

Abstract
Incorporation of fluorescent nucleoside analogs into duplex DNA usually leads to a reduction in quantum yield, which significantly limits their potential use and application. We have identified two pentamer DNA sequences containing 6-methylisoxanthopterin (6-MI) (ATFAA and AAFTA; where F=6-MI) that exhibit significant enhancement of fluorescence upon formation of duplex DNA with quantum yields close to that of monomeric 6-MI. The enhanced fluorescence dramatically increases the utility and sensitivity of the probe, and is used to study protein-DNA interactions of nanomolar specificity in this work. The increased sensitivity of 6-MI enables anisotropy binding measurements to be performed at concentrations of 1 nM DNA and fluorescence intensity measurements at 50 pM DNA. The ATFAA sequence was incorporated into DNA constructs to measure the binding affinity of four different protein-DNA interactions that exhibit sequence-specific and non sequence-specific recognition. In all cases, the Kd values obtained were consistent with previously reported values measured by other methods. Time-resolved and steady-state fluorescence measurements demonstrate that 6-MI fluorescence is very sensitive to local distortion and reports on different degrees of protein-induced perturbations with single base resolution, where the largest changes occur at the site of protein binding.
Many cellular processes including the regulation of replication, transcriptional regulation, recombination and DNA compaction require the action of DNA-binding proteins.1 A commonly used and powerful method for studying these interactions is fluorescence spectroscopy.2, 3 Typically, the fluorescent probes used for studying these interactions are covalently attached to the DNA and can be divided into two categories: external or internal.4 External fluorescent probes are extremely useful for investigating binding interactions and have the benefit of exhibiting high quantum yields, long lifetimes, and the ease of incorporation onto an oligomer.4, 5 External fluorescent probes are widely used in imaging and single molecule experiments where binding is measured. Disadvantages of using an external probe may arise from the molecular properties of the probe, such as the chemical nature of the probe, size, charge, and method of attachment to DNA.4, 5 For example, attachment to DNA through a 6-carbon linker limits the ability of the probe to report on DNA local structure and dynamics and may cause the probe to be a poor reporter of protein-induced structural perturbations upon binding.4
Internal probes, in many cases fluorescent nucleoside analogs, have greatly increased the amount of information obtained regarding local DNA structure and dynamics.4, 6 These probes can be viewed as providing complementary information to external probes, as they are more sensitive to local interactions. Nucleoside analogs minimally distort DNA structure and are able to base stack with adjacent bases.7, 8 This structural similarity of internal probes to nucleic acid bases and the ability to hydrogen bond with their cognate base have made these probes ideal for examining protein-DNA interactions.9–12 In addition, the fluorescent properties of internal probes are highly sensitive to changes in DNA structure and can be used to monitor local proteininduced perturbations.13–15 For example, when coupled with time-resolved fluorescence measurements the internal probe can provide dynamic resolution of DNA structure on the single base scale.6
A deterrent to using internal probes arises because base stacking interactions associated with incorporation into single-stranded DNA often lead to a significant reduction in the quantum yield, which is either maintained or further decreased upon formation of duplex DNA.2, 4 This reduction in quantum yield lowers sensitivity and introduces a concentration limit at which internally labeled oligomers can be used. Many DNA-binding proteins exhibit dissociation constants in the low nanomolar range, which is below the signal to noise threshold for many fluorescent nucleoside analogs. Thus, the study of these interactions using internal fluorescent probes has remained a challenging problem.
This study focuses on the nucleoside analog, 6-methylisoxanthopterin (6-MI), a guanosine analog that forms hydrogen bonds with cytosine. Previous work probing DNA and RNA dynamics has demonstrated that duplex DNA is minimally distorted upon introduction of 6-MI and that the fluorescent properties of 6-MI are sensitive to local environment.16 6-MI has been used in a number of studies of protein-nucleic acid interactions including investigations of the cleavage activity of HIV-1 integrase,17 probing the binding and destabilization of G tetrad structures by unwinding protein (UP1),18 formation of the RecA-DNA filament,11 and probing RNA dynamics in the Tetrahymena group I ribozyme.19
We herein report on the identification of a specific DNA sequence that enhances 6-MI fluorescence upon duplex formation and demonstrate its utility in examining protein-DNA interactions. The enhanced 6-MI fluorescence was initially observed in the recognition sequence of the E. coli protein Integration Host Factor (IHF) when studying IHF-DNA interactions (Figure 1; Table 1). The local sequences (ATFAA and AAFTA) give rise to the enhanced fluorescence, in which adenine and thymine are the nearest neighbors of the 6-MI probe. A sequence-dependent enhancement of 6-MI fluorescence upon duplex formation was previously observed by Knutson and co-workers, when the 6-MI was flanked by thymine residues (i.e. TFT).20 The fluorescent pentamers identified in this study are significantly brighter than the TFT sequence identified by Knutson and co-workers (Φrel = 0.27 vs. 0.75 for ATFAA) (Tables 1 and 2). Lifetime and quantum yield measurements point to a decrease in dynamic, collisional quenching of the excited state by adjacent bases as a plausible mechanism for the increased fluorescence. The significantly increased fluorescence of the ATFAA sequence allows measurement of binding at DNA concentrations as low as 50 pM and anisotropy measurements at a 1 nM concentration of DNA. The increased sensitivity of the ATFAA sequence context is used to investigate sequence-specific and non-sequence-specific protein-DNA interactions. We further demonstrate that the enhanced 6-MI fluorescence coupled with time-resolved lifetime and anisotropy measurements provides unique insight into DNA structure in the context of protein- DNA interactions.
Figure 1.

The IHF-DNA X-ray co-crystal structure depicted with the positions of 6-MI in the ATFAA and AAFTA sequences highlighted (cAFT10 = magenta, TFA15 = green, TFA07 = navy blue). The IHF consensus binding sequence is shown in yellow. Figure was generated with Pymol using coordinates from pdbid, 1IHF.
Table 1.
Sequences of 6-MI-containing oligomers used in this study
| Sequence name |
Sequence1 | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| AFT 07 | ||||||||||||
| TFA15 | ||||||||||||
| cAFT10 | ||||||||||||
| TFA07 | ||||||||||||
| TFT32 | ||||||||||||
| AFA_15 | ||||||||||||
| JX2 | ||||||||||||
| Msh_TFA15 |  |
|||||||||||
| Msh_TFA09 | 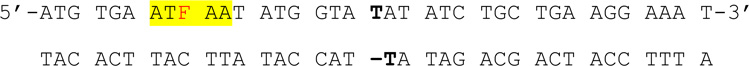 |
|||||||||||
| TBP_TFA23 |  |
|||||||||||
| HU_TFA15 |  |
|||||||||||
The 6-MI position is indicated in red ( ) and the pentamer sequences containing 6-MI are highlighted in yellow. Boldface highlights the protein-binding recognition sequence or +T insertion.
) and the pentamer sequences containing 6-MI are highlighted in yellow. Boldface highlights the protein-binding recognition sequence or +T insertion.
JX is the sequence used to form the 4WJ as shown in Supporting Information, Figure S8.
Table 2.
Parameters1 derived from fitting of time-resolved fluorescence decay curves
| DNA molecule | α(1)2 (±0.01) |
τ1 (ns) (±0.12) |
α(2) (±0.01) |
τ2 (ns) (±0.2) |
α(3) (±0.01) |
τ3 (ns) (±0.04) |
Mean lifetime (ns) (±0.03) |
Φrel SS/ Φrel TR (±0.05) |
Solvent Accessible Fraction (±0.06) |
|---|---|---|---|---|---|---|---|---|---|
| 6-MI | 1 | 6.57 | 6.57 | 1/1 | 1 | ||||
| AFT07ss | 0.62 | 0.45 | 0.21 | 2.59 | 0.17 | 7.27 | 5.14 | 0.19/0.31 | 0.32 |
| AFT07ds | 0.81 | 1.05 | 0.16 | 1.97 | 0.04 | 8.04 | 2.65 | 0.24/ 0.23 | 0.5 |
| TFA15ss | 0.49 | 0.61 | 0.31 | 2.9 | 0.2 | 6.78 | 4.72 | 0.20/0.39 | 0.46 |
| TFA15ds | - | - | 0.05 | 3.62 | 0.95 | 7.19 | 7.1 | 0.82/1.06 | 0.17 |
| TFA15ds + IHF |
- | - | 0.1 | 3.74 | 0.9 | 6.99 | 6.82 | ||
| TFA07ss | 0.61 | 0.52 | 0.23 | 2.91 | 0.17 | 6.96 | 4.76 | 0.15/0.33 | 0.31 |
| TFA07ds | - | - | 0.05 | 3.12 | 0.95 | 6.93 | 6.84 | 0.73/1.02 | 0.2 |
| TFA07ds + IHF |
- | - | 0.07 | 2.85 | 0.93 | 6.87 | 6.75 | ||
| cAFT10ss | 0.65 | 0.66 | 0.26 | 2.27 | 0.09 | 7.38 | 3.71 | 0.11/0.25 | 0.68 |
| cAFT10ds | - | - | 0.07 | 2.79 | 0.93 | 7.82 | 7.7 | 0.7/1.14 | 0.14 |
| cAFTlOds +IHF |
0.07 | 1.77 | 0.17 | 4.73 | 0.76 | 7.7 | 7 | ||
| Msh_TFA15ds | 0.14 | 1.24 | 0.43 | 4.02 | 0.43 | 6.65 | 5.56 | 0.7/0.75 | |
|
Msh TFA15ds +Msh2-Msh6 |
0.18 | 1.24 | 0.48 | 3.86 | 0.34 | 6.65 | 6.65 | ||
| Msh_TFA09ds | - | - | 0.08 | 2.89 | 0.92 | 7.37 | 7.22 | 0.78/1.06 | |
| Msh TFA09, +Msh2-Msh6 |
- | - | 0.09 | 2.87 | 0.91 | 7.37 | 7.2 | ||
Fluorescence decay curves were fit to a sum of exponentials:
For all parameters standard deviations are given in parentheses.
