar]
- Hoeller D, Hecker C-M, Dikic I. Ubiquitin and ubiquitin-like proteins in cancer pathogenesis. Nat Rev Cancer. 2006;6:776–788. doi: 10.1038/nrc1994. [DOI] [PubMed] [Google Scholar]
- Jin W, Liu H, Zhang Y, Otta SK, Plon SE, Wang LL. Sensitivity of RECQL4-deficient fibroblasts from Rothmund-Thomson syndrome patients to genotoxic agents. Hum Genet. 2008;123:643–653. doi: 10.1007/s00439-008-0518-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitao S, Lindor NM, Shiratori M, Furuichi Y, Shimamoto A. Rothmund-thomson syndrome responsible gene, RECQL4: genomic structure and products. Genomics. 1999a;61:268–276. doi: 10.1006/geno.1999.5959. [DOI] [PubMed] [Google Scholar]
- Kitao S, Shimamoto A, Goto M, Miller RW, Smithson WA, Lindor NM, Furuichi Y. Mutations in RECQL4 cause a subset of cases of Rothmund-Thomson syndrome. Nat Genet. 1999b;22:82–84. doi: 10.1038/8788. [DOI] [PubMed] [Google Scholar]
- Korhonen JA, Pham XH, Pellegrini M, Falkenberg M. Reconstitution of a minimal mtDNA replisome in vitro. The EMBO journal. 2004;23:2423–2429. doi: 10.1038/sj.emboj.7600257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kutay U, Guttinger S. Leucine-rich nuclear-export signals: born to be weak. Trends Cell Biol. 2005;15:121–124. doi: 10.1016/j.tcb.2005.01.005. [DOI] [PubMed] [Google Scholar]
- Larizza L, Magnani I, Roversi G. Rothmund-Thomson syndrome and RECQL4 defect: splitting and lumping. Cancer Lett. 2006;232:107–120. doi: 10.1016/j.canlet.2005.07.042. [DOI] [PubMed] [Google Scholar]
- Liu Y. Rothmund-Thomson syndrome helicase, RECQ4: On the crossroad between DNA replication and repair. DNA Repair (Amst) 2010;9:325–330. doi: 10.1016/j.dnarep.2010.01.006. [DOI] [PubMed] [Google Scholar]
- Mann MB, Hodges CA, Barnes E, Vogel H, Hassold TJ, Luo G. Defective sister-chromatid cohesion, aneuploidy and cancer predisposition in a mouse model of type II Rothmund-Thomson syndrome. Hum Mol Genet. 2005;14:813–825. doi: 10.1093/hmg/ddi075. [DOI] [PubMed] [Google Scholar]
- Marciniak RA, Lombard DB, Johnson FB, Guarente L. Nucleolar localization of the Werner syndrome protein in human cells. Proceedings of the National Academy of Sciences of the United States of America. 1998;95:6887–6892. doi: 10.1073/pnas.95.12.6887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monnat RJ., Jr Human RECQ helicases: roles in DNA metabolism, mutagenesis and cancer biology. Seminars in cancer biology. 2010;20:329–339. doi: 10.1016/j.semcancer.2010.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nie L, Sasaki M, Maki CG. Regulation of p53 nuclear export through sequential changes in conformation and ubiquitination. J Biol Chem. 2007;282:14616–14625. doi: 10.1074/jbc.M610515200. [DOI] [PubMed] [Google Scholar]
- Ouyang KJ, Woo LL, Ellis NA. Homologous recombination and maintenance of genome integrity: cancer and aging through the prism of human RecQ helicases. Mech Ageing Dev. 2008;129:425–440. doi: 10.1016/j.mad.2008.03.003. [DOI] [PubMed] [Google Scholar]
- Payne CM, Weber C, Crowley-Skillicorn C, Dvorak K, Bernstein H, Bernstein C, Holubec H, Dvorakova B, Garewal H. Deoxycholate induces mitochondrial oxidative stress and activates NF-kappaB through multiple mechanisms in HCT-116 colon epithelial cells. Carcinogenesis. 2007;28:215–222. doi: 10.1093/carcin/bgl139. [DOI] [PubMed] [Google Scholar]
- Petit C, Mathez D, Barthelemy C, Leste-Lasserre T, Naviaux RK, Sonigo P, Leibowitch J. Quantitation of blood lymphocyte mitochondrial DNA for the monitoring of antiretroviral drug-induced mitochondrial DNA depletion. Journal of acquired immune deficiency syndromes. 2003;33:461–469. doi: 10.1097/00126334-200308010-00006. [DOI] [PubMed] [Google Scholar]
