

Abstract
Thymic T-cell selection mechanisms generate a cross-reactive, self-MHC restricted peripheral T-cell pool. Affinity and avidity are of profound influence on this selection and the generation of immunity. Autoreactive T cells can escape thymic deletion by lowering their avidity and retain this “tuned” state in the periphery. Upon activation, tuned T cells can cause autoimmunity, while immunotherapeutic strategies may be hampered by existing T-cell tolerance. The regulation of T-cell avidity and tuning therefore determines the balance between tolerance and autoimmunity and should be taken into account in the design of therapeutic strategies aimed at T-cell reactivity.
Keywords: Autoimmunity, avidity, T-cell, Tolerance, Tuning
INTRODUCTION
T cells are of profound importance to adaptive cellular immunity. These cells express a surface receptor of a given specificity and circulate between peripheral lymphoid tissues and the bloodstream, until they encounter their specific antigen presented by an antigen-presenting cell (APC) in the context of a major histocompatibility complex (MHC). Upon antigen recognition and proper co-stimulation, naïve T cells are activated and differentiate into effector T cells mediating the removal of pathogen-infected cells from the body.
Generating a T cell–mediated immune response involves events surrounding and following the recognition of antigen by the T-cell receptor (TCR). Besides TCR affinity, T-cell avidity (or antigen-triggering sensitivity) is essential in T-cell activation. Multiple aspects and mechanisms determine T-cell affinity and avidity and mediate the balance between T-cell tolerance and the generation of immunity and are therefore instrumental in the understanding of autoimmunity.
T-CELL RECEPTOR AFFINITY
An essential process in the adaptive immune response is the intimate contact between the T cell and APC. The TCR is of fundamental importance to this process. It interacts with the MHC expressed on the APC, for example a dendritic cell (DC), B cell or macrophage, which have potent co-stimulatory and antigen-presenting properties when activated [1]. The TCR is able to recognize specific peptide antigen fragments presented to the T cell in the peptide-binding cleft of the MHC. An APC can present many different antigen fragments derived from numerous peptide sources. Basically, intracellular derived antigens are presented in MHC class I to CD8 T cells and extracellular derived antigens in MHC class II to CD4 T cells. TCRs are restricted to recognizing antigen as presented by one specific self-MHC molecule, termed MHC restriction.
The TCR-MHC interaction is relatively weak, and several measures increase TCR affinity (or binding-energy) for the MHC-peptide complex. As to date, it is unclear if the peptide or the MHC molecule makes the major contribution to this affinity. The peptide contributes a smaller portion of the interacting surface than does the MHC molecule. Additionally, it is known that the TCR complementarity determining regions (CDRs) undergo minor conformational changes upon MHC ligation to adapt to its surface. It could well be that TCR-MHC contacts play a permissive role, while TCR-peptide contacts provide the majority of the binding energy [2,3].
Antigen presentation via MHC class I is not as strict as stated above. An alternative antigen presentation pathway exists in which APCs present extracellular derived antigen embedded in MHC class I molecules to CD8 cells. This process is called cross-presentation [4] and is crucial in generating CD8 T cell–mediated immunity against certain viral or tumor antigens and is important in the maintenance of self-tolerance [5]. Recent data shows that this cross-presentation takes place via direct transfer of proteasome substrates from the cell of origin, instead of antigen fragments being internalized by the APC [6]. It has also been reported that specific virus and tumor antigens may not be detected by CD8 T cells because of impaired cross-priming, an important finding for optimizing immunotherapy and vaccine design [7].
If thymic selection (further discussed below) aims at deleting those T cells that show too high affinity for autoantigens, the autoantigen-specific T-cell repertoire would be small and of limited reactivity, creating a functionally self-tolerant state. It appears that more factors than TCR affinity influence the T cell’s sensitivity to antigenic triggering, having consequences for thymic selection and the generation of immunity.
T-CELL AVIDITY
The availability of the TCR, coreceptors, and peptide-MHC complexes to interact with each other determine T-cell structural avidity (Figure 1). Additionally, the sensitivity of a T cell to triggering by its antigen is termed “functional avidity” or “avidity” and is defined as the concentration of peptide able to induce 50% activation of the antigen-specific T-cell pool. Basically this is a measure of passing activation thresholds and subsequent induction of signaling and biological function upon establishing a stable interaction with the APC [8–12].
FIGURE 1.
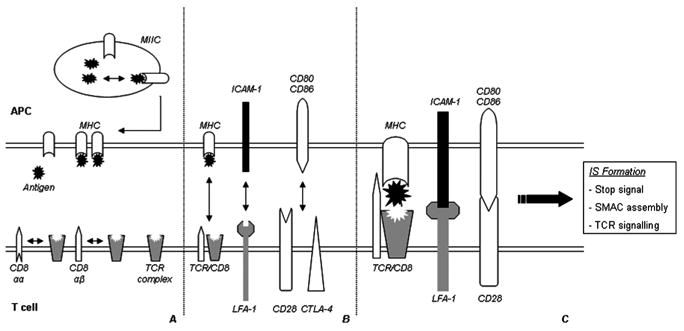
Several factors influence structural T-cell avidity: (A) Expression levels of TCR and MHC molecules, affinity of the peptide for the MHC molecule, and the affinity of the TCR for the peptide-MHC complex are basic factors of structural T-cell avidity. Furthermore, different coreceptor subtypes (such as CD8-αα or -αβ) display different affinities for the TCR complex, additionally influencing antigen recognition efficiency. (B) Besides availability of the MHC and TCR complexes, the expression levels of different co-stimulatory and adhesion molecules on the surface of the APC and T cell, and their ability to interact, are important determinants for proper activation and prolonged cell-cell contact. (C) Recognition of the peptide-MHC complex by the TCR, combined with co-stimulatory signaling and adhesion-molecule interaction, will induce a transient stop signal for the migrating T cell. This allows for prolonged TCR signaling and the assembly of an immunological synapse (IS), and eventually the formation of a supramolecular activation cluster (SMAC). Subsequent biological function depends on the T cell’s functional avidity, determined by the regulation of activation thresholds and induction of different signaling cascades.
Avidity relies on the availability of, and affinity by which the antigen is bound to the MHC molecule, combined with the affinity of the TCR for the MHC-peptide complex. An increase in one, or both, of these factors will increase the avidity of the interaction and thereby the T cell’s sensitivity for activation. As a consequence, TCR affinity for the peptide-MHC complex and the peptide to MHC affinity are compensatory. This means that the expression of a high-affinity TCR allows T-cell activation by poorly MHC-binding peptides, and vice versa [13]. This phenomenon is of essential importance when designing adoptive T-cell transfer therapies against a target antigen. The affinity by which the antigen is bound to the MHC complex directs the choice of T-cell avidity, as a low-affinity bound antigen requires a high-avidity T cell for a successful response. “Mismatching” a T cell with antigen in such a case would result in improper stimulation and T-cell unresponsiveness.
In addition to the TCR-MHC complex interaction, additional factors influence avidity. For example, coreceptor ligation (CD4 or CD8) influences the duration of contact between T cell and APC. The CD8 coreceptor plays a distinct role in this process. T cells expressing high levels of CD8 αβ heterodimeric coreceptors are of higher avidity than cells predominantly expressing CD8αα homodimers. This is likely caused by the αβ-dimer associating with the TCR more easily than does the αα-dimer [14]. Therefore presence, dimeric type, and mobility (availability to colocalize) of coreceptors has direct effects on T-cell avidity.
Besides coreceptors, different types of co-stimulatory and adhesion molecules also influence the T cell–APC interaction. The interaction half-lives of the molecular contacts themselves is of the order seconds, whereas T-cell activation requires interaction of the order hours. Therefore, the formation of a stable interaction is required [8,10,15]. This is provided by the formation of an immunological synapse (IS), which can materialize in several different manners depending on the circumstances of contact, extensively reviewed by Friedl et al. [10] and Jacobelli et al. [16].
The relevance of the IS to T-cell avidity becomes clearly evident when taking into account the different signaling cascades emanating from IS-mediated interaction (Figure 2). In the initial phases of formation, the TCR interacts with the peptide-MHC complex. Together with interactions between lymphocyte associated antigen-1 (LFA-1 on the T cell) and intercellular adhesion molecule-1 (ICAM-1 on the APC), this leads to a transient stop signal to the migrating lymphocyte [17]. Immediately after this stop signal, IS assembly and initial signaling events will start. Continuous TCR signaling upon antigen recognition is essential for the maintenance and further maturation of the IS [11].
FIGURE 2.
