

Primrose syndrome consists of cognitive and motor delay, characteristic facial dysmorphism with enlarged and calcified external ears, cataracts, hearing impairment, spastic paraparesis with joint contractures, and distal muscle wasting. To our knowledge, only 5 cases have been reported in the literature.1–5 We report the sixth case and only the second woman to fulfill all clinical elements of Primrose syndrome, who, in addition, manifested motor tics, hand stereotypies, and self-flagellating behaviors. This patient remained undiagnosed for over 4 decades.
Case report.
A 43-year-old woman, the product of a normal vaginal delivery and negative neurologic family history, was born with congenital heart disease (Ebstein malformation, mitral valve prolapse), congenital hip dysplasia, and agenesis of the corpus callosum. In early childhood, she was found to have hearing impairment, cataracts, a nondescript neoplasm of the bone in the palate, ossification of cartilage, cystic changes in bones, diabetes mellitus, and hypothyroidism. Walking began at the age of 6 years. The limited repertoire of her language included “mommy,” “daddy,” “bye-bye,” and “go to school,” before she lost vocalization altogether in her late 30s. She attended special education for about 18 years and worked at putting bakery projects together for several years thereafter. She had been able to run and roller skate before her gait deteriorated. She slowly lost the ability to ambulate independently. At age 35, she started using a walker, and by age 40 needed a wheelchair for most transportation needs. On examination, at 42 years, her facial dysmorphic features included micrognathia, repaired cleft palate, anteverted nares, prominent ears and nasal root, ptosis, and microphthalmia (figure, A). Review of prior family photographs revealed prognathism (instead of micrognathia), repaired cleft palate, anteverted nares, and prominent ears and nasal roots. On examination, her head circumference was 55 cm. She had spastic paraparesis with leg scissoring and contractures. Muscle stretch reflexes were absent. Associated movements included motor tics, hand stereotypies, and self-flagellating behaviors (see video on the Neurology® Web site at www.neurology.org). For the last 2 years, she has become unable to stand and bear weight without assistance. She is now mute, mumbling incomprehensible sounds, incontinent of urine and stools, having frequent painful spasms, and wheelchair-dependent.
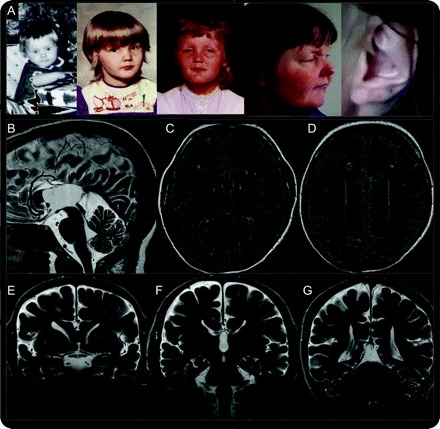
Figure Facial features and brain MRI
(A) Facial features of patient at age 1, 3, 7, and 43 years, with attention to the diagnostic clue, her enlarged and calcified external ear. Important early features include prognathism, repaired cleft palate, anteverted nares, prominent ears, and nasal roots. Early prognathism gave way to micrognathia in subsequent years. Ptosis was not apparent in early childhood. (B) Sagittal T2-weighted brain MRI demonstrates partial agenesis of the corpus callosum with “wandering” of the anterior cerebral arteries and high-riding third ventricle. (C–E) Increased fluid-attenuated inversion recovery signal is seen in patchy subcortical and diffuse periventricular distribution. (E–G) Coronal T2-weighted sequences demonstrate partial calcification of the putamen and globus pallidum and teardrop deformity of the lateral ventricles. The brainstem and cerebellum showed normal size and configuration. The patient's mother has given full informed consent for the publication of the video and images.
Her brain MRI revealed agenesis of the corpus callosum and partial calcification of the basal ganglia (figure, B–G). EEG was normal. Laboratory investigations showed increases in serum calcitonin (8 pg/mL, normal = 0.0–4.6 pg/mL) and phosphate (4.6 mg/dL, normal = 2.5–4.5 mg/dL).6
Since there is no known molecular cause for Primrose syndrome, we searched for a small duplication or deletion by expanded microarray analysis, using the Infinium Assay with the Illumina HumanCNV370-duo DNA Analysis BeadChip platform. Data from this SNP bead array analysis detected a small deletion on chromosome 11, and defined the region of deletion to be approximately 225.5 kb between markers rs12275693–>rs1442927 (linear position on chromosome 11 is approximately 26971983–27197527). The only annotated gene in this region is BBOX1, involved in carnitine biosynthesis. The deletion was not present in a maternal sample, but a paternal sample was not available.
Discussion.
