

Abstract
Autoimmune disorders are a complex and varied group of diseases that are caused by breakdown of self-tolerance. The aetiology of autoimmunity is multi-factorial, with both environmental triggers and genetically determined risk factors. In recent years, it has been increasingly recognized that genetic risk factors do not act in isolation, but rather the combination of individual additive effects, gene–gene interactions and gene–environment interactions determine overall risk of autoimmunity. The importance of gene–gene interactions, or epistasis, has been recently brought into focus, with research demonstrating that many autoimmune diseases, including rheumatic arthritis, autoimmune glomerulonephritis, systemic lupus erythematosus and multiple sclerosis, are influenced by epistatic interactions. This review sets out to examine the basic mechanisms of epistasis, how epistasis influences the immune system and the role of epistasis in two major autoimmune conditions, systemic lupus erythematosus and multiple sclerosis.
Keywords: autoimmunity, epistasis, genetic interaction, multiple sclerosis, systemic lupus erythematosus
In the era of systems biology, it is no longer possible to consider a gene in isolation: it is increasingly apparent that we must consider the interactions of a gene, including the interactions of its protein product, gene–environment interactions and gene–gene interactions. This is particularly relevant in complex human disease, where aetiology is multi-factorial. Autoimmune diseases are a varied group of complex disorders, with a strong genetic component, caused by breakdown of self-tolerance. This review sets out to examine the role of gene–gene interactions in autoimmunity: we review the known mechanisms of epistasis, and suggest where in the immune system they might act. We proceed to draw on disease-specific examples from two complex autoimmune conditions: multiple sclerosis (MS) and systemic lupus erythematosus (SLE).
Understanding epistasis: basic principles
The term epistasis has been used for over 100 years, being first used by Bateson to describe the masking of one disease-causing mutation by the co-inheritance of a mutation at a separate locus.1 The idea has since been expanded to include any statistical deviation from the additive combination of two loci, the definition that is most commonly used today.2,3 At first, this might seem a complex mathematical concept, but this is not the case. In simple terms, epistasis is the phenomenon whereby the effect of one genetic variant is altered by another. The term can be used interchangeably with gene–gene interaction, or genetic interactions.
It has been known for many years that disease-causing mutations show wide phenotypic variability, even within families, emphasizing that mutation outcome is dependent on the genetic background, as well as on environmental factors. There are many examples of simple Mendelian diseases showing phenotypic variability. An example in immunological disease comes from the study of X-linked lymphoproliferative disorder (XLP-1). Two families harbouring heterozygous deletion of SH2D1A demonstrated wide inter-familial and intra-familial variability of a clinical XLP-1 phenotype, despite identical disease mutations.4 The principle of epistasis is, however, just as relevant to complex diseases.
Complex genetic disorders are caused by polygenic inheritance of common variations, which are present within the normal population. Genome-wide association studies (GWAS) have begun to unravel the common traits underlying complex disease, with well over 1000 disease-linked loci identified.5,6 There is, however, a continuing mystery: despite identification of many important loci, only 20–30% of heritability can be explained, with the cause of the majority of heritability of complex disease remaining obscure.7 It has been argued that the missing heritability relies on the identification of novel variants associated with disease, which might be a large number of common alleles with small effects, or a few very rare alleles with large effects.8–14
It has been argued that this might not be the only answer: genetic interactions might explain a significant proportion of the missing heritability. For example, the inflammatory bowel disorder Crohn's disease has over 70 associated loci, which explain approximately 21·5% of the heritability of disease.15 The application of a relatively simple mathematical model that takes into account genetic interactions can, however, explain almost 80% of the current missing heritability.7 This is perhaps not surprising, as pervasive epistasis has been observed in several model organisms.16–18 In yeast, the effects of more than half of primary quantitative trait loci were shown to be affected by a secondary locus and, in both Caenorhabditis elegans and Saccharomyces cerevisiae, it was shown that nearly all disease-causing mutations were affected by variation at secondary loci.16,18–20 Furthermore, studies in yeast have revealed that it is not only binary epistatic interactions that need to be considered: the majority of phenotypic diversity seen within a yeast strain relies on complex multigenic epistatic interactions, involving at least five loci.7,21
This said, it is undeniable that there are risk-alleles associated with complex diseases that remain to be identified. Studies of phenotypic variance between monozygotic and dizygotic twins have emphasized that a large proportion of continuous traits (such as body mass index, height and blood pressure) can be explained by additive effects of genetic loci, and that non-additive (i.e. epistatic) effects play a relatively minor role.22 This effect might well be because quantitative traits, such as those studied in the twin pairs, are determined by a large number of genetic factors, each making a very small contribution to the quantitative measurement. In these instances, epistatic interactions play almost no role, and the effect becomes additive.23 We would argue, however, that in the cases of complex diseases there is a role for both additive effects and epistatic interactions. It is likely that for complex diseases there are a small number of major loci – many of which are likely to have been discovered already by GWAS – which interact with each other through epistasis, these interactions accounting for a substantial proportion of the missing heritability. In addition, some of the missing heritability will be caused by the additive effect of a large number of loci with individually very small contributions.