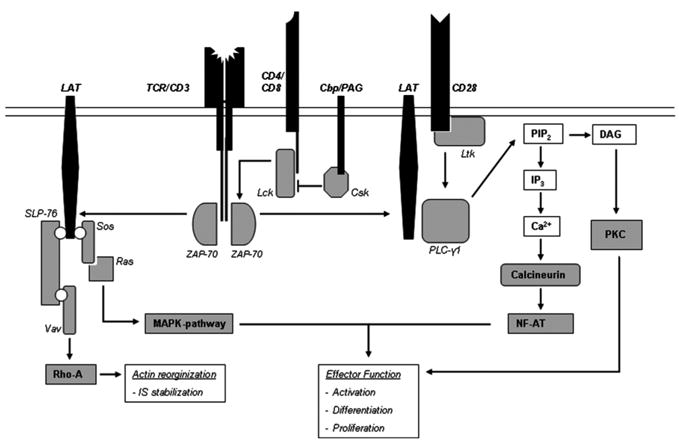
Schematic representation of the signaling pathways involved in T-cell activation. Antigen recognition by the TCR and coreceptor (CD4 or CD8) ligation releases Lck from Csk inhibition, leading to phosphorylation of the TCR ξ-chains and recruitment of ZAP-70. ZAP-70 phosphorylates LAT, which ligates PLCγ-1 and thereby amplifies and sustains the activation signal. PLCγ-1 can activate different signaling cascades via cleavage of PIP2, such as the PKC and calcineurin pathways leading to cellular activation and differentiation. The function of PLCγ-1 is further enhanced by CD28 ligation, via interaction with Ltk. Additionally, LAT can interact with several linker molecules, leading to activation of the MAPK-pathway via activation of Ras or the Rho A pathway leading to actin reorganization.
Upon initial TCR engagement, the tyrosine residues in the immunoreceptor tyrosine-based activation motifs (ITAMs) of the TCR-associated ξ-chain dimer and CD3 complex (γ, δ, and ε subunits) become phosphorylated and subjected to binding by different molecules. This phosphorylation is mediated by two protein tyrosine kinases (PTKs), namely Lck (released from Csk inhibition by CD4 or CD8 co-ligation with the TCR) and to a lesser degree Fyn. The most important event after ITAM phosphorylation is the binding of ZAP-70 to two phosphorylated ITAMs. ZAP-70, being a PTK on its own subsequently gets phosphorylated as well by Lck and is able to bind additional molecules [18,19]. High levels of coreceptor ligation will mediate high levels of C-terminal Src kinase (Csk) release, resulting in a more rapid T-cell activation. Coreceptors thereby have a dual role concerning T-cell avidity: they prolong the contactbetweenTCR andMHC complex and aid ZAP-70phosphorylation. T-cell tuning (described below) appears to involve the phosphorylation levels of the TCR-ξ chain and ZAP-70 on their inhibitory residues, thereby directly influencing activation thresholds.
Importantly, phosphorylation events are equally dependent upon the enzymes that subsequently remove the phosphates from the residues, such as protein tyrosine phosphatases (PTPases). There are more PTPases controlling T-cell activation than there are PTKs, nonetheless research has mainly focused on the PTKs. In normal cells, tyrosine phosphorylation is rapidly reversible, ensuring tight signal regulation [18]. It follows that subtle changes in PTK/PTPase balance can have major impacts on phosphorylation levels and hence T-cell avidity, activation, and proliferation.
Illustrating the importance of CD28 co-stimulation is the molecule Ltk. CD28 ligation will activate Ltk, thereby phosphorylating phospholipase-Cγ1 (PLC-γ). This molecule interacts with the linker for activation of T cells (LAT), and PLC-γ is essential in propagating and amplifying the TCR signal via cleavage of membrane phospholipid phosphatidylinositol biphosphate (PIP2) into its components inositol triphosphate (IP3) and diacylglycerol (DAG). Via complex signaling cascades involving protein kinase C (PKC) and calcineurin, this leads to the activation and differentiation of the T cell [19]. Consequently, T cells expressing high levels of CD28 will exhibit a high avidity.
LAT influences cytoskeletal rearrangements, essential in the formation and continuation of the IS as it stabilizes the cellular interaction and subsequent signaling events. Involving the protein SLP-76, LAT influences downstream actin reorganization via the Rho-A signaling cascade, allowing the T cell to rearrange and “spread out” its cytoskeleton to the site of contact with the APC [20,21]. Being downstream of TCR and CD28 ligation, LAT expression is essential in T-cell activation and avidity. LAT deficiency is known to cause impaired cellular interaction and disorganized IS formation [20].
When TCR signaling is sustained, these initial signaling events will lead up to the formation of a mature IS, characterized by the formation of a supramolecular activation cluster (SMAC) between the T cell and APC, setting the stage for full effector potential [10,15]. Additionally, the T cell can express other co-stimulatory molecules such as tumor necrosis factor receptor family members (TNFRs) like CD40, CD30, CD27, and 4-1BB having further influence on the outcome of interaction (reviewed by Croft [22]).
All signaling cascades emerging from interactions at the cell surface ultimately lead to the activation or inhibition of transcription factors, which play essential roles in T-cell activation and differentiation. During T-cell selection and the generation of immunity, these cascades will direct a T cell to its fate. Variations on the level of signal regulation, and the expression level of signaling surface receptors, are indispensably linked to the regulation of T-cell avidity. Defects or alterations can cause abnormal T-cell activation. For instance, it is known that polymorphisms in the cytotoxic T lymphocyte–associated antigen (CTLA)-4 molecule are associated with vitiligo in humans [23] and thyroid autoimmunity and type 1 diabetes in mice [24]. Expression of Human Leukocyte Antigen (HLA)-DR4 is also associated with vitiligo, which may suboptimally present certain autoantigens during thymic selection facilitating escape of autoreactive T cells [25,26]. Additionally, several mutations in transcription factors are known to cause autoimmunity, for example the IL-2 promoter binding factor T-bet in Crohn disease [27], and the SLC22A4/RUNX1 RUNX1 polymorphism proposed in rheumatoid arthritis [28]. Other defects appear to have a more widespread effect, for instance polymorphisms in the FoxP3 gene induce autoimmune disease such as the IPEX syndrome (immune dysregulation, polyendocrinopathy, enteropathy, X-linked syndrome). Defective FoxP3 inevitably results in defective regulatory T cell (Treg) development, allowing unrestrained generation of immune responses [29,30]. Additionally, there are defects that are able to influence the avidity to autoantigens of the entire T-cell pool. Defects in the autoimmune regulator (AIRE) can influence thymic selection and reactivity of the entire T-cell pool and like FoxP3 cause widespread autoimmunity.
T-CELL SELECTION
The generation of the peripheral T-cell repertoire is dependent upon thymic selection, and selection thereby predisposes the diversity and availability of the T-cell repertoire. Insight into central selection is important in understanding the basis of T cell–mediated autoimmunity. For a major part, T-cell affinity and functional avidity determine a cell’s fate during selection.
There are two main models describing T-cell selection: the qualitative or peptide model, and the quantitative or avidity-based model. The current focus is on the role of peptides during selection, and preferentially the avidity-based model is used to explain the selection of T cells. The main issue in thymic selection is the nature of the selecting ligand; do qualitatively distinct ligands promote positive or negative selection, or do specific TCR interactions promote intracellular signals to meet quantitative signaling thresholds for T cell survival [13,31]?
Immature T cells (thymocytes) express a broad range of TCR specificities and affinities, and the key in successful selection is to lose those T cells that recognize self-peptide–self-MHC complexes with either a too low or too high affinity. This produces mature T cells recognizing self-MHC with moderate affinity, while not being reactive to self-peptides [13]. Importantly, the interactions leading to negative selection are much weaker than those required to induce T-cell activation. In this way, negatively selected T cells are deleted as a result of interactions that would not lead to activation in the periphery. Possibly this provides an important safety margin, allowing some fluctuations in MHC and self-peptide expression in the periphery [32].
The qualitative or peptide model predicts that qualitatively different peptides presented in the thymus mediate selection. Thereby, the thymocyte is positively selected for when its TCR interacts with low or intermediate affinity to the presented self-peptide (termed antagonist peptide). The cell is deleted through activation-induced cell death, when it interacts with high affinity to this peptide-MHC complex (termed agonist peptide). Experiments using peptides having amino acid substitutions at residues known to contact the TCR when presented by an MHC molecule, known as altered peptide ligands (APLs), have shown that any given peptide and its variants can form a gradient from antagonist to agonist. Furthermore, for any given TCR a gradient of ligands can be described in terms of agonist qualities [33]. Thus, peptides do not fall within absolute categories, and thymocyte selection probably requires more than these all-or-none interactions [31].