- Petkovic M, Dietschy T, Freire R, Jiao R, Stagljar I. The human Rothmund-Thomson syndrome gene product, RECQL4, localizes to distinct nuclear foci that coincide with proteins involved in the maintenance of genome stability. J Cell Sci. 2005;118:4261–4269. doi: 10.1242/jcs.02556. [DOI] [PubMed] [Google Scholar]
- Rossi ML, Ghosh AK, Kulikowicz T, Croteau DL, Bohr VA. Conserved helicase domain of human RecQ4 is required for strand annealing-independent DNA unwinding. DNA Repair (Amst) 2010;9:796–804. doi: 10.1016/j.dnarep.2010.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sage JM, Gildemeister OS, Knight KL. Discovery of a novel function for human Rad51: maintenance of the mitochondrial genome. J Biol Chem. 2010;285:18984–18990. doi: 10.1074/jbc.M109.099846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sangrithi MN, Bernal JA, Madine M, Philpott A, Lee J, Dunphy WG, Venkitaraman AR. Initiation of DNA replication requires the RECQL4 protein mutated in Rothmund-Thomson syndrome. Cell. 2005;121:887–898. doi: 10.1016/j.cell.2005.05.015. [DOI] [PubMed] [Google Scholar]
- Santos C, Martinez M, Lima M, Hao YJ, Simoes N, Montiel R. Mitochondrial DNA mutations in cancer: a review. Current topics in medicinal chemistry. 2008;8:1351–1366. doi: 10.2174/156802608786141151. [DOI] [PubMed] [Google Scholar]
- Santos JH, Mandavilli BS, Van Houten B. Measuring oxidative mtDNA damage and repair using quantitative PCR. Methods in molecular biology. 2002;197:159–176. doi: 10.1385/1-59259-284-8:159. [DOI] [PubMed] [Google Scholar]
- Schurman SH, Hedayati M, Wang Z, Singh DK, Speina E, Zhang Y, Becker K, Macris M, Sung P, Wilson DM, 3rd, Croteau DL, Bohr VA. Direct and indirect roles of RECQL4 in modulating base excision repair capacity. Hum Mol Genet. 2009;18:3470–3483. doi: 10.1093/hmg/ddp291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh DK, Ahn B, Bohr VA. Roles of RECQ helicases in recombination based DNA repair, genomic stability and aging. Biogerontology. 2009;10:235–252. doi: 10.1007/s10522-008-9205-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh DK, Karmakar P, Aamann M, Schurman SH, May A, Croteau DL, Burks L, Plon SE, Bohr VA. The involvement of human RECQL4 in DNA double-strand break repair. Aging Cell. 2010;9:358–371. doi: 10.1111/j.1474-9726.2010.00562.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki T, Kohno T, Ishimi Y. DNA helicase activity in purified human RECQL4 protein. J Biochem. 2009;146:327–335. doi: 10.1093/jb/mvp074. [DOI] [PubMed] [Google Scholar]
- Taagepera S, McDonald D, Loeb JE, Whitaker LL, McElroy AK, Wang JY, Hope TJ. Nuclear-cytoplasmic shuttling of C-ABL tyrosine kinase. Proceedings of the National Academy of Sciences of the United States of America. 1998;95:7457–7462. doi: 10.1073/pnas.95.13.7457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thangavel S, Mendoza-Maldonado R, Tissino E, Sidorova JM, Yin J, Wang W, Monnat RJ, Jr, Falaschi A, Vindigni A. Human RECQ1 and RECQ4 helicases play distinct roles in DNA replication initiation. Mol Cell Biol. 2010;30:1382–1396. doi: 10.1128/MCB.01290-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Werner SR, Prahalad AK, Yang J, Hock JM. RECQL4-deficient cells are hypersensitive to oxidative stress/damage: Insights for osteosarcoma prevalence and heterogeneity in Rothmund-Thomson syndrome. Biochem Biophys Res Commun. 2006;345:403–409. doi: 10.1016/j.bbrc.2006.04.093. [DOI] [PubMed] [Google Scholar]
- Woo LL, Futami K, Shimamoto A, Furuichi Y, Frank KM. The Rothmund-Thomson gene product RECQL4 localizes to the nucleolus in response to oxidative stress. Exp Cell Res. 2006;312:3443–3457. doi: 10.1016/j.yexcr.2006.07.023. [DOI] [PubMed] [Google Scholar]