Gene–gene interactions within the immune system
The immune system shows marked complexity of phenotype and genotype and, as such, is a rich environment in which to observe epistasis. Networks of cell subsets – within and between which signalling molecules interact in cascades leading to many functional outcomes – give rise to a system in which there are many opportunities for epistasis. The understanding of these interactions at a functional level, and concomitantly at a genetic level, is key to understanding risk and susceptibility to autoimmune disease. The specific study of epistasis with regard to autoimmunity has so far been limited.24 The wealth of basic research into autoimmunity can, however, be probed to identify candidate interactions, which might be subject to between-molecule epistasis.3 Comparison of these potential epistatic players with genes which have been implicated in autoimmune disease (by GWAS) paves the way for an integrated and functionally supported analysis of the genetic basis of autoimmunity, as has begun to occur in other fields such as cardiovascular disease.25–27 Furthermore, genetic modelling that directly aims to detect epistasis might maximize the likelihood of identifying the susceptibility effects of individual genes.28
At the most basic level, epistasis operates at direct interfaces between proteins, wherein alterations in the genetic code of either partner have the capacity to alter the character and affinity of their physical interaction.3 Direct molecular recognition is also the basis of immune function and so interactions between cytokines and their receptors, signalling molecules in cell activation cascades, and antigen receptors and their binding partners are all potentially subject to epistasis. Functional redundancy is another simple cause of epistasis and, again, is known to be rife in the immune system. A prime example is the MHC, in which functional redundancy is common, and epistasis is most likely to be crucial in the functioning of these multi-allelic, polymorphic genes.29 The killer-cell immunoglobulin-like receptor system has similar functional redundancy between its members, and also directly binds MHC molecules.30 It is clear, then, that there is ample scope for a multitude of within-molecule and between-molecule genetic interactions involving these two systems, with wide-reaching implications for immune disease.31–33
As well as conceptually simple forms of epistasis between pairs of genes, or pairs of gene groups, more complex forms of epistasis operate within and between signalling pathways. Immune cell function is largely controlled by receptor-mediated activation of intracellular signalling pathways, which initiate transcriptional and non-transcriptional processes that affect the cell state and so the immune response. Epistasis might strongly govern the functional outcomes of pathway activation. In uni-directional signalling pathways, such as the mitogen-activated protein (MAP) kinase cascades (which are key to the function of both innate and adaptive immune cells), mutations in single genes can have adverse consequences for normal activity of the entire pathway.34 Conversely, functionally opposing mutations in different pathway members can have masking or relieving consequences for potentially deleterious genetic variations.3 For example, it is plausible that increased enzymatic activity of a MAP-kinase-kinase could compensate for reduced activity of an upstream MAP-kinase. It is also theoretically possible – and perhaps inevitable – that epistatic interactions will occur that involve regulatory components of pathways, as true pathway linearity is rare. In immunity, this means consideration of intracellular regulatory components, such as phosphatase and ubiquitinase enzymes, as well as the immune-regulatory system formed by T-regulatory cells and immunosuppressive cytokines, such as IL-10 and TGF-β.
When it is considered that functional redundancy and co-operation are observed between entire systems of molecules – not simply individual molecules – it is clear that many more epistatic interactions are possible. In the immune system, this might arise when there are multiple pro-inflammatory and anti-inflammatory inputs to a cell; for example, the synergistic functioning of the NF-κB pathway, the MAP kinases and the phosphatidyl inositol 3 kinase pathway in activation of innate sentinel cells.34–36 Another such possible source of epistasis is the stimulation of T cells through multiple cytokine receptors and their cogent Janus kinase/signal transducer and activator of transcription pathways, which occurs after recruitment of T cells by the cytokine milieu of an inflammatory focus.37,38 These buffering interactions, in which epistasis can compensate for mutation in one pathway, are not confined to signalling pathways but might also occur between functional modules, such as the inflammasome and, as such, these functional units must be considered as candidates for epistasis.39
The meta-interactions that are known to exist between pathways can be used to predict sets of epistatic partners for genes, and these ‘seed sets’ might prove invaluable in mapping epistatic networks onto the immunological genome. The mapping and modelling of epistasis has been proposed as a method which might prove invaluable in extracting useful hypotheses from GWAS datasets.40 One important aspect of this approach, and another key tenet of epistasis that must be considered in research focused as such, is the presence of genetic hubs, which are known to act in most investigated cell systems.19 Genetic hubs are infrequent genes that have numerous epistatic interactions – vastly in excess of most genes in the system – and consequently, in which minor mutations have large effects on the overall phenotype. The molecular functions of these genes are often unexpectedly specialized, and this lack of clarity as to what predisposes a gene to being an epistatic hub means that candidates are hard to predict. The elucidation of which immune genes are hubs will, therefore, have far-reaching consequences for understanding the genetic basis of immunity, and must be a priority.
Epistasis in systemic lupus erythematosus
Systemic lupus erythematosus is a multi-system autoimmune disease, characterized by B-cell autoreactivity and deposition of immune complexes in tissues, including the muscles, joints, kidney and heart. The disease prevalence has been estimated at 25/100 000 in the UK, and the incidence is approximately eight times higher in women than men.41 Familial aggregation of cases has been noted since the early study of disease, long supporting a strong genetic aetiology.42–45 A recent large survey confirmed that first-degree relatives of patients with SLE had an increased risk of SLE (odds ratio = 16·98), as well as of other autoimmune disorders.46 Studies using twins showed that approximately 25% of monozygotic twins share an SLE phenotype, with a significantly higher level of concordance between monozygotic than dizygotic twins, supporting the importance of genetic factors.47–49 The overall heritability of SLE was estimated at > 66%.50
The recent advances in genomic technology have begun to unravel the complex genetic aetiology of SLE. There are currently more than 30 loci associated with increased SLE risk, identified (or confirmed) by seven large GWAS in different ethnic populations.51–58 The two major loci associated with SLE are the HLA region on chromosome 6p and the FcγR gene cluster on chromosome 1q.51–60
The HLA region is a well-characterized gene cluster at chromosome 6p21.4, containing over 200 genes with immunological roles. There has been consistent linkage of two HLA class II genes (HLA-DR2 and HLA-DR3) and two HLA class III genes (C4, C2) with SLE susceptibility, and there have also been reports of other associated genes, that have yet to be independently replicated (e.g. TNF, SKIV2L and MSH5).50,52,59,60 A recent comprehensive study of epistatic interactions with the HLA region has confirmed that genetic interactions play an important role in SLE susceptibility.61 The two strongest epistatic effects were observed between CTLA4 and the HLA region. First, CTLA4 was shown to interact with rs3131379, a single nucleotide polymorphism in an intronic region of the MSH5 gene in the HLA Class 3 region, with a disease odds ratio of 1·19. Second, CTLA4 interacted with rs1270942, a single nucleotide polymorphism in an intronic region of the complement factor B (CFB2) gene, also in the HLA Class 3 region, this interaction increases the odds of disease by approximately 20%. Four other epistatic interactions were detected that associated less strongly with disease risk, although they were still statistically significant.
Cytotoxic T lymphocyte antigen 4 (CTL4A, also known as CD152) is a T-cell transmembrane protein responsible for negative regulation of T-cell activity following T-cell activation by antigen-presenting cells.62 The protein is critical for the maintenance of T-cell homeostasis and self-tolerance, acting through interactions with both conventional effector T-cells and regulatory T-cells.63 The epistatic interaction between the HLA locus and CTLA4 underlines the importance of inappropriate antigen presentation and T-cell activation in the breakdown of self-tolerance and the pathogenesis of SLE.