In contrast with the qualitative model, the avidity-based model proposes that low-avidity thymocyte interactions with self-peptide promotes positive selection, and if this recognition passes an avidity threshold, the cell will undergo apoptosis [13]. A large spectrum of functional avidities in the thymocyte population would interfere with thymocyte fate. However, avidity-influencing factors are more or less constant suggesting that the key factors in thymocyte selection are TCR affinity and thymocyte avidity. In contrast with the qualitative model, in which absolute TCR/MHC complex interactions govern the fate of a thymocyte, the avidity-based model integrates multiple TCR/MHC complex interactions to form a gradient defining cell fate. For example, numerous weak TCR binding events or limited high-affinity interactions provide sufficient avidity to induce thymocyte survival. Additionally, high-affinity interactions will provide high avidity and cause deletion. This model allows for a concentration-dependent overlap of positively and negatively selecting signals, as thymocyte signaling is an integrated process [31]. Convincing evidence for avidity-based selection comes from studies showing that low expression of a strong agonist peptide in the thymus mediates positive selection, while high expression of the same peptide causes clonal deletion of reactive thymocytes [34,35]. Recent data has again confirmed these results in favor of the avidity-based model and that emerging cells are possibly autoreactive [36].
Although the exact mechanisms of central T-cell selection remain elusive, it has become clear that thymic presentation of peripheral antigens is essential in generating a self-tolerant T-cell pool. For instance in mice, when positively selected thymocytes encounter medullary thymic epithelial cells (MTECs) functionally deficient of MHC molecules or B7 co-stimulatory receptors, all thymocytes will reach the periphery without negative selection [37,38]. These pathogenic T cells will subsequently cause extensive autoimmunity. Similar effects take place when there is a dysfunction of the AIRE protein. AIRE appears to optimize the presentation of peripheral antigens in the thymus. Deficiency of AIRE causes autoimmune polyendocrinopathy (APECED) characterized by defective negative selection of effector T cells [39]. AIRE deficiency does not inevitably mean deficiency of autoantigen presentation by the thymus. In some cases, the antigen is still presented while autoimmunity initiates. This suggests AIRE also influences thymic selection beyond the antigen expression level, likely by influencing chemokine expression or antigen processing and presentation by MTECs [40]. Absence of AIRE lowers the MTEC’s stimulatory state, resulting in weak avidity interactions for negative selection, allowing high-avidity self-reactive cells to escape to the periphery. Likewise is the case for the myelin basic protein Ac1-9 antigen in EAE [41]. This autoantigen on itself has extremely low affinity for the MHC molecule. Alternatively, an autoantigen being absent from the thymus also causes escape from deletion, for example due to alternative splicing variants having differential expression of isoforms throughout the body, as is the case with the myelin proteolipid protein-antigen in EAE [42].
As the T-cell repertoire is shaped by the numerous interactions that take place during thymocyte development, the thymus can establish susceptibility to or protection from autoimmune disease. Inevitably, some T cells do escape deletion, and these will encounter additional measures such as peripheral tuning in absence of danger signals. Even “successfully selected” T cells may react to autoantigens as antigen recognition is not all-or-none. Greatly enlarging the specificity of the T-cell repertoire, and hence the autoreactive T-cell pool, the cross-reactivity of the TCR gives additional flexibility to T-cell antigen recognition.
TCR CROSS-REACTIVITY
In the past decade, new light has been shed on the specificity of T-cell immunity. Although the TCR remains of given specificity, and is unable to undergo affinity maturation, it is apparent that there is considerable flexibility in TCR antigen recognition. Mathematical models have shown that the T-cell pool is not large enough to give rise to immunity against all possible foreign antigens on the basis of the classical one-TCR–one-epitope model. It has been estimated that for sufficient protection, each T cell should be able to react to as much as 106 structurally similar peptides. This is probably an overestimate, nonetheless it illustrates that most (if not all) TCRs will react to more than one ligand including numerous self-antigens [43,44]. Not only are single T-cell clones able to react to an array of different peptide-MHC complexes, but more T-cell clones are also able to react to one single peptide-MHC complex, indicating substantial cross-reactivity within the T-cell pool [13,45]. Highly variable junctional residues add diversity to the flexible CDRs, able to adapt to the peptide-MHC surface. These processes highly contribute to TCR cross-reactivity and diversity in the T-cell pool [2].
Multiple studies have defined TCR cross-reactivity with the use of APLs [46–48]. Currently, an APL describes any peptide that has one or more residues modified from the wild-type peptide of interest [13]. APL studies have shed new light on TCR antigen recognition. Early studies of molecular mimicry involved a foreign peptide provoking an immune response cross-reactive with a self-antigen, and this peptide often showed obvious sequence homology to a self-peptide [49]. Later studies indicated that only a few amino acid residues of a given peptide were required in TCR/peptide-MHC complex interaction. This insight practically supported the view that different antigens could stimulate the same T cell, as long as they showed similarity on these critical residues [50,51]. This cross-reactivity based on minimal residue similarity furthermore underscores the importance of the CD4 and CD8 coreceptors in providing prolonged T cell/APC contact [45].
Similar studies have also demonstrated that different T-cell subsets exhibit different levels of TCR cross-reactivity. TCRs of CD4 T cells show higher cross-reactivity than do TCRs of CD8 T cells. This could be explained by the generally higher affinity of the CD8 TCR for the peptide-MHC complex compared with the CD4 TCR, possibly associated with the monomer-dimer state of the CD8 coreceptor resulting in a more stringent T-cell activation threshold [52]. Additionally, the more constrained molecular interaction of peptide with MHC class I molecules in comparison with class II MHC molecules could be another cause of a less cross-reactive CD8 T cell pool.
Because T cells are able to respond to antigen over an antigen-concentration range, two effects should be considered concerning T-cell affinity and avidity: First, T cells have quantitatively different responses to related antigens, meaning that T cells will react to low ligand density if their avidity is high, but only to a high ligand density if their avidity for that antigen is low. In addition, the TCR repertoire responding to a single antigen is variable. Those cells expressing high-affinity TCRs will respond to low ligand densities while low affinity TCRs will react to high antigen concentrations [13]. Taken together, this means that a single T cell is able to respond to different related antigens to which it has various affinities on the basis of TCR cross-reactivity. Functional avidity for those individual antigens will determine the response threshold to that antigen.
Next to optimizing T-cell responses, cross-reactivity can also prevent pathogen escape by mutation. Upon secondary antigenic challenge, T cells respond to antigens shared with the original pathogen (due to memory cell expansion). In repeated infection, immunity is thereby optimized for specificity and speed against non-mutating pathogen antigens. This phenomenon is referred to as “original antigenic sin.” However, regularly mutating pathogens such as influenza pose a problem. Repeated mutation can put the pathogen outside the view of T-cell memory, now requiring a primary response for clearance. This is a disadvantage for the host, and TCR cross-reactivity can help the memory cells to cross-react with related, mutated epitopes of the same pathogen, or even between epitopes of related pathogens [53]. Less favorable aspect of cross-reactivity is so-called molecular mimicry: T cells initially directed to a pathogen could develop a destructive response to cross-reactive autoantigens [54]. However, this theory is under strong debate as convincing evidence is lacking [55]. With rapidly mutating pathogens, cross-reactivity offers no solution. HIV for instance mutates so rapidly that a T-cell response never is completely specific for the virus. As numerous viral subtypes exist in one patient, antigenic sin forces the induction of primary responses. Unable to keep pace with the virus, the immune system eventually fails through clonal exhaustion [9].
The cross-reactivity of TCRs illustrates that T cells exhibit flexibility in antigen recognition, and one T cell can react to many ligands (or vice versa) including self-antigens. It has been estimated that up to 50% of TCRs are able to bind autoantigen with dangerous efficiency [26]. Add to this the existence of dual αβTCR+ T cells displaying auto-reactivity and escape from deletion [56–58], the question arises how this situation is controlled in so many of us. Recent research has shown that CD4+ CD25+ Treg limit the risk of autoimmunity caused by cross-reactive TCRs [59]. As Treg cells impose their regulatory control at antigen levels lower than those required to activate effector cells, it is believed that Treg exert dominant control on autoreactive cells with lower sensitivity while still allowing high-avidity reactions to develop. In this setting, it is known that high antigen levels and strong co-stimulation (as present in infection) are factors that can overrule Treg suppression, which possibly focuses on inhibiting CD4 helper cells [60–62]. Additionally, it has been shown that Treg home to the target organ of the autoimmune response, which coincides with recovery from autoimmunity [63].
Cross-reactivity illustrates that besides individual deletion of autoreactive T cells in the thymus, more mechanisms probably govern tolerance. It appears that upon activation, T cells can undergo extensive functional maturation and modification. These phenomena are of special importance, as it poses problems and solutions for autoimmunity.
AVIDITY MATURATION AND SELECTIVE EXPANSION
During secondary antigen encounter, approximately two- to fourfold increases in TCR affinity occur compared with initial antigen encounter. This probably happens via selective expansion of memory cells expressing the highest affinity TCR [64]. In contrast with B cell–mediated antibody production, which shows considerable affinity maturation, there may be a functional “ceiling” to T-cell affinity. T cells expressing high-affinity TCRs may become ineffective if serial TCR-triggering and MHC sampling is impaired by extended TCR binding or become unresponsive when activated without co-stimulation. If affinity maturation were the only way of fine-tuning an immune response, the T-cell response would become limited by its own restraints. Therefore, T cells have evolved avidity maturation as an optimization method.
