

Abstract
Bacteria that colonize the mammalian intestine collectively possess a far larger repertoire of degradative enzymes and metabolic capabilities than their hosts. Microbial fermentation of complex non-digestible dietary carbohydrates and host–derived glycans in the human intestine has important consequences for health. Certain dominant species, notably among the Bacteroidetes, are known to possess very large numbers of genes that encode carbohydrate active enzymes and can switch readily between different energy sources in the gut depending on availability. Nevertheless, more nutritionally specialized bacteria appear to play critical roles in the community by initiating the degradation of complex substrates such as plant cell walls, starch particles and mucin. Examples are emerging from the Firmicutes, Actinobacteria and Verrucomicrobium phyla, but more information is needed on these little studied groups. The impact of dietary carbohydrates, including prebiotics, on human health requires understanding of the complex relationship between diet composition, the gut microbiota and metabolic outputs.
Keywords: dietary fiber, human intestine, microbiota, nutrition, polysaccharides, prebiotics, resistant starch
Introduction
Mammalian genomes do not encode most of the enzymes needed to degrade the structural polysaccharides present in plant material. Instead a complex mutual dependence has developed between the mammalian host and symbiotic gut microorganisms that do possess the ability to access this abundant source of energy. Herbivorous mammals rely on resident gut microorganisms to gain energy from their main food sources, and this has entailed major changes in digestive anatomy and physiology that allow efficient microbial fermentation to take place alongside the recovery of dietary energy by the host.1 Ruminants (foregut fermentors) benefit from microbial protein as well as the absorption of energy that is released by anaerobic microorganisms in the form of fermentation acids. Other herbivores and omnivores derive varying amounts of energy from microbial fermentation in the hind gut of those carbohydrates that are not digested in the upper gut. Interestingly, molecular profiles for the gut microbiota have been shown to group together for animal species that share similar nutrition and digestive anatomy.2 While humans derive a relatively small fraction (perhaps 10%) of their dietary energy through the activities of intestinal microorganisms,3 the microbial communities of the human intestine have important consequences for health and their composition and activities are known to be strongly influenced by the carbohydrate content of the diet.4,5
Most of the plant-derived polysaccharides that enter the rumen and large intestine are in the form of insoluble structures, in particular plant cell wall fragments and starch particles. Early work on the rumen established that only a small subset of rumen microorganisms, that include cellulolytic bacteria, fungi and protozoa, have the capacity to initiate degradation of plant cell walls.6 The most numerous groups of rumen microorganisms however are non-cellulolytic bacteria, many of which possess the ability to grow on soluble polysaccharides that are released by the primary degraders.7,8 Stratification of particle-associated microbial communities is evident from microscopic and fractionation studies both in the rumen and in human colon.9-11 It is reasonable to assume that the most closely adherent organisms will include the primary degraders, but also that more loosely adherent organisms within the consortium will contribute to polysaccharide degradation and utilization. Some primary colonizers are known to be nutritionally highly specialized; many rumen cellulolytic bacteria for example utilize breakdown products of cellulose, but fail to utilize products of xylan breakdown despite possessing a battery of hemicellulases and pectinases that are presumably required to degrade the plant cell wall matrix surrounding the cellulose fibrils.12 Solubilisation of the matrix polysaccharides therefore results in cross-feeding to other groups of bacteria. Metabolic cross-feeding is a central feature in anaerobic microbial communities that involves products of fermentation such as hydrogen and lactate as well as partial substrate degradation products.13,14 On the other hand, many other dominant gut bacteria show remarkable nutritional flexibility. The human intestinal species Bacteroides thetaiotaomicron for example encodes a huge repertoire of carbohydrate degrading activities15 and has the ability to switch between diet- and host-derived carbohydrates.16 The expression of genes involved in the degradation of complex carbohydrates by many gut bacteria is tightly regulated not only in response to the availability of specific substrates, but also in response to the host and other bacteria within the gut community.17
This review will focus mainly on the microbial ecology of carbohydrate degradation in the human large intestine, but for comparison also considers the degradation of plant structural polysaccharides in the rumen where this process is both more efficient and better studied. We also consider briefly some of the consequences of carbohydrate fermentation for human health.
Enzyme Families, Genomics and Metagenomics
In total 130 families of glycoside hydrolases (GH), 22 of polysaccharide lyases (PL), and 16 of carbohydrate esterases (CE) have now been described from all life forms and a high proportion of these are found to be encoded in microbial genomes (www.cazy.org).18 These include catalytic domains that degrade plant structural polysaccharides (cellulose, β-glucan, xylan, mannan and pectin) and storage carbohydrates (Fig. 1) and a wide variety of host-derived glycans. In addition there are currently 64 families of carbohydrate binding modules (CBMs) that are frequently found to be associated with the catalytic domains of extracellular degradative enzymes. Draft genomes are now available for several rumen bacteria and for 50- 100 species of commensal human intestinal bacteria (with more projected) and these provide important information on the potential polysaccharide-degrading enzyme repertoire of each strain (Table 1). Metagenomic approaches have the potential to identify novel enzymes and enzyme families involved in carbohydrate breakdown through functional screening19-21 as well as cataloguing the abundance of known genes via high-throughput sequencing.22 Metagenomic sequencing applied to the human gut microbiota has detected a large panel of carbohydrate active enzymes (CAZymes).23 However, the great majority of these potential enzymes remain to be characterized and their regulation studied. It is important to keep in mind also that organisms depend on complex interacting systems of degradative enzymes, transport functions and regulatory circuits in order to utilize complex carbohydrate substrates. For this reason the following sections will concentrate on examining function-based information that has so far been obtained mainly from cultured anaerobic gut bacteria.

Figure 1. Major diet-derived polysaccharides and microbial carbohydrate-degrading enzyme activities. The enzyme families most associated with particular activities in gut bacteria are indicated as follows: GH, glycoside hydrolase; PL, polysaccharide lyase; CE, carbohydrate esterase; G, glucose; F, fructose; X, xylose; A, arabinose; GalU, galacturonic acid; GluU, glucuronic acid. [For details refer to the CAZY website (/www.cazy.org/)].
Table 1. Predicted CAZymes encoded by the genomes of selected fibrolytic gut bacteria. [GH glycoside hydrolases, GT glycosyl transferases, PL polysaccharide lyases, CE carbohydrate esterases, CBM carbohydrate binding modules].
| Ecosystem | Phylum (family) |
Bacterium | Total CAZymes |
GH | GT | PL | CE | Total CBMs |
|---|---|---|---|---|---|---|---|---|
| Human colon |
Bacteroidetes |
Bacteroides thetaiotaomicron VPI-5482 |
386 |
263 |
87 |
16 |
20 |
31 |
| |
|
B. xylanisolvens XB1A* |
349 |
224 |
81 |
22 |
22 |
26 |
| |
|
B. vulgatus ATCC-8482 |
279 |
177 |
78 |
7 |
17 |
18 |
| |
|
B. fragilis 638R |
223 |
138 |
78 |
1 |
6 |
26 |
| |
Firmicutes: (Lachnospiraceae) (Ruminococcaceae) Actinobacteria |
Roseburia intestinalis XB6B4* Butyrivibrio fibrisolvens 16/4* Ruminococcus champanellensis 18P13* Bifidobacterium adolescentis ATCC15703 |
175 115 87 94 |
115 75 54 54 |
46 37 12 37 |
0 0 9 0 |
14 3 12 3 |
11 31 34 6 |
| Rumen |
Fibrobacteres/ Acidobacteria |
Fibrobacter succinogenes S85 |
183 |
100 |
54 |
12 |
17 |
73 |
| |
Bacteroidetes |
Prevotella ruminicola 23 |
215 |
133 |
60 |
3 |
19 |
16 |
| |
|
P. bryantii B14 |
203 |
107 |
53 |
14 |
19 |
un |
| Firmicutes: (Ruminococcaceae) |
Ruminococcus albus 7 R. flavefaciens FD1 |
145 140 + |
96 101 |
24 un |
7 13 |
18 26 |
128 68 |
For these strains, data were provided by the Pathogen Genomics group at the Wellcome Trust Sanger Institute and can be obtained from http://www.sanger.ac.uk/resources/downloads/bacteria/metahit/
Plant Cell Wall Degradation by Rumen Bacteria
Plant cell walls consist of cellulose fibrils embedded in a matrix of hemicellulose (xylan, mannan, xyloglucan and β-glucan) and pectin, with lignin also present in secondary walls (Fig. 2). Cellulose consists of linear chains of β(1,4)-linked glucose units that form microfibrils through hydrogen bonding. Highly crystalline cellulose is particularly recalcitrant to enzymatic degradation, whereas amorphous forms are more accessible. Xylan is a heterogenous polymer of β(1,4)-linked xylose residues substituted with acetyl, arabinosyl and 4-O-methyl-glucuronyl residues; ester cross linkages can also occur between arabinosyl substituents and ferulic acid present in lignin. Pectins are a family of complex polysaccharides that contain α(1,4)-linked D-galacturonic acid or rhamnogalacturonan backbones. Plant cell wall composition and structure, and consequently its digestibility and fermentability in the gut, however varies considerably between plant species, varieties and tissues.

