

Summary
Both mitochondria, which are metabolic powerhouses, and telomeres, which help maintain genomic stability, have been implicated in cancer and aging. However, the signaling events that connect these two cellular structures remain poorly understood. Here we report that the canonical telomeric protein TIN2 is also a regulator of metabolism. TIN2 is recruited to telomeres and associates with multiple telomere regulators including TPP1. TPP1 interacts with TIN2 N-terminus, which contains overlapping mitochondrial and telomeric targeting sequences, and controls TIN2 localization. We have found that TIN2 is post-translationally processed in mitochondria, and regulates mitochondria oxidative phosphorylation. Reducing TIN2 expression by RNAi knockdown inhibited glycolysis and reactive oxygen species (ROS) and production, and enhanced ATP levels and oxygen consumption in cancer cells. These results suggest a link between telomeric proteins and metabolic control, providing an additional mechanism by which telomeric proteins regulate cancer and aging.
Introduction
Telosome/shelterin, a six-protein complex consisting of TRF1, TRF2, TPP1, RAP1, TIN2, and POT1, has been shown to associate with mammalian telomeres and regulate telomere function (de Lange, 2005; Liu et al., 2004a). For example, TPP1 binds to POT1 to recruit and stimulate telomerase activity (Abreu et al., 2010; Tejera et al., 2010; Wang et al., 2007; Xin et al., 2007). And TIN2 bridges double- and single-stranded DNA binding activities by interacting with TRF1, TRF2, and TPP1 (Hockemeyer et al., 2007; Houghtaling et al., 2004; Kim et al., 2004; Kim et al., 1999; Liu et al., 2004a; Liu et al., 2004b; Santos et al., 2004; Ye et al., 2004). Mutations in TIN2 have been linked to diseases with premature aging phenotypes and bone marrow failures such as dyskeratosis congenita, ataxia-pancytopenia, and aplastic anemia (Du et al., 2009; Sasa et al., 2011; Savage et al., 2008; Tsangaris et al., 2008; Walne et al., 2008; Yang et al., 2011a).
Recent evidence has suggested extra-telomeric function for several core telomere proteins, such as TRF2, TRF1, and RAP1 (Mao et al., 2007; Martinez et al., 2010; Simonet et al., 2011; Yang et al., 2011b; Zhang et al., 2011; Zhang et al., 2008). In particular, the human telomerase reverse transcriptase (hTERT) was shown to also localize to mitochondria (Santos et al., 2004). In hTERT-overexpressing cells, increased mitochondrial membrane potential was accompanied by decreased superoxide production and peroxide levels (Ahmed et al., 2008). Moreover, telomerase deficiency in mice appeared to repress master mitochondrial transcription regulators such as PGCs and alter metabolic pathways (Sahin et al., 2011). The links between telomere and mitochondrial function signal the existence of additional players with dual roles in regulating genome integrity and cellular metabolism.
Previously, we found TIN2, TPP1, and POT1 to localize and interact in both the nucleus and cytoplasm (Chen et al., 2007; Liu et al., 2004b). TIN2-TPP1 interaction helps to promote TPP1 nuclear targeting (Chen et al., 2007). Here we report findings that TIN2 can in fact localize in mitochondria, a process also regulated by TIN2-TPP1 interaction. Expression of TIN2 proteins that were predominantly mitochondria-targeted led to altered mitochondrial structure. In addition, RNAi-mediated TIN2 depletion led to enhanced mitochondrial oxidative phosphorylation and reduced aerobic glycolysis in cancer cells. These results reveal pathways that TIN2 utilizes to modulate metabolism in human cells, and highlight the molecular links between telomere maintenance and metabolism.
Results
TIN2 can localize to mitochondria
TIN2 nuclear localization and telomeric function has long been established (Houghtaling et al., 2004; Kim et al., 1999; Liu et al., 2004a; Ye et al., 2004). Using a polyclonal antibody against the C-terminus of TIN2 (a.a. 197–354)(Chen et al., 2007), we found TIN2 to co-localize with TRF2 in the nucleus as previously reported (Liu et al., 2004a; Ye et al., 2004)(Figure 1A). Interestingly, we also observed tubular structures in the cytoplasm with anti-TIN2 staining in many of the cells. This cytoplasmic staining pattern overlapped with that of the mitochondrial maker ATP5A1 (Figure 1A), suggesting that TIN2 may be targeted to mitochondria as well. To confirm this finding, we performed immunogold labeling of endogenous TIN2 in HT1080 cells using anti-TIN2 antibodies. As expected, transmission electron microscopy (TEM) revealed the presence of gold particles in the cytoplasm and nucleus (data not shown). Furthermore, we could observe gold particles in the mitochondria where they appeared to cluster close to the mitochondrial membrane (Figure 1B). This immunogold particle distribution was greatly reduced in cells expressing TIN2 shRNAs (Figures 1B and S1). Similar results were also obtained using U2OS cells (Figures 1C and S1), supporting mitochondrial localization of human TIN2.
Figure 1. TIN2 localizes to both the nucleus and mitochondria.
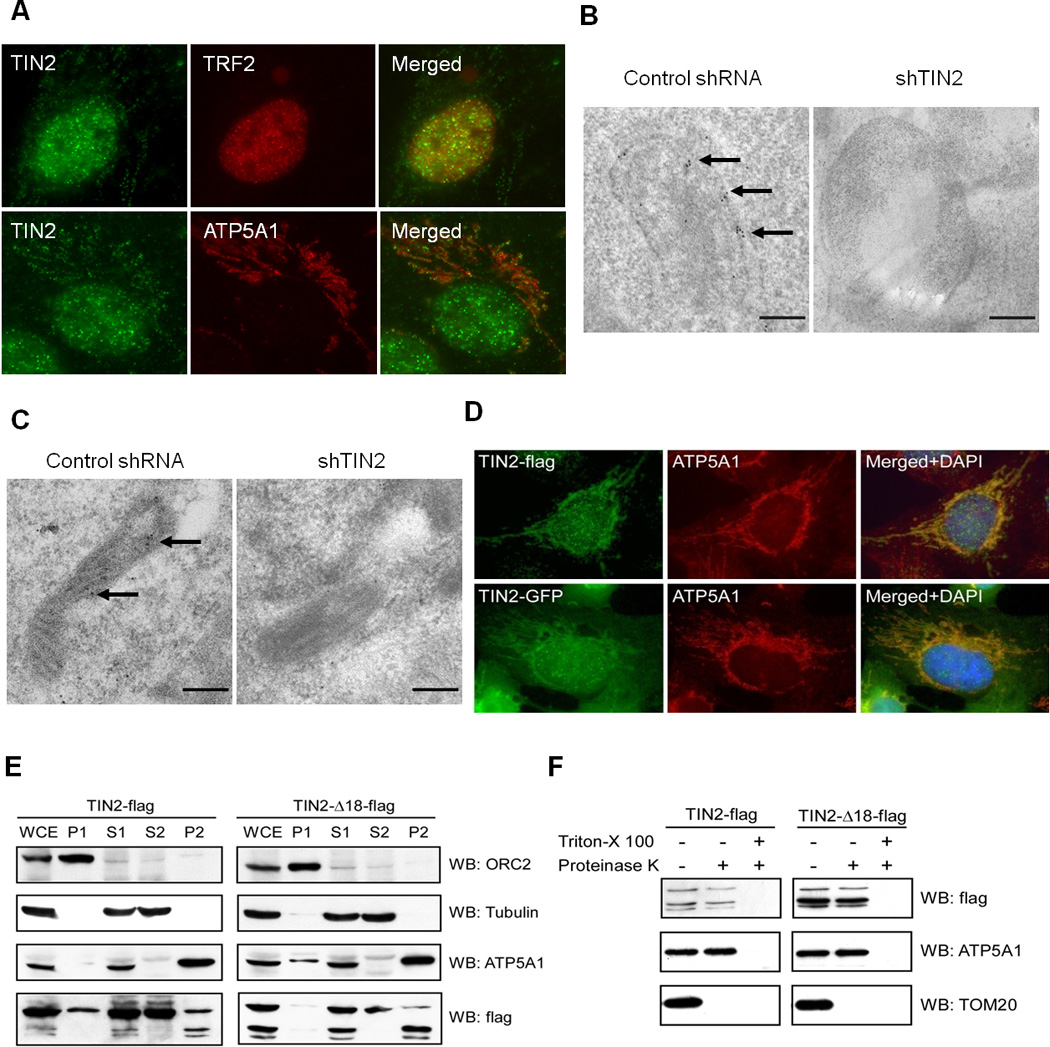
A) HTC75 cells were immunostained with anti-TIN2 antibodies. Telomeres and mitochondria were marked with anti-TRF2 and ATP5A1 antibodies, and nuclei stained with DAPI. Immunogold analysis in HT1080 (B) and U2OS (C) cells expressing control or TIN2-targeting shRNA. Cells were probed with anti-TIN2 antibodies. Arrows indicate gold particles. Scale bars: 200 nm. D) Immunostaining of HTC75 cells expressing C-terminally tagged TIN2 proteins. TIN2-flag was probed with anti-flag antibodies. E) HTC75 cells expressing flag-tagged full-length (TIN2-flag) or mutant TIN2 (TIN2-Δ18-flag) were fractionated by differential centrifugation. Whole-cell extract (WCE) and cellular fractions (nuclear P1, cytoplasmic S1, soluble cytoplasmic/light membrane S2, and heavy membrane P2) were analyzed. ORC2, tubulin and ATP5A1 were used as nuclear, cytoplasmic and mitochondrial markers. F) P2 fractions from (E) were digested with proteinase K in the presence or absence of detergent before western analysis.