The Fc receptors are a group of cell surface glycoproteins that bind the Fc portion of antibodies, thereby providing a link between humoral and cellular immune responses.64 They regulate a variety of immune responses, including phagocytosis, mast cell degranulation, antibody-dependent cellular toxicity, B-cell activation and immune complex clearance.65 The Fc receptors are encoded by a gene cluster at chromosome 1q21–24, which includes the Fc γ receptors (FcγR). Deficiency in FcγR has been implicated in several autoimmune diseases, including SLE, autoimmune glomerulonephritis and rheumatoid arthritis.66,67 A recent study in SLE-prone mice defined two regions associated with defective self-tolerance, and epistatic relationships within one of these regions were further explored.66 It was shown that a genetic interaction between FcγRIIB and slam genes was important in the development of SLE phenotype.68
FcγRIIB is a negative regulator of B-cell receptor-mediated B-cell activation.69 Deficiency of FcγRIIB in mice has been associated with the development of autoantibodies and autoimmune glomerulonephritis, whereas dysregulation of the human FcγrIIB during B-cell development has been implicated in SLE pathogenesis.66,70,71 The slam gene cluster, located at chromosome 1q in both mice and humans encodes a family of transmembrane immune cell receptors. Slam gene family members participate in cell–cell interactions among many cell lineages in the adaptive and innate immune systems.71 The slam genes play an important role in the development of central tolerance in both B- and T-cells, predominantly through modulation of apoptosis, anergy and cell-cycle progression.72 Congenic mice containing autoimmune-type slam alleles were shown to produce autoantibodies – indicative of a failure to maintain self-tolerance – and the mice developed an SLE-like phenotype.73,74
The discovery of Fcgr2b-slam interaction strengthens the argument that breakdown of induction of self-tolerance in lymphocytes is one of the key processes in autoimmune pathology. Despite the description of an epistatic interaction between Fcgr2b and slam, the study68 did not explore other potential genetic interactions between the myriad of strong candidate genes that resided within the linked region (such as Fcgr3, Fcrla, Fcrlb) and, as such, it is likely that further experimental work will define more epistatic interactions between Fc receptors that are relevant to SLE pathogenesis.24
Epistasis in multiple sclerosis
Multiple sclerosis is an inflammatory disease of the central nervous system in which activation of CD4+ T cells, predominantly of T helper type 1 polarization, leads to an influx of inflammatory cells, eventually causing demyelination, neuronal pathology and neurological dysfunction.75 It has long been clear that development of the disease must involve both a genetic predisposition and an environmental contribution or trigger. The genetic susceptibility is indicated by high disease frequencies in particular ethnic groups such as northern Europeans,76 and this is confirmed by twin studies, which show that monozygotic twins have a concordance rate of around 25%.75 The environmental trigger is widely suggested to be Epstein–Barr virus infection, whereas the roles of exposure to sunlight and dietary factors (both previously implicated) have yet to be established.77,78 The majority of research into the genetic basis of MS has been focused on identifying predisposing HLA alleles – notably variants at the HLA-DRB1 locus, particularly DRB1*15, as well as HLA-C.79–81 The exponentially increased power of genomics in the past decade has, however, vastly expanded the list of disease-associated genes. The National Institutes of Health Catolog of Published GWAS82 lists 14 studies83–96 that examined MS susceptibility; these studies implicated upwards of 100 distinct genes, although there was limited overlap of implicated loci between studies. Many of the identified genes – including HLA loci, which feature strongly – are immune-related. These include cytokines, chemokines and their receptors (such as IL-7, IL-12A, IL-12RA and CXCR4), signal transduction molecules and transcription factors (such as signal transducer and activator of transcription 3 and interferon-induced transcription factor 8) and co-stimulatory molecules (such as CD80, CD86 and CLECL1). Interestingly, molecules in the vitamin D metabolic pathway, such as CYP27B1, feature, whereas very few molecules involved in neurological pathways were associated.
Although GWAS have been invaluable in indicating potential contributors to the genetic risk for MS, and many loci have since been further established as risk factors in additional focused genetic studies, they have not been all-encompassing. First, identified loci account for < 50% of the known heritability of MS, meaning that there is substantial missing heritability. Second, direct testing and validation of implicated genes in vitro or in vivo has been limited, although models describing how they could act in scenarios such as CD4 T-cell maturation have been suggested, if not experimentally characterized.96
Recent work has made progress in the understanding of the role that epistasis plays in MS. Combined with genome-based advances, this work has the potential to functionally characterize GWAS-identified risk loci, thereby accounting for a proportion of the missing heritability associated with MS. The majority of studies into the role of epistasis have been focused on understanding the interactions between risk-associated HLA alleles. A seminal study in humanized mice showed that linkage disequilibrium between DRB risk alleles might be the result of modifying effects of one allele on the T-cell response provoked by the other, highlighting the importance of balance in the immune response and providing insight into the classic relapsing and remitting phenotype of MS.97
Further studies in mice have added to the understanding of how MHC alleles modify risk significance of co-expressed alleles.98,99 Population studies have established the risk contribution of DQA and DQB HLA Class II alleles in concert with DRB1,100 and have also defined the DRB1*15 risk contribution as being fundamentally subject to its overall HLA haplotype and hence potentially epistasis.101 Epistasis between HLA alleles has been identified in several ethnic populations, further cementing the importance HLA-region gene–gene interactions.102–104
It is firmly established that HLA alleles play a major role in MS, and although studying the role of HLA-mediated epistasis in MS is valuable in understanding the complexities of inherited risk, it is unlikely to yield new therapeutic targets – the discovery of novel therapeutic targets in MS, and other complex diseases, will require the integration of genomic, transcriptional and functional research (Fig. 1). GWAS provides a vital list of genes that might play a role in the pathogenesis of complex disorders – the answer to the ‘what is involved?’ question. With the wealth of data available in the post-GWAS era, it is essential that we move from what? to how? and why?, or the resources used for the GWAS will not have been explored to their fullest potential.
Figure 1.