Figure 2. Plant cell wall structure. Diagrammatic representation of the major structural polysaccharide components of a "typical" primary plant cell wall.
Only actively cellulolytic rumen species have been found to cause extensive solubilisation of plant cell wall material in pure culture.6 The two main groups of cellulose-degrading bacteria that have been isolated, Gram-positive ruminococci and Gram-negative Fibrobacter spp, possess contrasting fibrolytic enzyme systems. Ruminococcus flavefaciens is the only gut bacterium so far shown to produce a cellulosome-type enzyme complex, where the assembly of protein subunits depends on specific interactions between dockerin and cohesin modules found in the protein subunits.24 The genome of R. flavefaciens FD1 encodes 220 dockerin-containing proteins that are potential cellulosome subunits together with multiple cohesin-containing scaffolding proteins, four of which are encoded by the sca gene cluster.25-27 The dockerin- containing proteins include diverse GH, CE and PL domains as well as CBMs and peptidases, but the functions of around 30% of the associated domains remain unknown.27The cellulosomal xylanases in this species display remarkably complex structures28,29 with as many as five distinct catalytic domains and CBMs (Fig. 3), and include some that are upregulated more than 50 fold by growth on cellulose.27,30 The whole complex is anchored to a small protein that is bound to the bacterial cell surface by a sortase-mediated linkage.31 Dockerins, but not cohesins, have been found in the related R. albus, leaving it unclear how enzymes are organized in that species. Multidomain organization is also seen for non-cellulosomal xylanases of R. albus 832 however and both of these species of ruminococci produce prominent GH48 enzymes33 that are assumed to play a key role in cellulose hydrolysis, as related enzymes function as exo-acting cellobiohydrolases in Clostridium spp.24 Adhesion to the insoluble plant cell wall substrate involves multiple CBMs within enzyme subunits, together with cellulose binding pili in R. albus34 and a specific attachment protein in R. flavefaciens.35 In contrast, F. succinogenes is highly unusual among anaerobic cellulolytic bacteria in lacking dockerin sequences and in lacking a GH48 exo-cellulase, although processive GH9 cellulases may fulfil the same role.12,36 The organization of fibrolytic enzymes in this species, which achieves highly efficient degradation of crystalline cellulose, remains unclear.

Figure 3. Examples of cell surface organization of carbohydrate-degrading enzymes in anaerobic Gram-positive gut bacteria. A and C show the domain structures and organization of two major cell-surface anchored amylases from two human intestinal anaerobes (Numbering refers to the enzyme family (as in Figure 1) or carbohydrate binding module (CBM) family). B shows the domain structures of six examples of cellulosomal polysaccharidases from the rumen bacterium Ruminococcus flavefaciens FD1. D shows the likely organization of the cellulosome in R. flavefaciens FD1; scaE, scaB, scaA and scaC are structural proteins encoded by the sca gene cluster that interact with each-other and with the cellulosomal enzyme subunits via a series of specific, non-covalent dockerin:cohesin pairings (shown, in gray). The arrows in C and D indicate sortase-mediated anchoring to the bacterial cell wall (also indicated by cross-hatching in A).
Prevotella spp are among the most abundant bacteria within the rumen community; while none is cellulolytic, other plant cell wall polysaccharides can be utilized by many species. P. bryantii (formerly P. ruminicola) B14 grows well on water-soluble, but not on water-insoluble, xylans.37 Two gene clusters are now known to play an important role in xylan-utilizing Prevotella spp One includes a GH10 xylanase and GH43 β−xylosidase that contribute most of the assayable xylanase activity in cell extracts and whose expression is induced in response to xylo-oligosaccharides by a linked hybrid two component regulator (HTCS).37-39 Subsequent transcriptomic studies have revealed more than 50 genes whose expression is significantly higher during growth on xylans as compared with xylose and arabinose in P. bryantii B14.40 The most highly induced genes belonged to a second cluster (xus) that includes two susC and two susD paralogs (discussed further below) in tandem, and an endoxylanase gene (xyn10C). Xyn10C is unusual in carrying CBM sequences within the catalytic domain41,42 and is thought to be responsible for cleavage of xylan molecules at the cell surface. The related P. ruminicola 23 has been shown to encode at least 16 esterases that are involved in de-acetylation and de-methylation of xylans and pectins, as well as removing ester-linked phenolic acids.43,44
Metagenome surveys of rumen contents have tended to detect a low number of CBMs and a high % of GH domains that are typically associated with the utilization of soluble polysaccharides (e.g., 32% of glycoside hydrolases detected in rumen fiber fractions by Brulc et al.22 were related to GH2 or GH3 glycosidases). It is not clear whether this primarily reflects the difficulty of recovering DNA from tightly adherent cellulolytic species, or low populations of those bacteria that have so far been identified as fiber-degraders. A recent metatranscriptome analysis of the muskox rumen that targeted mRNA of eukaryotic origin, however, yielded very high numbers of glycosyl hydrolase genes.45 This emphasizes the important and distinctive contribution that is made by anaerobic eukaryotic microorganisms, fungi and protozoa, to fiber degradation in the rumen.
Degradation of Complex Carbohydrates by the Human Intestinal Microbiota
Microbial diversity in the human colon
Recent analyses of directly amplified 16S rRNA genes4,46 together with metagenomic surveys47 have helped to define those phylotypes (species defined by sequencing) that are most abundant within the human fecal microbiota. Perhaps not surprisingly, many of the dominant phylotypes correspond to cultured species, whereas only around 30% of the less abundant phylotypes are represented by cultures4 (Fig. 4). The dominant bacterial phyla in healthy subjects are the Bacteroidetes, Firmicutes and Actinobacteria, together with Verrucomicrobia and Proteobacteria. The composition of the human fecal microbiota responds to dietary carbohydrate intake in the short-term4 and apparently also in the longer term.5,48 De Filippo et al.5 ascribed the differences they observed in fecal microbiota composition between two groups of children to different intakes of fiber and starch, with Bacteroidetes, especially Prevotella spp, being favored in a group of rural African children and Firmicutes in Italian children. An association between a Prevotella–dominated microbiota and fiber intake, and between a Bacteroides-dominated microbiota and protein intake, has also been noted in adults.48 Major inter-individual variation in microbiota composition is also evident however and this can strongly affect individual responses to dietary carbohydrate.4,49 It has recently been proposed that inter-individual variation in the microbiota can be classified into three discrete enterotypes across the healthy human population.50

Figure 4. Dominant bacterial species identified by analysis of 16S rRNA sequences in fecal samples from six individuals. Data are from Walker et al. (2011)4 and represent the mean of 26 fecal samples from six obese male volunteers (4, or in one case 6, samples per person) taken during a 12 week controlled dietary study. Phylotypes corresponding to the 25 most abundant cultured bacterial species, that accounted for almost 50% of all sequences, are shown in descending order of abundance on the right hand side. The gray area on the left represents the 295 additional phylotypes (both cultured and uncultured organisms) that were detected.
There is limited information on the spatial distribution of bacteria in the human intestine, with most information being available from fecal samples that are assumed mainly to reflect events in the distal colon. In human fecal material, Firmicutes, in particular certain Ruminococcaceae, have been shown to be enriched in the particulate fraction, with Bacteroides more prevalent in the liquid phase.10 This is likely to reflect different ecological niches and roles in substrate breakdown. Relatively little is known about the small intestinal microbiota in humans, but passage rates are more rapid and microbial concentrations lower than in the large intestine, making it unlikely that this is a significant site for microbial fiber degradation. Some reports indicate that the distal ileum harbours a community somewhat similar in composition to that of the proximal colon,51 but the major energy sources appear to be simple carbohydrates.52
Early phenotypic surveys revealed that members of the Bacteroides genus harbor very broad saccharolytic potential, with some strains able to target dozens of different complex glycans.53,54 Gram-positive bacteria (especially the Firmicutes) have received far less attention and their importance in polysaccharide breakdown is only now beginning to emerge. 16S rRNA sequences from human colonic bacteria attaching to wheat bran, resistant starch and mucin in a fermentor system were shown to include high proportions of Firmicutes (75%, 51% and 44%, respectively).55
Degradation of diet-derived carbohydrates
It is estimated that around 20–60 g of dietary carbohydrates reach the colon each day56,57 having escaped digestion by host enzymes. The main categories are resistant starches, plant cell wall polysaccharides and non-digestible oligosaccharides, although some di- and mono-saccharides (e.g., sugar alcohols) also show limited digestion and/or absorption.