In our earlier studies, N-terminally tagged TIN2 (with GFP or Myc) appeared to localize to the telomeres, nucleus, and cytoplasm, but not the mitochondria (Chen et al., 2007). When we tagged TIN2 at its C-terminus, these proteins (TIN2-flag and TIN2-GFP) exhibited both punctate (in the nucleus) and tubular (in the cytoplasm) immunostaining patterns (Figure 1D). The nuclear signals overlapped with TRF2 (data not shown), indicating telomeric localization. The cytoplasmic tubular signals were superimposable with those of ATP5A1, consistent with mitochondrial targeting of TIN2. Again, we found no tubular distribution of anti-TIN2 signals in cells expressing TIN2 tagged with GFP or Flag on the N-terminus (data not shown), suggesting blocked TIN2 mitochondrial localization by N-terminal tagging.
To further investigate TIN2 subcellular distribution, we performed cell fractionation experiments using HTC75 cells stably expressing TIN2-flag. Of the nuclear (P1), total cytoplasmic (S1), soluble cytoplasmic (S2), and heavy membrane (P2) fractions, fraction P2 was enriched for mitochondria. As shown in Figure 1E, successful fractionation was indicated by the relative enrichment of the respective proteins in various fractions. Consistent with our immunostaining results, full-length TIN2 proteins could be detected in both the nuclear (P1) and cytoplasmic (S1 and S2) fractions (Figure 1E, left panel). Importantly, a small amount of full-length TIN2 (as well as two fast-migrating TIN2 products) co-fractionated with ATP5A1 in fraction P2, adding further support to mitochondrial localization of TIN2.
Mitochondrial proteins such as ATP5A1 are targeted to the inner compartments of the mitochondria and resistant to proteolysis in the absence of detergent, whereas proteins such as TOM20 are localized to the mitochondria surface and sensitive to protease digestion even without detergent (Figure 1F). When we treated fraction P2 of TIN2-flag cells with proteinase K, TIN2 appeared to be degradable only in the presence of detergent (Figure 1F), indicative of its residing inside the mitochondria and consistent with our TEM experiment (Figure 1B). Taken together, our data suggest that TIN2 localizes to multiple cellular compartments including the telomeres and the mitochondria.
TIN2 N-terminal signal sequences mediate its mitochondrial localization
The N-termini of mitochondrial proteins often contain targeting sequences (Chacinska et al., 2009). The failure of N-terminally tagged TIN2 to localize to the mitochondria underscores the importance of its N-terminus in mitochondria targeting. To further explore this aspect, we generated GFP-tagged TIN2 proteins with serial N-terminal truncations. As shown in Figure 2A, removal of the first 90 residues led to a complete loss of mitochondrial GFP signals, demonstrating that this region of TIN2 is required for its mitochondrial localization. Indeed, the N-terminal 89 amino acids were sufficient to target GFP to the mitochondria (Figure 2B), indicating that the N-terminus of TIN2 contains bona fide mitochondrial targeting sequences. In fact, deletion analysis revealed the presence of two mitochondria targeting peptides, residues 1–58 and 59–89 (Figure 2A and 2B). Our results thus far indicate that the N-terminus of TIN2 is both necessary and sufficient to target TIN2 to mitochondria.
Figure 2. TIN2 N-terminus contains mitochondrial targeting signals.
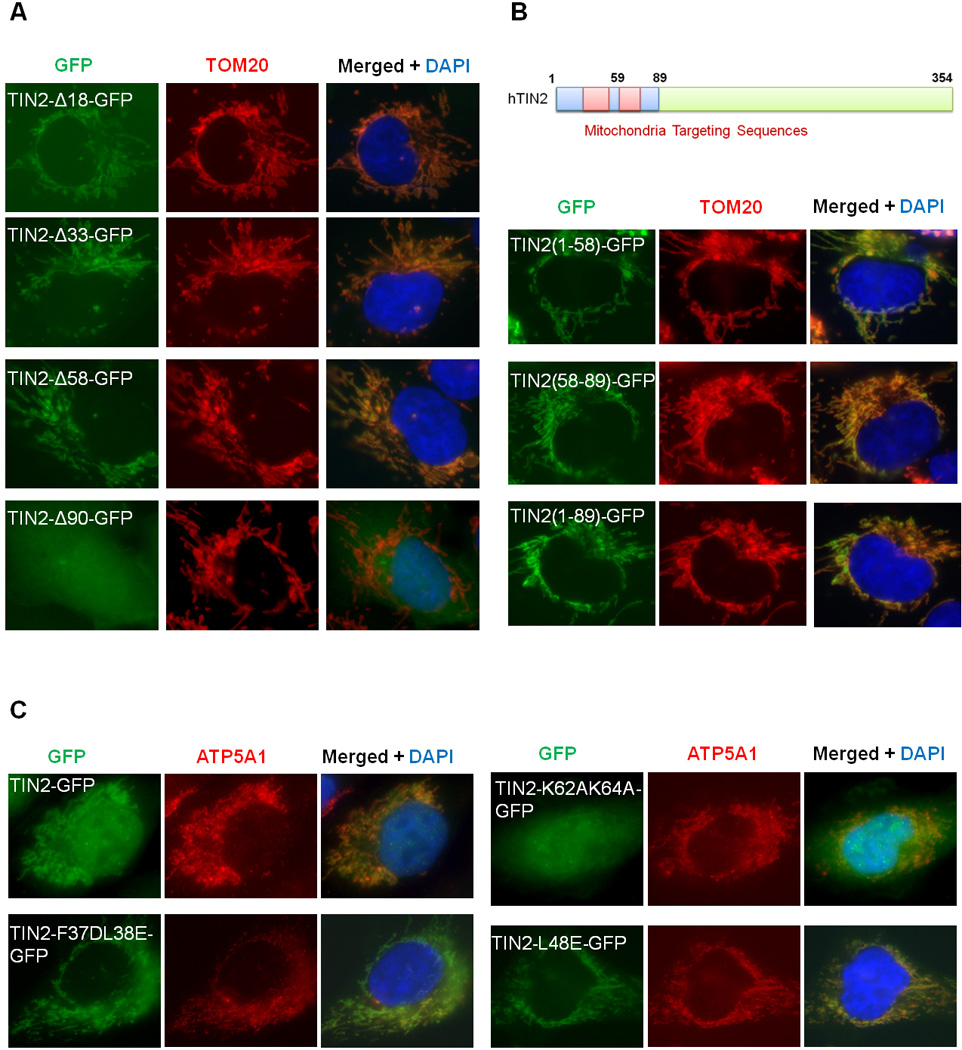
Immunostaining studies were performed in HTC75 cells expressing GFP-tagged wildtype and N-terminal truncation mutants of TIN2 (A), GFP proteins tagged with TIN2 N-terminal fragments (B), or GFP-tagged wildtype and point mutants of TIN2 (C). Anti-TOM20 or ATP5A1 antibodies were used to mark the mitochondria.
N-terminal mitochondrial targeting sequences frequently adopt the conformation of amphipathic helices, where hydrophobic and positively charged resides are clustered on either side (Chacinska et al., 2009; Moberg et al., 2004; Saitoh et al., 2007; Yamano et al., 2008). Secondary structure prediction and helical wheel analysis of residues 1–90 of TIN2, which are enriched with hydrophobic and positively charged amino acids, point to two potential amphipathic helices spanning residues 31–76. To determine whether these amphipathic helices mediate mitochondrial targeting of TIN2, we generated a series of full-length TIN2-GFP constructs with point mutations in the region of residues 31–89, where positively charged residues were replaced with alanine and conserved hydrophobic residues with glutamate or aspartate (Table S1). Interestingly, mutations of K62 and K64 (TIN2-K62A/K64A-GFP) decreased mitochondrial localization of TIN2 (Figure 2C, Table S1). In contrast, the TIN2-F37D/L38E and TIN2-L48E mutants exhibited increased GFP signals in the mitochondria and a concurrent loss of nuclear and telomeric signals (Figure 2C, Table S1). Together, these results demonstrate that TIN2 mitochondrial and nuclear localization is mediated by distinct and separable targeting sequences at its N-terminus.
TIN2 is post-translationally processed in the mitochondria
N-terminal targeting sequences of mitochondrial proteins can be proteolytically removed following mitochondrial import (Chacinska et al., 2009; Saitoh et al., 2007; Taylor et al., 2001). Neither of the two TIN2 lower molecular weight forms (~30 kDa) in mitochondria enriched fractions (Figures 1E and 1F) was affected by the mutation of Met59 (a potential translation start site)(data not shown), indicating that they were not generated by alternative internal translation initiation. We therefore postulated that these smaller TIN2 forms might be products of proteolytic cleavage.
To test this idea and rule out the possibility that overexpression alone accounted for mitochondria targeting and processing of TIN2, we established HT1080 cells expressing TIN2 shRNA sequences as well as SFB-tagged TIN2 that was under the control of a tetracycline-inducible promoter. Addition of doxycycline (10ng/ml) led to TIN2-SFB expression at a level similar to endogenous TIN2 (Figure 3A). Under these conditions, the two fast-migrating forms of TIN2-SFB remained apparent in the mitochondria fraction (Figure 3B), indicating that mitochondria targeted TIN2 is processed into two shorter fragments. In addition, we were able to detect a small percentage of endogenous TIN2 that co-fractionated with the mitochondria (Figure 3C). Importantly, the levels of both full-length and shorter forms of TIN2 were reduced with TIN2 knockdown, further supporting the notion that endogenous TIN2 is processed in the mitochondria.