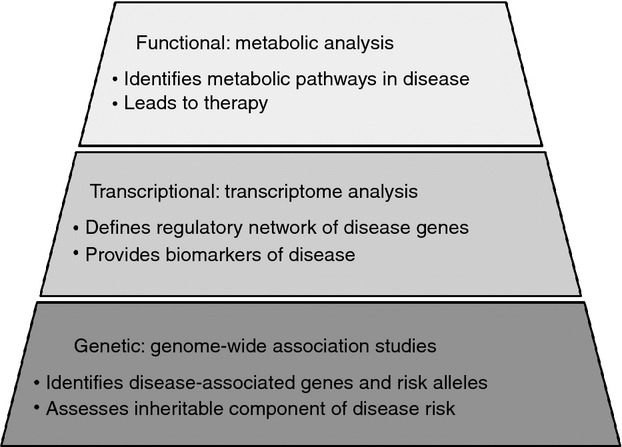
Approaches to research: one of the ‘pinnacles’ of medical research is the identification of novel therapeutic targets. In polygenic diseases, the most basic level (‘the foundation’) is genetic studies, which should include analysis of epistatic interactions. Genetic studies support transcriptional analysis, which provides a more comprehensive view of the gene within the cellular system. Finally, the top level of research is functional studies, which can lead to the discovery of novel therapeutic targets.
The first step post-GWAS is to analyse the epistatic interactions of the candidate genes identified, as this in itself might explain missing heritability in complex disorders. Furthermore, the interactions identified can be used to construct hypotheses and to guide future experimental work. For example, it might highlight important transcriptional or functional phenotypes, which could be explored further. This has been achieved in several studies that have used knowledge-driven analysis of genetic data to identify disease-associated non-HLA gene–gene interactions. Such strategies have highlighted previously unreported roles for T helper type 2 cytokines and complement factors in MS, a paradigmatically T helper type 1 disease, as well as highlighting the importance of dendritic cells in pathogenesis.105,106 A further example of how GWAS data can be used in a constructive manner comes from a study into the genetics of osteoarthritis.107 The study identified a gene, DOT1L, which was strongly linked to the osteoarthritis phenotype. Further studies were subsequently performed in mice, that demonstrated relevant tissue-specific expression of this gene (transcriptional/expression studies), and a new role for DOT1L in the process of chondrogenesis and connective tissue morphology (functional studies). The study concluded that DOT1L will be an interesting new target for osteoarthritis therapy. Another informative strategy is likely to be the integration of pathway and gene ontology databases with GWAS data, and this has recently provided the first genetic evidence implicating variants in neurological pathways in MS risk.108
Genetic data might, furthermore, be used in this way to study autoimmunity as a paradigm. Genetic commonalities between conditions might be found by examining genes and groups of genes that are associated with several autoimmune diseases (outlined in Table 1). Predictably, genes associated with antigen presentation, the interferon pathways and T-cell signalling were found to be associated with several autoimmune conditions. Understanding the ways in which such genes interact epistatically, and the functional consequences of these interactions, might pave the way to a more global understanding of the development of autoimmune pathology and loss of self-tolerance that occurs in these debilitating conditions.
Table 1.
Common gene associations identified by genome-wide association studies in six major autoimmune disorders – rheumatoid arthritis (RA), inflammatory bowel disease (Crohn's disease and ulcerative colitis; IBD), systemic lupus erythematosus (SLE), multiple sclerosis (MS), type I diabetes mellitus (DM-I) and systemic sclerosis (SS). The Catalog of published genome-wide association studies (http://www.genome.gov/gwastudies/) was used to identify all genes that were associated with three or more of the autoimmune diseases of interest
| Associated gene | Functions | RA | IBD | SLE | MS | DM-I | SS |
|---|---|---|---|---|---|---|---|
| Class II HLA alleles | Antigen presentation | x | x | x | x | x | x |
| IL2RA/B | Interleukin-2 receptor | x | x | x | x | ||
| IRF5 | Interferon-induced transcription factors | x | x | x | x | ||
| IRF8 | x | x | x | x | |||
| ETS1 | Transcription factor, some roles in immune function shown | x | x | x | |||
| PTPN2 | Apoptosis regulator | x | x | x | |||
| PTPN22 | T-cell receptor signalling | x | x | x | |||
| TNFA1P3 | Anti-inflammatory signalling molecule | x | x | x | |||
| TNFRSF14 | T-cell signalling receptor. Herpes simplex virus entry receptor | x | x | x | |||
| AFF family members | Transcription factor expressed in lymphoid tissue | x | x | x | |||
| STAT4 | Interleukin-2 induced transcription factor | x | x | x | |||
| BACH2 | Transcription factor, some roles in immune function shown | x | x | x | |||
| IL7R | Interleukin-7 receptor | x | x | x |
In summary, the phenomenon of epistasis is a crucial concept in the understanding of autoimmune disease. The abundant molecular interactions, functional redundancy within gene clusters, complex signalling pathways and genetic hubs mean that the immune system is a rich source of epistatic interactions. We are beginning to observe the extent to which gene–gene interactions alter the phenotypic outcome of risk-alleles for common autoimmune disorders, such as MS and SLE, although a great deal of further work remains to be done. It is highly likely that consideration of epistatic interactions will solve the paradigm of missing heritability in complex immune disorders, and further research in this field that uses genetic data as the basis for functional studies will, no doubt, clarify our understanding of the complex processes involved in autoimmune disorders.
Acknowledgments
The authors would like to thank Prof. Avrion Mitchison for helpful ideas, feedback and comments.
Disclosures
The authors declare no conflict of interest.