The combination of a neural tube congenital malformation (corpus callosum agenesis), facial dysmorphism (micrognathia, repaired cleft palate, anteverted nares, prominent ears and nasal root, ptosis, and microphthalmia), deafness, cataracts, congenital heart disease (mitral valve prolapse, Ebstein malformation), diabetes, and premature degenerative joint disease are classic constituents of Primrose syndrome. The calcified enlargement of the external ears was the telltale sign, which along with basal ganglia calcification and known skeletal cystic changes suggest an underlying disorder in calcium homeostasis, supported by the increased serum calcitonin in our patient. Motor tics, hand stereotypies, and self-flagellating behaviors are novel elements to the phenotype. However, the deletion identified in chromosome 11 may not have any relevance to the phenotype and may be a benign copy number variant. Additional effort to identify a genetic cause will require study of additional patients using evolving genetic tools.
The prior 5 cases reported, including Primrose's original description in 1982, are summarized in table e-1 on the Neurology® Web site at www.neurology.org. Long-term historical information is only available for the individual reported by one of the authors (N.M.L.) in 1996, who also had schizophrenia. He became progressively stiffer and had increasing difficulty moving at all. His hands became clenched fists. He had progressive difficulty swallowing food and secretions and required a feeding tube. He was admitted to the hospital with abdominal distention and pain and died of his illness at age 45. No autopsy was performed.
Movement disorders have been reported in Primrose syndrome in the form of parkinsonism (masked facies, low-amplitude tremor)3 and ataxia.4 Our case adds tics, stereotypes, and self-flagellation to the phenotypic spectrum. Primrose syndrome may be added to the growing list of secondary tics and stereotypies, which includes drug-induced (amphetamines, pemoline, cocaine, neuroleptics), encephalitis, schizophrenia, and neurodegenerative diseases.6,7
Supplementary Material
Footnotes
Supplemental data at www.neurology.org
Received December 18, 2009. Accepted in final form March 19, 2010.
Disclosure
P. Dalal reports no disclosures. Dr. Leslie has received research support from the Centers for Disease Control (R18DD00344 [Co-I]). Dr. Lindor receives research support from the NIH (R24/U24CA 74800-11 [PI], R01CA 104132-5 [PI], and P50 CA 116201-5 [Co-I]). Dr. Gilbert serves on the editorial board of the American Academy of Pediatrics (PREP Self Assessment); serves on the medical advisory board for the Tourette Syndrome Association; and receives research support from the NIH (NIMH R01 MH078160 [Co-I], NIMH R01 MH08185 [Co-I], and NINDS NS056276 [Co-I]) and the Tourette Syndrome Association. Dr. Espay has served/serves on scientific advisory boards for Boehringer Ingelheim and Solvay Pharmaceuticals, Inc.; serves on the editorial advisory board of The European Neurological Journal; has received speaker honoraria from UCB, Medtronic, Inc., and Novartis; has served/serves on speakers' bureaus for UCB and Novartis; and has received/receives research support from Medtronic, Inc., Allergan, Inc., Cleveland Medical Devices Inc., the Davis Phinney Foundation, the Michael J. Fox Foundation, and the KL2 Research Scholars mentored career development award through the NIH Institutional Clinical and Translational Science Award.
References
- 1.Primrose DA. A slowly progressive degenerative condition characterized by mental deficiency, wasting of limb musculature and bone abnormalities, including ossification of the pinnae. J Ment Defic Res 1982;26:101–106. [DOI] [PubMed] [Google Scholar]
- 2.Collacott RA, O'Malley BP, Young ID. The syndrome of mental handicap, cataracts, muscle wasting and skeletal abnormalities: report of a second case. J Ment Defic Res 1986;30:301–308. [DOI] [PubMed] [Google Scholar]
- 3.Lindor NM, Hoffman AD, Primrose DA. A neuropsychiatric disorder associated with dense calcification of the external ears and distal muscle wasting: ‘Primrose syndrome.’ Clin Dysmorphol 1996;5:27–34. [DOI] [PubMed] [Google Scholar]
- 4.Battisti C, Dotti MT, Cerase A, et al. The Primrose syndrome with progressive neurological involvement and cerebral calcification. J Neurol 2002;249:1466–1468. [DOI] [PubMed] [Google Scholar]
- 5.Mathijssen IB, van Hasselt-van der Velde, Hennekam RC. Testicular cancer in a patient with Primrose syndrome. Eur J Med Genet 2006;49:127–133. [DOI] [PubMed] [Google Scholar]
- 6.Bharucha KJ, Sethi KD. Tardive tourettism after exposure to neuroleptic therapy. Mov Disord 1995;10:791–793. [DOI] [PubMed] [Google Scholar]
- 7.Edwards MJ, Dale RC, Church AJ, et al. Adult-onset tic disorder, motor stereotypies, and behavioural disturbance associated with antibasal ganglia antibodies. Mov Disord 2004;19:1190–1196. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.