For example, CD8 T-cell activation caused by acute viral infection can induce a 50- to 70-fold increase in the T cell’s functional avidity without selection of a higher affinity TCR in vivo [65]. Maximal T-cell avidity in this case was reached 8 days after initial infection and was passed to memory T cells for the life of the host. These results are supported by in vitro data using T-cell clones [66] and shows that T cells undergo profound functional maturation upon activation.
It is known that stimulation of low-avidity T cells requires co-ligation of CD8, however this is often dispensable in high-avidity cells [67,68]. T cells are very susceptible to CD8 co-ligation at the initiation of an antiviral response, while 8 days later following avidity maturation, this ligation is expendable [65]. Furthermore, no changes in TCR affinity or expression of adhesion or co-stimulatory molecules were found, which could explain the enhanced antigen responsiveness. Instead, they identified a marked increase in Lck expression. This kinase is associated with the cytoplasmic domain of the CD4 and CD8 coreceptors, mediating ITAM phosphorylation on accessory chains of the TCR complex. Additionally, changes in the TCR-surrounding plasma membrane were found to aid increased avidity after T-cell activation [69]. It appears that T cells will gradually reorganize and cholesterol-enrich their membrane lipid rafts upon activation, in which the TCR and many co-stimulatory molecules are embedded. This reorganization enhances TCR cross-linking and the formation of the SMAC, allowing T cells to bind multimeric MHC complexes with improved efficiency, enhancing intra-cellular signaling. This allows more efficient recognition of low-density peptide-MHC complexes.
Besides the avidity maturation observed on the single cell level, the T-cell response as a whole can display progressive maturation. Refinement of the TCR repertoire takes place via selection of optimal avidity clones [64,70,71]. At the initiation of a primary response, high-avidity T cells, expressing high-affinity TCRs, will dominate over low-affinity cells. During the response, intermediate- and high-affinity T cells have a proliferative advantage and the population will express an increasingly optimal TCR range of “narrowing” affinity, thereby exhibiting avidity maturation. The loss of low-affinity T cells may be obvious in this process, nonetheless the high-affinity T cells expanding at the initiation of the response can become deleted when the response progresses. Leaving room for intermediate-affinity cells to dominate, this is mediated by the increasing antigen dose being present during the course of infection [72]. This mechanism ensures the selective deletion of high-avidity T cells from the reactive pool and guarantees a consistent optimization of the activation threshold. Constant selection mediates avidity maturation and maintains control over T-cell reactivity. However, in some cases avidity maturation can cause the progression from local inflammation to autoimmune disease. This shows important in the progressive pathology of autoimmune diabetes. Local inflammation will cause an accumulation of T cells while avidity maturation subsequently drives the inflammation progressively to diabetes. As illustrated in non-obese diabetic (NOD) mice, timely treatment of prediabetic mice with relevant antigenic peptide will cause deletion of high-avidity clones, slowing down the avidity maturation and inhibiting the progression from insulitis to diabetes [73,74].
This pathogenesis is probably important in the development of many autoimmunities originating from (chronic) inflammation and can explain the progressive severity of many autoimmunities. Evidently, this phenomenon differs from molecular mimicry as the causative inflammation is not caused by a pathogen mediating auto-immunity via cross-reactivity. Instead, inflammation is likely caused by low-avidity autoreactive T cells accumulating on site, gradually increasing in efficiency via avidity maturation. Certainly, the inflammation could very well be initiated by a pathogen or trauma, causing a temporary rise in local autoantigen presentation via tissue-damage, facilitating the activation of the low-avidity T cells. Subsequently, as avidity maturation progresses, the reactive T-cell pool can react to the physiological autoantigen levels present when the trauma has subsided, and the inflammation can spread.
T-CELL TUNING
Based on the avidity-based model of thymocyte selection, autoreactive T cells entering the periphery are of low avidity and hence insensitive to the autoantigen levels presented here. These T cells may still be reactive to foreign antigen to which they may exhibit sufficient cross-reactive avidity when co-stimulatory conditions are optimal. During thymic development, T cells are able to desensitize (tune) their avidity, altering their survival and future reaction to antigens via modifying avidity-regulating mechanisms.
Although the phenomenon remains elusive, a classic example involves CD5. T cell–expressed CD5 can negatively influence TCR signaling and promote thymocyte positive selection [31,75]. Additionally, thymocytes are able to up- or downregulate their CD4 or CD8 expression (and thus the recruitment of the Lck signaling molecule to the CD3 complex) thereby directly influencing their maturation by modulating their avidity [76,77]. Furthermore, tuning can also involve the phosphorylation state of the regulating signal cascades. Recently, it has been shown that ZAP-70 gain-of-function transgenic mice, exhibiting enhanced basal phosphorylation of ZAP-70 and LAT, display reduced TCR and CD5 levels together with decreased cytokine production and activation. This hyporesponsive state could be explained by the reduced TCR and CD5 levels. Nonetheless, it appeared that TCR-induced activation signals were also more rapidly downregulated, indicating a role for increased dephosphorylation by PTPases. Furthermore, this tuned state could be inverted by inhibiting TCR interactions with autoantigen for several days. This indicates that peripheral tuning requires constant interaction of the TCR with the autoantigen and that constant exposure will tolerize T cells [78].
Additionally, studies using altered peptide ligands have shown an added phenomenon of tuning. T cells matured in the presence of endogenous peptide are able to respond to a weak-agonist APL of that antigen, as well as to the endogenous peptide itself. However, T cells matured in the presence of the APL lost the ability to respond to it but retained their ability to react to the endogenous peptide. This is in line with reports that APLs that fail to induce T-cell activation in the periphery can nevertheless be used as ligands in thymic positive selection. This tuning process by non-deleting self-antigens induces mature T cells that have high avidity for foreign antigens while having lost self-reactivity that was present in early stages of development [13,79].
Besides tuning in the thymus, mature T cells can also undergo tuning in the periphery, as proposed by the tunable activation-threshold model by Grossman and Paul [80]. Autoantigen presented by resting APCs in the periphery appears to be essential in maintaining T-cell self-tolerance. Low-level presentation of autoantigen directs to a state of tolerance to those antigens. It has been shown that CD4 TCR transgenic T cells adoptively transferred into a lymphopenic host expressing their specific antigen in the periphery will undergo an initial limited expansion to “populate” the host, after which these cells became increasingly insensitive to antigenic stimulation. Within 38 days, the T cells required 30-fold greater amounts of antigen to become activated, with 90% reduction in cytokine production and a clear desensitization of proliferation while TCR levels remained normal. Surprisingly, transfer of these hyporesponsive cells into a second lymphopenic host (not bearing the antigen), induced a gradual regain of T-cell responsiveness and cytokine production with gradual expansion after 7 days. If this second host did express the antigen, the hyporesponsive cells underwent a slow repopulation expansion as seen in the original host, hereafter entering an even more profound hyporesponsive state [81,82]. These results form strong evidence that the autoantigen tolerant state of T cells in the periphery is dependent upon constant autoantigen presentation intensity. Disrupting the steady-state of autoantigen presentation intensity will disturb the tuned equilibrium and set off a T-cell response. It remains to be confirmed if the same mechanisms of tuning apply to T cells being activated by their ligand under immunizing conditions, subsequently undergoing avidity-maturation.
These means of tuning the immune response prove important in maintaining peripheral tolerance and make it more difficult to induce an autoimmune response. In this way, a “reserve” of autoantigen-specific cells remains available when antigen levels suddenly rise. Tumors can cause a sudden rise in self-antigen presentation, and triggering tuned cells can induce potent antitumor immunity.
In order for autoimmunity to develop, it is likely that the tolerance of tuned T cells needs to be overcome. There are several mechanisms by which this could take place. Most obvious would be a sudden rise in antigen levels, under conditions not favoring additional tuning. In this case, infection can induce the necessary upregulation of co-stimulation on the local APCs while also providing autoantigens by causing tissue damage [83]. Furthermore, this inflammatory environment could induce alterations in antigen-processing by the APC. New autoantigens could also be presented as a result of changes in the cellular environment, inducing changes in protein processing or synthesis [84]. This could introduce antigens the T cell was not tuned for. In addition, there could also be a role for reduced expression of self-antigen in the periphery [81]. As constant expression of antigen will tune T cells, a prolonged decrease in the levels of antigen presented causes “resensitization” of these cells. Caused by a physiological disruption, a subsequent reestablishment of antigenic load could prove sufficient to induce activation of these now sensitized T cells. Nonetheless, all these changes in the surroundings of the T cell will likely induce further tuning, if there is no activated APC to supply essential activation signals. As suggested in the danger model, an APC needs to sense a threat in order to be activated and induce an immune response. As proposed by Matzinger [85], the factors controlling the generation of immunity are possibly the tissues themselves. Under healthy conditions, tissues will maintain tolerance; under stress they will promote the generation of immunity. The end point being a stress-induced tissue-specific response, in all immune reactions the activated APC plays a key role. Combining an activated APC with the above-mentioned mechanisms could result in tuned T cells becoming activated to react against autoantigens. From there on, cross-reactivity, selective expansion, and avidity maturation will ensure an optimal and progressive response. It appears that the same mechanisms could be considered to terminate the initiated response.