Experimental Procedures
DNA Preparation
DNA strands containing the nucleoside analogue 6MI were obtained from Fidelity Inc. (Gaithersburg MD.) in HPLC-purified form (Table 1). Crude complementary strands were purchased from Integrated DNA Technologies (IDT, Coralville IA), and were purified using a denaturing 7 M urea 20% polyacrylamide gel. DNA bands were identified and isolated from the gel using UV shadowing. DNA was electroeluted from the gel (Schlechier and Schuell, Dassel, Germany) and dialyzed against ddH2O prior to lyophilization and storage. Samples were resuspended in ddH2O and sample concentrations were determined by UV absorption spectroscopy at 260 nm using molar extinction coefficients calculated using standard methods for modified or unmodified oligonucleotide.21, 22 Duplexes were prepared by adding equal moles of the probe strand to a complementary strand in a 50 mM sodium phophate buffer, pH 7.5. The samples were heated in a water bath at 90 °C for 5 min and then allowed to cool slowly to room temperature in the bath. Annealed samples were stored at −20 °C.
Steady State Fluorescence Measurements
The steady state quantum yields were determined relative to 6-MI monomer for each single-and double-stranded oligomer containing 6-MI. The quantum yield was determined from an average of at least three experiments and was calculated using the equation given below:1
where A is the absorbance of the sample at the excitation wavelength, I is the intensity of the exciting light at the excitation wavelength, n is the index of refraction of the solution, and D is the integrated fluorescence emission. In this case, x refers to the single- and double-stranded oligomers containing 6MI, r is the reference or 6MI monomer. Absorbance measurements were performed with a Beckman Coulter DU-650 at a concentration of 15 µM 6-MI monomer in the strands to ensure an absorbance of at least 0.1 at 340 nm. For fluorescence measurements, oligomers were diluted either 1:25 or 1:50 and the 6-MI monomer was diluted 1:200. Fluorescent emission measurements were made from 390 nm to 550 nm at 1 nm/pt, with a 1 s integration time and with the excitation polarizer set to 0° and emission polarizer set to 54.7°, using a Horiba SPEX Fluoromax-4 spectrofluorometer (Edison, NJ). Samples were analyzed in 3 mm square quartz cuvettes, while being stirred continuously. Spectral analyses were performed with Grams AI ver. 8 (Thermo Electron Corp.).
Fluorescence Quenching
Collisional quenching measurements of fluorescence intensity were performed by titrating 0 to 200 mM KI into a 400 nM solution of DNA. The ionic strength of the solution was held constant with 200 mM KCl. The concentration of 400 nM DNA was held constant during the titration. Single point emission was measured at 430 nm with an excitation of 340 nm using a Horiba Fluoromax-2 fluorimeter using slits of 2 and a 4 nm band-pass. The intensity was corrected for background and buffer contributions. For 6-MI monomer, quenching was measured by titrating potassium iodide from 0 to 200 mM into a solution of 400 nM 6-MI monomer. The 6-MI monomer quenching data were evaluated using the Stern-Volmer equation:
where I0 and I are the steady-state intensities, KSV is the Stern-Volmer constant and τ0 and τ are the amplitude-weighted fluorescence lifetimes in the absence and presence of quencher Q, respectively. In ss DNA and ds DNA the solvent accessible fraction was determined from a non-linear least squares fit of the data using the following equation:
where I represents the intensity, I0 is the initial intensity, Ifa is the intensity from fluorophores that are accessible to quencher, and Ksv is the Stern-Volmer constant. The fraction accessible to quencher (fa) was determined from a minimum of at least three separate fluorescence-quenching experiments.
DNA binding experiments
The anisotropy binding experiments for Msh2-Msh6, IHF and HU were performed by titrating protein into a constant 1 nM concentration of DNA. The TBP binding experiment was performed by preparing individual samples containing 1nM DNA and varying concentrations of TBP. A sample volume of 1 ml was used in quartz cuvettes measuring 4 mm × 10 mm. The Msh2-Msh6 binding buffer contained 5 mM MgCl2, 5 mM Tris-HCl pH 8.0, and 50 mM NaCl. The IHF and HU binding buffer contained 5 mM Tris-HCl pH 7.5, 0.1 mM EDTA, and 70 mM KCl. TBP binding buffer contained 10% ethylene glycol, 5 mM Hepes, pH 8.0, 0.04% Triton X-100, 0.5 mM EDTA, and 60 mM KCl. Msh2-Msh6 was incubated for 3 minutes at 298K for each titration point; for IHF and HU each titration point was incubated for 10 minutes at 283K and for TBP the samples were incubated at 298K for 2 hours. Steady state fluorescence measurements were performed as described above. The excitation wavelength was 340 nm and emission was monitored at 430 nm, with slits of 5 and 15 nm bandpass, respectively. Samples were monitored for a total time of 30 s. All anisotropy measurements were corrected for background and buffer contributions and reported values result from at least 3 independent measurements. Anisotropy was calculated using the following equation:
where III is vertically polarized light, I┴ refers to horizontally polarized light and G refers to the G-factor determined for each experiment.
The intensity binding experiments for Msh2-Msh6, HU and IHF were performed under the same conditions used for anisotropy experiments, except that the polarizers were removed. Proteins were titrated into a 500 pM concentration of DNA (or 50 pM for IHF titrations).
Anisotropy and intensity binding curves were analyzed assuming a 1:1 binding interaction with the following equation:
where r refers to either the anisotropy or the intensity at a certain protein concentration, ri and rf refer to the initial and final anisotropy or intensity values respectively, [D ] is the molar concentration of DNA, [P] is the protein concentration; and Kd is the dissociation constant. Fits were generated and evaluated using Origin 6.0 (MicroCal).
Fluorescent Lifetime Measurements
The Photon Technology International (PTI) TimeMaster instrument was used for time-correlated single photon counting (TCSPC). Samples containing 50 nM DNA were excited with a Becker & Hickl (BDL-375-SMC) 375nm pulsed picosecond- laser diode (rep rate=1MHz, <1mW average power). Emission was detected at 460 nm with emission slits of 15 nm using a 450 nm cut-off filter. Intensity decay data was collected to 65000 in the peak channel with the emission polarizer set to 54.7°. The vertical/vertical (V/V) polarization data used for anisotropy lifetime analyses were collected to 65000 in the peak channel and vertical/horizontal (V/H) polarization data were collected for the same amount of time as the V/V data. Stoichiometric amounts of DNA and protein (50 nM for IHF experiments, and 200 nM for Msh2-Msh6) were incubated for 10 min and data were collected using the same conditions as described above.
Time-resolved fluorescence decays were analyzed using the following equation:
where I is the intensity at time t and αi represents the fractional subpopulation with a lifetime of τi. The fluorescence decay curves were fit to a multi-exponential model using an iterative reconvolution method. All fitting and analysis were performed with Globals Unlimited.23. To reconvolve the instrument response function (IRF) with fluorescence lifetime data, the response function of the system was acquired by measuring scattered light from a colloidal suspension of non-dairy creamer to give a full width at half-maximum of 14 channels corresponding to ~200 psec. The multi-exponential decay model I'(t) was used to describe the change in fluorescence intensity over time by fitting to the measured fluorescence decay data:
where L(t) is the IRF and I(t) is the intensity at time, t. To determine the necessity for an additional decay term we evaluated the goodness of fit through examination of the residuals for systematic oscillations, reduction of the reduced chi-squared upon addition of the decay term and if the addition of a new decay term lead to a unique lifetime. The mean lifetime τm is defined as follows:
The experimentally measured anisotropy decay, r(t) is modeled as the sum of one or more exponential decay terms:
where r0 is the limiting anisotropy, βi are the weighting factors for each contributing exponential and θi is the rotational correlation time for component i. The weighting factors βi are normalized such that . Two correlation times are used to define the experimentally observed time-dependent anisotropy decay. The molecular motions, which result in anisotropy decay, can be defined as fast local motion of the chromophore with a rotational correlation time defined as θL and a slower motion due to overall rotation due to DNA tumbling with a rotational correlation time defined as θR . The time dependent anisotropy decay is defined in terms of the molecular parameters as follows:
This can be rearranged as:
The relationship between the experimental expression and the molecular motion is defined as:
This expression for the anisotropy decay can then be rearranged recognizing that:
so that the slow component of the observed decay can be equated directly to loss of anisotropy from the overall tumbling motion. The fast component contains contribution from both the local chromophore motion as well as the tumbling but the fast molecular motion, θL, can be extracted from the fit of the experimental data as follows:
The experimental anisotropy decay curves, r(t) , were fit to a multi-exponential model using an iterative reconvolution method as described above. The lifetime values determined from fitting the excited state decay experimental data were fixed during the analysis of time dependent anisotropy decay. The typical value for χ2 ranged from 1.05 to 1.32. The standard deviations for lifetime and anisotropy decay values were determined from a minimum of three separate experiments.