Figure 3. TIN2 is proteolytically processed in the mitochondria.
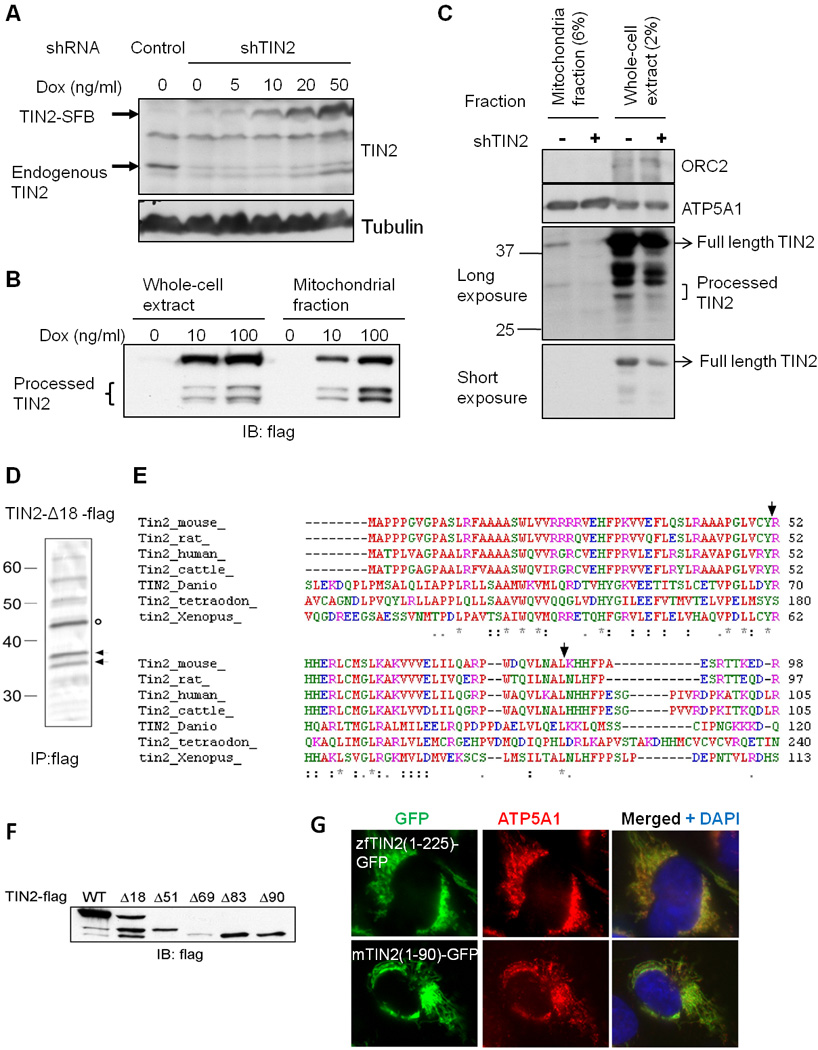
A) Induction of TIN2 expression in HT1080 cells co-expressing TIN2 shRNA (shTIN2) and doxycycline (Dox)-inducible SFB-tagged wildtype TIN2. Cells expressing shRNA sequences against GFP served as control. B) Whole cell extracts and mitochondrial fractions from cells in (A) were western blotted following doxycycline induction (48hr). C) Whole cell extract and mitochondria fractions from U2OS cells expressing control or TIN2 shRNA were were western blotted. D) HTC75 cells expressing TIN2-Δ18-flag (indicated by circle) were immunoprecipitated with anti-flag antibodies. Arrows indicate TIN2 processed forms that were excised for sequencing. E) Sequence alignment of TIN2. Arrows indicate human TIN2 processing sites. F) Western analysis of HEK-293 cells transiently expressing flag-tagged wildtype and N-terminal deletion mutants of TIN2. G) Immunostaining of GFP proteins tagged with the N-terminus of zebrafish (a.a.1-225) or mouse (a.a. 1-90) TIN2 in HTC75 cells.
Next, we decided to take advantage of the fact that deletion of the first 18 amino acids resulted in higher levels of the two smaller TIN2 fragments in fraction P2 (Figures 1E and 1F), and purified TIN2 fragments from TIN2-Δ18-flag expressing HTC75 cells (Figure 3D) for sequencing by Edman degradation. N-terminal protein sequencing indicated that cleavage of mitochondrial TIN2 occurred immediately after residues Y51 and L83 (Figure 3E). When truncation mutants TIN2-Δ51-flag and TIN2-Δ83-flag were expressed in cells, they migrated to positions similar to the two fragments in wildtype TIN2 expressing cells (Figure 3F). Residues Y51 and L83 are highly conserved among vertebrates (Figure 3E), raising the possibility that TIN2 orthologues may also localize to the mitochondria. Indeed, the N-terminal regions of mouse and zebrafish TIN2 were capable of targeting GFP to the mitochondria as well (Figure 3G), indicating that mitochondrial targeting and processing of TIN2 may be conserved through evolution.
TPP1-TIN2 interaction controls TIN2 nuclear vs. mitochondria localization
The N-terminal 196 residues of TIN2 encompass the mitochondrial targeting sequences and are required for its interaction with TPP1 in both the cytoplasm and nucleus (Chen et al., 2007; Liu et al., 2004b). Disruption of the region reduced TIN2 nuclear/telomeric localization while promoting its cytoplasmic/mitochondrial targeting (Figures 2A and 2C). These observations emphasize the importance of TPP1-TIN2 interaction in determining TIN2 nuclear localization. To test the notion that disrupting TPP1-TIN2 interaction would block nuclear localization of TIN2, we carried out co-immunoprecipitation experiments using HEK-293T cells co-expressing flag-TPP1 and GFP-tagged TIN2 deletion mutants. Indeed, an intact N-terminus appeared essential for TIN2-TPP1 interaction, as deleting just the first 18 amino acids was sufficient to abolish TIN2-TPP1 interaction (Figure 4A) and prevent nuclear localization of TIN2 (Figure 2A).
Figure 4. The N-terminus of TIN2 mediates TIN2-TPP1 interaction.
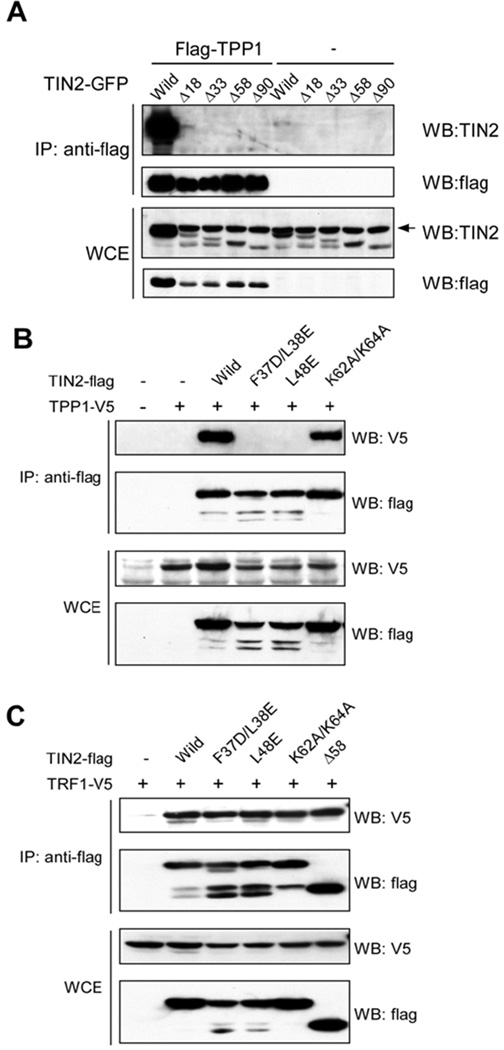
A) Wildtype (Wild) and mutant TIN2-GFP proteins were transiently expressed with or without flag-TPP1 in HEK-293 cells, immunoprecipitated (IP) with anti-flag antibodies, and western blotted as indicated. Arrow indicates a non-specific band. B) Similar analysis of flag-tagged wildtype (Wild) and point mutants of TIN2 proteins in HEK-293 cells transiently co-expressing TPP1-V5. C) Analysis of cells co-expressing TRF1-V5 with flag-tagged wildtype (Wild) or mutant TIN2 proteins.
The point mutants TIN2-F37D/L38E and TIN2-L48E localized predominantly to the mitochondria (Figure 3A). We therefore asked whether such changes occurred as a result of disrupted TIN2-TPP1 interaction by examining TIN2-flag (wildtype or mutant) interactions with V5-tagged TPP1 and TRF1 in HEK-239T cells. TPP1 could only co-immunoprecipitate with wildtype TIN2 and TIN2-K62A/K64A, both of which maintained telomeric localization. In contrast, TIN2-F37D/L38E and TIN2-L48E failed to bring down TPP1 (Figure 4B). All TIN2 proteins examined were expressed at comparable levels, and could interact with TRF1 (Figure 4C). Collectively, these results demonstrate that TIN2-TPP1 interaction mediates nuclear and telomeric targeting of TIN2.