IMPLICATIONS FOR AUTOIMMUNITY AND IMMUNOTHERAPY
The classic view of T-cell selection proposes that the balance between autoreactivity and tolerance is regulated by the individual exclusion of self-reactive cells from the circulation. This appears to be only partially the case, and recent findings have shed new light on mechanisms shaping the T-cell repertoire. The regulation of T-cell tolerance shows flexibility, providing a window of opportunity for the treatment of autoimmune disease or the induction of successful immunotherapy.
Vitiligo and melanoma are two conditions in which the state of the immune system is clearly opposed. In both conditions, melanocyte antigen-specific cytotoxic T cells are present. Nevertheless, in vitiligo these cells mediate destruction of melanocytes and cause the characteristic depigmented lesions. In contrast, melanoma-infiltrating T cells will often not achieve any effective response. Recent research has shown that T cells in both pathologies are directed against identical melanocyte antigens and exhibit the same activation status. However, melanoma-infiltrating T cells display a profound reduction in the affinity for their antigens. Additionally, T cells from vitiligo patients were capable of efficient IFN-γ production and TCR downregulation in response to HLA-matched melanoma cells, whereas melanoma-derived T cells were not [86]. These findings suggest that in melanoma the slow rise in peripheral antigen presentation of melanocyte antigens, resulting from progressive tumor necrosis, most likely tunes responsive T cells. Especially in the first phases of growth, a melanoma does not provide sufficient danger signals to induce proper APC activation. In later phases of progression and metastasis, it is likely that the minor APC activation induced by local tissue damage is unable to overrule the tuned T-cell activation thresholds. Moreover, it is believed that antigen-shedding from progressing tumors activates natural Treg, thereby impeding development of efficient T-cell responses [62].
Recent research has shown that vaccination of autologous mela-noma cells transduced with GM-CSF can induce successful anti-mela-noma immunity. This is illustrated by the long-term survival of 6 out of 14 patients, and the additional development of vitiligo in 2 of these patients. Remarkably, vitiligo and long-term survival were only observed in patients experiencing nonevaluable disease [87]. This suggests that lowered presentation of melanocyte-antigens under tolerizing conditions in the periphery could result in a resensitized and higher avidity T-cell repertoire. This could ensure better vaccine efficacy and illustrates that supplying proper activation signals to an antigen-sensitive T-cell population can result in successful and specific immunotherapy. Besides the induction of immunity, future immunotherapy should also focus on breaking the tolerance of tuned T cells. As this tolerant state often impairs an efficient response, combining the two could drive tolerance over the edge. The most fit strategy here is probably the use of adoptive cell transfer (ACT), as reviewed by Overwijk [88]. The isolation and subsequent culture of T cells under resensitizing conditions could provide an expanded, antigen-sensitive T-cell population to be transferred into the donor. Furthermore, it allows for the genetic optimization of these cells before their use in therapy. In order to promote the activation of these cells and cross-reactive residual T cells, drive avidity maturation, and provide the necessary cytokines, the activation of local APCs via vaccination is absolutely essential. Vaccination will only be optimal though upon transfer of antigen-sensitive T cells and the use of strong adjuvants combined with sufficient immunogenic antigen to bypass the inhibition of natural Treg. Additionally, it has recently been shown that the success of adoptive transfer can depend on the presence of CD4 helper T cells providing IL-2, especially in the absence of Treg. When limited amounts of antigen-specific CD8 cells are transferred, sustained immunity is dependent upon this IL-2 and lost in the presence of Treg cells [62]. Moreover, exogenous IL-2 therapy failed in the presence of regulatory cells, illustrating that Tregs are able to suppress helper T cells and prevent autoreactivity under the tested circumstances. Depletion of Treg could therefore boost immunotherapy by uninhibited T-cell help during the activation and effector phase. Additionally, lymphodepletion of the host before adoptive transfer enhances subsequent autoimmunity [89], possibly by depleting the suppressive Treg pool. Novel immunotherapy designs should aim at combining the use of resensitized T cells with activation under noninhibited conditions when pursuing an optimal effect.
On the contrary, the restraints in immunotherapy constitute the excess in T cell–mediated autoimmune disease. Therefore, therapies should aim at re-establishing tolerance via T-cell tuning. For auto-immune diabetes, it has been shown that treating prediabetic NOD mice with agonistic APL peptide of the causative antigen protects mice from developing the disease. Unexpectedly, reactive T cells were not deleted in these mice due to overtriggering by the APL. Instead, the cells exhibited a significantly less cytotoxic phenotype while still infiltrating the islets in great numbers [73]. Indicative of T-cell tuning, this illustrates how autoimmunity could be prevented when the causative antigen is known. Use of APLs presented on peripheral immature APCs before disease onset could prove useful in tuning reactive T cells and preventing disease. Besides this possible preventive strategy, stopping progressive autoimmune disease will probably require the influence of Treg as these are able to suppress autoreactive T cells and mediate recovery from autoimmune disease [63]. Isolation and expansion of Treg, followed by an adoptive transfer of these cells could provide recovery from autoimmune disease. Importantly, Treg seem to influence CD4 T helper cells in their stimulatory function. In this context, recent data shows that antibody-mediated autoimmune responses can also be interrupted by Treg [90]. Because these cells appear to be powerful regulators of the humoral and adaptive immune responses, they could be useful targets in novel therapeutic approaches. Increasing Treg presence or function could thereby weaken co-stimulation by APCs and induce T-cell tuning.
Taken together, future immunotherapeutic designs should exploit these insights into the regulation of T-cell activity. These designs need to take into account the fact that the balance between tolerance and autoimmunity is flexible, and this will have implications for the course and outcome of therapy.
Contributor Information
Jasper G. van den Boorn, Netherlands Institute for Pigment Disorders and Department of Dermatology, AMC, University of Amsterdam, Amsterdam, The Netherlands