Results and Discussion
Duplex formation leads to enhanced fluorescence of 6-MI in distinct sequences
We have examined the fluorescence behavior of the nucleoside analogue, 6-methylisoxanthopterin (6-MI) in several different DNA molecules. The incorporation of this analogue minimally perturbs DNA duplex stability as measured by thermal melting (Supporting Information: Figure S1). In our studies, fluorescent enhancement of nucleoside analog 6-MI in duplex DNA was observed in oligomers containing the consensus sequence for the DNA-binding protein Integration Host Factor (IHF) (Figure 1). 6-MI was incorporated at several unique positions flanking the IHF consensus sequence to study the effect of IHF binding on local DNA dynamics (Table 1). In previous studies of 6-MI and other fluorescent nucleoside analogs,6, 24, 25 fluorescence intensity remains quenched upon duplex formation as observed with the AFT07 oligomer (Φrel,ds = 0.24 ± 0.05, Φrel,ss = 0.19 ± 0.04) (Figure 2; Table 2). In contrast to the previously observed quenching, 6-MI displayed an ~4-fold or greater increase in fluorescence intensity upon duplex formation within the TFA15 oligomer (Φrel, ds =0.82 ± 0.03, Φrel, ss =0.20 ± 0.05) and cAFT10 oligomer (Φrel, ds =0.73 ± 0.03, Φrel, ss =0.15 ± 0.05) (Figure 2, Table 2). Examination of local sequence demonstrates that the identity of the n ± 2 base differed between the highly fluorescent duplex sequences and the quenched duplex sequence. For the fluorescent duplexes, TFA15 (with the following sequence surrounding the 6-MI: ATFAA) and cAFT10 (AAFTA) the n ± 2 positions were adenine whereas for the quenched duplex, AFT07 (CAFTC) the n ± 2 positions were cytosine (Table 1). The highly fluorescent duplexes, TFA15 and cAFT10, contained a single purine adjacent to the 6-MI with adenine located at the n+1 (TFA15) and the n-1 position (cAFT10). As the base composition of the ATFAA and the AAFTA sequences are the same, these findings suggest that subtle changes in base sequence can lead to altered fluorescence properties as has been previously observed for the fluorescent nucleoside analog, 6MAP.24 Finally, to determine whether duplex context influences the fluorescence properties, we replaced the CAFTC sequence with the ATFAA sequence (TFA07) in our original duplex sequence. As shown in Figure 2, formation of the TFA07 duplex leads to enhanced 6-MI fluorescence similar to the TFA15 and cAFT10 duplexes and in distinct contrast to the AFT07 duplex (Table 2).
Figure 2.

Fluorescence spectra of ss DNA and ds DNA containing 6-MI are shown based on their quantum yields measured relative to 6-MI monomer (black) using an excitation wavelength of 340 nm. The Φrel of 6-MI in ssDNA (teal, pink, brown, dark green) is highly quenched (Φrel < 0.2). The dsDNA molecules containing either the AFTAA or the AAFTA sequences exhibit significantly higher quantum yields (Φrel ≥ 0.7) (blue, magenta, green). While formation of the AFT07 duplex has only a slight effect on the quantum yield.
Previous studies, focused on sequence-dependent quenching of 6-MI in DNA, showed that the presence of purines adjacent to 6-MI (or 3-methylisoxanthopterin (3-MI)) resulted in greater fluorescence quenching than pyrimidines.16, 26, 27 In a recent study, Knutson and co-workers reported on another sequence which exhibited enhanced 6-MI fluorescence upon incorporation into duplex DNA. Specifically, they observed a 70% increase in quantum yield from 0.12 to 0.19 when 6-MI was flanked by thymine residues at the n ± 1 position (i.e. TFT).20 The enhanced fluorescence observed for 6-MI in the TFT sequence, was attributed to restricted dynamic motion within the duplex.20 Unlike this study, the effect of bases extending beyond the n ± 1 position was not considered; although we note the n ± 2 residues are G for this sequence. In this study, we address whether the mechanism of enhanced fluorescence observed in the ATFAA (TFA15) and AAFTA (cAFT10) sequences arises from a unique, constrained conformation of 6-MI, as observed in the TFT sequence of Knutson and co-workers20 or from a largely extrahelical population of 6-MI as a consequence of the surrounding sequence (Supporting Information: Figure S2).
Sequence-dependent solvent exposure of 6-MI
To parse out these effects, the relative levels of solvent exposure of 6-MI in different sequence contexts were measured by fluorescence quenching. Quenching of 6-MI fluorescence intensity was measured through titration of KI, which quenches the fluorescence through collisional or dynamic quenching of the excited state. Stern-Volmer analysis of time-resolved and steady-state fluorescence quenching of 6-MI monomer, reveals linear relationships between the fluorescence intensity and quencher demonstrating that the process is predominately dynamic (Supporting Information: Figure S3). When 6-MI was incorporated into DNA oligomers and duplexes, the Stern-Volmer plots deviate from linearity indicative of a population not accessible to solvent (Supporting Information: Figure S4). In ssDNA, solvent exposure of 6-MI was reduced by 40% or more for all sequences tested as compared to the free monomer (Figure 3, Table 2). Interestingly, for the AFT07ds duplex the solvent accessible fraction of 6-MI increases to 50 ± 6% from 30% in the single strand. In contrast, the sequences that exhibit enhanced fluorescence experience a decrease in solvent exposure upon duplex formation: 17 ± 2% for TFA15ds, 20 ± 7% for cAFT10ds, and 14 ± 3% for TFA07ds (Figure 3, Table 2). Similar behavior has been observed for 2-aminopurine (2AP), where steric constraints introduced by specific ds DNA sequences led to a reduction in probe solvent exposure. For example, when 2AP was placed in the TATA duplex sequence it exhibited a slower rotational correlation time and reduced solvent exposure, consistent with a restrained motion of the probe.28 Likewise, 3-MI a close structural analog of 6-MI, is minimally quenched by acrylamide, when incorporated into duplex DNA. 26
Figure 3.

The solvent accessibility in ssDNA and dsDNA measured relative to 6-MI monomer by fluorescence collisional quenching with KI. For duplexes containing either the ATFAA or the AAFTA sequence, the formation of dsDNA leads to a reduction in 6-MI solvent accessibility relative to ssDNA and 6-MI monomer, which suggests 6-MI is not extrahelical in these sequences (TFA15ds, cAFT10ds, TFA07ds). Solvent exposure is increased in the quenched duplex, AFT07ds.
Our findings are consistent with hydroxyl radical footprinting methods, which also point to differences in base solvent exposure as a consequence of sequence context. This method requires that the OH• accesses the DNA backbone for cleavage. The hydroxyl radical cleavage intensity for the IHF consensus sequence used in this study were predicted using the OH Radical Cleavage Intensity Database,29 which determines hydroxyl radical cleavage propensities at a particular position using an algorithm based on experimentally observed sequence dependent cleavage patterns.30, 31 The predicted cleavage intensity for the IHF consensus sequence at the 7th position (AFT07) is 1.6 while for the 15th position (TFA15) it is only 0.68 (Supporting Information: Figure S5). Similarly, predicted cleavage intensity for the IHF complementary sequence at the 10th position (cAFT10) is only 0.78. Thus, experimental fluorescence quenching results are in good agreement with predicted OH• cleavage propensities and further suggest that 6-MI in the ATFAA and AAFTA sequence contexts is relatively protected from solvent and unlikely to have a large population that is extrahelical.
The excited state lifetime of 6-MI is sequence dependent
To further probe how local sequence influences 6-MI fluorescence properties, we have examined the excited state lifetime of 6-MI in the different sequence contexts and compared that to the monomer. In aqueous solution 6-MI monomer exhibits a mono-exponential excited state lifetime of ~6.5 ns. Upon incorporation of 6-MI into ssDNA, much shorter lifetimes are observed, characterized by a complex, multiexponential decay of the excited state (Figure 4, Table 2).6, 16, 25 The decay rates are attributed either directly to the rate of quenching for a particular conformation or to the rate of change to an efficiently quenched conformation.
Figure 4.

Fluorescence lifetime decay curves (ex=375, em=460) of 6-MI containing ssDNA and dsDNA molecules compared to monomer (black). Dynamic quenching of 6-MI occurs upon incorporation into ssDNA oligomers, as revealed by significantly faster decays (brown, dark green, pink, teal). The dsDNA molecules containing either the ATFAA or AAFTA sequences (TFA15ds-green, TFA07ds-blue, and cAFT10ds-magenta) exhibit fluorescence decays similar to 6-MI monomer (black), indicative of a lack of quenching. Notably, the AFT07 duplex exhibits a fast decay (red). The instrument response function is shown in gray.