References
- 1.Bateson W. Mendel's Principles of Heredity. Cambridge: Cambridge [Eng.] University Press; 1909. [Google Scholar]
- 2.Fisher RA. The correlation between relatives on the supposition of Mendelian inheritance. Trans R Soc Edinb. 1918;52:399–433. [Google Scholar]
- 3.Lehner B. Molecular mechanisms of epistasis within and between genes. Trends Genet. 2011;27:323–31. doi: 10.1016/j.tig.2011.05.007. [DOI] [PubMed] [Google Scholar]
- 4.Mejstríková E, Janda A, Hrusák O, et al. Skin lesions in a boy with X-linked lymphoproliferative disorder: comparison of 5 SH2D1A deletion cases. Pediatrics. 2011;129:e523–8. doi: 10.1542/peds.2011-0870. [DOI] [PubMed] [Google Scholar]
- 5.Lander ES. Initial impact of the sequencing of the human genome. Nature. 2011;470:187–97. doi: 10.1038/nature09792. [DOI] [PubMed] [Google Scholar]
- 6.Manolio TA, Brooks LD, Collins FS. A HapMap harvest of insights into the genetics of common disease. J Clin Invest. 2008;118:1590–605. doi: 10.1172/JCI34772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zuk O, Hechter E, Sunyaev SR, Lander ES. The mystery of missing heritability: genetic interactions create phantom heritability. Proc Natl Acad Sci U S A. 2012;109:1193–8. doi: 10.1073/pnas.1119675109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hirschorn JN. Genomewide association studies – illuminating biologic pathways. N Engl J Med. 2009;360:1699–701. doi: 10.1056/NEJMp0808934. [DOI] [PubMed] [Google Scholar]
- 9.Manolio TA, Collins FS, Cox NJ, et al. Finding the missing heritability of complex diseases. Nature. 2009;461:747–53. doi: 10.1038/nature08494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Eichler EE, Flint J, Gibson G, et al. Missing heritability and strategies for finding the underlying causes of complex disease. Nat Rev Genet. 2010;11:446–50. doi: 10.1038/nrg2809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Goldstein DB. Common genetic variation and human traits. N Engl J Med. 2009;360:1696–8. doi: 10.1056/NEJMp0806284. [DOI] [PubMed] [Google Scholar]
- 12.McClellan J, King MC. Genetic heterogeneity in human disease. Cell. 2010;141:210–7. doi: 10.1016/j.cell.2010.03.032. [DOI] [PubMed] [Google Scholar]
- 13.Slatkin M. Epigenetic inheritance and the missing heritability problem. Genetics. 2009;182:845–50. doi: 10.1534/genetics.109.102798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Maher B. Personal genomes: the case of the missing heritability. Nature. 2008;456:18–21. doi: 10.1038/456018a. [DOI] [PubMed] [Google Scholar]
- 15.Franke A, McGovern DP, Barrett JC, et al. Genome-wide meta-analysis increases to 71 the number of confirmed Crohn's disease susceptibility loci. Nat Genet. 2010;42:1118–25. doi: 10.1038/ng.717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gerke J, Lorenz K, Cohen B. Genetic interactions between transcription factors cause natural variation in yeast. Science. 2009;323:498–501. doi: 10.1126/science.1166426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Brem RB, Storey JD, Whittle J, Kruglyak L. Genetic interactions between polymorphisms that affect gene expression in yeast. Nature. 2005;436:701–3. doi: 10.1038/nature03865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Byrne AB, Weirauch MT, Wong V, et al. A global analysis of genetic interactions in Caenorhabditis elegans. J Biol. 2007;6:8. doi: 10.1186/jbiol58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Costanzo M, Baryshnikova A, Bellay J, et al. The genetic landscape of a cell. Science. 2010;327:425–31. doi: 10.1126/science.1180823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lehner B, Crombie C, Tischler J, et al. Systematic mapping of genetic interactions in Caenorhabditis elegans identifies common modifiers of diverse signaling pathways. Nat Genet. 2006;38:896–903. doi: 10.1038/ng1844. [DOI] [PubMed] [Google Scholar]
- 21.Dowell RD, Ryan O, Jansen A, et al. Genotype to phenotype: a complex problem. Science. 2010;328:469. doi: 10.1126/science.1189015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hill WG, Goddard ME, Visscher PM. Data and theory point to mainly additive genetic variance for complex traits. PLoS Genet. 2007;4:e1000008. doi: 10.1371/journal.pgen.1000008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Crow JF. On epistasis: why it is unimportant in polygenic directional selection. Philos Trans R Soc Lond B Biol Sci. 2010;365:1241–4. doi: 10.1098/rstb.2009.0275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mitchison NA, Rose AM. Epistasis: the key to understanding immunological disease? Eur J Immunol. 2011;41:2152–4. doi: 10.1002/eji.201141811. [DOI] [PubMed] [Google Scholar]
- 25.Marchini J, Donnelly P, Cardon LR. Genome-wide strategies for detecting multiple loci that influence complex diseases. Nat Genet. 2005;37:413–7. doi: 10.1038/ng1537. [DOI] [PubMed] [Google Scholar]
- 26.Ritchie MD. Using biological knowledge to uncover the mystery in the search for epistasis in genome-wide association studies. Ann Hum Genet. 2011;75:172–82. doi: 10.1111/j.1469-1809.2010.00630.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Basson J, Simino J, Rao DC. Between candidate genes and whole genomes: time for alternative approaches in blood pressure genetics. Curr Hypertens Rep. 2012;14:46–61. doi: 10.1007/s11906-011-0241-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cordell HJ. Genome-wide association studies: detecting gene–gene interactions that underlie human diseases. Nat Rev Genet. 2009;10:392–404. doi: 10.1038/nrg2579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Traherne JA. Human MHC architecture and evolution: implications for disease association studies. Int J Immunogenet. 2008;35:179–92. doi: 10.1111/j.1744-313X.2008.00765.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Middleton D, Gonzelez F. The extensive polymorphism of KIR genes. Immunology. 2010;129:8–19. doi: 10.1111/j.1365-2567.2009.03208.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Trowsdale J. The MHC, disease and selection. Immunol Lett. 2011;137:1–8. doi: 10.1016/j.imlet.2011.01.002. [DOI] [PubMed] [Google Scholar]
