

Abstract
Summary: With the rapid advances and prevalence of high-throughput genomic technologies, integrating information of multiple relevant genomic studies has brought new challenges. Microarray meta-analysis has become a frequently used tool in biomedical research. Little effort, however, has been made to develop a systematic pipeline and user-friendly software. In this article, we present MetaOmics, a suite of three R packages MetaQC, MetaDE and MetaPath, for quality control, differentially expressed gene identification and enriched pathway detection for microarray meta-analysis. MetaQC provides a quantitative and objective tool to assist study inclusion/exclusion criteria for meta-analysis. MetaDE and MetaPath were developed for candidate marker and pathway detection, which provide choices of marker detection, meta-analysis and pathway analysis methods. The system allows flexible input of experimental data, clinical outcome (case–control, multi-class, continuous or survival) and pathway databases. It allows missing values in experimental data and utilizes multi-core parallel computing for fast implementation. It generates informative summary output and visualization plots, operates on different operation systems and can be expanded to include new algorithms or combine different types of genomic data. This software suite provides a comprehensive tool to conveniently implement and compare various genomic meta-analysis pipelines.
Availability: http://www.biostat.pitt.edu/bioinfo/software.htm
Contact: ctseng@pitt.edu
Supplementary Information: Supplementary data are available at Bioinformatics online.
1 INTRODUCTION
Many high-throughput genomic technologies have advanced dramatically in the past decade. Microarray experiment is one example that has evolved into maturity with generally consensus experimental protocols and data analysis strategies. Its extensive application in the biomedical field has led to an explosion of gene expression profiling studies publicly available. Meta-analysis methods for combining multiple microarray studies have been widely applied to increase statistical power and provide validated conclusions (Tseng et al., 2012). In this article, we present the ‘MetaOmics’ software suite that contains three unified R packages—MetaQC, MetaDE and MetaPath—for systematic microarray meta-analysis pipeline. The MetaQC (Kang et al., 2012) package provides a quantitative and objective tool for determining the inclusion/exclusion criteria for meta-analysis. MetaDE contains many state-of-the-art genomic meta-analysis methods to detect differentially expressed genes. Finally, the MetaPath package (Shen and Tseng, 2010) provides a unified meta-analysis framework and inference to detect enriched pathways associated with outcome.
2 THE THREE R PACKAGES
The three R packages in MetaOmics allow flexible input format of experimental data and four different types of outcome variables (case–control, multi-class, continuous and survival). They also allow missing values in the individual experimental study or missing values caused by mismatched genes across studies (i.e. genes covered in one study but not covered in another study). For some computationally intensive routines, the packages allow usage of multi-core parallel computing for timely implementation. Detailed help files, tutorial and a case study are available in an online supplementary document as well as in the R packages. Below, we briefly describe features and functionality of the three packages.
2.1 MetaQC
MetaQC calculates the following six quantitative quality control (QC) measures: internal homogeneity of co-expression structure among studies (IQC), external consistency of co-expression pattern with pathway database (EQC) and accuracy and consistency of differentially expressed gene detection (AQCg and CQCg) or enriched pathway identification (AQCp and CQCp). Each QC index is defined as the minus log-transformed P-values from formal hypothesis testing in each QC criterion. Principal component analysis (PCA) biplots and standardized mean ranks are finally generated to assist visualization and decision. The identified problematic studies are suggested for further inspection to detect potential technical or biological causes of their low quality and to determine their exclusion from meta-analysis.
2.2 MetaDE
MetaDE package implements 12 major meta-analysis methods for differential expression (DE) analysis: Fisher (Rhodes et al., 2002), Stouffer, adaptively weighted statistic (AW) (Li and Tseng, 2011), minimum P-value (minP), maximum P-value (maxP), rth ordered P-value (rOP) (Song and Tseng, 2012), fixed effects model (FEM), random effects model (REM) (Choi et al., 2003), rank product (rankProd), rank sum (rankSum) (Hong et al., 2006), naive sum of ranks and naive product of ranks. Detailed algorithms, pros and cons of different methods have been discussed in a recent review article (Tseng et al., 2012). Two additional considerations are involved in the implementation: (i) different choices of test statistics are available for different outcome variables, for example t-statistics, F-statistics, minimum multi-class correlation (Lu et al., 2010), linear regression, correlation coefficient and log-rank test; (ii) one-sided test correction may be needed to exclude genes with discordant DE direction (e.g. up-regulation in one study but down-regulation in another study). MetaDE also provides options for gene matching across studies and gene filtering before meta-analysis. Outputs of the meta-analysis results include DE gene lists with corresponding raw P-values, q-values and various visualization tools. Heatmaps can be plotted across studies.
2.3 MetaPath
MetaPath implements three meta-analysis framework for pathway enrichment analysis: MAPE_G, MAPE_P and MAPE_I (Shen and Tseng, 2010). Meta-analyses for pathway enrichment are integrated either at the gene level (MAPE_G) or at the pathway level (MAPE_P). For MAPE_G, information across studies is combined at the gene level and then pathway enrichment analysis is applied. Conversely, for MAPE_P, pathway analysis is first performed in each study independently. The information across studies is then combined at the pathway level. Since MAPE_G and MAPE_P have been found with complementary advantages under different data structure, a hybrid framework (MAPE_I) has been developed. Similar to MetaDE, MetaPath also provides multiple options of gene matching, gene filtering, meta-analysis methods and test statistics to associate with outcomes.