The excited state lifetimes of all the 6-MI-containing ssDNA molecules are similar to one another and can be understood in terms of the observed decay components and fractional subpopulations (Figure 5, Table 2). The largest fractional subpopulations (α1 = 0.5–0.8) typically correspond to the shortest lifetime ranging from 0.45–0.66 ns. The next largest fractional subpopulations (α1 = 0.21–0.31) are associated with an intermediate lifetime of 2.7 ns on average. The smallest fractional subpopulation (α3 = 0.09–0.20) is associated with the longest lifetime of ~7.0 ns (Figure 5, Table 2).
Figure 5.

Fractional populations determined from time-resolved fluorescence decay curves (shown in Figure 4) of 6-MI-containing ssDNA and dsDNA molecules relative to monomer. Duplex formation leads to a shift in the peak population to the longest lifetime component (α3) for sequences containing either the ATFAA or AAFTA sequence (TFA15ds, cAFT10ds, TFA07ds). For ssDNA oligomers, the largest fractional population is associated with the shortest- lived component (α1). The AFT07 duplex exhibits a population distribution similar to the single strands where the largest population is associated with the shortest-lived component (α1).
An examination of the excited state lifetimes of the dsDNA sequences that exhibit enhanced fluorescence upon duplex formation reveal pronounced changes in excited state dynamics relative to the single strand. A significant shift in the fractional subpopulations for dsDNA is observed compared to ssDNA; where the largest subpopulation for the dsDNA sequences (α3 = ~0.9) is associated with the longest-lived component (τ3 = ~7.0 ns), while the largest population for ssDNA (α1 = ~0.55) is associated with the shortest lifetime component (τ1 = ~0.5 ns) (Figure 5, Table 2). Importantly, this shift in populations is not observed for the quenched duplex, AFT07. In addition, the excited state lifetimes for TFA15ds, cAFT10ds, and TFA07ds are predominately characterized by a single exponential decay with a lifetime similar to that of 6-MI monomer (τ = ~6.5ns). The mean lifetime of dsDNA containing the ATFAA sequences is ~3 ns longer than for ssDNA, which is attributed to decreased quenching interactions with adjacent bases. This finding can be interpreted in terms of either a homogenous population or a continuum of conformations; in both cases interbase quenching interactions are minimized.
The multi-exponential decay observed for 6-MI in ssDNA is similar to that detected for 2AP and suggests that the mechanism of dynamic quenching is probably comparable for both probes.6, 16, 25, 32–34 Studies reporting on the composition of excited state subpopulations for 2AP are consistent with discrete populations existing in thermal equilibrium with an energy barrier between states.35, 36 The multiexponential decay for 6-MI in ssDNA is interpreted similarly and is suggestive of the presence of discrete subpopulations for each decay component. Importantly, overlap between these populations is unlikely due to the large differences between the decay times (Table 2).
We also determined relative quantum yields based on the time-resolved measurements. Importantly, these quantum yields only reflect the radiative component of the fluorescence. Similar to the steady-state data, a significant increase in quantum yield is observed upon duplex formation for the TFA15, TFA07 and cAFT10 sequences. For these sequences, the steady-state quantum yields are approximately 30% lower than those determined by time-resolved measurements and are strongly suggestive of a static quenching mechanism. Interestingly, this effect is more pronounced in the single strands, where steady-state quantum yields are more than 50% lower than those determined by time-resolved methods and thus suggest that static quenching mechanisms are even greater than in ds DNA (Table 2). It is also possible that the discrepancy between absolute values of the steady-state and time-resolved quantum yields results from a fast decay rate that is not resolved with the current instrumentation.
Local motion of 6-MI is decreased in ATFAA sequence
The mobility of 6-MI within the different sequence contexts was directly assessed by time-resolved fluorescence anisotropy measurements. A two-component exponential decay was found to correctly model the time-dependent anisotropy decay for DNA ss and ds molecules containing 6-MI. All of the ssDNA molecules exhibit a fast rotational correlation time that ranges from 0.13–0.42 ns, indicative of a considerable amount of local motion of 6-MI in the ssDNA (Table 3, Supporting Information: Figure S6). Interestingly, the cAFT10 sequence exhibits the fastest anisotropic decay of the four single strands. The local motion reflects the stability of the probe, which arises in part from base stacking interactions and has been shown to be sequence dependent.28, 32 In the AFT07 duplex the rotational correlation time associated with 6-MI internal motion (θL) is increased to 0.61 ns, consistent with reduced rotational mobility of the 6-MI due to base stacking and base pairing interactions (Figure 6, Table 3). The ds DNA molecules that give rise to enhanced fluorescence all exhibit a markedly different behavior in which the internal motion of the probe is dramatically reduced and the fastest anisotropic decay observed is ~1.2 ns or greater (Table 3). We consider this rotational time too long to be attributed solely to the local motion of 6-MI and suggest this decay also reflects rotation of the duplex or segmental motion.28, 32 The longer correlation times observed (θR) are attributed to the overall tumbling or rotational motion of the DNA molecule.32 The average tumbling anisotropic decay of the ss DNA is 4.1 ns and this relatively short correlation time suggests that the ss DNA does not behave as a linear rod (Table 3). The θR rotational correlation time of the ds DNA is approximately four times longer than that observed for ss DNA and reflects the increased rigidity of the DNA molecule upon duplex formation (Figure 6, Table 3). The decreased internal motion of 6-MI is consistent with a model in which duplex formation leads to restrained motion of the probe that causes reduced solvent accessibility and decreased collisional quenching of the excited state with adjacent bases.
Table 3.
Parameters1 derived from fitting of time-resolved anisotropy decay curves
| DNA molecule | β12 (±0.02) |
θL (ns) (±0.06) |
β2 (±0.02) |
θR (ns) (±0.9) |
|---|---|---|---|---|
| 6-MI | - | - | - | - |
| AFT07ss | 0.25 | 0.3 | 0.75 | 3.68 |
| AFT07ds | 0.2 | 0.61 | 0.8 | 8.8 |
| TFA15ss | 0.22 | 0.32 | 0.78 | 4.42 |
| TFA15ds | 0.09 | 1.66 | 0.91 | 16.81 |
| TFA15ds + IHF | 0.04 | 1.68 | 0.96 | 26.25 |
| TFA07ss | 0.33 | 0.13 | 0.67 | 3.27 |
| TFA07ds | 0.16 | 1.16 | 0.84 | 15.6 |
| TFA07ds + IHF | 0.1 | 1.18 | 0.91 | 25.99 |
| cAFT10ss | 0.22 | 0.42 | 0.78 | 5.24 |
| cAFT10ds | 0.13 | 1.41 | 0.87 | 17 |
| cAFT10ds + IHF | 0.1 | 0.9 | 0.9 | 24.78 |
| Msh_TFA15ds | 0.14 | 0.73 | 0.86 | 14.64 |
| Msh_TFA15ds +Msh2-Msh6 |
0.16 | 2.11 | 0.84 | 20.73 |
| Msh_TFA09ds | 0.16 | 2.33 | 0.84 | 18.08 |
| Msh_TFA09 +Msh2-Msh6 |
0.2 | 2.81 | 0.8 | 25.51 |
Fitting of anisotropy decay curves was done with the following expression:
For all parameters errors are given in parentheses.
Figure 6.

Time-resolved fluorescence anisotropy decay curves of 6-MI-containing ssDNA and dsDNA molecules. The local motion of 6-MI in the duplex containing the ATFAA sequence (TFA15ds-green) is reduced compared to the AFT07 duplex (red). In general, duplex formation (green, red) leads to slower overall motion of the DNA. The time-resolved anisotropy of ssDNA (dark green, brown) exhibits a bi-exponential decay consisting of significant local motion of 6-MI (~200ps) and overall tumbling of the DNA (~4 ns) (Table 3).
Use of 6-MI-enhanced fluorescence to examine protein-DNA interactions
Given the high fluorescence quantum yields exhibited by these sequences (TFA15ds and cAFT10ds) we investigated the utility of these sequences for studying protein-DNA interactions in fluorescence binding experiments. We also specifically incorporated the ATFAA sequence into four different DNA sequences (TFA07ds, JX, Msh_TFA09ds, TBP_TFA23ds) (Table 1) Additionally, the JX sequence was used to form a four-stranded DNA four way junction (4WJ) (Supporting Information: Figure S8), which allowed us to assess the utility of the ATFAA sequence in unconventional DNA structures. Even within the 4WJ construct 6-MI exhibited enhanced fluorescence similar to that observed in duplex sequences (Φrel = 0.95). In general, the ATFAA sequence, when incorporated into different DNA constructs, retained the enhanced fluorescence (Φrel ≥ 0.7) (Table 2, Supporting Information: Figure S7), making it ideal for investigating structural and dynamic perturbations of DNA in protein-DNA complexes and other structural contexts.
We specifically exploited the enhanced fluorescence to examine four different high affinity protein-DNA binding interactions at DNA concentrations of 1 nM and less (Figures 7 and 8). DNA binding interactions were studied with two non sequence-specific DNA binding proteins (HU and MutS Homolog (Msh) 2–6) and two sequence-specific DNA-binding proteins (Integration Host Factor, IHF and TATA-box Binding Protein, TBP). These proteins represent good model systems for our measurements as their binding interactions have been well characterized by several methods.37–43 For all the protein-DNA interactions examined, saturable binding curves are observed when measured by fluorescence anisotropy (Figure 7). As the 6-MI is located within the DNA duplex, the starting anisotropy is in the range of 0.16–0.18 reflecting the anisotropy of the free duplex. Addition of protein leads to anisotropy increases of 0.06–0.10. These changes are well outside the error range of our measurements and the increase in anisotropy observed is consistent with protein binding. The magnitudes of the anisotropy changes are not uniform as the masses of the proteins are different and in some cases a profound change in shape accompanies protein binding, such as with the IHF protein.44
Figure 7.