- 32.Carrington M, Martin MP. The impact of variation at the KIR gene cluster on human disease. Curr Top Microbiol Immunol. 2006;298:225–57. doi: 10.1007/3-540-27743-9_12. [DOI] [PubMed] [Google Scholar]
- 33.Rajagopalan S, Long EO. Understanding how combinations of HLA and KIR genes influence disease. J Exp Med. 2005;201:1025–9. doi: 10.1084/jem.20050499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Dong C, Davis RJ, Flavell RA. MAP kinases in the immune response. Annu Rev Immunol. 2002;20:55–72. doi: 10.1146/annurev.immunol.20.091301.131133. [DOI] [PubMed] [Google Scholar]
- 35.Vallabhapurapu S, Karin M. Regulation and function of NF-κB transcription factors in the immune system. Annu Rev Immunol. 2009;27:693–733. doi: 10.1146/annurev.immunol.021908.132641. [DOI] [PubMed] [Google Scholar]
- 36.Hazeki K, Nigorikawa K, Hazeki O. Role of phosphoinositide 3-kinase in innate immunity. Biol Pharm Bull. 2007;30:1617–23. doi: 10.1248/bpb.30.1617. [DOI] [PubMed] [Google Scholar]
- 37.O'Shea JJ, Lahesmaa R, Vahedi G, et al. Genomic views of STAT function in CD4+ T helper cell differentiation. Nat Rev Immunol. 2011;11:239–50. doi: 10.1038/nri2958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ghoreschi K, Laurence A, O'Shea JJ. Janus kinases in immune cell signaling. Immunol Rev. 2009;228:273–87. doi: 10.1111/j.1600-065X.2008.00754.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Davis BK, Wen H, Ting JP-Y. The inflammasome NLRs in immunity, inflammation, and associated diseases. Annu Rev Immunol. 2011;29:707–35. doi: 10.1146/annurev-immunol-031210-101405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.McKinney BA, Pajewski NM. Six degrees of epistasis: statistical network models for GWAS. Front Genet. 2011;2:109. doi: 10.3389/fgene.2011.00109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Drug and Therapeutics Bulletin. Systemic lupus erythematosus – an update. Drug Ther Bull. 2011;49:81–4. doi: 10.1136/dtb.2011.02.0044. [DOI] [PubMed] [Google Scholar]
- 42.Sestak AL, Shaver TS, Moser KL, et al. Familial aggregation of lupus and autoimmunity in an unusual multiplex pedigree. J Rheumatol. 1999;26:1495–9. [PubMed] [Google Scholar]
- 43.Buckman KJ, Moore SK, Ebbin AJ, et al. Familial systemic lupus erythematosus. Arch Intern Med. 1978;138:1674–6. [PubMed] [Google Scholar]
- 44.Lawrence JS, Martins CL, Drake GL. A family survey of lupus erythematosus. 1. Heritability. J Rheumatol. 1987;14:913–21. [PubMed] [Google Scholar]
- 45.Michel M, Johanet C, Meyer O, et al. Familial lupus erythematosus. Clinical and immunologic features of 125 multiplex families. Medicine (Baltimore) 2001;80:153–8. doi: 10.1097/00005792-200105000-00001. [DOI] [PubMed] [Google Scholar]
- 46.Arora-Singh RK, Assassi S, del Junco DJ, et al. Autoimmune diseases and autoantibodies in the first degree relatives of patients with systemic sclerosis. J Autoimmun. 2010;35:52–7. doi: 10.1016/j.jaut.2010.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Deapen D, Escalante A, Weinrib L, et al. A revised estimate of twin concordance in systemic lupus erythematosus. Arthritis Rheum. 1992;35:311–8. doi: 10.1002/art.1780350310. [DOI] [PubMed] [Google Scholar]
- 48.Reichlin M, Harley JB, Lockshin MD. Serologic studies of monozygotic twins with systemic lupus erythematosus. Arthritis Rheum. 1992;35:457–64. doi: 10.1002/art.1780350416. [DOI] [PubMed] [Google Scholar]
- 49.Grennan DM, Parfitt A, Manolios N, et al. Family and twin studies in systemic lupus erythematosus. Dis Markers. 1997;13:93–8. [PubMed] [Google Scholar]
- 50.Deng Y, Tsao BP. Genetic susceptibility to systemic lupus erythematosus in the genomic era. Nat Rev Rheumatol. 2010;6:683–92. doi: 10.1038/nrrheum.2010.176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hom G, Graham RR, Modrek B, et al. Association of systemic lupus erythematosus with C8orf13-BLK and ITGAM-ITGAX. N Engl J Med. 2008;358:900–9. doi: 10.1056/NEJMoa0707865. [DOI] [PubMed] [Google Scholar]
- 52.International Consortium for Systemic Lupus Erythematosus Genetics (SLEGEN) Harley JB, Alarcón-Riquelme ME, et al. Genome-wide association scan in women with systemic lupus erythematosus identifies susceptibility variants in ITGAM, PXK, KIAA1542 and other loci. Nat Genet. 2008;40:204–10. doi: 10.1038/ng.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kozyrev SV, Abelson AK, Wojcik J, et al. Functional variants in the B-cell gene BANK1 are associated with systemic lupus erythematosus. Nat Genet. 2008;40:211–6. doi: 10.1038/ng.79. [DOI] [PubMed] [Google Scholar]
- 54.Graham RR, Cotsapas C, Davies L, et al. Genetic variants near TNFAIP3 on 6q23 are associated with systemic lupus erythematosus. Nat Genet. 2008;40:1059–61. doi: 10.1038/ng.200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Gateva V, Sandling JK, Hom G, et al. A large-scale replication study identifies TNIP1, PRDM1, JAZF1, UHRF1BP1 and IL10 as risk loci for systemic lupus erythematosus. Nat Genet. 2009;41:1228–33. doi: 10.1038/ng.468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Han JW, Zheng HF, Cui Y, et al. Genome-wide association study in a Chinese Han population identifies nine new susceptibility loci for systemic lupus erythematosus. Nat Genet. 2009;41:1234–7. doi: 10.1038/ng.472. [DOI] [PubMed] [Google Scholar]
- 57.Yang W, Shen N, Ye DQ, et al. Genome-wide association study in Asian populations identifies variants in ETS1 and WDFY4 associated with systemic lupus erythematosus. PLoS Genet. 2010;6:e1000841. doi: 10.1371/journal.pgen.1000841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Okada Y, Shimane K, Kochi Y, et al. A genome-wide association study identified AFF1 as a susceptibility locus for systemic lupus eyrthematosus in Japanese. PLoS Genet. 2012;8:e1002455. doi: 10.1371/journal.pgen.1002455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Walport MJ. Complement. Second of two parts. N Engl J Med. 2001;344:1140–4. doi: 10.1056/NEJM200104123441506. [DOI] [PubMed] [Google Scholar]