The findings that TIN2 mutants with compromised abilities to interact with TPP1 showed increased mitochondrial localization (Figures 2A, 2C, 4A and 4B), coupled with the existence of overlapping sequences for TPP1 interaction and mitochondrial targeting on TIN2, suggest a role of TIN2-TPP1 interaction in determining TIN2 nuclear vs. mitochondrial localization. Indeed, when co-expressed with TPP1-flag, wildtype TIN2 showed decreased mitochondrial localization, whereas the TPP1 interaction mutant TIN2-F37D/L38E maintained its mitochondrial localization (Figures 5A and 5B). In addition, the TIN2-F37D/L38E mutant, but not TIN2-K62A/K64A, could be processed in mitochondria (Figure 5C). In fact, co-expression of TPP1-V5 diminished mitochondrial processing of wildtype TIN2-flag, but had little effect on TIN2-F37D/L38E or TIN2-Δ18 (Figure 5D). These data combined underline the importance of TIN2-TPP1 interaction in targeting TIN2 to different cellular compartments.
Figure 5. TPP1 inhibits TIN2 mitochondrial localization and processing.
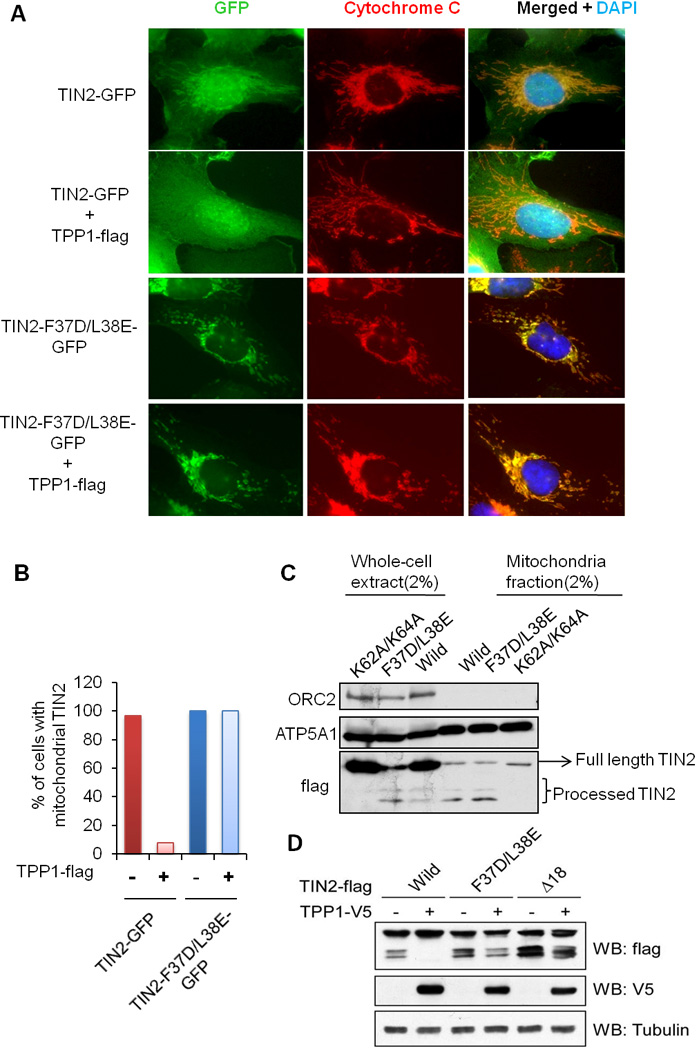
A) HTC75 cells expressing GFP-tagged wildtype TIN2 or TIN2-F37D/L38E alone or with TPP1-flag were analyzed. Anti-cytochrome c antibodies were used to stain the mitochondria. B) Percentages of cells with mitochondrial TIN2 signals from (A) were graphed (n≥100). C) Whole cell extract and mitochondria fractions from cells expressing flag-tagged wildtype (Wild) and mutant TIN2 were western blotted. D) Western analysis of HEK-293 cells transiently co-expressing TPP1-V5 with flag-tagged wildtype (Wild) or mutant TIN2.
TIN2 regulates mitochondrial morphology and function
The finding of TIN2 mitochondrial localization points to the intriguing possibility that TIN2 may participate in metabolic regulation, in addition to its well-established roles in telomere maintenance. As cellular powerhouses for energy production and metabolic control, mitochondria have also been implicated in aging and cancer development (Finkel et al., 2007; Guarente, 2008; Sahin and Depinho, 2010). Mitochondria generate ATP through oxidative phosphorylation, a process that consumes oxygen and requires mitochondrial membrane potential. Morphological changes of mitochondria often reflect alterations in mitochondrial function (Chen et al., 2005; Detmer and Chan, 2007; Lodi et al., 2004). When we examined the mitochondria in HTC75 cells expressing wildtype or mutant TIN2, we found the mitochondria in control cells to take on an extended tubular structure (Figure 6A). In contrast, cells expressing either wildtype TIN2 or TIN2-F37D/L38E exhibited a marked increase in giant spherical mitochondria (Figures 6A and 6B). Such morphology changes were not observed in cells expressing TIN2-K62A/K64A, suggesting that they were direct results of mitochondrial processing of wildtype TIN2 and TIN2-F37D/L38E (Figures 6A and 6B). Moreover, GFP proteins tagged with the N-terminal 58 residues of TIN2 (TIN2-(1-58)-GFP) were targeted to the mitochondria and these cells exhibited similar tubular mitochondrial morphology as control cells expressing untagged GFP (Figures 6A and 6B), indicating that GFP overexpression in the mitochondria did not impact on the mitochondrial protein transport machinery.
Figure 6. TIN2 expression regulates mitochondria morphology.
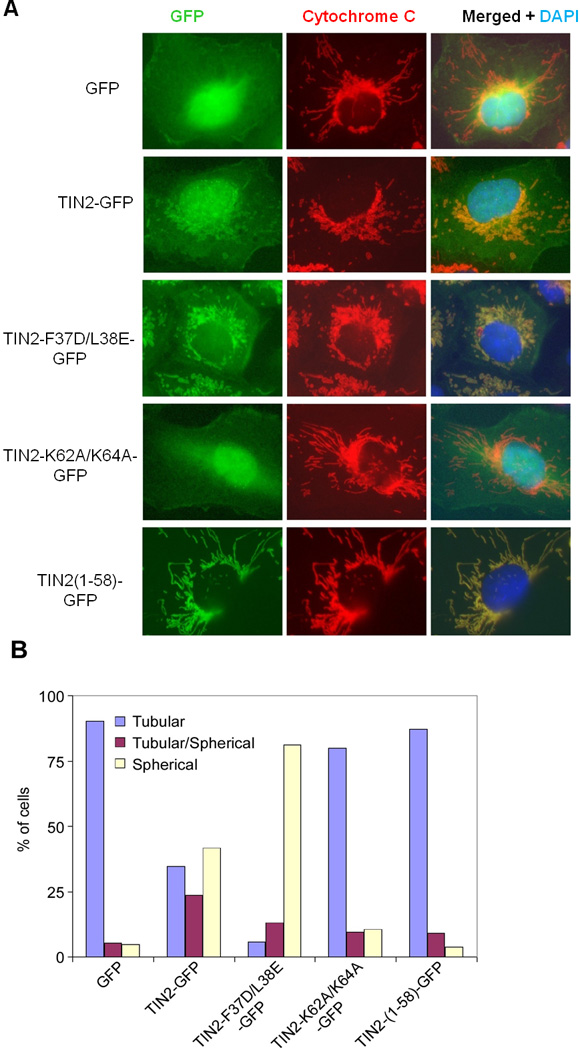
A) HTC75 cells stably expressing GFP-tagged wildtype and mutant TIN2 proteins were immunostained with anti-cytochrome C antibodies. B) Quantification of data from (A) (n≥300).
To understand the function of TIN2 in mitochondria, we generated TIN2 knockdown cells to assess mitochondria function. Using two different shRNAs, we achieved significant inhibition of endogenous TIN2 (>70%) (Figure 7A), which led to minimal telomeric DNA damage responses as measured by telomere dysfunction-induced foci (TIF) formation (data not shown). Surprisingly, these TIN2 knockdown cells produced ≥25% more total ATP (Figure 7B, base), and displayed higher mitochondria membrane potential (Figure 7C). Compared with control cells, basal oxygen consumption in TIN2 knockdown cells also increased by ≥40% (Figure 7D, base). Next, we utilized the mitochondrial ATP synthase inhibitor oligomycin. Adding oligomycin to cells leads to decreased ATP levels, which is indicative of the contribution of mitochondrial oxidative phosphorylation to total ATP production. Interestingly, oligomycin addition to TIN2 knockdown cells abrogated the elevated ATP synthesis and oxygen consumption that we observed when mitochondria oxidative phosphorylation was intact (Figures 7B and 7D, right panels). These data suggest that TIN2 depletion resulted in an increase in energy metabolism in the mitochondria. In support of this idea, overexpression of shRNA-resistant TIN2 restored basal oxygen consumption to a level comparable to control cells (Figure S2). Collectively, these results support a role of mitochondrial TIN2 in modulating mitochondrial oxidative phosphorylation.
Figure 7. TIN2 modulates essential mitochondria function and glucose metabolism.