The equilibrium fluorescence anisotropy binding curves show the versatility and sensitivity of the ATFAA sequence, where only 1 nM DNA is needed to accurately measure equilibrium binding affinity of four different protein-DNA complexes. Proteins investigated include sequence specific DNA binding proteins (IHF, TBP) and non-sequence specific DNA binding proteins (Msh2-Msh6, HU). The 6-MI was excited at 340 nm and emission was detected at 440 nm. Representative error bars are shown on the TBP-DNA binding curve; the other curves have similar error.
Figure 8.

Equilibrium binding curves measured by fluorescence intensity with decreasing concentrations of 6-MI-containing DNA (exc = 340; em = 440 nm). DNA concentrations as low as 50 pM can accurately measure protein binding affinity using fluorescence intensity. Even at the lowest DNA concentration, binding is well determined as shown by the consistent dissociation constants obtained, (Kd = 2 ± 0.5nM) (Table 4).
The incorporated 6-MI minimally perturbs DNA structure and protein-DNA interactions even when a portion of the pentamer sequence is contained within the consensus region as in the IHFDNA complex formed with TFA15ds (Table 1). The binding affinity measured was comparable to previously reported values (Table 4).45 Similarly, minimal perturbations were observed when the TFA15ds sequence was used to investigate the HU-DNA interaction, as shown by the Kd value (Table 4). The HU_TFA15ds construct consisted of a single-stranded and duplex portion where the pentamer sequence is located at the junction of the duplex and single strand - the point at which HU putatively binds with nanomolar affinity.37 Even in this altered structural context, enhanced fluorescence is observed and protein binding is measured with high sensitivity (Table 4).39
Table 4.
Equilibrium Protein-DNA Dissociation constants (Kd) measured with 6-MI containing duplexes
| DNA construct | Kd values | Kd values | Kd values | Kd values | Literature Kd |
|---|---|---|---|---|---|
| (nM)1 | (nM)2 | (nM)3 | (nM)4 | Values (nM) | |
| TFA15ds | 1.2 ± 0.8 | 3.7 ± 0.7 | 2.4 ± 0.8 | 1.2 ± 0.4 | 9±15 |
| TFA07ds | 1.5± 1.0 | ||||
| cAFTlOds | 1.1 ± 0.3 | ||||
| MSHTFA15ds | 10.2 ± 3.5 | 28±1.66 | |||
| MSH TFA09ds | 11 ± 1.9 | 13±3 | 28±1.66 | ||
| TBP TFA23ds | 10.6 ± 3.1 | 5±27 | |||
| HUTFA15ds | 4.6 ± 2.2 | 1.5±0.3 | 168 | ||
Measured with constant [DNA] = 2000 pM using fluorescence anisotropy.
Measured with constant [DNA]=1000 pM using fluorescence anisotropy.
Measured with constant [DNA]=500 pM using fluorescence intensity.
Measured with constant [DNA]=50 pM using fluorescence intensity.
From Vitko et al.45
From Zhai and Hingorani.43
From Perez-Howard et al.41
Apparent Kd measured by GMSA.37
For all the protein-DNA interactions investigated, the presence of 6-MI minimally perturbed the investigated binding interactions as shown by the measured Kd values (Table 4), which are either in good agreement with or below previously reported values.37–43 Small discrepancies with reported values are not surprising given that some of the reported values were measured using DNA concentrations higher than the Kd value, which can lead to an elevation of the measured Kd value through non-specific binding. For example, Msh2-Msh6 binds to both pentamer-containing DNA substrates with a slightly higher affinity (Figure 7, Table 4) relative to the previously reported value (10 nM vs. 28 nM).43 This difference in Kd values is attributed to the 25-fold lower concentration used in this study (1 nM vs. 25 nM).
The lowest concentrations used in this study are based on analysis of signal to noise ratios (S/N) for 6-MI fluorescence in dsDNA. The point at which the signal does not track linearly with concentration is estimated to be the limiting usable concentration for experiments (Supporting Information: Figure S9). We compare these measurements with the AFT07ds because of the similarity in overall sequence; although we note that for AFT07ds Φrel = 0.24, which is higher than 6-MI in other sequence contexts6, 16, 20 (Supporting Information: Figure S7). From our S/N analysis, we find that in the presence of polarizers the concentration after which the signal is not linear with concentration is approximately 1nM for ATFAA-containing duplexes, TFA15ds and TFA07ds, and is 5nM for AFT07ds (Supporting Information S9). A comparison of 1 nM TFA15ds with 1 nM AFT07ds in IHF binding experiments demonstrates the benefit of the higher S/N, as binding is not detected with the AFT07ds (Supporting Information S10). Thus, we estimate that a minimum concentration of 1 nM is needed for fluorescence anisotropy measurements when using duplexes containing the ATFAA sequence.
Greater sensitivity in binding experiments was obtained by monitoring fluorescence intensity instead of anisotropy, which allows for the use of sub-nanomolar DNA concentrations (Figure 8, Table 4). Intensity data obtained using 500 pM of DNA substrates TFA15ds, HU_TFA15ds, and MSH_TFA09ds yielded Kd values that are consistent with the anisotropy data (Table 4; Supporting Information: Figure S11). To fully explore the sensitivity of the pentamer sequence, we further decreased the TFA15ds concentration to 50 pM. At this DNA concentration, we detected IHF binding and measured a Kd value of 1.2 ± 0.3 nM, consistent with measurements performed at 500 pM and 1.0 nM DNA (Figure 8, Table 4). We employed the same S/N analysis as described above to estimate the lowest usable concentration for the ATFAA-containing sequences (Supporting Information: Figure S9). Of note, IHF binding to the quenched AFT07ds induced a 130% increase in fluorescence intensity when a 1 nM concentration was used, which is larger than the 30–40% change detected for the ATFAA- or AAFTA-containing sequences (Figure 9). However, IHF binding experiments performed with 50 pM AFT07ds do not detect binding because of the relatively low signal (Supporting Information S12). This finding illustrates that even though the relative intensity change may be smaller for the pentamer-containing sequences the overall sensitivity is better because of the higher signal of the initial point. Thus, the advantages of the 6-MI enhanced fluorescence are notable as it affords the opportunity to examine protein-DNA interactions using low nanomolar to sub-nanomolar concentrations of DNA (Figures 7 and 8). Typically, 2-AP10, 46–50 and 6-MI in other sequence contexts18 are used at nanomolar concentrations and so, the sensitivity afforded by the enhanced 6-MI fluorescence potentially leads to more accurate determinations of high affinity binding interactions at low DNA concentrations.
Figure 9.

The increase in 6-MI fluorescence intensity upon protein binding is a sensitive indicator of structural distortions near the protein-binding site. In the Msh2-Msh6-DNA binding interaction (top), when the probe is located adjacent to the +T insertion loop (■) the intensity change was larger (Msh_TFA15ds, 45%) than when located 10 bp away (Msh_TFA09, 20%). In the IHF-DNA binding interaction (bottom), the intensity increase is greatest when 6-MI is located within the IHF consensus sequence (cAFT10ds, 43%), and is decreased when located adjacent to the consensus sequence (TFA15ds, 35%) or 8bp away (TFA07ds, 26%). The location of 6-MI in the IHF or Msh2-Msh6 duplexes had little to no effect on the Kd values (Table 4).
Influence of ATFAA Sequence Location on Binding Measurements
The enhanced fluorescence is detected regardless of the location of the pentamer sequence in the DNA duplex and thus can be used effectively to investigate protein-DNA interactions. In the case of TATA-Box binding protein (TBP), the ATFAA sequence was located 3' to the TATA recognition sequence (Table 1). The titration of TBP yielded an increase in anisotropy with a Kd consistent with previous measurements demonstrating that the ATFAA sequence reports on TBP binding without interfering with it (Figure 7, Table 4).41 The effect of pentamer location was further investigated with the Msh2-Msh6 and IHF binding interactions. As shown in figure 9, similar Kd values were obtained with different sequences, illustrating that the location of the 6-MI does not influence the measurement. Specifically, the ATFAA sequence was located either directly next to the protein-binding site (Msh_TFA15ds) or 10 base pairs away from the +T insertion loop on the 5' side (Msh_TFA09ds) (Figure 10). In the IHF duplex sequence the ATFAA sequence was placed in two distinct positions: adjacent to the binding site on the 5’ side (TFA15ds), and 8 base pairs away on the 5’ side (TFA07ds) (Figure 1). As noted above, the AAFTA sequence, which is also highly fluorescent, is located on the complementary strand in the region of the consensus sequence (Figure 1, Table 1). For all three duplexes, similar Kd values were obtained despite the difference in position on the duplex (Table 4; Figure 9). These findings demonstrate that the pentamer sequence does not need to be located at the proteinbinding site to accurately measure Kd values confirming the utility and versatility of the pentamer sequence for studying high-affinity protein-DNA interactions by intensity or anisotropy (Supporting Information: Figure S13).