Resistant starch
While the majority of ingested dietary starch is completely digested in the small intestine, a variable fraction survives to reach the large intestine.56-58 This fraction is referred to as "resistant starch" and for most diets it is estimated to provide the single largest source of diet-derived energy for colonic bacteria.59 Dietary starch can be resistant because of protection from plant cell wall polymers (type 1), granular structure (type 2), retrogradation (resulting from heating and cooling) (type 3) or chemical cross-linking (type 4). It is also likely however that more rapid oro-cecal transit, and perhaps meals that provide a particularly high starch intake, may result in more digestible starch reaching the large intestine. The fraction of dietary starch that is resistant will therefore vary with diet composition and intake, cooking methods and even between individuals. Resistant starch has been suggested to confer a number of human health benefits that may result from its fermentation and stimulation of microbial growth in the colon.59 Dietary starch typically comprises a mixture of amylose [linear chains of α(1,4)-linked glucose residues] and amylopectin [amylose chains connected by α(1,6)-linked side branches] (Fig. 1). Cereal starches that have a higher content of amylose often show greater resistance to host amylases than those with more amylopectin.60 Pullulan, a repeat polymer comprising α(1,6)-linked maltotriose residues, provides a useful test substrate for enzymatic activity. Catalytic domains that hydrolyze α(1,4) linkages (mainly α amylases) and α(1,6)-linkages (e.g., type 1 pullulanases) in starches are mostly found within GH family 13, while binding domains belonging to several different families can be responsible for binding starch molecules. It is important to note that the preparation of starches both in cooking and in laboratory experimentation strongly influences their fermentability, as well as digestibilility, with autoclaved starches generally being more fermentable by amylolytic human gut bacteria than boiled, or raw, starches.61
Plant cell wall polysaccharides
By comparison with the rumen, discussed above, understanding of the fibrolytic microbial community of the human large intestine remains somewhat limited. The digestibility of cellulose and hemicellulose in a group of seven women on a standardised diet was estimated at 70% and 72% respectively,62showing that there is extensive degradation of these polysaccharides in dietary plant cell wall material during passage through the human intestine. The type of cellulose appears to be critical, however, since in the same study only 8% of an added refined cellulose (Solka Floc) was digested.62 Whereas bacteria able to grow on sources of hydrated, amorphous cellulose, such as spinach cell walls, can apparently be isolated from most individuals, bacteria able to degrade largely crystalline cellulose substrates, such as milled filter paper, are not always recoverable.63-65 Cellulolytic strains isolated from human feces have been classified as Ruminococcus sp, Clostridium sp, Eubacterium sp and Bacteroides sp.63-66 Interestingly, it has been suggested that the structure and activity of the cellulose-degrading community varies according to the methanogenic-status of the individual; thus among cellulolytic isolates, Ruminococcus sp were predominant in methane excretors and Bacteroides in the non-methane excretors.65 It was hypothesized that these differences might be linked to H2 transfer between H2-producing cellulolytic bacteria (the ruminococci) and methanogenic archaea65 although gut transit also tends to be slower in methanogenic individuals.67
Inulin, oligosaccharides and prebiotics
There is a strong interest in optimising the colonic microbiota through dietary manipulation. A prebiotic has been defined as “a selectively fermented ingredient that allows specific changes, both in the composition and/or activity in the gastrointestinal microflora that confers benefits upon host well-being and health”68,69. Currently used prebiotics are mainly low digestible carbohydrates that are found naturally in foods. These include xylo-oligosaccharides (XOS), galacto-oligosaccharides (GOS) and fructans, including inulin and fructo-oligosaccharides (FOS).70,71 Any dietary substrate that remains undigested in the upper GIT, and that may have beneficial effects, is however a potential prebiotic. The health benefits attributed to various prebiotics, including FOS and GOS, have been extensively reviewed (eg.70,71).
Most of the available information on prebiotics has focused on fructans which were the first carbohydrates to be used to increase the abundance of bifidobacteria in the human colon. The inulin type fructans are present in foods such as onions, garlic and bananas that are linear polymers of β (2,1)-linked fructose residues, with terminal glucose residues (Fig. 1). Oligofructose has a DP (degree of polymerisation) of between two and eight units and inulin has a DP of up to 65. Bacterial utilization of fructans is dependent on the presence of β-fructofuranosidases. Different bacterial β-fructofuranosidases vary in their ability to cleave the β(2,1) bonds in sucrose, FOS and inulin.72 Galacto-oligosaccharides (GOS) are chains of galactose residues (DP 3–10) with a terminal glucose residue. GOS can be formed by treating lactose with β-galactosidase, and the final GOS product has a range of linkages (β-(1,2); β(1,3); β(1,4)) depending on the production conditions. One of the most abundant natural sources of GOS is human milk, and this has led to the development of GOS-enriched formula milk.
Prebiotics are also used in conjunction with probiotics, the so-called “synbiotic” approach. In a group of elderly patients given a double probiotic mixture of Bifidobacterium bifidum and B. lactis, the inclusion of an inulin/FOS prebiotic enhanced the survival of the introduced bifidobacteria, and increased numbers of native bifidobacterial populations in some volunteers.73 Whereas earlier work to investigate the effects of prebiotics on the gut microbiota tended to focus entirely on the intended target organisms (normally Bifidobacterium spp), it is now possible, and certainly desirable, to monitor the response of the whole community so as to assess the selectivity of different prebiotics. Utilization of prebiotic carbohydrates is proving to be more widespread among phylogenetically diverse bacteria than was originally considered.74
Utilization of host-derived glycans
From birth most infants are exposed to oligosaccharides, present at concentrations of around 10 g/L in human breast milk, that consist mainly of L-fucose, D-glucose or D-galactose residues. Bifidobacterium spp usually dominate in the feces of breast fed babies and this is thought to be due to their abilities to utilize oligosaccharides in breast milk.75 In total, human milk contains around 200 different oligosaccharides, with as many as 130 in milk from a single mother.76 Since none of these can be metabolised by infant digestive enzymes, the reason for their production is assumed to be the selective stimulation of particular bacteria.
The major host-derived source of glycans entering the gut throughout life is mucin, a group of glycoproteins that are produced continuously in large amounts by the gut epithelium. A limited number of microbial species appear able to digest mucin; these include the recently described bacterium Akkermansia muciniphila, a member of the Verrucomicrobium phylum, which can comprise as much as 3% of gut bacteria detected in feces of adults.77 Comparing the genome sequence of A. muciniphila with those of other Verrucomicrobia reveals the presence of relatively more genes involved in carbohydrate transport and metabolism.78 Signal peptides were detected in 26% of the predicted proteome from A. muciniphila, indicating a high proportion of exported products that include many with a likely role in mucin degradation.78 Sugars are also present on gut epithelial surface glycoconjugates. Bacterial cells able to use endogenously derived substrates as an energy source are likely to have a competitive advantage during periods of reduced dietary intake.
Human Colonic Bacteroides
Early work showed that human Bacteroides species were able to degrade diverse plant polysaccharides, including pectin, galactomannan, arabinogalactan, alginate, laminarin and xylans,53,79,80 while more recent work has extended this to include xyloglucan, rhamnogalacturonans I and II, β-glucans and glucomannan.81 Bacteroides ovatus, B. thetaiotaomicron and B. uniformis ferment a particularly wide range of polysaccharides, and this versatility may help to explain their prevalence as dominant species in the colon.4,46,47 The xylanolytic microbiota was recently re-investigated, yielding new isolates belonging to B. intestinalis, B. ovatus, B. dorei, B. cellulosilyticus and B. xylanisolvens.82 Furthermore, the direct cloning of xylanase genes has suggested that other as yet uncultivated xylanolytic Bacteroides and Prevotella exist in the human intestine.83 The main cellulose-degrading bacteria isolated recently from non-methane-excreting subjects belonged to the new species B. cellulosilyticus, which is the only cellulolytic Bacteroides described to date.65,84 Cellulases have not yet been characterized however from any human Bacteroides strain.
Among hemicellulose-degrading activities, enzymes involved in the hydrolysis of xylans, mannans and galactomannans were characterized in B. ovatus.85,86 Polygalacturonases from B. thetaiotaomicron were also characterized.79 All of the enzymes or activities identified in these Bacteroides species were found cell-associated rather than extracellular, and their production appeared highly regulated by the substrate. For most of them, the cellular location was either in the outer membrane or in the periplasm.79,85 Xylan utilization has been studied more recently in B. xylanisolvens, which has proved to be the most active of several newly described xylanolytic Bacteroides species.87,88
Starch utilization and the Sus paradigm
The organization of starch-degrading enzymes was first studied in B. thetaiotaomicron, which can utilize various forms of starch, including amylose, amylopectin and pullulan as well as the corresponding malto-oligosaccharides.53 Salyer’s group showed that the starch degradation enzymes are cell-associated and that the binding of the polysaccharide to the cell surface is the first step in the degradation process.89 The starch binding activity and the degradative enzymes are maltose inducible, with these functions encoded by an operon of eight genes (susRABCDEFG).90 Gene-disruption analysis enabled roles to be assigned to the proteins encoded by the different genes. Salyers proposed the original model for the highly efficient « Starch Utilization System » of B. thetaiotaomicron, that has recently been reviewed91 (Fig. 5).