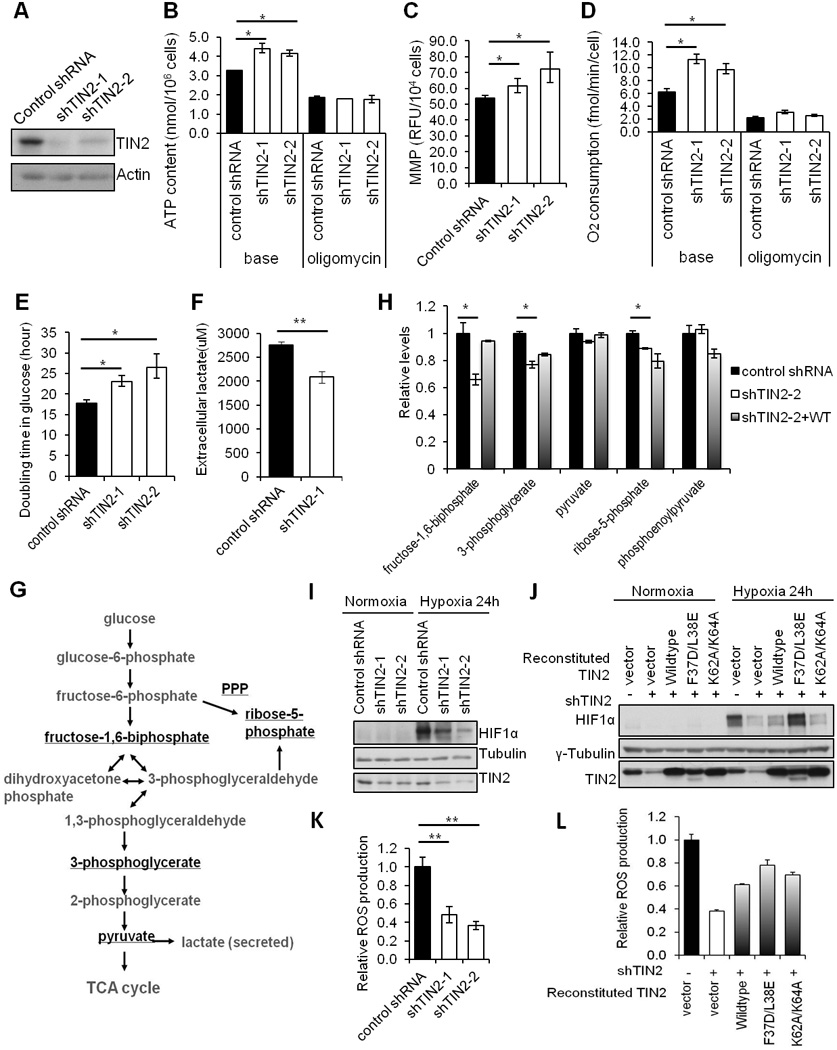
A) Knockdown efficiency of two TIN2 shRNAs in HTC75 cells. B) Total cellular ATP (base) and oligomycin-resistant ATP content were determined using cells from (A). C) Quantification of mitochondrial membrane potential (MMP) of cells from (A). D) Quantification of oxygen consumption in intact cells (base) and following oligomycin treatment. E) Growth analysis of cells from (A) in glucose-containing DMEM media. F) Measurement of media lactate levels of cells from (A). G) Schematic representation of glycolysis. H) Mass spectrometry analysis of glycolytic metabolites in HT1080 cells expressing control shRNA, TIN2 shRNA alone or with RNAi-resistant wildtype TIN2. Relative levels of the underlined metabolites from (G) were plotted. I) TIN2 knockdown MCF-7 cells were cultured under normoxia (21% O2) or hypoxia (1% O2) conditions and western blotted. J) HT1080 cells expressing TIN2 shRNA alone, or with RNAi-resistant wildtype or mutant TIN2 proteins were assessed under normoxia vs. hypoxia conditions. K) Measurement of reactive oxygen species (ROS) in TIN2 knockdown cells. L) Measurement of reactive oxygen species (ROS) in HT1080 cells expressing TIN2 shRNA alone or together with untagged RNAi-resistant wildtype or mutant TIN2. For all panels, error bars represent S.E. (n=3). p values were calculated using Student’s t test (*p<0.05; **p <0.01).
Mammalian cells depend on both oxidative phosphorylation and glycolysis to generate ATP. In normal differentiated cells, the energy is derived primarily from mitochondria oxidative phosphorylation. In most cancer cells, however, glucose is metabolized mainly through cytoplasmic aerobic glycolysis instead of mitochondria oxidative phosphorylation (Warburg, 1956). While this generates fewer ATP per molecule of glucose, it conserves more carbon molecules for the synthesis of other carbohydrates necessary for cell growth (Gatenby and Gillies, 2004; Lunt and Vander Heiden, 2011; Vander Heiden et al., 2009; Vander Heiden et al., 2010). If reducing TIN2 levels in cancer cells promotes oxidative phosphorylation while slowing down aerobic glycolysis, these cells should have slower proliferation due to a decrease of glucose-derived metabolites. We went on to compare the growth rates of control and TIN2 knockdown cells in glucose-containing media. As shown in Figure 7E, TIN2 knockdown cells showed decelerated growth with glucose as the sole carbon source, suggesting that TIN2 inhibition may slow down glycolysis and accelerate oxidative phosphorylation (Figure 7E). The level of lactate in the media also dropped in these cells (Figure 7F), again suggesting decreased glycolysis. When we compared the levels of glycolytic metabolites by mass spectrometry in control HT1080 cells vs. cells that expressed TIN2 shRNA alone or TIN2 shRNA plus RNAi-resistant TIN2, we observed decreases of several glucose-derived metabolites that are essential for biosynthesis, including 3-phosphoglycerate and fructose-1,6-biphosphate, in TIN2 knockdown cells compared to control cells (Figures 7G and 7H). Our data thus far lend further support to the notion that TIN2 depletion promotes mitochondrial oxidative phosphorylation and inhibits aerobic glycolysis in cancer cells.
The in vivo tumor microenvironment is usually low in oxygen (hypoxia), which activates signaling cascades that involve the HIF1 transcription factor complex, a heterodimer of HIF1α and HIF1β (Kim et al., 2006; Papandreou et al., 2006; Zhong et al., 1999). Under hypoxic conditions, HIF1α is stabilized and promotes transcription of many genes to support glycolysis, and cell survival and growth (Kaelin and Ratcliffe, 2008). Changes in glycolysis as a result of TIN2 knockdown led us to examine how TIN2 changes impact on HIF1α stability when TIN2 knockdown cells were cultured in hypoxia conditions. As expected, HIF1α was rapidly stabilized in control MCF-7 and HT1080 cells grown in the presence of 1% O2 (Figures 7I and 7J). Stabilization of HIF1α in TIN2 knockdown cells, on the other hand, appeared slower and to a lesser extent (Figures 7I and 7J). In addition, restoring TIN2 expression with untagged wild-type TIN2 or the TIN2 mutant F37E/L38D, led to HIF1α stabilization comparable to that of control cells (Figure 7J). These results indicate that loss of TIN2 deceases HIF1α stability.
In addition to oxygen, several intracellular signals also regulate the stability of HIF1α. For example, mitochondria-generated ROS is essential for hypoxia activation of HIF1α (Bell et al., 2007a; Guzy et al., 2005; Mansfield et al., 2005). Given our observations that TIN2 modulates HIF1α stability under hypoxic conditions, we next examined the effect of TIN2 loss on ROS. As shown in Figure 7K and 7L, TIN2 knockdown led to reduced ROS levels and restoring TIN2 expression partially rescued ROS production, which is consistent with our findings of deceased HIF1α stabilization in TIN2-depleted cells (Figures 7I and 7J). The effect of TIN2 on ROS levels and the slower proliferation of TIN2 knockdowns cells are also consistent with published reports that ROS generation supports cell proliferation and tumorigenecity (Weinberg et al., 2010). Taken together, these results suggest that TIN2 functions as an additional regulator of glucose metabolism, possibly through regulating ROS production and HIF1α stability.
Discussion
We present findings that the core telomeric protein TIN2 can not only localize to mitochondria, but also play an important role in regulating mitochondrial function. Both endogenous and epitope-tagged TIN2 could be detected in mitochondria and proteolytically processed. N-terminal tagging blocks TIN2 mitochondria targeting, possibly explaining why we had failed to detect TIN2 in the mitochondria previously. Knocking down TIN2 led to minimal change in TIF formation or the expression of several telomeric proteins (data not shown), indicating that mitochondrial phenotypes in TIN2 knockdown cells were not a result of telomere dysfunction.
The N-terminus of TIN2 contains two potential mitochondrial targeting motifs. Having multiple mitochondrial targeting motifs is a feature shared by several proteins that associate with the mitochondrial inner membrane or matrix (Gakh et al., 2002; Glick et al., 1992; Jan et al., 2000), suggesting possible targeting of TIN2 to these compartments. Sequences flanking the proteolytic cleavage sites on TIN2 appear highly conserved, indicating an ancient mechanism in TIN2 mitochondria targeting and processing. In zebrafish, several proteins harbor regions homologous to TIN2 N-terminal domain (TND) and long C-terminal zinc finger repeats that are distinct from TIN2 orthologues (data not shown), suggesting that TND structures may serve as mitochondrial targeting sequences for other proteins as well.