Figure 10.

The X-ray co-crystal structure of Msh2-Msh6 bound to DNA containing a single +Tloop (yellow) with the positions of 6-MI highlighted. The location of 6-MI in the Msh_TFA15 duplex is shown in blue and in the Msh_TFA09 duplex is shown in red. The Phe residue (green) inserts at the +T loop, distorts the DNA backbone and is proximal to the 6-MI in the Msh_TFA15 duplex. Figure was generated with pymol using the coordinates from pdbid, 2O8F.
Detection of DNA Structural Perturbations with 6-MI in Protein-DNA interactions
A distinct advantage of the pentamer sequence relative to external probes is the ability to examine more than just protein binding affinities; 6-MI fluorescence intensity also monitors any perturbations or distortions to the DNA structure. Any protein-induced structural changes in base stacking or base pairing results in an increase in 6-MI fluorescence intensity.14 Specifically, we observe that 6-MI reports on structural perturbations according to location within the duplex and the largest intensity changes are associated with the regions of greatest perturbation. In this study all four of the proteins used distort the DNA to varying extents upon binding as determined by x-ray crystallography and all four protein-DNA complexes exhibit an increase in fluorescence intensity upon protein binding (Figure 9, Supporting Information: Figure S11).44, 51–53
This effect on 6-MI fluorescence is explored in detail with the Msh2-Msh6 and IHF proteins. In the case of Msh2-Msh6 binding to the two DNA constructs, the smallest intensity increase was observed for Msh2-Msh6 binding 10 bp away from the 6-MI (Msh_TFA09ds) (25%), while an increase of 45% is observed if 6-MI is located at the +T insertion loop (Msh_TFA15ds) (Figure 9). The crystal structure of the Msh2-Msh6-DNA complex reveals that perturbation of DNA structure occurs largely at the mismatch binding site, with a reported bend angle of 42° (Figure 10).54 Importantly, the smaller intensity change observed for the Msh_TFA09ds complex suggests that the distortion induced by the protein is substantively weakened 10 bp away from the binding site (Figure 9); however, a shorter DNA construct was used in the crystal structure so a direct correlation with bend angle cannot be made for this site.
Similarly, IHF, which has been shown to bend DNA by >160°, also induces an increase in fluorescence intensity that is dependent on the location of 6-MI relative to the binding site (Figure 9).44 The greatest intensity increase (cAFT10ds; 43%) occurs when 6-MI is located within the consensus sequence on the complementary strand. The intensity increase is reduced when 6-MI is located adjacent to the 5' side of the consensus sequence (TFA15ds; 35%) and an even smaller intensity change is observed, when 6-MI is located 8 bases away from the consensus sequence (TFA07ds; 26%) (Figure 9). As shown by the IHF-DNA crystal structure, protein binding induces large changes to the DNA structure and the 6-MI intensity changes reveal these are greatest in the consensus sequence (Figure 1). These observations demonstrate that 6-MI is a sensitive reporter of local conformation and protein-induced distortion of the DNA helix.
These effects are further explored using time-resolved fluorescence lifetime and anisotropy measurements of 6-MI in the protein-DNA complexes, which provide greater detail of local DNA structural changes that occur upon protein binding. For all of the complexes studied, we observe that the mean lifetime decreases upon protein binding and the magnitude of the change is dependent on location of 6-MI relative to protein binding site. Not surprisingly, the largest changes are associated with those sequences where the probe is located directly in the binding region. For the IHF and Msh2-Msh6 complexes protein binding leads to an increase in dynamic quenching, as reflected by a redistribution of the subpopulations to shorter decay components (Table 2). Dynamic quenching upon IHF binding is the greatest for cAFT10ds, where a shortening of the mean lifetime by 700 ps is observed (Table 2; Supporting Information: Figure S14). The larger subpopulation associated with the shorter decay component is attributed to increased collisional quenching with adjacent bases as a consequence of increased flexibility of 6-MI upon IHF binding. As the distance between 6-MI and the protein binding site increases (Figure 1) the amount of dynamic quenching decreases, where IHF binding to TFA15ds leads to a 200 ps decrease in mean lifetime, while only a 90 ps decrease in mean lifetime is detected for the TFA07ds duplex (Table 2; Supporting Information: Figure S14). The position specific dynamic quenching in the IHF-DNA complex demonstrates the high sensitivity of 6-MI and the ATFAA sequence to local structural perturbations.
In the case of the Msh2-Msh6-DNA complex, when 6-MI is located adjacent to the +T loop - the putative site of protein binding, the greatest change in excited state dynamics is detected. Specifically, the mean lifetime of Msh_TFA15ds decreases by ~500 ps upon protein binding, which contrasts with the Msh_TFA09ds duplex where no appreciable dynamic quenching is observed and the probe is 10 bp away from the protein binding site (Table 2; Supporting Information: Figure S15).
For both protein-DNA complexes, the increase in dynamic quenching and corresponding decrease in mean lifetime observed upon protein binding would suggest that the fluorescence intensity decreases. As noted above, steady state fluorescence measurements show an increase in intensity upon binding (Figure 9). These observations are strongly suggestive that the increase in fluorescence intensity detected in the steady state measurements results from a decrease in static quenching of the 6-MI, as previously suggested by Knutson and co-workers.20 One source of the decrease could be a reduction in the amount of hypochromicity as base stacking interactions are disrupted as a consequence of the protein-induced distortions.
Time-resolved anisotropy measurements are used to further understand the effect of protein binding on the local motion of 6-MI, as reflected in the rotational correlation time. As expected protein binding (either IHF or Msh2-Msh6) increases the correlation time associated with DNA tumbling (θR) by 7–10 ns reflecting the larger size and mass of the protein-DNA complex (Supporting Information: Figures S16 and S17). This increase in correlation time is readily detected in the time-dependent anisotropy decay curves as a shallower slope and is a good indicator of protein binding.
As described above, the shorter correlation time is attributed to local motion of 6-MI, and similar to the lifetime measurements, the largest changes in local motion are detected when the probe is located in the consensus sequence (Figure 1). Binding of IHF to the cAFT10ds leads to increased flexibility of 6-MI as detected by a shorter rotational correlation time of the probe (θL) of ~900ps, which is not seen for the other IHF duplexes (Table 3; Supporting Information: Figure S16). Significantly, in the X-ray crystal structure, salt bridge and van der Waals contacts are observed between Arg 42 and Arg 46 and the DNA backbone of the residues adjacent to 6- MI in the cAFT10 sequence. Additionally, replacement of the adenine next to 6-MI with either 2-AP or diaminopurine reduces binding affinity 10–20 fold,44, 55 indicating that these protein-backbone interactions in the IHF-DNA complex are key to recognition and binding, and probably give rise to the increased mobility observed.
Interestingly, binding of Msh2-Msh6 to the +T-insertion loop results in a longer correlation time for 6-MI local motion (θL) (2.11 ns bound vs. 0.73 ns free) suggesting that protein binding stabilizes the probe when located adjacent to the +T (Table 3: Supporting Information: Figure S17). This decrease in motion is attributed to the stacking of residues Phe 432 and Met 452 with the +T insertion as shown in the x-ray co-crystal structure (Figure 10).56 The 6-MI result further suggests that stabilization of the +T loop, also leads to a stabilization of the surrounding bases. Similar to the IHF duplexes, the effect is spatially dependent as no change in local motion of 6- MI is detected for the Msh_TFA09 duplex, where 6-MI is 10 bases away from the +T loop (Table 3, Supporting Information: Figure S17). Thus, local motion of the probe as detected by time-resolved anisotropy is a sensitive indicator of protein-DNA interactions and can provide unique insight into the nature of the interaction.
Summary
We report on the discovery of two pentamer sequences, ATFAA and AAFTA, which lead to enhanced fluorescence of nucleoside analog 6-MI upon formation of duplex DNA. The ATFAA sequence was incorporated into a number of different DNA duplexes and retained the enhanced fluorescence. This observation of enhanced fluorescence regardless of DNA structure suggests that the ATFAA sequence construct is a good tool for any experiment where high sensitivity and minimal substrate perturbation is desired and we demonstrate that concentrations as low as 50 pM can be used effectively. These sequences were used to measure binding interactions with four distinct DNA-binding proteins. We show that the ATFAA sequence works equally well with sequence-specific and non sequence-specific DNA-binding proteins. Furthermore, time-resolved and steady-state fluorescence measurements demonstrate that 6-MI fluorescence is very sensitive to local distortion and can report on different degrees of distortion at unique sites within one protein-DNA complex. Given the high sensitivity, versatility and overall utility in measuring binding, we anticipate that these sequence constructs, ATFAA and AAFTA, will be broadly used for investigating protein-DNA interactions.
Supplementary Material
Acknowledgments
We thank Manju Hingorani for the Msh2-Msh6 protein and Michael Brenowitz for the TBP protein. We are grateful to Roger McMacken for the HU over-expressing strain, RLM1078 and Steve Goodman for the IHF over-expressing strain. We thank Li Yan for help in acquiring some of the data.
Funding:
This work was supported by the National Science Foundation (MCB-0316625; MCB-0843656 awarded to IM). A NIH training grant in molecular biophysics T32GM08271 supported AM.