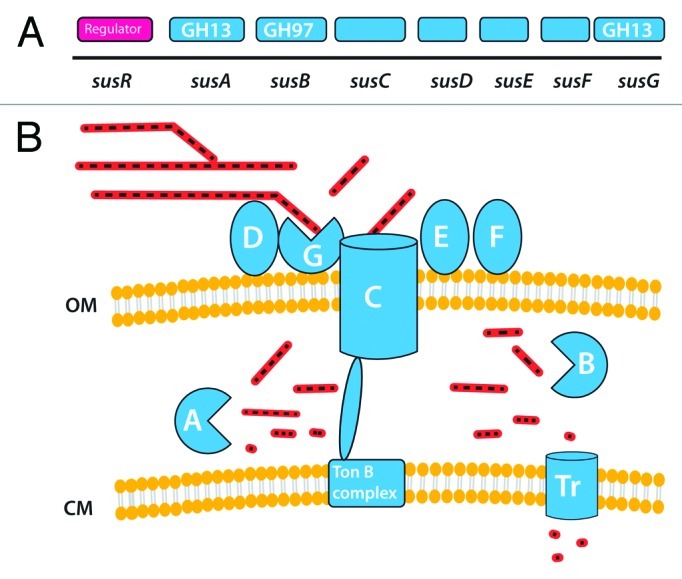
Figure 5.Bacteroides thetaiotaomicron sus system. A shows the order of genes in the sus cluster that is responsible for starch utilization in this species. B shows the inferred organization of gene products on or near the bacterial cell surface (OM outer membrane, CM cytoplasmic membrane). Starch molecules are shown as sugar chains, at various stages of hydrolysis.
SusCDEFG are localized at the cell surface and bind, degrade and import soluble starch molecules.92-94 SusDEFG are lipoproteins anchored at the outer membrane of the cell. SusD is responsible for the binding of starch to the cell surface, and this binding appears to be driven by recognition of the overall three-dimensional shape of the starch molecule.95 SusE and SusF are also likely to be involved in starch binding.91,92 SusC is a pivotal protein in the system: it is a TonB-dependent transporter (TBDT), a group of outer-membrane-spanning β-barrel proteins that sense and transport various molecules in Gram negative bacteria.96 Unlike the other TBDT characterized to date, SusC cannot bind the ligand alone and requires the starch-binding protein SusD for starch import. Therefore, SusD likely plays a critical role in targeting polymeric starch to the Sus complex and may facilitate movement of linear oligosaccharides to SusC. SusG is a GH13 α-amylase that may have evolved to work as part of a carbohydrate- processing/import complex rather than just as an outer-membrane amylase. susG deletion mutants could still bind starch at the cell surface, but could not grow on starch,92,93 suggesting that SusG is essential for the metabolism of starch, despite the presence of four other predicted amylases in B. thetaiotaomicron genome. The proposed mechanism is as follows91: starch molecules are held on the surface of the bacteria through multiple interactions with SusD proteins. This anchors the polysaccharide in close proximity to SusG, and enables the enzyme to hydrolyze the starch. The cleaved maltooligosaccharides still bound to SusD are then presented to the entrance of the SusC porin (Fig. 5). The maltooligosaccharides are translocated by SusC and released in the periplasm where they are broken down by SusA and SusB, a periplasmic GH13 α-amylase and a GH 97 α-glucosidase, respectively.97 The small saccharides produced can then be transported into the cytoplasm (Fig. 4). The sus cluster is regulated by the transcriptional regulator SusR in response to maltooligosaccharides, amylose, amylopectin, and pullulan but is also controlled by the regulatory protein MalR.98 The Sus system thus appears to be a very efficient and well controlled selfish system for the capture, sequestration and degradation of starch, giving B. thetaiotaomicron an ecological advantage in a very competitive ecosystem.
Polysaccharide Utilization Loci
Since the discovery of the Sus complex, 88 similar Sus-like Polysaccharide Utilization Loci (PULs) have been identified, representing 18% of the B. thetaiotaomicron genome.16,99,100 35 of these have been reported to degrade mucin or other host-derived glycans and include enzymes that target glycan decorations such as sulfatases and acetyl-esterases.99,100 The other PULs are probably involved in the degradation of plant polysaccharides, ten of them being dedicated to pectins.81 PULs have also been identified for the utilization of FOS and levans.101 Recent studies using B. thetaiotaomicron-associated gnotobiotic mice and bacterial genome transcriptional profiling have shown that this species has evolved mechanisms to adapt glycan utilization to nutrient availability within the ecosystem. When dietary polysaccharides were supplied to the mice, B. thetaiotaomicron expanded its niche from host derived glycans to accommodate the additional diet-derived nutrients.99 When the dietary polysaccharides became less available, the bacterium turned to the utilization of the host mucins. In addition, in the gut of suckling mice, B. thetaiotaomicron relied on host-derived mucosal polysaccharides in addition to mono and oligosaccharides present in mother’s milk, but after weaning, the bacterium expanded its metabolism to exploit abundant, plant-derived dietary polysaccharides.102 All these mechanisms of adaptation are based on the regulation of the expression of the different PULs, which very efficiently sense the substrates available.
More generally, Sus-like complexes appear as a paradigm for glycan uptake in bacteria belonging to the phylum Bacteroidetes, and have been identified in other human gut Bacteroides as well as in ruminal Prevotella and in environmental Bacteroidetes.103,104 More than 50 Bacteroides genomes are currently available in the NCBI database, and these confirm that gut-associated Bacteroidetes possess an extensive repertoire of genes predicted to encode CAZymes. PULs have been identified that target pectins and hemicelluloses such as xylans (the xus cluster) and galactomannans.41,103,104 Although it was first thought that PULs were adapted only to soluble or well-hydrated polysaccharides, PULs have also been found associated with genes coding CAZymes targeting insoluble polysaccharides.105
Although different PULs encode different repertoires of proteins involved in the utilization of specific polysaccharides, they are all organized in a manner similar to the sus operon of B. thetaiotaomicron (Fig. 5), and comprise SusC-like TBDT and SusD paralogs, as well as CAZymes adapted to the substrate, located at the cell surface and in the periplasm. The susC-like and susD-like genes are the central units of substrate-specific PULs. For example, B. thetaiotaomicron possesses 108 paralogs of susC, of which 101 are paired to a susD-like gene, and 88 of these pairs are associated with CAZyme genes.15,99,103 Pairs of susC/susD-like genes often appear in tandem, possibly as a result of gene duplication. In conclusion, the PUL system appears to be a generic feature of carbohydrate nutrient acquisition by gut and environmental Bacteroidetes.
Regulation
PULs also include regulators belonging to the hybrid two-component histidine kinase response regulators and Extra Cytoplasmic Function (ECF-type) sigma factors and anti-sigma factors, which participate in trans-envelope signaling.106 In B. thetaiotaomicron, at least 18 of the 88 susC/susD clusters contain ECF-sigma factors and adjacent anti-sigma factors.103 Analysis of the TBDT sequences in B. fragilis genome indicated that most of them were associated to CAZymes and possessed an N-terminal extension identifying them as putative transducers.106 Protein contacts are made in the periplasm (TBDT/anti-σ interaction) and then at the cytoplasmic face of the inner membrane (anti-σ/ECF-σ interaction). The signal is transduced across the entire bacterial cell envelope and results in activation of the ECF-σ transcription factor which activates the PULs. The protein interactions were experimentally demonstrated for some of the B. thetaiotaomicron PULs, in particular mucin Ο-glycan signaling.104 B. thetaiotaomicron also contains an expanded collection of 32 hybrid two-component systems (HTCS), of which 17 are adjacent to susC/susD genes.107 HTCS are proteins that incorporate all domains found in classical two-component environmental sensors into one polypeptide. Twenty eight of the 32 B. thetaiotaomicron HTCS reside within loci that are induced transcriptionally in response to modified polysaccharide content of the host mice diet, suggesting the contribution of these systems to glycan sensing.99 The various HTCS and anti-σ/ECF-σ factors might sense specifically the glycan ligand released in the cytoplasm by their associated TBDTs allowing the induction of the corresponding genes or operons. This allows a sophisticated integrated control of the carbohydrate hydrolytic and metabolic machinery of the cell in response to the availability of nutrients in the gut environment.
Evidence for horizontal transfer of polysaccharide utilization genes
Horizontal transfer within the gut microbiota, but also from microbes living outside the gut and ingested with the food, has probably played an important role in the diversification of the degrading-activity of each species.21,108 The plasticity of Bacteroidetes genomes appears to be driven by frequent genetic rearrangements, gene duplications, and horizontal gene transfers (HGT) between species. Using a phylogenetic approach, around 5.5% of the genes in gut Bacteroidetes genomes were inferred to be laterally acquired from non gut-associated bacteria, among which glycosyltransferases (GT) where significantly over-represented.103 In addition, the convergence of GT and GH repertoires in gut Bacteroidetes was due mainly to massive HGT rather than gene duplications.109 Recently, a porphyran/agar degradation locus was discovered and characterized in a member of the marine Bacteroidetes.108 Surprisingly, homologs were found in the human gut bacterium B. plebeius that was isolated from Japanese individuals who are used to eat seaweeds.108 Comparative gut metagenome analysis show that porphyranases and agarases are frequent in the Japanese population while they are absent in metagenomic data from North Americans.108 The authors concluded that gut bacteria were able to acquire new functions via transfer of a complete degradation pathway from food-associated environmental bacteria.108 Thus, acquisition of selectively advantageous genes by successive HGT events could explain how gut symbionts acquired CAZymes involved in the degradation of plant polysaccharides.