I. Caroline Le Poole, Department of Pathology, Loyola University Medical Center, Maywood, Illinois, USA.
Rosalie M. Luiten, Netherlands Institute for Pigment Disorders and Department of Dermatology, AMC, University of Amsterdam, Amsterdam, The Netherlands
References
- 1.Chambers CA, Kuhns MS, Egen JG, Allison JP. CTLA-4-mediated inhibition in regulation of T cell responses: mechanisms and manipulation in tumor immunotherapy. Annu Rev Immunol. 2001;19:565–594. doi: 10.1146/annurev.immunol.19.1.565. [DOI] [PubMed] [Google Scholar]
- 2.Garcia KC, Teyton L, Wilson IA. Structural basis of T cell recognition. Annu Rev Immunol. 1999;17:369–397. doi: 10.1146/annurev.immunol.17.1.369. [DOI] [PubMed] [Google Scholar]
- 3.van der Merwe PA, Davis SJ. Molecular interactions mediating T cell antigen recognition. Annu Rev Immunol. 2003;21:659–684. doi: 10.1146/annurev.immunol.21.120601.141036. [DOI] [PubMed] [Google Scholar]
- 4.Toes RE, Schoenberger SP, van d V, Offringa R, Melief CJ. CD40-CD40Li-gand interactions and their role in cytotoxic T lymphocyte priming and anti-tumor immunity. Semin Immunol. 1998;10:443–448. doi: 10.1006/smim.1998.0147. [DOI] [PubMed] [Google Scholar]
- 5.Heath WR, Carbone FR. Cross-presentation in viral immunity and self-tolerance. Nat Rev Immunol. 2001;1:126–134. doi: 10.1038/35100512. [DOI] [PubMed] [Google Scholar]
- 6.Norbury CC, Basta S, Donohue KB, Tscharke DC, Princiotta MF, Berglund P, Gibbs J, Bennink JR, Yewdell JW. CD8+ T cell cross-priming via transfer of proteasome substrates. Science. 2004;304:1318–1321. doi: 10.1126/science.1096378. [DOI] [PubMed] [Google Scholar]
- 7.Wolkers MC, Brouwenstijn N, Bakker AH, Toebes M, Schumacher TN. Antigen bias in T cell cross-priming. Science. 2004;304:1314–1317. doi: 10.1126/science.1096268. [DOI] [PubMed] [Google Scholar]
- 8.Alegre ML, Frauwirth KA, Thompson CB. T-cell regulation by CD28 and CTLA-4. Nat Rev Immunol. 2001;1:220–228. doi: 10.1038/35105024. [DOI] [PubMed] [Google Scholar]
- 9.Nikolich-Zugich J, Slifka MK, Messaoudi I. The many important facets of T-cell repertoire diversity. Nat Rev Immunol. 2004;4:123–132. doi: 10.1038/nri1292. [DOI] [PubMed] [Google Scholar]
- 10.Friedl P, den Boer AT, Gunzer M. Tuning immune responses: Diversity and adaptation of the immunological synapse. Nat Rev Immunol. 2005;5:532–545. doi: 10.1038/nri1647. [DOI] [PubMed] [Google Scholar]
- 11.Huppa JB, Gleimer M, Sumen C, Davis MM. Continuous T cell receptor signaling required for synapse maintenance and full effector potential. Nat Immunol. 2003;4:749–755. doi: 10.1038/ni951. [DOI] [PubMed] [Google Scholar]
- 12.Margulies DH. TCR avidity: It’s not how strong you make it, it’s how you make it strong. Nat Immunol. 2001;2:669–670. doi: 10.1038/90601. [DOI] [PubMed] [Google Scholar]
- 13.Anderton SM, Wraith DC. Selection and fine-tuning of the autoimmune T-cell repertoire. Nat Rev Immunol. 2002;2:487–498. doi: 10.1038/nri842. [DOI] [PubMed] [Google Scholar]
- 14.Cawthon AG, Alexander-Miller MA. Optimal colocalization of TCR and CD8 as a novel mechanism for the control of functional avidity. J Immunol. 2002;169:3492–3498. doi: 10.4049/jimmunol.169.7.3492. [DOI] [PubMed] [Google Scholar]
- 15.Bromley SK, Burack WR, Johnson KG, Somersalo K, Sims TN, Sumen C, Davis MM, Shaw AS, Allen PM, Dustin ML. The immunological synapse. Annu Rev Immunol. 2001;19:375–396. doi: 10.1146/annurev.immunol.19.1.375. [DOI] [PubMed] [Google Scholar]
- 16.Jacobelli J, Andres PG, Boisvert J, Krummel MF. New views of the immunological synapse: Variations in assembly and function. Curr Opin Immunol. 2004;16:345–352. doi: 10.1016/j.coi.2004.03.008. [DOI] [PubMed] [Google Scholar]
- 17.Dustin ML, Bromley SK, Kan Z, Peterson DA, Unanue ER. Antigen receptor engagement delivers a stop signal to migrating T lymphocytes. Proc Natl Acad Sci U S A. 1997;94:3909–3913. doi: 10.1073/pnas.94.8.3909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mustelin T, Tasken K. Positive and negative regulation of T-cell activation through kinases and phosphatases. Biochem J. 2003;371:15–27. doi: 10.1042/BJ20021637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Samelson LE. Signal transduction mediated by the T cell antigen receptor: The role of adapter proteins. Annu Rev Immunol. 2002;20:371–394. doi: 10.1146/annurev.immunol.20.092601.111357. [DOI] [PubMed] [Google Scholar]
- 20.Bunnell SC, Kapoor V, Trible RP, Zhang W, Samelson LE. Dynamic actin polymerization drives T cell receptor-induced spreading: A role for the signal transduction adaptor LAT. Immunity. 2001;14:315–329. doi: 10.1016/s1074-7613(01)00112-1. [DOI] [PubMed] [Google Scholar]
- 21.Dustin ML, Cooper JA. The immunological synapse and the actin cytoskeleton: Molecular hardware for T cell signaling. Nat Immunol. 2000;1:23–29. doi: 10.1038/76877. [DOI] [PubMed] [Google Scholar]
- 22.Croft M. Co-stimulatory members of the TNFR family: Keys to effective T-cell immunity? Nat Rev Immunol. 2003;3:609–620. doi: 10.1038/nri1148. [DOI] [PubMed] [Google Scholar]
- 23.Kemp EH, Ajjan RA, Waterman EA, Gawkrodger DJ, Cork MJ, Watson PF, Weetman AP. Analysis of a microsatellite polymorphism of the cytotoxic T-lymphocyte antigen-4 gene in patients with vitiligo. Br J Dermatol. 1999;140:73–78. doi: 10.1046/j.1365-2133.1999.02610.x. [DOI] [PubMed] [Google Scholar]
- 24.Ueda H, Howson JM, Esposito L, Heward J, Snook H, Chamberlain G, Rainbow DB, Hunter KM, Smith AN, Di GG, Herr MH, Dahlman I, Payne F, Smyth D, Lowe C, Twells RC, Howlett S, Healy B, Nutland S, Rance HE, Everett V, Smink LJ, Lam AC, Cordell HJ, Walker NM, Bordin C, Hulme J, Motzo C, Cucca F, Hess JF, Metzker ML, Rogers J, Gregory S, Allahabadia A, Nithiyananthan R, Tuomilehto-Wolf E, Tuomilehto J, Bingley P, Gillespie KM, Undlien DE, Ronningen KS, Guja C, Ionescu-Tirgoviste C, Savage DA, Maxwell AP, Carson DJ, Patterson CC, Franklyn JA, Clayton DG, Peterson LB, Wicker LS, Todd JA, Gough SC. Association of the T-cell regulatory gene CTLA4 with susceptibility to autoimmune disease. Nature. 2003;423:506–511. doi: 10.1038/nature01621. [DOI] [PubMed] [Google Scholar]
- 25.Foley LM, Lowe NJ, Misheloff E, Tiwari JL. Association of HLA-DR4 with vitiligo. J Am Acad Dermatol. 1983;8:39–40. doi: 10.1016/s0190-9622(83)80279-5. [DOI] [PubMed] [Google Scholar]
- 26.Goodnow CC, Sprent J, Fazekas de St GB, Vinuesa CG. Cellular and genetic mechanisms of self tolerance and autoimmunity. Nature. 2005;435:590–597. doi: 10.1038/nature03724. [DOI] [PubMed] [Google Scholar]
- 27.Neurath MF, Weigmann B, Finotto S, Glickman J, Nieuwenhuis E, Iijima H, Mizoguchi A, Mizoguchi E, Mudter J, Galle PR, Bhan A, Autschbach F, Sullivan BM, Szabo SJ, Glimcher LH, Blumberg RS. The transcription factor T-bet regulates mucosal T cell activation in experimental colitis and Crohn’s disease. J Exp Med. 2002;195:1129–1143. doi: 10.1084/jem.20011956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tokuhiro S, Yamada R, Chang X, Suzuki A, Kochi Y, Sawada T, Suzuki M, Nagasaki M, Ohtsuki M, Ono M, Furukawa H, Nagashima M, Yoshino S, Mabuchi A, Sekine A, Saito S, Takahashi A, Tsunoda T, Nakamura Y, Yamamoto K. An intronic SNP in a RUNX1 binding site of SLC22A4, encoding an organic cation transporter, is associated with rheumatoid arthritis. Nat Genet. 2003;35:341–348. doi: 10.1038/ng1267. [DOI] [PubMed] [Google Scholar]
- 29.Bennett CL, Christie J, Ramsdell F, Brunkow ME, Ferguson PJ, Whitesell L, Kelly TE, Saulsbury FT, Chance PF, Ochs HD. The immune dysregulation, polyendocrinopathy, enteropathy, X-linked syndrome (IPEX) is caused by mutations of FOXP3. Nat Genet. 2001;27:20–21. doi: 10.1038/83713. [DOI] [PubMed] [Google Scholar]
- 30.Ziegler SF. FOXP3: Of Mice and Men. Annu Rev Immunol. 2005;24:209–226. doi: 10.1146/annurev.immunol.24.021605.090547. [DOI] [PubMed] [Google Scholar]