- 60.Fernando MM, Stevens CR, Sabeti PC, et al. Identification of two independent risk factors for lupus within the MHC in United Kingdom families. PLoS Genet. 2007;23:e192. doi: 10.1371/journal.pgen.0030192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Hughes T, Adler A, Kelly JA, et al. Evidence for gene–gene epistatic interactions among susceptibility loci for systemic lupus erythematosus. Arthritis Rheum. 2012;64:485–92. doi: 10.1002/art.33354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Eagar TN, Karandikar NJ, Bluestone JA, Miller SD. The role of CTLA-4 in induction and maintenance of peripheral T cell tolerance. Eur J Immunol. 2002;32:972–81. doi: 10.1002/1521-4141(200204)32:4<972::AID-IMMU972>3.0.CO;2-M. [DOI] [PubMed] [Google Scholar]
- 63.Karman J, Jiang JL, Gumlaw N, et al. Ligation of cytotoxic T lymphocyte antigen-4 to the TCR inhibits T cell activation and directs differentiation into FOXP3+ regulatory T cells. J Biol Chem. 2012;287:11098–107. doi: 10.1074/jbc.M111.283705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Fanciulli M, Vyse TJ, Aitman TJ. Copy number variation of Fcγ receptor genes and disease predisposition. Cytogenet Genome Res. 2008;123:161–8. doi: 10.1159/000184704. [DOI] [PubMed] [Google Scholar]
- 65.Brown EE, Edberg JC, Kimberly RP. Fc receptor genes and the systemic lupus erythematosus diathesis. Autoimmunity. 2007;40:567–81. doi: 10.1080/08916930701763710. [DOI] [PubMed] [Google Scholar]
- 66.Bolland S, Ravetch JV. Spontaneous autoimmune disease in FcγRIIB-deficient mice results from strain-specific epistasis. Immunity. 2000;13:277–85. doi: 10.1016/s1074-7613(00)00027-3. [DOI] [PubMed] [Google Scholar]
- 67.Sato-Hayashizaki A, Ohtsuji M, Lin Q, et al. Presumptive role of 129 strain-derived Sle16 locus in rheumatoid arthritis in a new mouse model with Fcγ receptor type IIb-deficient C57BL/6 genetic background. Arthritis Rheum. 2011;63:2930–8. doi: 10.1002/art.30485. [DOI] [PubMed] [Google Scholar]
- 68.Fujii T, Hou R, Sato-Hayashizaki A, et al. Susceptibility loci for the defective foreign protein-induced tolerance in New Zealand Black mice: implication of epistatic effects of Fcgr2b and Slam family genes. Eur J Immunol. 2011;41:2333–40. doi: 10.1002/eji.201141552. [DOI] [PubMed] [Google Scholar]
- 69.Nimmerjahn F, Ravetch JV. Fcγ receptors: old friends and new family members. Immunity. 2006;24:19–28. doi: 10.1016/j.immuni.2005.11.010. [DOI] [PubMed] [Google Scholar]
- 70.Mackay M, Stanevsky A, Wang T, et al. Selective dysregulation of the FcγIIB receptor on memory B cells in SLE. J Exp Med. 2006;203:2157–64. doi: 10.1084/jem.20051503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Su K, Yang H, Li X, et al. Expression profile of FcγRIIb on leukocytes and its dysregulation in systemic lupus erythematosus. J Immunol. 2007;178:3272–80. doi: 10.4049/jimmunol.178.5.3272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Wang A, Batteux F, Wakeland EK. The role of SLAM/CD2 polymorphisms in systemic autoimmunity. Curr Opin Immunol. 2010;22:706–14. doi: 10.1016/j.coi.2010.10.014. [DOI] [PubMed] [Google Scholar]
- 73.Morel L, Blenman KR, Croker BP, Wakeland EK. The major murine systemic lupus erythematosus susceptibility locus, Sle1, is a cluster of functionally related genes. Proc Natl Acad Sci U S A. 2001;98:1787–92. doi: 10.1073/pnas.031336098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Wandstrat AE, Nguyen C, Limaye N, et al. Association of extensive polymorphisms in the SLAM/CD2 gene cluster with murine lupus. Immunity. 2004;21:769–80. doi: 10.1016/j.immuni.2004.10.009. [DOI] [PubMed] [Google Scholar]
- 75.Sospedra M, Martin R. Immunology of multiple sclerosis. Annu Rev Immunol. 2005;23:683–747. doi: 10.1146/annurev.immunol.23.021704.115707. [DOI] [PubMed] [Google Scholar]
- 76.Rosati G. The prevalence of multiple sclerosis in the world: an update. Neurol Sci. 2001;22:117–39. doi: 10.1007/s100720170011. [DOI] [PubMed] [Google Scholar]
- 77.Pender MP. The essential role of Epstein–Barr virus in the pathogenesis of multiple sclerosis. The Neuroscientist. 2011;17:351–67. doi: 10.1177/1073858410381531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Coo H, Aronson KJ. A systematic review of several potential non-genetic risk factors for multiple sclerosis. Neuroepidemiology. 2004;23:1–12. doi: 10.1159/000073969. [DOI] [PubMed] [Google Scholar]
- 79.Lincoln MR, Montpetit A, Cader MZ, et al. A predominant role for the HLA class II region in the association of the MHC region with multiple sclerosis. Nat Genet. 2005;37:1108–12. doi: 10.1038/ng1647. [DOI] [PubMed] [Google Scholar]
- 80.Barcellos LF, Sawcer S, Ramsay PP, et al. Heterogeneity at the HLA-DRB1 locus and risk for multiple sclerosis. Hum Mol Genet. 2006;15:2813–24. doi: 10.1093/hmg/ddl223. [DOI] [PubMed] [Google Scholar]
- 81.Yeo TW, De Jager PL, Gregory SG, et al. A second major histocompatibility complex susceptibility locus for multiple sclerosis. Ann Neurol. 2007;61:228–36. doi: 10.1002/ana.21063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Hindorff LA, MacArthur J, Wise A, et al. A catalog of published genome-wide association studies. http://www.genome.gov/gwastudies [accessed on 8 March 2011]
- 83.Hafler DA, Compston A, Sawcer S, et al. Risk alleles for multiple sclerosis identified by a genomewide study. N Engl J Med. 2007;357:851–62. doi: 10.1056/NEJMoa073493. [DOI] [PubMed] [Google Scholar]
- 84.Comabella M, Craig DW, Camiña-Tato M, et al. Identification of a novel risk locus for multiple sclerosis at 13q31.3 by a pooled genome-wide scan of 500,000 single nucleotide polymorphisms. PLoS ONE. 2008;3:e3490. doi: 10.1371/journal.pone.0003490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Aulchenko YS, Hoppenbrouwers IA, Ramagopalan SV, et al. Genetic variation in the KIF1B locus influences susceptibility to multiple sclerosis. Nat Genet. 2008;40:1402–3. doi: 10.1038/ng.251. [DOI] [PubMed] [Google Scholar]