Actinobacteria
This phylum of high % G+C Gram –positive bacteria includes the highly abundant Collinsella aerofaciens and Atopobium spp, but by far the greatest amount of work has been focused on Bifidobacterium spp Many bifidobacterial genes are conserved between species, creating a "core genome" and of the conserved genes, 6.5% are concerned with carbohydrate metabolism.110 Approximately 8% of the genome of B. longum subsp longum is dedicated to carbohydrate metabolism, with many genes organized into clusters containing a LacI-type of repressor protein.111 These sugar-responsive regulators, which carry sugar-binding motifs, presumably permit a rapid response to changes in the availability of different substrates.111 B. longum subsp infantis contains more unique genes than other sequenced Bifidobacterium spp,112 many of which are located in a large cluster of carbohydrate utilization genes with both enzymatic and transport activities. Many of these genes were found to be specific for mammalian derived carbohydrates and were absent in bifidobacterial species normally associated with adults.113
Starch utilization
Bifidobacterium spp have been reported to be particularly effective degraders of high amylose starches58 and some strains are known to be able to attach to starch particles.55,114 Degradation of RS and of pullulan appears to be associated with particular strains and species, especially B. breve and B. adolescentis.115 Detailed work on B. breve has identified a major cell surface anchored enzyme that comprises distinct α(1,4) amylase and type 1 pullulanase domains together with multiple CBMs (Fig. 3). Deletion of this one gene abolished growth on starch, pullulan and glycogen116 although multiple GH13 genes are found in the genomes of B. adolescentis and B. breve.
Other plant polysaccharides and prebiotics
Tannock et al.117 reported that, in addition to Bifidobacterium spp, C. aerofaciens increased in fecal samples from volunteers consuming FOS, and another human study found that numbers of Bifidobacterium and Atopobium were increased by long chain inulin (average DP > 55).118 Two types of β-fructofuranosidase have been identified in Bifidobacterium spp: those that are more active against the β(2,1) glucose–fructose bonds, releasing only the terminal glucose residue for growth119 and those that are more active against β(2,1) fructose–fructose links.120 Both types of β-fructofuranosidase however had only low activities against long-chain inulin molecules.119-121 Only eight out of 55 Bifidobacterium strains tested, from five different species, were able to grow on long chain inulin, although all grew well on FOS.122 It appears that bifidobacteria can be split into clusters: those unable to use any fructan (B. bifidum and B. breve); those able to use only short chain FOS (7 species); and those able to use scFOS and short chain inulin.123 Fructan-utilizing ability was not species-specific, with strains of B. longum for instance falling into different clusters.123 The chain length of the substrate is therefore likely to be critical to its subsequent effect on the composition of the microbial community. None of the Bifidobacterium sp tested were active against inulin chains longer than 20 units long.123 Complex cross-feeding interactions have been demonstrated for co-cultures between human colonic B. thetaiotaomicron and Bifidobacterium spp that have different abilities to utilize fructan molecules of different chain length.124 Meanwhile other bifidobacteria can be involved in cross-feeding with butyrate-producing bacteria either by releasing oligo- and mono-saccharides from complex substrates, or via the utilization of acidic fermentation products.14
Studies in adults consuming GOS (5 g per day) revealed that there was more than a 100-fold increase in abundance of Bifidobacterium populations in fecal samples. Only 50% of the subjects were responders however, thereby revealing a considerable degree of inter-individual variation.125 Doses below 5 g GOS per day were not sufficient to induce a response while 10 g per day gave an even greater increase in bifidobacteria in some volunteers suggesting a dose-response effect.126 Analysis of the pyrosequencing data revealed that GOS enriched for particular Bifidobacterium-related OTUs. B. bifidum possesses four distinct β-galactosidases, which seem to act in complementary ways on different substrate bonds, thus contributing to efficient substrate degradation.127
It has also been shown that substrate responses occur at a species-specific level. B. adolescentis was elevated in response to FOS/inulin in humans74 and in humanised rats.128 Bifidobacterium spp are also reported to have some ability to utilize arabinoxylans and arabinogalactan, but benefit from initial substrate breakdown of the complex polymers by other bacteria.129 Supplementation with the novel prebiotic long-chain arabinoxylan significantly increased numbers of bifidobacteria in humanised rats, particularly boosting B. longum.128
Host-derived carbohydrates
The plethora of genes specific for degradation of mammalian derived carbohydrates in B. longum subsp infantis presumably reflects the adaptation of this species to use human milk oligosaccharides (HMOs) for growth. Only B. bifidum and B. longum subsp infantis were able to grow well on HMOs, with other Bifidobacterial species having variable abilities.130 B. longum subsp infantis expresses specific genes in direct response to the composition of the milk; with fucosidases only detected during growth on HMOs.131 The B. longum subsp infantis genome is also enriched in family 1 solute binding proteins (F1SBPs) that are particularly associated with oligosaccharide uptake. Different classes of F1SBPs were induced specifically during growth on different substrates.132 In a separate study B. breve was found to be prevalent in breast-fed babies and not in those fed on formula milk.133 The genome sequence of B. bifidum also contains many genes involved in the degradation of host-derived glycans, in particular the O-linked glycans attached to mucin, which appear to be co-regulated.134
Firmicutes
Two families of Firmicutes, Lachnospiraceae and the Ruminococcaceae, are particularly abundant in the human large intestine, typically accounting for 50–70% of bacteria in fecal samples from healthy human adults based on 16S rRNA analyses. These include some highly oxygen-sensitive organisms and are seriously underrepresented by available cultured isolates, but they are responsible for some of the key metabolic conversions within the intestinal community.13 They include for example the major butyrate-producing species,49,135,136 as well as species that convert lactate to butyrate or propionate137 and species that perform reductive acetogenesis.138,139 Emerging evidence suggests that these and other Firmicutes play key roles in polysaccharide degradation.
Starch utilization
Three recent studies have reported an increase in Ruminococcus bromii-related bacteria in volunteers consuming diets enriched with RS.4,140,141 This group was also prominent among fecal bacteria shown by stable isotope probing to utilize 13C labeled starch in vitro.142 Walker et al.4 saw a mean increase of > 4 fold (from 3.8% to 17%) in the overall proportion of cluster IV Ruminococcus-related 16S rRNA sequences detected by qPCR in 14 obese volunteers when consuming a diet containing 26 g/day of type 3 RS compared with a low RS, wheat bran-enriched diet. The fractional increase was greater for sequences > 98% related to R. bromii (0.4% to 5%). qPCR analysis indicated that further uncultured phylotypes among the Ruminococcaceae also responded to the RS diet. Remarkably, two of the 14 individuals showed no detectable ruminococci in their fecal samples and these were the only two individuals to give low estimates for starch fermentation.4 Additional evidence has now been obtained that supports the view that R. bromii –related organisms may indeed play a ‘keystone’ role in the initial stages of breakdown of particulate resistant starch.61 R. bromii showed a much greater ability to degrade raw or boiled RS2 and RS3 starches than B. thetaiotaomicron, and non-growing R. bromii cells were found to greatly enhance the utilization of these starches by three other prominent human amylolytic species, B. thetaiotaomicron, E. rectale or B. adolescentis.61 By contrast, a second, highly abundant group of Ruminococcaceae related to Faecalibacterium prausnitzii apparently does not utilize starch, based on the cultured strains currently available.143The enzyme systems that allow R. bromii to efficiently utilize particulate starch have not yet been fully investigated. In contrast to other abundant amylolytic bacteria found in the human colon R. bromii fails to grow on glucose, and grows more rapidly on malto-oligosaccharides than on maltose.61
Among the Lachnospiraceae, the ability to utilize starch has been reported for most members of the Roseburia/ Eubacterium rectale group of butyrate-producing bacteria.144 Furthermore the population of this group in fecal samples has been found to increase on average in human volunteers on RS-enriched diets4 and to decrease on diets low in total carbohydrate.145 Roseburia spp produce a major, high molecular weight (> 180kDa) amylase that is detectable by zymogram analysis. The enzyme from R. inulinivorans, Amy13A, includes a GH13 amylase and two or more CBMs and is able to cleave α(1,4) linkages in amylose, amylopectin and pullulan.146 The pre-protein carries an N-terminal signal peptide and a C-terminal sortase-mediated anchoring sequence indicating that it becomes anchored to the cell wall but extrudes into the extracellular matrix (Fig. 3). R. inulinivorans Amy13A is induced by growth on starch, along with expression of flagella that are characteristic of this group of bacteria and that may perhaps help cells to migrate toward particulate substrates.147 Genome sequences indicate 9 to13 GH13 genes in Roseburia spp and in the related species E. rectale, but the roles of the different gene products have not been elucidated. E. rectale was less active against boiled or raw RS than R. bromii.61 Although GH13 genes are present in most sequenced representatives of the Lachnospiraceae, the contribution of other species to starch degradation in the colon is currently unknown.