- 31.Sebzda E, Mariathasan S, Ohteki T, Jones R, Bachmann MF, Ohashi PS. Selection of the T cell repertoire. Annu Rev Immunol. 1999;17:829–874. doi: 10.1146/annurev.immunol.17.1.829. [DOI] [PubMed] [Google Scholar]
- 32.Ohashi PS. T-cell signalling and autoimmunity: Molecular mechanisms of disease. Nat Rev Immunol. 2002;2:427–438. doi: 10.1038/nri822. [DOI] [PubMed] [Google Scholar]
- 33.Bachmann MF, Speiser DE, Zakarian A, Ohashi PS. Inhibition of TCR triggering by a spectrum of altered peptide ligands suggests the mechanism for TCR antagonism. Eur J Immunol. 1998;28:3110–3119. doi: 10.1002/(SICI)1521-4141(199810)28:10<3110::AID-IMMU3110>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- 34.Ashton-Rickardt PG, Bandeira A, Delaney JR, Van Kaer L, Pircher HP, Zinkernagel RM, Tonegawa S. Evidence for a differential avidity model of T cell selection in the thymus. Cell. 1994;76:651–663. doi: 10.1016/0092-8674(94)90505-3. [DOI] [PubMed] [Google Scholar]
- 35.Sebzda E, Wallace VA, Mayer J, Yeung RS, Mak TW, Ohashi PS. Positive and negative thymocyte selection induced by different concentrations of a single peptide. Science. 1994;263:1615–1618. doi: 10.1126/science.8128249. [DOI] [PubMed] [Google Scholar]
- 36.Laurie KL, La Gruta NL, Koch N, van Driel IR, Gleeson PA. Thymic expression of a gastritogenic epitope results in positive selection of self-reactive pathogenic T cells. J Immunol. 2004;172:5994–6002. doi: 10.4049/jimmunol.172.10.5994. [DOI] [PubMed] [Google Scholar]
- 37.Gao JX, Zhang H, Bai XF, Wen J, Zheng X, Liu J, Zheng P, Liu Y. Perinatal blockade of b7-1 and b7-2 inhibits clonal deletion of highly pathogenic autoreactive T cells. J Exp Med. 2002;195:959–971. doi: 10.1084/jem.20011948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Laufer TM, DeKoning J, Markowitz JS, Lo D, Glimcher LH. Unopposed positive selection and autoreactivity in mice expressing class II MHC only on thymic cortex. Nature. 1996;383:81–85. doi: 10.1038/383081a0. [DOI] [PubMed] [Google Scholar]
- 39.Anderson MS, Venanzi ES, Chen Z, Berzins SP, Benoist C, Mathis D. The cellular mechanism of Aire control of T cell tolerance. Immunity. 2005;23:227–239. doi: 10.1016/j.immuni.2005.07.005. [DOI] [PubMed] [Google Scholar]
- 40.Kuroda N, Mitani T, Takeda N, Ishimaru N, Arakaki R, Hayashi Y, Bando Y, Izumi K, Takahashi T, Nomura T, Sakaguchi S, Ueno T, Takahama Y, Uchida D, Sun S, Kajiura F, Mouri Y, Han H, Matsushima A, Yamada G, Matsumoto M. Development of autoimmunity against transcriptionally unrepressed target antigen in the thymus of Aire-deficient mice. J Immunol. 2005;174:1862–1870. doi: 10.4049/jimmunol.174.4.1862. [DOI] [PubMed] [Google Scholar]
- 41.Liu GY, Fairchild PJ, Smith RM, Prowle JR, Kioussis D, Wraith DC. Low avidity recognition of self-antigen by T cells permits escape from central tolerance. Immunity. 1995;3:407–415. doi: 10.1016/1074-7613(95)90170-1. [DOI] [PubMed] [Google Scholar]
- 42.Anderson AC, Nicholson LB, Legge KL, Turchin V, Zaghouani H, Kuchroo VK. High frequency of autoreactive myelin proteolipid protein-specific T cells in the periphery of naive mice: Mechanisms of selection of the self-reactive repertoire. J Exp Med. 1995;191:761–770. doi: 10.1084/jem.191.5.761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kuchroo VK, Anderson AC, Waldner H, Munder M, Bettelli E, Nicholson LB. T cell response in experimental autoimmune encephalomyelitis (EAE): Role of self and cross-reactive antigens in shaping, tuning, and regulating the autopathogenic T cell repertoire. Annu Rev Immunol. 2002;20:101–123. doi: 10.1146/annurev.immunol.20.081701.141316. [DOI] [PubMed] [Google Scholar]
- 44.Mason D. A very high level of crossreactivity is an essential feature of the T-cell receptor. Immunol Today. 1998;19:395–404. doi: 10.1016/s0167-5699(98)01299-7. [DOI] [PubMed] [Google Scholar]
- 45.Wilson DB, Wilson DH, Schroder K, Pinilla C, Blondelle S, Houghten RA, Garcia KC. Specificity and degeneracy of T cells. Mol Immunol. 2004;40:1047–1055. doi: 10.1016/j.molimm.2003.11.022. [DOI] [PubMed] [Google Scholar]
- 46.Gundlach BR, Wiesmuller KH, Junt T, Kienle S, Jung G, Walden P. Specificity and degeneracy of minor histocompatibility antigen-specific MHC-restricted CTL. J Immunol. 1996;156:3645–3651. [PubMed] [Google Scholar]
- 47.Hemmer B, Vergelli M, Pinilla C, Houghten R, Martin R. Probing degeneracy in T-cell recognition using peptide combinatorial libraries. Immunol Today. 1998;19:163–168. doi: 10.1016/s0167-5699(97)01217-6. [DOI] [PubMed] [Google Scholar]
- 48.Udaka K, Wiesmuller KH, Kienle S, Jung G, Walden P. Self-MHC-restricted peptides recognized by an alloreactive T lymphocyte clone. J Immunol. 1996;157:670–678. [PubMed] [Google Scholar]
- 49.Fujinami RS, Oldstone MB. Amino acid homology between the encephalitogenic site of myelin basic protein and virus: Mechanism for autoimmunity. Science. 1985;230:1043–1045. doi: 10.1126/science.2414848. [DOI] [PubMed] [Google Scholar]
- 50.Albert LJ, Inman RD. Molecular mimicry and autoimmunity. N Engl J Med. 1999;341:2068–2074. doi: 10.1056/NEJM199912303412707. [DOI] [PubMed] [Google Scholar]
- 51.Evavold BD, Sloan-Lancaster J, Wilson KJ, Rothbard JB, Allen PM. Specific T cell recognition of minimally homologous peptides: evidence for multiple endogenous ligands. Immunity. 1995;2:655–663. doi: 10.1016/1074-7613(95)90010-1. [DOI] [PubMed] [Google Scholar]
- 52.Davis MM, Boniface JJ, Reich Z, Lyons D, Hampl J, Arden B, Chien Y. Ligand recognition by alpha beta T cell receptors. Annu Rev Immunol. 1998;16:523–544. doi: 10.1146/annurev.immunol.16.1.523. [DOI] [PubMed] [Google Scholar]
- 53.Kim SK, Cornberg M, Wang XZ, Chen HD, Selin LK, Welsh RM. Private specificities of CD8 T cell responses control patterns of heterologous immunity. J Exp Med. 2005;201:523–533. doi: 10.1084/jem.20041337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Fujinami RS, Oldstone MB. Molecular mimicry as a mechanism for virus-induced autoimmunity. Immunol Res. 1989;8:3–15. doi: 10.1007/BF02918552. [DOI] [PubMed] [Google Scholar]
- 55.Benoist C, Mathis D. Autoimmunity provoked by infection: How good is the case for T cell epitope mimicry? Nat Immunol. 2001;2:797–801. doi: 10.1038/ni0901-797. [DOI] [PubMed] [Google Scholar]
- 56.Davodeau F, Peyrat MA, Romagne F, Necker A, Hallet MM, Vie H, Bonneville M. Dual T cell receptor beta chain expression on human T lymphocytes. J Exp Med. 1995;181:1391–1398. doi: 10.1084/jem.181.4.1391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Paterson RK, Bluethmann H, Tseng P, Dunlap A, Finkel TH. Development and function of autospecific dual TCR+ T lymphocytes. Int Immunol. 1999;11:113–119. doi: 10.1093/intimm/11.1.113. [DOI] [PubMed] [Google Scholar]
- 58.von BH, Kirberg J, Rocha B. An unusual lineage of alpha/beta T cells that contains autoreactive cells. J Exp Med. 1991;174:1001–1008. doi: 10.1084/jem.174.5.1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Stephens LA, Gray D, Anderton SM. CD4+ CD25+ regulatory T cells limit the risk of autoimmune disease arising from T cell receptor crossreactivity. Proc Natl Acad Sci U S A. 2005;102:17418–17423. doi: 10.1073/pnas.0507454102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.George TC, Bilsborough J, Viney JL, Norment AM. High antigen dose and activated dendritic cells enable Th cells to escape regulatory T cell-mediated suppression in vitro. Eur J Immunol. 2003;33:502–511. doi: 10.1002/immu.200310026. [DOI] [PubMed] [Google Scholar]
- 61.Pasare C, Medzhitov R. Toll pathway-dependent blockade of CD4+ CD25+ T cell-mediated suppression by dendritic cells. Science. 2003;299:1033–1036. doi: 10.1126/science.1078231. [DOI] [PubMed] [Google Scholar]