The TIN2 mitochondrial targeting sequences overlap with its TPP1 interaction domain. Interaction with TPP1 is essential for TIN2 nuclear localization, and inhibits TIN2 mitochondrial localization, presumably through blocking recognition of TIN2 by the mitochondrial transport machinery. These data indicate that TIN2 subcellular distribution may be determined by TPP1 abundance.
Our data thus far suggest that TIN2 may modulate mitochondria morphology and activity. It will be interesting to determine whether morphological changes in TIN2 overexpression cells (Figures 6A and 6B) were due to differences in mitochondrial fusion and fission. TIN2 appears to negatively regulate ATP synthesis, because depleting TIN2 enhanced ATP production. This change was oligomycin sensitive, suggesting that TIN2 most likely regulates mitochondrial ATP synthesis. Furthermore, TIN2 inhibition led to higher mitochondria membrane potential and oxygen consumption, but lower ROS generation. These observations support a role of TIN2 in modulating ATP production by limiting efficient electron transport and decreasing mitochondria membrane potential. Here, mitochondrial rather than nuclear TIN2 most likely plays a key role, because TIN2-F37D/L38E, a mutant not targeted to telomeres, was able to rescue mitochondrial phenotypes in TIN2-depleted cells. In addition, TIN2 processing is not required for modulating mitochondria function, given that TIN2-K62A/K64A, which was partially defective for mitochondria localization and not processed, still rescued TIN2 knockdown phenotypes.
Enhanced mitochondrial oxidative phosphorylation coupled with slower growth of cancer cells upon TIN2 knockdown implicates TIN2 in regulating glucose metabolism in human cells. Indeed, TIN2 knockdown led to a decrease of several metabolites in the glycolytic pathway. Mitochondria convert carbohydrates into ATP, a process that also produces ROS. TIN2 depletion repressed ROS production, and resulted in reduced stabilization of HIF1α under hypoxic conditions. These results are consistent with previous observations that ROS production in mitochondria is critical for hypoxia activation of HIF1α and cell proliferation (Bell et al., 2007a; Guzy et al., 2005; Mansfield et al., 2005; Weinberg et al., 2010), and point to TIN2 as a regulator of glucose metabolism and a potential factor to combat the Warburg Effect.
Mitochondrial integrity is critical for cell survival and longevity as well (Kirkwood, 2005; Wallace, 2005). Cells gradually accumulate mitochondrial DNA mutations that can increase ROS, which in turn upregulates HIF1α activity, leading to beneficial effects on lifespan in certain situations (Bell et al., 2007b; Hekimi et al., 2011; Lee et al., 2010) or detrimental effects including DNA damage, genomic instability, and ultimately aging (Balaban et al., 2005). Recent studies suggest crosstalks between mitochondria and telomeres. Progressive telomere loss limits the replicative capacity of dividing cells such as stem cells (Cong et al., 2002), and uncaps chromosomal ends and induces p53 activation in aging animals (Karlseder et al., 1999; Smogorzewska and de Lange, 2002; van Steensel et al., 1998), leading to cellular senescence, apoptosis, and ultimately contributing to the pathogenesis of aging-related diseases. Telomere dysfunction was reported to repress mitochondrial biogenesis and function through the telomere-p53-PGC axis, thereby linking telomere biology to mitochondrial regulation (Sahin et al., 2011). In addition, hTERT has been found in mitochondria to protect cells against oxidative stress and apoptosis (Ahmed et al., 2008; Passos et al., 2007). Whether loss of TIN2 can trigger aging-related phenotypes warrants further investigation.
Experimental Procedures
Immunogold labelling
105~106 cells were fixed in 4% paraformaldehyde for 2 h at 4°C before being dehydrated through a series of graded ethanol solutions at −20°C, transferred into Lowicryls K4M resin (SPI Supplies Inc.), and polymerized under 360 nm ultraviolet at − 20°C as previously described (Liu et al., 2011; Peters and Pierson, 2008). Subsequently, ultrathin sections were collected on carbon-coated nickel grids and floated on a drop of ddH2O for 5 min. Following blocking (1% bovine serum albumin, 0.02% polyethylene glycol 20000, 100 mM NaCl, and 1% NaN3) and antibody incubation, the sections were incubated with 12 nm colloidal gold-affinipure goat anti-rabbit IgG (H+(Jackson ImmunoResearch) for 60 min, air dried, and then stained for 15 minutes in aqueous 1% uranyl acetate followed by 0.2% lead citrate. Samples were analyzed with a JEM-100CX/II transmission electron microscope at an accelerating voltage of 100 KV.
Subcellular fractionation
Subcellular fractionation was performed using the MITOISO2 kit (Sigma) with minor modifications. Briefly, cells were resuspended in Extraction buffer (50mM HEPES, 1M mannitol, 350mM sucrose, 5mM EGTA, and protease inhibitor cocktail), homogenized, and then centrifuged at 300×g for 5 min to yield pellet P1 fraction and S1 supernatant. S1 fraction was further centrifuged at 11,000×g for 10 min to yield pellet P2 and supernatant S2 fractions. P2 fraction was then resuspended in hypotonic buffer (10 mM HEPES-KOH, pH 7.4, and 1 mM EDTA) at 4°C, followed by centrifugation at 10,000×g for 6 min. The mitochondria enriched pellet was then resuspended in hypotonic buffer and digested on ice with proteinase K (200µg/ml) ±1% Triton X-100 for 20 min. The digest was then precipitated with tricholoroacetic acid and analyzed by SDS-PAGE and immunoblotting.
Measuring ATP production
The ATP Determination Kit (Invitrogen, A22066) was used to assess ATP production. Cells grown to ~40% confluence (2×106 in 10cm dish) were washed with Tris-based TD buffer (Mg2+ and Ca2+-deficient) (0.137 M NaCl, 5 mM KCl, 0.7 mM Na2HPO4, and 25 mM Tris-HCl, pH 7.4), and boiled in Boiling Buffer (100mM Tris, 4mM EDTA) for 5 min before centrifugation. The supernatant was used in luciferase assays and measured in a Synergy™ HT Multi-Detection Microplate Reader (Bio-Tek Instruments Inc.). Oligomycin (Sigma) was added (15µg/ml) 30 min before harvesting.
Measuring mitochondrial respiration
Cells were grown to 60%–80% confluence with media change the day before. O2 consumption was determined using ~5×106 cells harvested in TD buffer (pH7.4 at 25°C) as previously described (Bai and Attardi, 1998). Measurements were taken in two chambers of an YSI Model 5300 Biological Oxygen Monitor. Oligomycin (2.5µg/ml) was added to measure uncoupled respiration.
Analysis of mitochondrial membrane potential
Mitochondrial membrane potential was assessed by tetramethylrhodamine, methyl ester, and perchlorate (TMRM) (Invitrogen, Cat. No. T-668). Cells were washed with TD buffer, incubated with 50 nm TMRM in TD buffer at 37 °C for 15 min, and then scraped off and plated in 96-well plates (1×106 cells (200µl) per well) for measurement at 540nm/620nm using a Synergy HT Microplate Reader (Bio-Tek Instruments Inc.).
Measurement of reactive oxygen species (ROS)
Harvested cells were washed in TD buffer and then resuspended in TD buffer containing 5µM MitoSOX™ (red mitochondrial superoxide indicator)(Invitrogen, M36008) at 37 °C for 15 min. After washing with TD buffer, the cells were plated on 96-well plates and measured in a Synergy HT Microplate Reader at 485/590 nm.
Lactate measurement
Extracellular lactate levels were determined with the Lactate Assay Kit (University at Buffalo, NY, USA). Cells were seeded in 6cm dishes (1×105 cells/dish) in triplicates and the media collected and diluted 30-fold with ddH2O after 2 days. Reactions were carried out in 96-well plates (20µl of the diluted media and 50µl of lactate assay solution) in a humidified chamber at 37°C for 30 min, followed by the addition of 50µl of 3% acetic acid. Absorbance was measured at 490 nm. Standard curves were generated using 20µl of lactate solutions with varying concentrations and 50µl of lactate assay solution.
Supplementary Material
Highlight.
The telomeric core protein TIN2 can be specifically targeted to the mitochondria
TIN2 is processed in the mitochondria
Mitochondrially localized TIN2 regulates mitochondria function
Acknowledgements
We thank Drs. Fengtao Shi and Dong Yang for technical help, and Dr. Dan Liu for critical reading of this manuscript. This work is supported by National Basic Research Program (973 Program 2010CB945400 and 2012CB911200), National Natural Science Foundation of China (NSFC 91019020), NCI CA133249, NIGMS GM081627, GM095599, the Welch Foundation Q-1673, and the Leukemia and Lymphoma Society. This work is also supported by NIH grants DK058242 (JX) and 1R21NS072777 (YB), the Dan L. Duncan Cancer Center (P30CA125123), and the Administrative and Genomewide RNAi Screens Cores (IDDRC P30HD024064).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
The authors declare no conflicts of interest.