Plant cell wall polysaccharides
Salyers et al.53,54 reported finding a lower frequency of plant polysaccharide utilizers among Gram-positive anaerobes than among the Bacteroides spp tested. Subsequent evidence, both from molecular studies and new isolations, has however suggested that Firmicutes play a significant role in the degradation of complex plant carbohydrates. In particular, Ruminococcus champanellensis, a new species related to R. flavefaciens, is the only human colonic bacterium so far reported to degrade microcrystaline cellulose.148 Human colonic strains related to R. albus were reported to utilize galactomannan.54 A shortage of cultured organisms from the Ruminococcaceae means that information on this group remains limited, especially for the human gut, but many appear to be closely associated with particulate material.10 Among the Lachnospiraceae, cellulolytic activity was reported in the acetogenic bacterium Bryantella formatexigens on first isolation, but apparently proved unstable.149 Xylan-utilization has been reported for Roseburia intestinalis82,150 and also for the human B. fibrisolvens strain 16/4, which was isolated from a wheat bran enrichment.151 The distribution of the two main families of endoxylanases (GH10 and GH11) appears limited among other human intestinal Firmicutes for which draft genome sequences are available. The highly abundant species F. prausnitzii is now known to include strains able to utilize apple pectin for growth.143 The only other pectin-utilizing Firmicutes species identified so far from the human colon are Eubacterium eligens54 and Lachnospira pectinoschiza.
Prebiotics
Relatively little attention has been paid to the utilization of prebiotic oligosaccharides by Firmicutes. It is clear however that many species can utilize FOS, while some utilize long chain inulin. F. prausnitzii, E. rectale and R. inulinivorans for example are abundant butyrate-producing species4,49 that include strains able to grow on inulin and FOS in pure culture. R. inulinivorans encodes a β-fructofuranosidase that acts against short and long chain length molecules.147 The genes for the major β-fructofuranosidase and a linked ABC sugar transport system in R. inulinivorans are upregulated during growth on inulin compared with starch.147
In a human study where a FOS/inulin mixture was supplemented to the diet, numbers of both Bifidobacteria spp and Faecalibacterium prausnitzii were significantly increased.74 GOS consumption has also been reported to increase F. prausnitzii.126 In human flora-associated rats, Lachnospiraceae numbers increased following dietary supplementation with either an inulin/FOS mixture, or inulin alone, but no effect was observed with FOS alone.152 Increases have also been reported in the numbers of Lachnospiraceae upon in vitro fermentation of both pectic oligosaccharides and FOS153 while inulin enhanced the survival of R. inulinivorans against a background of total fecal bacteria in a fermentor system designed to simulate the proximal colon.154
Increased production of butyric acid has been noted with FOS supplementation, although the intended targets, bifidobacteria, do not produce butyrate.153 This may be explained in part by reduced pH favoring butyrogenic bacteria155,156 and in part by the ability of butyrate-producing bacteria such as R. inulinivorans and F. prausnitzii to metabolise fructans including long chain inulins.74,143,147,154 Humanised rats fed with a FOS/inulin mixture had increased cecal concentrations of butyrate, which correlated with a higher incidence of butyrate producing bacteria in the Roseburia/E. rectale group.128 In addition, bifidobacteria form acetate and lactate as major end products and these products can be co-metabolised by cross-feeders including Anaerostipes spp and Eubacterium hallii to form butyrate.137
Host-derived carbohydrates
Certain Lachnospiraceae, notably R. torques, have been identified as mucin-degraders.157 Strains of F. prausnitzii were recently shown to utilize N-acetyl glucosamine for growth, although none was able to utilize mucin.143 The sugar fucose is found extensively in host glycoconjugates and can be utilized by R. inulinivorans as a growth substrate.158 Interestingly B. thetaiotaomicron can only partially utilize fucose for growth,159 whereas R. inulinivorans can convert the propane-1,2-diol intermediate into propionate and propanol via the toxic propionaldehyde intermediate.158 A similar metabolic route for fucose metabolism has been described in the gut pathogen Salmonella serovar Typhimurium LT2. The fucose utilization genes in R. inulinivorans A2–194 are strongly upregulated during growth on fucose.158 Genome searching indicates that other species of Lachnospiraceae normally found in the human colon, R. obeum and R. gnavus, also possess homologs of the key R. inulinivorans fucose utilization genes, including those involved in the synthesis of a polyhedral body required for propane-1,2-diol metabolism. This indicates that these bacteria may employ a similar pathway for fucose utilization.
Metabolic Consequences of Carbohydrate Fermentation in the Human Colon
Impact on the gut environment
Addition of any non-digestible but fermentable, carbohydrate to the diet will increase fermentative activity, especially in the proximal colon, resulting in increased acid production. This tends to decrease luminal pH, with important consequences for the composition of the microbiota and the balance of microbial metabolites. In vitro studies indicate that Bacteroides populations are likely to be curtailed, while butyrate-producing Firmicutes are favored, within the community at mildly acidic pH.155,160 Reduced overall intake of complex dietary carbohydrates by obese subjects on weight loss diets was found to decrease short chain fatty acid formation, with a disproportionate decrease in fecal butyrate.145,161,162 Interestingly, the major butyrate-producing bacteria detected on high carbohydrate diets were the starch-utilizing Roseburia spp and E. rectale and the decrease in fecal butyrate on diets very low in carbohydrates was associated with a major decrease in this group, with F. prausnitzii becoming the main butyrate producer. Dietary complex carbohydrates also decrease the levels of potentially harmful metabolites that arise from proteolytic activity in the colon.162 Separately De Preter et al.163 demonstrated in in vitro studies that there was a dose dependent stimulation of saccharolytic fermentation when fructans were included in their growth medium concomitant with a decrease in toxic peptide fermentation metabolites.
Increased SCFA concentrations may also increase the solubility of certain minerals such as calcium, and enhance absorption and expression of calcium-binding proteins.164 Changes in intestinal microbial metabolism following the consumption of inulin fructans have also been shown to benefit bone health by increasing calcium absorption while β-glucans may lower total cholesterol levels.165 High fiber diets increase fecal bulking, short chain fatty acid production and transit rates along the large intestine.166,167 Slow transit rates will encourage growth of the slower growing microorganisms such as some of the hydrogen-utilizers including methanogens, that are present in approximately 50% of the population. Methane has been shown to slow gut transit in animal studies168 and the presence of methanogens is also associated with slower gut transit in humans.67 Digestion of plant fiber also results in the release of phenolic compounds. Epidemiological studies suggest that there is an inverse association between the intake of polyphenol-rich diets and the incidence of cardiovascular disease, diabetes and cancer169 but it is unclear at present what proportion of absorbed bioactive phenolic compounds can be ascribed to microbial activity.
Recovery of energy from dietary carbohydrates: consequences for obesity, weight loss and metabolic health
A high proportion of the SCFA produced by microbial fermentation of indigestible carbohydrates in the large intestine is absorbed by the host. Thus microbial activity contributes energy to the host (estimated to be around 10% of calories obtained from the diet3) that would otherwise be lost through excretion of undegraded substrate in the feces. On the other hand, the calories that are obtained from a sugar via fermentation, followed by absorption and metabolism of SCFA, are estimated to be less than half the amount that would be gained by direct absorption of the same amount of sugar in the small intestine.170 The net effect of replacing consumption of a digestible carbohydrate in the diet with consumption of the same amount of fermentable, non-digestible carbohydrate is therefore to reduce the calories acquired from the diet.
The possible involvement of the gut microbiota in the development of obesity is proving far more complex than was first proposed. Variation in microbiota composition has the potential to influence “energy harvest” from fiber171 if it affects key groups involved in energy release and recovery, but factors such as gut transit and absorption seem likely to be more important172 (Fig. 6). Phylum level differences in the gut microbiota in obese vs. lean individuals have been reported in some studies, but not in others, and it appears that differences in dietary intake are mainly responsible for microbiota changes.173,174 Interestingly, however, microbiota composition also has the potential to influence satiety (and thus dietary intake) and energy expenditure. The finding that germ-free animals were apparently protected from developing diet-induced obesity175 has recently been balanced by a study reporting the opposite effect.176 These effects were thus shown to be highly dependent on the type of high-fat diet fed to germ-free mice, and were also found to be linked to differences in energy expenditure.176 Potential links between the gut microbiota and metabolic disease have also been under intense investigation in recent years.177-179 Serum levels of lipopolysaccharide (LPS), derived from Gram-negative bacteria, are reported to increase in obese, diabetic or high-fat fed subjects, and reproduction of similar LPS levels by chronic injection lead to a loss of insulin sensitivity in animals.178 The increased LPS levels may result from a decrease in the gut barrier function. It was shown that the administration of prebiotics (FOS) improved gut barrier function, which was strongly correlated with reduced portal plasma LPS levels. The effect seems to be mediated by the gut hormone glucacon-like peptide-2 178.