Abbreviations
- 6-MI
6-methylisoxanthopterin
- 2AP
2-aminopurine
- 3-MI
3-methylisoxanthopterin
- ds
double stranded
- IRF
instrument response function
- IHF
Integration Host Factor
- Msh
MutS homolog
- ss
single stranded
- TBP
TATA-Box Binding Protein
- TCSPC
time-correlated single photon counting
Footnotes
Supporting Information Available:
Thermal stability of DNA duplexes containing 6-MI (S1), The proposed models for 6-MI in dsDNA (S2), the comparison of TR and SS fluorescence KI quenching of 6-MI monomer (S3), Stern-Volmer plots of KI quenching of dsDNA (S4), Orchid OH radical cleavage intensity plot of dsDNA (S5), the time-dependent anisotropy decay curves of ssDNA and dsDNA (S6), the relative quantum yield of dsDNA used in protein binding experiments (S7), diagram of 4WJ containing ATFAA (S8), Relative sensitivity of different 6-MI containing sequences (S9), Comparison of AFT07ds and TFA15ds measuring IHF binding at 1 nM concentration (S10), Protein binding curves generated with 500 pM DNA (S11), IHF binding curves measured with 50 pM and 1nM AFT07ds (S12), Anisotropy binding curves of IHF to 6-MI-containing duplexes (S13), Time-resolved fluorescence decay curves of IHF-DNA complexes (S14), Time-resolved fluorescence decay curves of Msh2-Msh6-DNA complexes (S15), Time-resolved anisotropy decay curves of IHF-DNA complexes (S16), Time-resolved anisotropy decay curves of Msh2-Msh6-DNA complexes (S17). This material is available free of charge via the Internet at http://pubs.acs.org
References
- 1.Stormo GD, Zhao Y. Determining the specificity of protein–DNA interactions. Nat Rev Genet. 2010;11:751–760. doi: 10.1038/nrg2845. [DOI] [PubMed] [Google Scholar]
- 2.Anderson BJ, Larkin C, Guja K, Schildbach JF. Using Fluorophore- Labeled Oligonucleotides to Measure Affinities of Protein-DNA Interactions. Methods Enzymol. 2008;450:253–272. doi: 10.1016/S0076-6879(08)03412-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lakowicz JR. Principles of Fluorescence Spectroscopy. 2 ed. New York: Kluwer Academic; 1999. [Google Scholar]
- 4.Wilhelmsson LM. Fluorescent nucleic acid base analogues. Q. Rev. Biophys. 2010;43:159–183. doi: 10.1017/S0033583510000090. [DOI] [PubMed] [Google Scholar]
- 5.Royer CA, Scarlata SF. Fluorescence Approaches to Quantifying Biomolecular Interactions. Methods Enzymol. 2008;450:79–106. doi: 10.1016/S0076-6879(08)03405-8. [DOI] [PubMed] [Google Scholar]
- 6.Hawkins ME. Fluorescent Pteridine Probes for Nucleic Acid Analysis. Methods Enzymol. 2008;450:201–231. doi: 10.1016/S0076-6879(08)03410-1. [DOI] [PubMed] [Google Scholar]
- 7.Zang H, Fang Q, Pegg AE, Guengerich FP. Kinetic Analysis of Steps in the Repair of Damaged DNA by Human O6-Alkylguanine-DNA Alkyltransferase. J. Biol. Chem. 2005;280:30873–30881. doi: 10.1074/jbc.M505283200. [DOI] [PubMed] [Google Scholar]
- 8.Berry DA, Jung K-Y, Wise DS, Sercel AD, Pearson WH, Mackie H, Randolph JB, Somers RL. Pyrrolo-dC and pyrrolo-C: fluorescent analogs of cytidine and 2'-deoxycytidine for the study of oligonucleotides. Tetrahedron Lett. 2004;45:2457–2461. [Google Scholar]
- 9.Magde D, Wong R, Seybold PG. Fluorescence Quantum Yields and Their Relation to Lifetimes of Rhodamine 6G and Fluorescein in Nine Solvents: Improved Absolute Standards for Quantum Yields. Photochem. Photobiol. 2002;75:327–334. doi: 10.1562/0031-8655(2002)075<0327:fqyatr>2.0.co;2. [DOI] [PubMed] [Google Scholar]
- 10.Lemay J-F, Penedo JC, Tremblay R, Lilley DMJ, Lafontaine Daniel A. Folding of the Adenine Riboswitch. Chem. Biol. 2006;13:857–868. doi: 10.1016/j.chembiol.2006.06.010. [DOI] [PubMed] [Google Scholar]
- 11.Singleton SF, Roca AI, Lee AM, Xiao J. Probing the structure of RecA-DNA filaments. Advantages of a fluorescent guanine analog. Tetrahedron. 2007;63:3553–3566. doi: 10.1016/j.tet.2006.10.092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wojtuszewski K, Hawkins ME, Cole JL, Mukerji I. HU Binding to DNA: Evidence for Multiple Complex Formation and DNA Bending. Biochemistry. 2001;40:2588–2598. doi: 10.1021/bi002382r. [DOI] [PubMed] [Google Scholar]
- 13.Mukherjee S, Feig M. Conformational Change in MSH2-MSH6 upon Binding DNA Coupled to ATPase Activity. Biophys. J. 2009;96:L63–L65. doi: 10.1016/j.bpj.2009.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hawkins ME, Pfieiderer W, Mazumder A, Pommier YG, Balis FM. Incorporation of a fluorescent guanosine analog into olignoucleotides and its application to a real time assay for the HIV-1 integrase 3-Processing reaction. Nucl. Acids Res. 1995;23:2872–2880. doi: 10.1093/nar/23.15.2872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yang K, Stanley RJ. The Extent of DNA Deformation in DNA Photolyase– Substrate Complexes: A Solution State Fluorescence Study. Photochem. Photobiol. 2008;84:741–749. doi: 10.1111/j.1751-1097.2007.00251.x. [DOI] [PubMed] [Google Scholar]
- 16.Hawkins ME, Pfleiderer W, Balis FM, Porter D, Knutson JR. Fluorescence Properties of Pteridine Nucleoside Analogs as Monomers and Incorporated into Oligonucleotides. Anal. Biochem. 1997;244:86–95. doi: 10.1006/abio.1996.9879. [DOI] [PubMed] [Google Scholar]
- 17.Joyce CM, Potapova O, DeLucia AM, Huang X, Basu VP, Grindley NDF. Fingers-Closing and Other Rapid Conformational Changes in DNA Polymerase I (Klenow Fragment) and Their Role in Nucleotide Selectivity. Biochemistry. 2008;47:6103–6116. doi: 10.1021/bi7021848. [DOI] [PubMed] [Google Scholar]
- 18.Myers JC, Moore SA, Shamoo Y. Structure-based Incorporation of 6- Methyl-8-(2-deoxy-Î2-ribofuranosyl)isoxanthopteridine into the Human Telomeric Repeat DNA as a Probe for UP1 Binding and Destabilization of G-tetrad Structures. J. Biol. Chem. 2003;278:42300–42306. doi: 10.1074/jbc.M306147200. [DOI] [PubMed] [Google Scholar]
- 19.Shi X, Mollova ET, PljevaljcÌŒicÌ G, Millar DP, Herschlag D. Probing the Dynamics of the P1 Helix within the Tetrahymena Group I Intron. J. Am. Chem. Soc. 2009;131:9571–9578. doi: 10.1021/ja902797j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wojtuszewski Poulin K, Smirnov AV, Hawkins ME, Balis FM, Knutson JR. Conformational Heterogeneity and Quasi-Static Self-Quenching in DNA Containing a Fluorescent Guanine Analogue, 3MI or 6MI. Biochemistry. 2009;48:8861–8868. doi: 10.1021/bi9003414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Evans K, Xu D, Kim Y, Nordlund TM. 2-Aminopurine optical spectra: Solvent, pentose ring, and DNA helix melting dependence. J. Fluoresc. 1992;2:209–216. doi: 10.1007/BF00865278. [DOI] [PubMed] [Google Scholar]
- 22.Xu D, Evans KO, Nordlund TM. Melting and Premelting Transitions of an Oligomer Measured by DNA Base Fluorescence and Absorption. Biochemistry. 1994;33:9592–9599. doi: 10.1021/bi00198a027. [DOI] [PubMed] [Google Scholar]
- 23.Beechem JM, Gratton E. Fluorescence Spectroscopy Data Analysis Enviroment A Second Generation Global Analysis Program Vol. 909. Bellingham, WA: Society of Photo Optical; 1988. [Google Scholar]
- 24.Augustyn KE, Wojtuszewski K, Hawkins ME, Knutson JR, Mukerji I. Examination of the premelting transition of DNA A-tracts using a fluorescent adenosine analogue. Biochemistry. 2006;45:5039–5047. doi: 10.1021/bi0518343. [DOI] [PubMed] [Google Scholar]
- 25.Hawkins M. Fluorescent pteridine nucleoside analogs. Cell Biochem. Biophys. 2001;34:257–281. doi: 10.1385/CBB:34:2:257. [DOI] [PubMed] [Google Scholar]
- 26.Driscoll SL, Hawkins ME, Balis FM, Pfleiderer W, Laws WR. Fluorescence properties of a new guanosine analog incorporated into small oligonucleotides. Biophys. J. 1997;73:3277–3286. doi: 10.1016/S0006-3495(97)78352-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hawkins ME, Pfleiderer W, Jungmann O, Balis FM. Synthesis and Fluorescence Characterization of Pteridine Adenosine Nucleoside Analogs for DNA Incorporation. Anal. Biochem. 2001;298:231–240. doi: 10.1006/abio.2001.5399. [DOI] [PubMed] [Google Scholar]