- 62.Antony PA, Piccirillo CA, Akpinarli A, Finkelstein SE, Speiss PJ, Surman DR, Palmer DC, Chan CC, Klebanoff CA, Overwijk WW, Rosenberg SA, Restifo NP. CD8+ T cell immunity against a tumor/self-antigen is augmented by CD4+ T helper cells and hindered by naturally occurring T regulatory cells. J Immunol. 2005;174:2591–2601. doi: 10.4049/jimmunol.174.5.2591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.McGeachy MJ, Stephens LA, Anderton SM. Natural recovery and protection from autoimmune encephalomyelitis: contribution of CD4+ CD25+ regulatory cells within the central nervous system. J Immunol. 2005;175:3025–3032. doi: 10.4049/jimmunol.175.5.3025. [DOI] [PubMed] [Google Scholar]
- 64.Busch DH, Pamer EG. T cell affinity maturation by selective expansion during infection. J Exp Med. 1999;189:701–710. doi: 10.1084/jem.189.4.701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Slifka MK, Whitton JL. Functional avidity maturation of CD8(+) T cells without selection of higher affinity TCR. Nat Immunol. 2001;2:711–717. doi: 10.1038/90650. [DOI] [PubMed] [Google Scholar]
- 66.Hesse MD, Karulin AY, Boehm BO, Lehmann PV, Tary-Lehmann M. A T cell clone’s avidity is a function of its activation state. J Immunol. 2001;167:1353–1361. doi: 10.4049/jimmunol.167.3.1353. [DOI] [PubMed] [Google Scholar]
- 67.Bachmann MF, Gallimore A, Linkert S, Cerundolo V, Lanzavecchia A, Kopf M, Viola A. Developmental regulation of Lck targeting to the CD8 coreceptor controls signaling in naive and memory T cells. J Exp Med. 1999;189:1521–1530. doi: 10.1084/jem.189.10.1521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Kerry SE, Buslepp J, Cramer LA, Maile R, Hensley LL, Nielsen AI, Kavathas P, Vilen BJ, Collins EJ, Frelinger JA. Interplay between TCR affinity and necessity of coreceptor ligation: high-affinity peptide-MHC/TCR interaction overcomes lack of CD8 engagement. J Immunol. 2003;171:4493–4503. doi: 10.4049/jimmunol.171.9.4493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Fahmy TM, Bieler JG, Edidin M, Schneck JP. Increased TCR avidity after T cell activation: A mechanism for sensing low-density antigen. Immunity. 2001;14:135–143. [PubMed] [Google Scholar]
- 70.Fasso M, Anandasabapathy N, Crawford F, Kappler J, Fathman CG, Ridgway WM. T cell receptor (TCR)-mediated repertoire selection and loss of TCR vbeta diversity during the initiation of a CD4(+) T cell response in vivo. J Exp Med. 2000;192:1719–1730. doi: 10.1084/jem.192.12.1719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Savage PA, Boniface JJ, Davis MM. A kinetic basis for T cell receptor repertoire selection during an immune response. Immunity. 1999;10:485–492. doi: 10.1016/s1074-7613(00)80048-5. [DOI] [PubMed] [Google Scholar]
- 72.Anderton SM, Radu CG, Lowrey PA, Ward ES, Wraith DC. Negative selection during the peripheral immune response to antigen. J Exp Med. 2001;193:1–11. doi: 10.1084/jem.193.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Amrani A, Verdaguer J, Serra P, Tafuro S, Tan R, Santamaria P. Progression of autoimmune diabetes driven by avidity maturation of a T-cell population. Nature. 2000;406:739–742. doi: 10.1038/35021081. [DOI] [PubMed] [Google Scholar]
- 74.Han B, Serra P, Yamanouchi J, Amrani A, Elliott JF, Dickie P, Dilorenzo TP, Santamaria P. Developmental control of CD8 T cell-avidity maturation in autoimmune diabetes. J Clin Invest. 2005;115:1879–1887. doi: 10.1172/JCI24219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Azzam HS, DeJarnette JB, Huang K, Emmons R, Park CS, Sommers CL, El Khoury D, Shores EW, Love PE. Fine tuning of TCR signaling by CD5. J Immunol. 2001;166:5464–5472. doi: 10.4049/jimmunol.166.9.5464. [DOI] [PubMed] [Google Scholar]
- 76.Robey EA, Ramsdell F, Kioussis D, Sha W, Loh D, Axel R, Fowlkes BJ. The level of CD8 expression can determine the outcome of thymic selection. Cell. 1992;69:1089–1096. doi: 10.1016/0092-8674(92)90631-l. [DOI] [PubMed] [Google Scholar]
- 77.Zuniga-Pflucker JC, McCarthy SA, Weston M, Longo DL, Singer A, Kruisbeek AM. Role of CD4 in thymocyte selection and maturation. J Exp Med. 1989;169:2085–2096. doi: 10.1084/jem.169.6.2085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Marquez ME, Ellmeier W, Sanchez-Guajardo V, Freitas AA, Acuto O, Di B. V: CD8 T cell sensory adaptation dependent on TCR avidity for self-antigens. J Immunol. 2005;175:7388–7397. doi: 10.4049/jimmunol.175.11.7388. [DOI] [PubMed] [Google Scholar]
- 79.Sebzda E, Kundig TM, Thomson CT, Aoki K, Mak SY, Mayer JP, Zamborelli T, Nathenson SG, Ohashi PS. Mature T cell reactivity altered by peptide agonist that induces positive selection. J Exp Med. 1996;183:1093–1104. doi: 10.1084/jem.183.3.1093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Grossman Z, Paul WE. Adaptive cellular interactions in the immune system: The tunable activation threshold and the significance of subthreshold responses. Proc Natl Acad Sci U S A. 1992;89:10365–10369. doi: 10.1073/pnas.89.21.10365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Tanchot C, Barber DL, Chiodetti L, Schwartz RH. Adaptive tolerance of CD4+ T cells in vivo: multiple thresholds in response to a constant level of antigen presentation. J Immunol. 2001;167:2030–2039. doi: 10.4049/jimmunol.167.4.2030. [DOI] [PubMed] [Google Scholar]
- 82.Singh NJ, Schwartz RH. The strength of persistent antigenic stimulation modulates adaptive tolerance in peripheral CD4+ T cells. J Exp Med. 2003;198:1107–1117. doi: 10.1084/jem.20030913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Kissler S, Anderton SM, Wraith DC. Antigen-presenting cell activation: a link between infection and autoimmunity? J Autoimmun. 2001;16:303–308. doi: 10.1006/jaut.2000.0498. [DOI] [PubMed] [Google Scholar]
- 84.Manoury B, Gregory WF, Maizels RM, Watts C. Bm-CPI-2, a cystatin homolog secreted by the filarial parasite Brugia malayi, inhibits class II MHC-restricted antigen processing. Curr Biol. 2001;11:447–451. doi: 10.1016/s0960-9822(01)00118-x. [DOI] [PubMed] [Google Scholar]
- 85.Matzinger P. The danger model: a renewed sense of self. Science. 2002;296:301–305. doi: 10.1126/science.1071059. [DOI] [PubMed] [Google Scholar]
- 86.Palermo B, Garbelli S, Mantovani S, Scoccia E, Da Prada GA, Bernabei P, Avanzini MA, Brazzelli V, Borroni G, Giachino C. Qualitative difference between the cytotoxic T lymphocyte responses to melanocyte antigens in melanoma and vitiligo. Eur J Immunol. 2005;35:3153–3162. doi: 10.1002/eji.200535110. [DOI] [PubMed] [Google Scholar]
- 87.Luiten RM, Kueter EW, Mooi W, Gallee MP, Rankin EM, Gerritsen WR, Clift SM, Nooijen WJ, Weder P, van de Kasteele WF, Sein J, van den Berk PC, Nieweg OE, Berns AM, Spits H, de Gast GC. Immunogenicity, including vitiligo, and feasibility of vaccination with autologous GM-CSF-transduced tumor cells in metastatic melanoma patients. J Clin Oncol. 2005;23:8978–8991. doi: 10.1200/JCO.2005.01.6816. [DOI] [PubMed] [Google Scholar]
- 88.Overwijk WW. Breaking tolerance in cancer immunotherapy: Time to ACT. Curr Opin Immunol. 2005;17:187–194. doi: 10.1016/j.coi.2005.01.011. [DOI] [PubMed] [Google Scholar]
- 89.Dudley ME, Wunderlich JR, Robbins PF, Yang JC, Hwu P, Schwartzentruber DJ, Topalian SL, Sherry R, Restifo NP, Hubicki AM, Robinson MR, Raffeld M, Duray P, Seipp CA, Rogers-Freezer L, Morton KE, Mavroukakis SA, White DE, Rosenberg SA. Cancer regression and autoimmunity in patients after clonal repopulation with antitumor lymphocytes. Science. 2002;298:850–854. doi: 10.1126/science.1076514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Fields ML, Hondowicz BD, Metzgar MH, Nish SA, Wharton GN, Picca CC, Caton AJ, Erikson J. CD4+ CD25+ regulatory T cells inhibit the maturation but not the initiation of an autoantibody response. J Immunol. 2005;175:4255–4264. doi: 10.4049/jimmunol.175.7.4255. [DOI] [PubMed] [Google Scholar]