Figure 6. Energy intake and expenditure. The diagram summarizes the potential of gut microorganisms to influence energy gain and expenditure in a mono-gastric animal such as man.168 The energy arising from microbial fermentation via the absorption of short chain fatty acids may be influenced by microbiota composition, but more especially by gut transit, affecting the efficiency of substrate breakdown, digestion and absorption. Other potentially important, but little understood, influences however include possible effects of microbial products on satiety and energy expenditure.
Physiological impact of SCFA
Besides supplying energy to the host, SCFA, along with other microbial metabolic products, have wider effects on host physiology. The gut microbiota may influence the expression of host peptides and hormones by production of short-chain fatty acids via their interaction with free fatty acid receptors FFA2 and FFA3, thus influencing host energy metabolism and appetite regulation.180 Propionate has been shown to increase satiety and improve glucose homeostasis also when taken orally.181 The effect of butyrate on the host has received much attention due to its anti-inflammatory and anti-carcinogenic effects, but it also appears to be involved in the regulation of other host functions.182 A recent study found that oral administration of butyrate to mice fed a high-fat diet prevented development of insulin resistance and obesity. This effect was not due to a reduced food intake, but to increased energy expenditure.183 Butyrate concentrations in plasma were only increased by approximately 1.6-fold compared with control animals, therefore manipulation of the microbiota to increase systemic butyrate levels via the colonic route could possibly be achieved, despite the fact that most bacterially produced butyrate is consumed in the colonic wall.
Another potential route linking microbial activity with the host is via the gut-brain axis, a bi-directional communication system based on neural, endocrine and immunological mechanisms. There is increasing evidence that there may indeed be a link between the gut microbiota and the brain.184 Recent rodent studies indicated that changes in the microbiota composition led to behavioral changes and altered levels of brain-derived neurotropic factor (BDNF) in different brain regions.185 These changes did not appear to be mediated by gut inflammation, specific enteric neurotransmitters or the autonomic nervous system, and it was hypothesized that microbial products acting on the central nervous system are likely to be involved, with butyrate being one potential candidate.185
The immune system is influenced by microbial metabolic products, but can also recognize a diverse range of microbial cell components. This leads to complex interactions between the species composition of the microbiota and the host’s innate and adaptive immune systems that are thought to underlie many probiotic effects.186
Conclusions
Bacteria that colonize the mammalian intestine collectively possess a far wider diversity of genes and a larger repertoire of degradative enzymes and metabolic capabilities than their hosts. Fermentation of complex carbohydrates in the intestine involves interactions between community members that include both nutritionally specialized and widely adapted species. Certain dominant species, notably among the Bacteroidetes, possess very large numbers of genes encoding carbohydrate active enzymes (CAZymes). This allows them to switch readily between different energy sources in the gut depending on availability, using sophisticated sensing and regulatory mechanisms to control gene expression. Other groups encode fewer CAZymes and are clearly more specialized, but some of these organisms appear to play critical roles in the community by initiating the degradation of complex substrates such as plant cell walls, starch particles and mucin. Identification of these ‘keystone’ groups and their roles, particularly among members of the under-investigated Firmicutes phylum, should be a priority for future research. Finally, the impact of dietary carbohydrates, including prebiotics, on health in man requires further progress in understanding of the relationship between diet composition, gut microbiota and metabolic outputs. This demands, in addition to mechanistic understanding, systems-based approaches187 to integrate and model the many complex interactions between functional groups.
Acknowledgments
The authors acknowledge support from the Scottish Government (RESAS).
Footnotes
Previously published online: www.landesbioscience.com/journals/gutmicrobes/article/19897
References
- 1.Van Soest PJ. Nutritional Ecology of the Ruminant. Second edition. 2004. Cornell Univ Press USA. [Google Scholar]
- 2.Ley RE, Hamady M, Lozupone C, Turnbaugh PJ, Ramey RR, Bircher JS, et al. Evolution of mammals and their gut microbes. Science. 2008;320:1647–51. doi: 10.1126/science.1155725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.McNeil NI. The contribution of the large intestine to energy supplies in man. Am J Clin Nutr. 1984;39:338–42. doi: 10.1093/ajcn/39.2.338. [DOI] [PubMed] [Google Scholar]
- 4.Walker AW, Ince J, Duncan SH, Webster LM, Holtrop G, Ze X, et al. Dominant and diet-responsive groups of bacteria within the human colonic microbiota. ISME J. 2011;5:220–30. doi: 10.1038/ismej.2010.118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.De Filippo C, Cavalieri D, Di Paola M, Ramazzotti M, Poullet JB, Massart S, et al. Impact of diet in shaping gut microbiota revealed by a comparative study in children from Europe and rural Africa. Proc Natl Acad Sci U S A. 2010;107:14691–6. doi: 10.1073/pnas.1005963107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Morris EJ, van Gylswyk NO. Comparison of the action of rumen bacteria on cell walls from Eragrostis tef. J Agric Sci Camb. 1980;95:313–23. doi: 10.1017/S0021859600039332. [DOI] [Google Scholar]
- 7.Dehority BA. Effects of microbial synergism on fibre digestion in the rumen. Proc Nutr Soc. 1991;50:149–59. doi: 10.1079/PNS19910026. [DOI] [PubMed] [Google Scholar]
- 8.Flint HJ, Bayer EA, Rincon MT, Lamed R, White BA. Polysaccharide utilization by gut bacteria: potential for new insights from genomic analysis. Nat Rev Microbiol. 2008;6:121–31. doi: 10.1038/nrmicro1817. [DOI] [PubMed] [Google Scholar]
- 9.McAllister TA, Bae HD, Jones GA, Cheng KJ. Microbial attachment and feed digestion in the rumen. J Anim Sci. 1994;72:3004–18. doi: 10.2527/1994.72113004x. [DOI] [PubMed] [Google Scholar]
- 10.Walker AW, Duncan SH, Harmsen HJM, Holtrop G, Welling GW, Flint HJ. The species composition of the human intestinal microbiota differs between particle-associated and liquid phase communities. Environ Microbiol. 2008;10:3275–83. doi: 10.1111/j.1462-2920.2008.01717.x. [DOI] [PubMed] [Google Scholar]
- 11.Swidsinski A, Loening-Baucke V, Verstraelen H, Osowska S, Doerffel Y. Biostructure of fecal microbiota in healthy subjects and patients with chronic idiopathic diarrhea. Gastroenterology. 2008;135:568–79. doi: 10.1053/j.gastro.2008.04.017. [DOI] [PubMed] [Google Scholar]
- 12.Suen G, Weimer PJ, Stevenson DM, Aylward FO, Boyum J, Deneke J, et al. The complete genome sequence of Fibrobacter succinogenes S85 reveals a cellulolytic and metabolic specialist. PLoS One. 2011;6:e18814. doi: 10.1371/journal.pone.0018814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Flint HJ, Duncan SH, Scott KP, Louis P. Interactions and competition within the microbial community of the human colon: links between diet and health. Environ Microbiol. 2007;9:1101–11. doi: 10.1111/j.1462-2920.2007.01281.x. [DOI] [PubMed] [Google Scholar]
- 14.Belenguer A, Duncan SH, Calder AG, Holtrop G, Louis P, Lobley GE, et al. Two routes of metabolic cross-feeding between Bifidobacterium adolescentis and butyrate-producing anaerobes from the human gut. Appl Environ Microbiol. 2006;72:3593–9. doi: 10.1128/AEM.72.5.3593-3599.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Xu J, Bjursell MK, Himrod J, Deng S, Carmichael LK, Chiang HC, et al. A genomic view of the human-Bacteroides thetaiotaomicron symbiosis. Science. 2003;299:2074–6. doi: 10.1126/science.1080029. [DOI] [PubMed] [Google Scholar]
- 16.Sonnenburg JL, Xu J, Leip DD, Chen CH, Westover BP, Weatherford J, et al. Glycan foraging in vivo by an intestine-adapted bacterial symbiont. Science. 2005;307:1955–9. doi: 10.1126/science.1109051. [DOI] [PubMed] [Google Scholar]