- Xu X, Liu Y. Dual DNA unwinding activities of the Rothmund-Thomson syndrome protein, RECQ4. EMBO J. 2009;28:568–577. doi: 10.1038/emboj.2009.13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin J, Kwon YT, Varshavsky A, Wang W. RECQL4, mutated in the Rothmund-Thomson and RAPADILINO syndromes, interacts with ubiquitin ligases UBR1 and UBR2 of the N-end rule pathway. Hum Mol Genet. 2004;13:2421–2430. doi: 10.1093/hmg/ddh269. [DOI] [PubMed] [Google Scholar]
- Ziech D, Franco R, Pappa A, Panayiotidis MI. Reactive oxygen species (ROS)--induced genetic and epigenetic alterations in human carcinogenesis. Mutation research. 2011;711:167–173. doi: 10.1016/j.mrfmmm.2011.02.015. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Figure 1. Subcellular distribution of RecQL4 with deletion of NLS motif by immunoflurescence staining analysis. Flag-tagged RecQL4_NLS(−) vector was transfected in HEK293 cells. After staining with MitoTracker Red, cells were fixed and examined by immunoflurescence staining using anti-Flag antibody followed by DyLight 488 horse anti-mouse IgG antibody. Cells were counterstained by DAPI and the fluorescent images were captured using the Leica Confocal microscope.
Supplemental Figure 2. Subcellular distribution of GFP after transient transfection of GFP-GFP-NES constructs including pNES4 & 6–12 in U2OS cells. No strong GFP fluorescence in the cytoplasm was observed after transfection with the above pNES-containing constructs.
Supplemental Figure 3. Cytoplasmic distribution of GFP in U2OS cells after transfection with either full length or truncated GFP-RecQL4 constructs without any potential pNES: RecQL4_N(1–475) or containing a weak pNES5: RecQL4_M(476–824).
Supplemental Figure 4. RecQL4 nuclear exporting mediated by pNES3. An significantly increased cytoplasmic proportion of RecQL4 was observed when putting pNES3 to the location of pNES2 (GFP-RecQL4_C-pNES2→3) or switching the locations of pNES2 and pNES3.
Supplemental Figure 5. Mitochondrial localization of endogenous RecQL4 and PolG in U2OS cells. Immunofluorescence staining was performed by incubating the cells with primary antibodies: goat anti-human PolG (Santa Cruz, catalog: SC-5931, 1:100 dilution) and rabbit anti-human RecQL4 (SDI, Catalog: 2547, 1:100 dilution)] at 4 °C overnight followed by fluorescence-conjugated secondary antibodies: Alexa Fluor 488 Chicken Anti-Goat IgG (H+L) (Invitrogen, Catalog: A-21467, 1:100 dilution) and Texas Red Goat Anti-Rabbit IgG antibodies (Vector laboratories, 1:100 dilution) at RT for 2h. After several washes with PBS, the cells were counterstained by DAPI and the fluorescent images were captured using the Leica confocal microscope. After quantitative analysis in 10 randomly selected cells by CoLocalize Express, the Pearson’s correlation coefficient value is 0.9117.
Supplemental Figure 6. Mitochondrial morphological change in U2OS cells after RecQL4 suppression. Cells were stained with 25 nM DiOC6 for 15 min, fixed and analyzed by confocal microscopy analysis. Mitochondria in RecQL4-suppressed cells stained with DiOC6 tend to be aggregate together when compared with control shRNA-transfected U2OS cells.
Supplemental Figure 7. Addition of mono-ubiquitin tail to C-terminus of RecQL4 protein promotes its nuclear exporting. U2OS cells were transfected with Flag-RecQL4, Flag-RecQL4+Ub or Flag-RecQL4ΔNES2&3+Ub expression vector. Subcellular distribution of individual protein was examined by western blot analysis. The membrane was reprobed with control antibodies: anti-lamin B (nulear marker), GAPDH (cytosolic marker) and VDAC (mitochondrial marker). The result showed that mitochondrial proportion is free of nucleoic and cytosolic contaminations.
Supplemental Figure 8. Western blot analysis of RecQL4 protein level in AG18375 RTS cells relative to AG18459 heterozygous cells.