- 86.Baranzini SE, Wang J, Gibson RA, et al. Genome-wide association analysis of susceptibility and clinical phenotype in multiple sclerosis. Hum Mol Genet. 2009;18:767–78. doi: 10.1093/hmg/ddn388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.De Jager PL, Jia X, Wang J, et al. Meta-analysis of genome scans and replication identify CD6, IRF8 and TNFRSF1A as new multiple sclerosis susceptibility loci. Nat Genet. 2009;41:776–82. doi: 10.1038/ng.401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Bahlo M, Booth DR, Broadley SA, et al. Genome-wide association study identifies new multiple sclerosis susceptibility loci on chromosomes 12 and 20. Nat Genet. 2009;41:824–8. doi: 10.1038/ng.396. [DOI] [PubMed] [Google Scholar]
- 89.Jakkula E, Leppä V, Sulonen AM, et al. Genome-wide association study in a high-risk isolate for multiple sclerosis reveals associated variants in STAT3 gene. Am J Hum Genet. 2010;86:285–91. doi: 10.1016/j.ajhg.2010.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Sanna S, Pitzalis M, Zoledziewska M, et al. Variants within the immunoregulatory CBLB gene are associated with multiple sclerosis. Nat Genet. 2010;42:495–7. doi: 10.1038/ng.584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Nischwitz S, Cepok S, Kroner A, et al. Evidence for VAV2 and ZNF433 as susceptibility genes for multiple sclerosis. J Neuroimmunol. 2010;227:162–6. doi: 10.1016/j.jneuroim.2010.06.003. [DOI] [PubMed] [Google Scholar]
- 92.Baranzini SE, Srinivasan R, Khankhanian P, et al. Genetic variation influences glutamate concentrations in brains of patients with multiple sclerosis. Brain. 2010;133:2603–11. doi: 10.1093/brain/awq192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Wang JH, Pappas D, De Jager PL, et al. Modeling the cumulative genetic risk for multiple sclerosis from genome-wide association data. Genome Med. 2011;3:3. doi: 10.1186/gm217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Briggs FB, Shao X, Goldstein BA, et al. Genome-wide association study of severity in multiple sclerosis. Genes Immun. 2011;12:615–25. doi: 10.1038/gene.2011.34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Patsopoulos NA, de Bakker PI, Esposito F, et al. Genome-wide meta-analysis identifies novel multiple sclerosis susceptibility loci. Ann Neurol. 2011;70:897–912. doi: 10.1002/ana.22609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Sawcer S, Hellenthal G, Pirinen M, et al. Genetic risk and a primary role for cell-mediated immune mechanisms in multiple sclerosis. Nature. 2011;476:214–9. doi: 10.1038/nature10251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Gregersen JW, Kranc KR, Ke X, et al. Functional epistasis on a common MHC haplotype associated with multiple sclerosis. Nature. 2006;443:574–7. doi: 10.1038/nature05133. [DOI] [PubMed] [Google Scholar]
- 98.Kaushansky N, Altmann DM, David CS, et al. DQB1*0602 rather than DRB1*1501 confers susceptibility to multiple sclerosis -like disease induced by proteolipid protein (PLP) J Neuroinflammation. 2012;9:29. doi: 10.1186/1742-2094-9-29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Luckey D, Bastakoty D, Mangalam AK. Role of HLA class II genes in susceptibility and resistance to multiple sclerosis: studies using HLA transgenic mice. J Autoimmun. 2011;37:122–8. doi: 10.1016/j.jaut.2011.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Lincoln MR, Ramagopalan SV, Chao MJ, et al. Epistasis among HLA-DRB1, HLA-DQA1, and HLA-DQB1 loci determines multiple sclerosis susceptibility. Proc Natl Acad Sci U S A. 2009;106:7542–7. doi: 10.1073/pnas.0812664106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Chao MJ, Barnardo MC, Lincoln MR, et al. HLA class I alleles tag HLA-DRB1*1501 haplotypes for differential risk in multiple sclerosis susceptibility. Proc Natl Acad Sci U S A. 2008;105:13069–74. doi: 10.1073/pnas.0801042105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Romero-Pinel L, Pujal JM, Martínez-Yélamos S, et al. Epistasis between HLA-DRB1 parental alleles in a Spanish cohort with multiple sclerosis. J Neurol Sci. 2010;298:96–100. doi: 10.1016/j.jns.2010.07.026. [DOI] [PubMed] [Google Scholar]
- 103.Wu JS, Qiu W, Castley A, et al. Modifying effects of HLA-DRB1 allele interactions on age at onset of multiple sclerosis in Western Australia. Multiple Sclerosis. 2010;16:15–20. doi: 10.1177/1352458509350312. [DOI] [PubMed] [Google Scholar]
- 104.Isobe N, Matsushita T, Yamasaki R, et al. Influence of HLA-DRB1 alleles on the susceptibility and resistance to multiple sclerosis in Japanese patients with respect to anti-aquaporin 4 antibody status. Mult Scler. 2010;16:147–55. doi: 10.1177/1352458509355067. [DOI] [PubMed] [Google Scholar]
- 105.Motsinger AA, Brassat D, Caillier SJ, et al. Complex gene–gene interactions in multiple sclerosis: a multifactorial approach reveals associations with inflammatory genes. NeuroGenetics. 2007;8:11–20. doi: 10.1007/s10048-006-0058-9. [DOI] [PubMed] [Google Scholar]
- 106.Brassat D, Motsinger AA, Caillier SJ, et al. Multifactor dimensionality reduction reveals gene–gene interactions associated with multiple sclerosis. Susceptibility in African Americans. Genes Immunity. 2006;7:310–5. doi: 10.1038/sj.gene.6364299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Castaño Betancourt MC, Cailotto F, Kerkhof HJ, et al. Genome-wide association and functional studies identify the DOT1L gene to be involved in cartilage thickness and hip osteoarthritis. Proc Natl Acad Sci U S A. 2012;109:8218–23. doi: 10.1073/pnas.1119899109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Bush WS, McCauley JL, DeJager PL, et al. A knowledge-driven interaction analysis reveals potential neurodegenerative mechanism of multiple sclerosis susceptibility. Genes Immun. 2011;12:335–40. doi: 10.1038/gene.2011.3. [DOI] [PMC free article] [PubMed] [Google Scholar]