- 28.Rai P, Cole TD, Thompson E, Millar DP, Linn S. Steady-state and time-resolved fluorescence studies indicate an unusual conformation of 2-aminopurine within ATAT and TATA duplex DNA sequences. Nucl. Acids Res. 2003;31:2323–2332. doi: 10.1093/nar/gkg339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Xu D-G, Nordlund TM. Sequence Dependence of Energy Transfer in DNA Oligonucleotides. Biophys. J. 2000;78:1042–1058. doi: 10.1016/S0006-3495(00)76663-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kalnik MW, Norman DG, Li BF, Swann PF, Patel DJ. Conformational transitions in thymidine bulge-containing deoxytridecanucleotide duplexes. Role of flanking sequence and temperature in modulating the equilibrium between looped out and stacked thymidine bulge states. J. Biol. Chem. 1990;265:636–647. [PubMed] [Google Scholar]
- 31.Coppel Y, Berthet N, Coulombeau C, Coulombeau C, Garcia J, Lhomme J. Solution Conformation of an Abasic DNA Undecamer Duplex d(CGCACXCACGC)·d(GCGTGTGTGCG): The Unpaired Thymine Stacks Inside the Helix. Biochemistry. 1997;36:4817–4830. doi: 10.1021/bi962677y. [DOI] [PubMed] [Google Scholar]
- 32.Guest CR, Hochstrasser RA, Sowers LC, Millar DP. Dynamics of mismatched base pairs in DNA. Biochemistry. 1991;30:3271–3279. doi: 10.1021/bi00227a015. [DOI] [PubMed] [Google Scholar]
- 33.Hall KB, Williams DJ. Dynamics of the IRE RNA hairpin loop probed by 2-aminopurine fluorescence and stochastic dynamics simulations. RNA. 2004;10:34–47. doi: 10.1261/rna.5133404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Nordlund TM, Andersson S, Nilsson L, Rigler R, Graeslund A, McLaughlin LW. Structure and dynamics of a fluorescent DNA oligomer containing the EcoRI recognition sequence: fluorescence, molecular dynamics, and NMR studies. Biochemistry. 1989;28:9095–9103. doi: 10.1021/bi00449a021. [DOI] [PubMed] [Google Scholar]
- 35.Somsen OJG, Keukens LB, de Keijzer MN, van Hoek A, van Amerongen H. Structural Heterogeneity in DNA: Temperature Dependence of 2-Aminopurine Fluorescence in Dinucleotides. ChemPhysChem. 2005;6:1622–1627. doi: 10.1002/cphc.200400648. [DOI] [PubMed] [Google Scholar]
- 36.Wu P, Nordlund TM, Gildea B, McLaughlin LW. Base stacking and unstacking as determined from a DNA decamer containing a fluorescent base. Biochemistry. 1990;29:6508–6514. doi: 10.1021/bi00479a024. [DOI] [PubMed] [Google Scholar]
- 37.Kamashev D, Rouviere-Yaniv J. The histone-like protein HU binds specifically to DNA recombination and repair intermediates. EMBO J. 2000;19:6527–6535. doi: 10.1093/emboj/19.23.6527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kuddus R, Schmidt MC. Effect of the non-conserved N-terminus on the DNA binding activity of the yeast TATA binding protein. Nucl. Acids Res. 1993;21:1789–1796. doi: 10.1093/nar/21.8.1789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lorenz M, Hillisch A, Goodman SD, Diekmann S. Global Structure similarities of intact and nicked DNA complexed with IHF measured in solution by fluorescence resonance energy transfer. Nucl. Acids Res. 1999;27:4619–4625. doi: 10.1093/nar/27.23.4619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Marsischky GT, Kolodner RD. Biochemical Characterization of the Interaction between theSaccharomyces cerevisiae MSH2-MSH6 Complex and Mispaired Bases in DNA. J. Biol. Chem. 1999;274:26668–26682. doi: 10.1074/jbc.274.38.26668. [DOI] [PubMed] [Google Scholar]
- 41.Perez-Howard GM, Weil PA, Beechem JM. Yeast TATA binding protein interaction with DNA: fluorescence determination of oligomeric state, equilibrium binding, on-rate, and dissociation kinetics. Biochemistry. 1995;34:8005–8017. doi: 10.1021/bi00025a006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Swinger KK, Rice PA. IHF and HU: flexible architects of bent DNA. Curr. Opin. Struct. Biol. 2004;14:28–35. doi: 10.1016/j.sbi.2003.12.003. [DOI] [PubMed] [Google Scholar]
- 43.Zhai J, Hingorani MM. Saccharomyces cerevisiae Msh2-Msh6 DNA binding kinetics reveal a mechanism of targeting sites for DNA mismatch repair. Proc. Natl Acad. Sci. 107:680–685. doi: 10.1073/pnas.0908302107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Rice PA, Yang S-w, Mizuuchi K, Nash HA. Crystal Structure of an IHF-DNA Complex: A Protein-Induced DNA U-Turn. Cell. 1996;87:1295–1306. doi: 10.1016/s0092-8674(00)81824-3. [DOI] [PubMed] [Google Scholar]
- 45.Vitko J, Rujan I, Androga L, Mukerji I, Bolton PH. Molecular Beacon-Equilibrium Cyclization Detection of DNA-Protein Complexes. Biophys. J. 2007;93:3210–3217. doi: 10.1529/biophysj.106.097642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Allan BW, Reich NO. Targeted Base Stacking Disruption by the EcoRI DNA Methyltransferase. Biochemistry. 1996;35:14757–14762. doi: 10.1021/bi9615708. [DOI] [PubMed] [Google Scholar]
- 47.Gilbert SD, Stoddard CD, Wise SJ, Batey RT. Thermodynamic and Kinetic Characterization of Ligand Binding to the Purine Riboswitch Aptamer Domain. J. Mol. Biol. 2006;359:754–768. doi: 10.1016/j.jmb.2006.04.003. [DOI] [PubMed] [Google Scholar]
- 48.Holz B, Weinhold E, Klimasauskas S, Serva S. 2-Aminopurine as a fluorescent probe for DNA base flipping by methyltransferases. Nucl. Acids Res. 1998;26:1076–1083. doi: 10.1093/nar/26.4.1076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zhang H, Cao W, Zakharova E, Konigsberg W, De La Cruz EM. Fluorescence of 2-aminopurine reveals rapid conformational changes in the RB69 DNA polymerase-primer/template complexes upon binding and incorporation of matched deoxynucleoside triphosphates. Nucl. Acids Res. 2007;35:6052–6062. doi: 10.1093/nar/gkm587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bandwar RP, Patel SS. Peculiar 2-Aminopurine Fluorescence Monitors the Dynamics of Open Complex Formation by Bacteriophage T7 RNA Polymerase. J. Biol.Chem. 2001;276:14075–14082. doi: 10.1074/jbc.M011289200. [DOI] [PubMed] [Google Scholar]
- 51.Larsen OFA, van Stokkum IHM, Gobets B, van Grondelle R, van Amerongen H. Probing the Structure and Dynamics of a DNA Hairpin by Ultrafast Quenching and Fluorescence Depolarization. Biophys. J. 2001;81:1115–1126. doi: 10.1016/S0006-3495(01)75768-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hieb AR, Halsey WA, Betterton MD, Perkins TT, Kugel JF, Goodrich JA. TFIIA Changes the Conformation of the DNA in TBP/TATA Complexes and Increases their Kinetic Stability. J. Mol. Biol. 2007;372:619–632. doi: 10.1016/j.jmb.2007.06.061. [DOI] [PubMed] [Google Scholar]
- 53.Swinger KK, Lemberg KM, Zhang Y, Rice PA. Flexible DNA bending in HU-DNA cocrystal structures. EMBO J. 2003;22:3749–3760. doi: 10.1093/emboj/cdg351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Warren JJ, Pohlhaus TJ, Changela A, Iyer RR, Modrich PL, Beese Lorena S. Structure of the Human MutS[alpha] DNA Lesion Recognition Complex. Mol Cell. 2007;26:579–592. doi: 10.1016/j.molcel.2007.04.018. [DOI] [PubMed] [Google Scholar]
- 55.Wang S, Cosstick R, Gardner JF, Gumport RI. The specific binding of Escherichia coli integration host factor involves both major and minor grooves of DNA. Biochemistry. 1995;34:13082–13090. doi: 10.1021/bi00040a020. [DOI] [PubMed] [Google Scholar]
- 56.Tonelli M, Ragg E, Bianucci AM, Lesiak K, James TL. Nuclear Magnetic Resonance Structure of d(GCATATGATAG)·d(CTATCATATGC): A Consensus Sequence for Promoters Recognized by σK RNA Polymerase. Biochemistry. 1998;37:11745–11761. doi: 10.1021/bi980481n. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.