References
- Abreu E, Aritonovska E, Reichenbach P, Cristofari G, Culp B, Terns RM, Lingner J, Terns MP. TIN2-tethered TPP1 recruits human telomerase to telomeres in vivo. Mol Cell Biol. 2010;30:2971–2982. doi: 10.1128/MCB.00240-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahmed S, Passos JF, Birket MJ, Beckmann T, Brings S, Peters H, Birch-Machin MA, von Zglinicki T, Saretzki G. Telomerase does not counteract telomere shortening but protects mitochondrial function under oxidative stress. J Cell Sci. 2008;121:1046–1053. doi: 10.1242/jcs.019372. [DOI] [PubMed] [Google Scholar]
- Bai Y, Attardi G. The mtDNA-encoded ND6 subunit of mitochondrial NADH dehydrogenase is essential for the assembly of the membrane arm and the respiratory function of the enzyme. Embo J. 1998;17:4848–4858. doi: 10.1093/emboj/17.16.4848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balaban RS, Nemoto S, Finkel T. Mitochondria, oxidants, and aging. Cell. 2005;120:483–495. doi: 10.1016/j.cell.2005.02.001. [DOI] [PubMed] [Google Scholar]
- Bell EL, Klimova TA, Eisenbart J, Moraes CT, Murphy MP, Budinger GR, Chandel NS. The Qo site of the mitochondrial complex III is required for the transduction of hypoxic signaling via reactive oxygen species production. J Cell Biol. 2007a;177:1029–1036. doi: 10.1083/jcb.200609074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bell EL, Klimova TA, Eisenbart J, Schumacker PT, Chandel NS. Mitochondrial reactive oxygen species trigger hypoxia-inducible factor-dependent extension of the replicative life span during hypoxia. Mol Cell Biol. 2007b;27:5737–5745. doi: 10.1128/MCB.02265-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chacinska A, Koehler CM, Milenkovic D, Lithgow T, Pfanner N. Importing mitochondrial proteins: machineries and mechanisms. Cell. 2009;138:628–644. doi: 10.1016/j.cell.2009.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H, Chomyn A, Chan DC. Disruption of fusion results in mitochondrial heterogeneity and dysfunction. J Biol Chem. 2005;280:26185–26192. doi: 10.1074/jbc.M503062200. [DOI] [PubMed] [Google Scholar]
- Chen LY, Liu D, Songyang Z. Telomere maintenance through spatial control of telomeric proteins. Mol Cell Biol. 2007;27:5898–5909. doi: 10.1128/MCB.00603-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cong YS, Wright WE, Shay JW. Human telomerase and its regulation. Microbiol Mol Biol Rev. 2002;66:407–425. doi: 10.1128/MMBR.66.3.407-425.2002. table of contents. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Lange T. Shelterin: the protein complex that shapes and safeguards human telomeres. Genes Dev. 2005;19:2100–2110. doi: 10.1101/gad.1346005. [DOI] [PubMed] [Google Scholar]
- Detmer SA, Chan DC. Functions and dysfunctions of mitochondrial dynamics. Nat Rev Mol Cell Biol. 2007;8:870–879. doi: 10.1038/nrm2275. [DOI] [PubMed] [Google Scholar]
- Du HY, Mason PJ, Bessler M, Wilson DB. TINF2 mutations in children with severe aplastic anemia. Pediatr Blood Cancer. 2009;52:687. doi: 10.1002/pbc.21903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finkel T, Serrano M, Blasco MA. The common biology of cancer and ageing. Nature. 2007;448:767–774. doi: 10.1038/nature05985. [DOI] [PubMed] [Google Scholar]
- Gakh O, Cavadini P, Isaya G. Mitochondrial processing peptidases. Biochim Biophys Acta. 2002;1592:63–77. doi: 10.1016/s0167-4889(02)00265-3. [DOI] [PubMed] [Google Scholar]
- Gatenby RA, Gillies RJ. Why do cancers have high aerobic glycolysis? Nat Rev Cancer. 2004;4:891–899. doi: 10.1038/nrc1478. [DOI] [PubMed] [Google Scholar]
- Glick BS, Brandt A, Cunningham K, Muller S, Hallberg RL, Schatz G. Cytochromes c1 and b2 are sorted to the intermembrane space of yeast mitochondria by a stop-transfer mechanism. Cell. 1992;69:809–822. doi: 10.1016/0092-8674(92)90292-k. [DOI] [PubMed] [Google Scholar]
- Guarente L. Mitochondria--a nexus for aging, calorie restriction, and sirtuins? Cell. 2008;132:171–176. doi: 10.1016/j.cell.2008.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guzy RD, Hoyos B, Robin E, Chen H, Liu L, Mansfield KD, Simon MC, Hammerling U, Schumacker PT. Mitochondrial complex III is required for hypoxia-induced ROS production and cellular oxygen sensing. Cell Metab. 2005;1:401–408. doi: 10.1016/j.cmet.2005.05.001. [DOI] [PubMed] [Google Scholar]
- Hekimi S, Lapointe J, Wen Y. Taking a "good" look at free radicals in the aging process. Trends Cell Biol. 2011;21:569–576. doi: 10.1016/j.tcb.2011.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hockemeyer D, Palm W, Else T, Daniels JP, Takai KK, Ye JZ, Keegan CE, de Lange T, Hammer GD. Telomere protection by mammalian Pot1 requires interaction with Tpp1. Nat Struct Mol Biol. 2007;14:754–761. doi: 10.1038/nsmb1270. [DOI] [PubMed] [Google Scholar]
- Houghtaling BR, Cuttonaro L, Chang W, Smith S. A dynamic molecular link between the telomere length regulator TRF1 and the chromosome end protector TRF2. Curr Biol. 2004;14:1621–1631. doi: 10.1016/j.cub.2004.08.052. [DOI] [PubMed] [Google Scholar]
- Jan PS, Esser K, Pratje E, Michaelis G. Som1, a third component of the yeast mitochondrial inner membrane peptidase complex that contains Imp1 and Imp2. Mol Gen Genet. 2000;263:483–491. doi: 10.1007/s004380051192. [DOI] [PubMed] [Google Scholar]
- Kaelin WG, Jr, Ratcliffe PJ. Oxygen sensing by metazoans: the central role of the HIF hydroxylase pathway. Mol Cell. 2008;30:393–402. doi: 10.1016/j.molcel.2008.04.009. [DOI] [PubMed] [Google Scholar]
- Karlseder J, Broccoli D, Dai Y, Hardy S, de Lange T. p53- and ATMdependent apoptosis induced by telomeres lacking TRF2. Science. 1999;283:1321–1325. doi: 10.1126/science.283.5406.1321. [DOI] [PubMed] [Google Scholar]
- Kim JW, Tchernyshyov I, Semenza GL, Dang CV. HIF-1-mediated expression of pyruvate dehydrogenase kinase: a metabolic switch required for cellular adaptation to hypoxia. Cell Metab. 2006;3:177–185. doi: 10.1016/j.cmet.2006.02.002. [DOI] [PubMed] [Google Scholar]
- Kim SH, Beausejour C, Davalos AR, Kaminker P, Heo SJ, Campisi J. TIN2 mediates functions of TRF2 at human telomeres. J Biol Chem. 2004;279:43799–43804. doi: 10.1074/jbc.M408650200. [DOI] [PubMed] [Google Scholar]
- Kim SH, Kaminker P, Campisi J. TIN2, a new regulator of telomere length in human cells. Nat Genet. 1999;23:405–412. doi: 10.1038/70508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirkwood TB. Understanding the odd science of aging. Cell. 2005;120:437–447. doi: 10.1016/j.cell.2005.01.027. [DOI] [PubMed] [Google Scholar]
- Lee SJ, Hwang AB, Kenyon C. Inhibition of respiration extends C. elegans life span via reactive oxygen species that increase HIF-1 activity. Curr Biol. 2010;20:2131–2136. doi: 10.1016/j.cub.2010.10.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu C, Ye L, Lang G, Zhang C, Hong J, Zhou X. The VP37 protein of Broad bean wilt virus 2 induces tubule-like structures in both plant and insect cells. Virus Res. 2011;155:42–47. doi: 10.1016/j.virusres.2010.08.013. [DOI] [PubMed] [Google Scholar]
- Liu D, O'Connor MS, Qin J, Songyang Z. Telosome, a mammalian telomere-associated complex formed by multiple telomeric proteins. J Biol Chem. 2004a;279:51338–51342. doi: 10.1074/jbc.M409293200. [DOI] [PubMed] [Google Scholar]
- Liu D, Safari A, O'Connor MS, Chan DW, Laegeler A, Qin J, Songyang Z. PTOP interacts with POT1 and regulates its localization to telomeres. Nat Cell Biol. 2004b;6:673–680. doi: 10.1038/ncb1142. [DOI] [PubMed] [Google Scholar]
- Lodi R, Tonon C, Valentino ML, Iotti S, Clementi V, Malucelli E, Barboni P, Longanesi L, Schimpf S, Wissinger B, et al. Deficit of in vivo mitochondrial ATP production in OPA1-related dominant optic atrophy. Ann Neurol. 2004;56:719–723. doi: 10.1002/ana.20278. [DOI] [PubMed] [Google Scholar]
- Lunt SY, Vander Heiden MG. Aerobic glycolysis: meeting the metabolic requirements of cell proliferation. Annu Rev Cell Dev Biol. 2011;27:441–464. doi: 10.1146/annurev-cellbio-092910-154237. [DOI] [PubMed] [Google Scholar]