Supplementary Figure S1 shows a workflow diagram of meta-analysis pipeline using the three packages. After data are preprocessed, MetaQC is applied to determine inclusion/exclusion criteria. MetaDE and MetaPath are then used to detect candidate markers or pathways associated with disease outcome.
3 PROSTATE CANCER EXAMPLE
To demonstrate application of MetaQC, MetaDE and MetaPath, we collected nine prostate cancer studies (Welsh, Yu, Lapointe, Varambally, Singh, Wallace, Nanni, Tomlins and Dhanasekaran), which contained normal and primary cancer samples. After gene matching by official gene symbols, preprocessing and filtering, 4441 genes were used for meta-analysis. Figure 1A shows result of the MetaQC PCA biplot. Three of the nine studies (Nanni, Tomlins and Dhanasekaran) were determined with lower quality and were removed from meta-analysis. Figure 1B shows the number of detected DE genes under different FDR threshold in the remaining six single study analysis and meta-analyses by Fisher, maxP, rOP (r = 4) and AW methods. It is clear that meta-analysis usually detects more candidate markers, except for maxP. Finally, Figure 1C and D shows a heatmap of detected pathways (q-value < 0.2 in any method) and Venn diagram of pathways detected by MAPE_P, MAPE_G and MAPE_I using MetaPath. The majority of the detected pathways appeared to be cancer related. Single-study analyses showed very weak pathway enrichment; MAPE_P and MAPE_G appeared to have complementary detection power (identified 23 and 15 pathways with only 5 in common). MAPE_I detected the largest number of pathways (34 pathways).
Fig. 1.
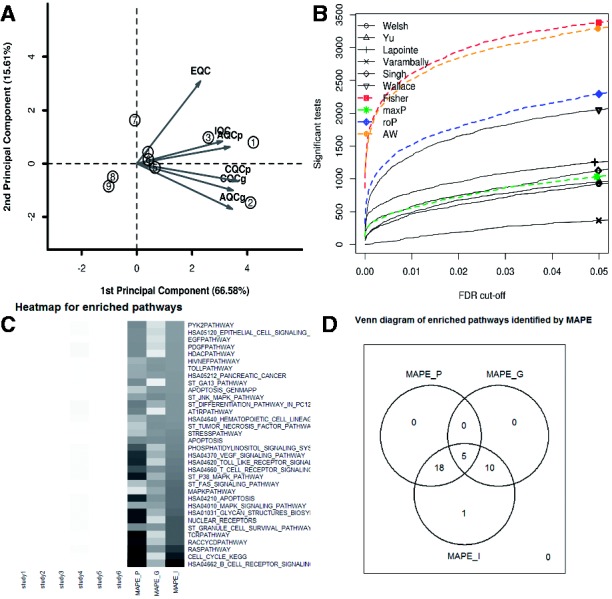
(A) PCA bi-plot from MetaQC. (B) Number of detected DE genes under different q-value threshold. (C) Heatmap showing minus logged q-values of detected pathways. (D) Venn diagram of detected pathways by the three MAPE methods
Funding: The National Institutes of Health (MH077159, MH094862, HL095397 and HL101715).
Conflict of inerest: None declared.
Supplementary Material
REFERENCES
- Choi JK, et al. Combining multiple microarray studies and modeling interstudy variation. Bioinformatics. 2003;19:i84–i90. doi: 10.1093/bioinformatics/btg1010. [DOI] [PubMed] [Google Scholar]
- Hong F, et al. RankProd: a bioconductor package for detecting differentially expressed genes in meta-analysis. Bioinformatics. 2006;22:2825–2827. doi: 10.1093/bioinformatics/btl476. [DOI] [PubMed] [Google Scholar]
- Kang DD, et al. MetaQC: objective quality control and inclusion/exclusion criteria for genomic meta-analysis. Nucleic Acids Res. 2012;40:e15. doi: 10.1093/nar/gkr1071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Tseng GC. An adaptively weighted statistic for detecting differential gene expression when combining multiple transcriptomic studies. Ann. Appl. Stat. 2011;5:994–1019. [Google Scholar]
- Lu S, et al. Biomarker detection in the integration of multiple multi-class genomic studies. Bioinformatics. 2010;26:333–340. doi: 10.1093/bioinformatics/btp669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhodes DR, et al. Meta-analysis of microarrays: interstudy validation of gene expression profiles reveals pathway dysregulation in prostate cancer. Cancer Res. 2002;62:4427–4433. [PubMed] [Google Scholar]
- Song,C. and Tseng,G.C. (2012) Hypothesis setting and order statistic for robust genomic meta-analysis. Annals of Applied Statistics. In press. [DOI] [PMC free article] [PubMed]
- Shen K, Tseng GC. Meta-analysis for pathway enrichment analysis when combining multiple genomic studies. Bioinformatics. 2010;26:1316–1323. doi: 10.1093/bioinformatics/btq148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tseng GC, et al. Comprehensive literature review and statistical considerations for microarray meta-analysis. Nucleic Acids Res. 2012;40 (90):3785–3799. doi: 10.1093/nar/gkr1265. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.