- Mansfield KD, Guzy RD, Pan Y, Young RM, Cash TP, Schumacker PT, Simon MC. Mitochondrial dysfunction resulting from loss of cytochrome c impairs cellular oxygen sensing and hypoxic HIF-alpha activation. Cell Metab. 2005;1:393–399. doi: 10.1016/j.cmet.2005.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mao Z, Seluanov A, Jiang Y, Gorbunova V. TRF2 is required for repair of nontelomeric DNA double-strand breaks by homologous recombination. Proc Natl Acad Sci U S A. 2007;104:13068–13073. doi: 10.1073/pnas.0702410104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez P, Thanasoula M, Carlos AR, Gomez-Lopez G, Tejera AM, Schoeftner S, Dominguez O, Pisano DG, Tarsounas M, Blasco MA. Mammalian Rap1 controls telomere function and gene expression through binding to telomeric and extratelomeric sites. Nat Cell Biol. 2010;12:768–780. doi: 10.1038/ncb2081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moberg P, Nilsson S, Stahl A, Eriksson AC, Glaser E, Maler L. NMR solution structure of the mitochondrial F1beta presequence from Nicotiana plumbaginifolia. J Mol Biol. 2004;336:1129–1140. doi: 10.1016/j.jmb.2004.01.006. [DOI] [PubMed] [Google Scholar]
- Papandreou I, Cairns RA, Fontana L, Lim AL, Denko NC. HIF-1 mediates adaptation to hypoxia by actively downregulating mitochondrial oxygen consumption. Cell Metab. 2006;3:187–197. doi: 10.1016/j.cmet.2006.01.012. [DOI] [PubMed] [Google Scholar]
- Passos JF, Saretzki G, von Zglinicki T. DNA damage in telomeres and mitochondria during cellular senescence: is there a connection? Nucleic Acids Res. 2007;35:7505–7513. doi: 10.1093/nar/gkm893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters PJ, Pierson J. Immunogold labeling of thawed cryosections. Methods Cell Biol. 2008;88:131–149. doi: 10.1016/S0091-679X(08)00408-1. [DOI] [PubMed] [Google Scholar]
- Sahin E, Colla S, Liesa M, Moslehi J, Muller FL, Guo M, Cooper M, Kotton D, Fabian AJ, Walkey C, et al. Telomere dysfunction induces metabolic and mitochondrial compromise. Nature. 2011;470:359–365. doi: 10.1038/nature09787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sahin E, Depinho RA. Linking functional decline of telomeres, mitochondria and stem cells during ageing. Nature. 2010;464:520–528. doi: 10.1038/nature08982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saitoh T, Igura M, Obita T, Ose T, Kojima R, Maenaka K, Endo T, Kohda D. Tom20 recognizes mitochondrial presequences through dynamic equilibrium among multiple bound states. Embo J. 2007;26:4777–4787. doi: 10.1038/sj.emboj.7601888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santos JH, Meyer JN, Skorvaga M, Annab LA, Van Houten B. Mitochondrial hTERT exacerbates free-radical-mediated mtDNA damage. Aging Cell. 2004;3:399–411. doi: 10.1111/j.1474-9728.2004.00124.x. [DOI] [PubMed] [Google Scholar]
- Sasa G, Ribes-Zamora A, Nelson N, Bertuch A. Three novel truncating TINF2 mutations causing severe dyskeratosis congenita in early childhood. Clin Genet. 2011 doi: 10.1111/j.1399-0004.2011.01658.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savage SA, Giri N, Baerlocher GM, Orr N, Lansdorp PM, Alter BP. TINF2, a component of the shelterin telomere protection complex, is mutated in dyskeratosis congenita. Am J Hum Genet. 2008;82:501–509. doi: 10.1016/j.ajhg.2007.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simonet T, Zaragosi LE, Philippe C, Lebrigand K, Schouteden C, Augereau A, Bauwens S, Ye J, Santagostino M, Giulotto E, et al. The human TTAGGG repeat factors 1 and 2 bind to a subset of interstitial telomeric sequences and satellite repeats. Cell Res. 2011;21:1028–1038. doi: 10.1038/cr.2011.40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smogorzewska A, de Lange T. Different telomere damage signaling pathways in human and mouse cells. Embo J. 2002;21:4338–4348. doi: 10.1093/emboj/cdf433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor AB, Smith BS, Kitada S, Kojima K, Miyaura H, Otwinowski Z, Ito A, Deisenhofer J. Crystal structures of mitochondrial processing peptidase reveal the mode for specific cleavage of import signal sequences. Structure. 2001;9:615–625. doi: 10.1016/s0969-2126(01)00621-9. [DOI] [PubMed] [Google Scholar]
- Tejera AM, Stagno d'Alcontres M, Thanasoula M, Marion RM, Martinez P, Liao C, Flores JM, Tarsounas M, Blasco MA. TPP1 is required for TERT recruitment, telomere elongation during nuclear reprogramming, and normal skin development in mice. Dev Cell. 2010;18:775–789. doi: 10.1016/j.devcel.2010.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsangaris E, Adams SL, Yoon G, Chitayat D, Lansdorp P, Dokal I, Dror Y. Ataxia and pancytopenia caused by a mutation in TINF2. Hum Genet. 2008;124:507–513. doi: 10.1007/s00439-008-0576-7. [DOI] [PubMed] [Google Scholar]
- van Steensel B, Smogorzewska A, de Lange T. TRF2 protects human telomeres from end-to-end fusions. Cell. 1998;92:401–413. doi: 10.1016/s0092-8674(00)80932-0. [DOI] [PubMed] [Google Scholar]
- Vander Heiden MG, Cantley LC, Thompson CB. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science. 2009;324:1029–1033. doi: 10.1126/science.1160809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vander Heiden MG, Locasale JW, Swanson KD, Sharfi H, Heffron GJ, Amador-Noguez D, Christofk HR, Wagner G, Rabinowitz JD, Asara JM, et al. Evidence for an alternative glycolytic pathway in rapidly proliferating cells. Science. 2010;329:1492–1499. doi: 10.1126/science.1188015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallace DC. A mitochondrial paradigm of metabolic and degenerative diseases, aging, and cancer: a dawn for evolutionary medicine. Annu Rev Genet. 2005;39:359–407. doi: 10.1146/annurev.genet.39.110304.095751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walne AJ, Vulliamy T, Beswick R, Kirwan M, Dokal I. TINF2 mutations result in very short telomeres: analysis of a large cohort of patients with dyskeratosis congenita and related bone marrow failure syndromes. Blood. 2008;112:3594–3600. doi: 10.1182/blood-2008-05-153445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang F, Podell ER, Zaug AJ, Yang Y, Baciu P, Cech TR, Lei M. The POT1-TPP1 telomere complex is a telomerase processivity factor. Nature. 2007;445:506–510. doi: 10.1038/nature05454. [DOI] [PubMed] [Google Scholar]
- Warburg O. On the origin of cancer cells. Science. 1956;123:309–314. doi: 10.1126/science.123.3191.309. [DOI] [PubMed] [Google Scholar]
- Weinberg F, Hamanaka R, Wheaton WW, Weinberg S, Joseph J, Lopez M, Kalyanaraman B, Mutlu GM, Budinger GR, Chandel NS. Mitochondrial metabolism and ROS generation are essential for Kras-mediated tumorigenicity. Proc Natl Acad Sci U S A. 2010;107:8788–8793. doi: 10.1073/pnas.1003428107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xin H, Liu D, Wan M, Safari A, Kim H, Sun W, O'Connor MS, Songyang Z. TPP1 is a homologue of ciliate TEBP-beta and interacts with POT1 to recruit telomerase. Nature. 2007;445:559–562. doi: 10.1038/nature05469. [DOI] [PubMed] [Google Scholar]
- Yamano K, Yatsukawa Y, Esaki M, Hobbs AE, Jensen RE, Endo T. Tom20 and Tom22 share the common signal recognition pathway in mitochondrial protein import. J Biol Chem. 2008;283:3799–3807. doi: 10.1074/jbc.M708339200. [DOI] [PubMed] [Google Scholar]
- Yang D, He Q, Kim H, Ma W, Songyang Z. TIN2 protein dyskeratosis congenita missense mutants are defective in association with telomerase. J Biol Chem. 2011a;286:23022–23030. doi: 10.1074/jbc.M111.225870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang D, Xiong Y, Kim H, He Q, Li Y, Chen R, Songyang Z. Human telomeric proteins occupy selective interstitial sites. Cell Res. 2011b doi: 10.1038/cr.2011.39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye JZ, Donigian JR, van Overbeek M, Loayza D, Luo Y, Krutchinsky AN, Chait BT, de Lange T. TIN2 binds TRF1 and TRF2 simultaneously and stabilizes the TRF2 complex on telomeres. J Biol Chem. 2004;279:47264–47271. doi: 10.1074/jbc.M409047200. [DOI] [PubMed] [Google Scholar]
- Zhang P, Casaday-Potts R, Precht P, Jiang H, Liu Y, Pazin MJ, Mattson MP. Nontelomeric splice variant of telomere repeat-binding factor 2 maintains neuronal traits by sequestering repressor element 1-silencing transcription factor. Proc Natl Acad Sci USA. 2011 doi: 10.1073/pnas.1106906108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang P, Pazin MJ, Schwartz CM, Becker KG, Wersto RP, Dilley CM, Mattson MP. Nontelomeric TRF2-REST interaction modulates neuronal gene silencing and fate of tumor and stem cells. Curr Biol. 2008;18:1489–1494. doi: 10.1016/j.cub.2008.08.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong H, De Marzo AM, Laughner E, Lim M, Hilton DA, Zagzag D, Buechler P, Isaacs WB, Semenza GL, Simons JW. Overexpression of hypoxiainducible factor 1alpha in common human cancers and their metastases. Cancer Res. 1999;59:5830–5835. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.