- 17.Mahowald MA, Rey FE, Seedorf H, Turnbaugh PJ, Fulton RS, Wollam A, et al. Characterizing a model human gut microbiota composed of members of its two dominant bacterial phyla. Proc Natl Acad Sci U S A. 2009;106:5859–64. doi: 10.1073/pnas.0901529106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cantarel BL, Coutinho PM, Rancurel C, Bernard T, Lombard V, Henrissat B. The Carbohydrate-Active EnZymes database (CAZy): an expert resource for Glycogenomics. Nucleic Acids Res. 2009;37(Database issue):D233–8. doi: 10.1093/nar/gkn663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hess M, Sczyrba A, Egan R, Kim T-W, Chokhawala H, Schroth G, et al. Metagenomic discovery of biomass-degrading genes and genomes from cow rumen. Science. 2011;331:463–7. doi: 10.1126/science.1200387. [DOI] [PubMed] [Google Scholar]
- 20.Ferrer M, Golyshina OV, Chernikova TN, Khachane AN, Reyes-Duarte D, Santos VA, et al. Novel hydrolase diversity retrieved from a metagenome library of bovine rumen microflora. Environ Microbiol. 2005;7:1996–2010. doi: 10.1111/j.1462-2920.2005.00920.x. [DOI] [PubMed] [Google Scholar]
- 21.Tasse L, Bercovici J, Pizzut-Serin S, Robe P, Tap J, Klopp C, et al. Functional metagenomics to mine the human gut microbiome for dietary fiber catabolic enzymes. Genome Res. 2010;20:1605–12. doi: 10.1101/gr.108332.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Brulc JM, Antonopoulos DA, Miller ME, Wilson MK, Yannarell AC, Dinsdale EA, et al. Gene-centric metagenomics of the fiber-adherent bovine rumen microbiome reveals forage specific glycoside hydrolases. Proc Natl Acad Sci U S A. 2009;106:1948–53. doi: 10.1073/pnas.0806191105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kurokawa K, Itoh T, Kuwahara T, Oshima K, Toh H, Toyoda A, et al. Comparative metagenomics revealed commonly enriched gene sets in human gut microbiomes. DNA Res. 2007;14:169–81. doi: 10.1093/dnares/dsm018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bayer EA, Lamed R, White BA, Flint HJ. From cellulosomes to cellulosomics. Chem Rec. 2008;8:364–77. doi: 10.1002/tcr.20160. [DOI] [PubMed] [Google Scholar]
- 25.Ding SY, Rincon MT, Lamed R, Martin JC, McCrae SI, Aurilia V, et al. Cellulosomal scaffoldin-like proteins from Ruminococcus flavefaciens. J Bacteriol. 2001;183:1945–53. doi: 10.1128/JB.183.6.1945-1953.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jindou S, Borovok I, Rincon MT, Flint HJ, Antonopoulos DA, Berg ME, et al. Conservation and divergence in cellulosome architecture between two strains of Ruminococcus flavefaciens. J Bacteriol. 2006;188:7971–6. doi: 10.1128/JB.00973-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rincon MT, Dassa B, Flint HJ, Travis AJ, Jindou S, Borovok I, et al. Abundance and diversity of dockerin-containing proteins in the fiber-degrading rumen bacterium, Ruminococcus flavefaciens FD-1. PLoS One. 2010;5:e12476. doi: 10.1371/journal.pone.0012476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Flint HJ, Martin JC, McPherson CA, Daniel AS, Zhang J-X. A bifunctional enzyme, with separate xylanase and beta(1,3-1,4)-glucanase domains, encoded by the xynD gene of Ruminococcus flavefaciens. J Bacteriol. 1993;175:2943–51. doi: 10.1128/jb.175.10.2943-2951.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Aurilia V, Martin JC, McCrae SI, Scott KP, Rincon MT, Flint HJ. Three multidomain esterases from the cellulolytic rumen anaerobe Ruminococcus flavefaciens 17 that carry divergent dockerin sequences. Microbiology. 2000;146:1391–7. doi: 10.1099/00221287-146-6-1391. [DOI] [PubMed] [Google Scholar]
- 30.Berg Miller ME, Antonopoulos DA, Rincon MT, Band M, Bari A, Akraiko T, et al. Diversity and strain specificity of plant cell wall degrading enzymes revealed by the draft genome of Ruminococcus flavefaciens FD-1. PLoS One. 2009;4:e6650. doi: 10.1371/journal.pone.0006650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rincon MT, Cepeljnik T, Martin JC, Lamed R, Barak Y, Bayer EA, et al. Unconventional mode of attachment of the Ruminococcus flavefaciens cellulosome to the cell surface. J Bacteriol. 2005;187:7569–78. doi: 10.1128/JB.187.22.7569-7578.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Moon YH, Iakiviak M, Bauer S, Mackie RI, Cann IK. Biochemical analyses of multiple endoxylanases from the rumen bacterium Ruminococcus albus 8 and their synergistic activities with accessory hemicellulose-degrading enzymes. Appl Environ Microbiol. 2011;77:5157–69. doi: 10.1128/AEM.00353-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Devillard E, Goodheart DB, Karnati SK, Bayer EA, Lamed R, Miron J, et al. Ruminococcus albus 8 mutants defective in cellulose degradation are deficient in two processive endocellulases, Cel48A and Cel9B, both of which possess a novel modular architecture. J Bacteriol. 2004;186:136–45. doi: 10.1128/JB.186.1.136-145.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rakotoarivonina H, Larson MA, Morrison M, Girardeau JP, Gaillard-Martinie B, Forano E, et al. The Ruminococcus albus pilA1-pilA2 locus: expression and putative role of two adjacent pil genes in pilus formation and bacterial adhesion to cellulose. Microbiology. 2005;151:1291–9. doi: 10.1099/mic.0.27735-0. [DOI] [PubMed] [Google Scholar]
- 35.Rincon MT, Čepeljnik T, Martin JC, Barak Y, Lamed R, Bayer EA, et al. A novel cell surface-anchored cellulose-binding protein encoded by the sca gene cluster of Ruminococcus flavefaciens. J Bacteriol. 2007;189:4774–83. doi: 10.1128/JB.00143-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wilson DB. Microbial diversity of cellulose hydrolysis. Curr Opin Microbiol. 2011;14:259–63. doi: 10.1016/j.mib.2011.04.004. [DOI] [PubMed] [Google Scholar]
- 37.Miyazaki K, Martin JC, Marinsek-Logar R, Flint HJ. Degradation and utilization of xylans by the rumen anaerobe Prevotella bryantii (formerly P. ruminicola subsp. brevis) B(1)4. Anaerobe. 1997;3:373–81. doi: 10.1006/anae.1997.0125. [DOI] [PubMed] [Google Scholar]
- 38.Gasparic A, Martin J, Daniel AS, Flint HJ. A xylan hydrolase gene cluster from Prevotella ruminicola B(1)4: sequence relationships, synergistic interactions, and oxygen sensitivity of a novel enzyme with exoxylanase and beta-(1,4) xylosidase activities. Appl Environ Microbiol. 1995;61:2958–64. doi: 10.1128/aem.61.8.2958-2964.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Miyazaki K, Miyamoto H, Mercer DK, Hirase T, Martin JC, Kojima Y, et al. Involvement of the multidomain regulatory protein XynR in positive control of xylanase gene expression in the ruminal anaerobe Prevotella bryantii B(1)4. J Bacteriol. 2003;185:2219–26. doi: 10.1128/JB.185.7.2219-2226.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Dodd D, Moon YH, Swaminathan K, Mackie RI, Cann IK. Transcriptomic analyses of xylan degradation by Prevotella bryantii and insights into energy acquisition by xylanolytic bacteroidetes. J Biol Chem. 2010;285:30261–73. doi: 10.1074/jbc.M110.141788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Dodd D, Mackie RI, Cann IKO. Xylan degradation, a metabolic property shared by rumen and human colonic Bacteroidetes. Mol Microbiol. 2011;79:292–304. doi: 10.1111/j.1365-2958.2010.07473.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Flint HJ, Whitehead TR, Martin JC, Gasparic A. Interrupted catalytic domain structures in xylanases from two distantly related strains of Prevotella ruminicola. Biochim Biophys Acta. 1997;1337:161–5. doi: 10.1016/S0167-4838(96)00213-0. [DOI] [PubMed] [Google Scholar]
- 43.Kabel MA, Yeoman CJ, Han Y, Dodd D, Abbas CA, de Bont JA, et al. Biochemical characterization and relative expression levels of multiple carbohydrate esterases of the xylanolytic rumen bacterium Prevotella ruminicola 23 grown on an ester-enriched substrate. Appl Environ Microbiol. 2011;77:5671–81. doi: 10.1128/AEM.05321-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Purushe J, Fouts DE, Morrison M, White BA, Mackie RI, Coutinho PM, et al. North American Consortium for Rumen Bacteria Comparative genome analysis of Prevotella ruminicola and Prevotella bryantii: insights into their environmental niche. Microb Ecol. 2010;60:721–9. doi: 10.1007/s00248-010-9692-8. [DOI] [PubMed] [Google Scholar]
- 45.Qi M, Wang P, O’Toole N, Barboza PS, Ungerfeld E, Leigh MB, et al. Snapshot of the eukaryotic gene expression in muskoxen rumen--a metatranscriptomic approach. PLoS One. 2011;6:e20521. doi: 10.1371/journal.pone.0020521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Tap J, Mondot S, Levenez F, Pelletier E, Caron C, Furet JP, et al. Towards the human intestinal microbiota phylogenetic core. Environ Microbiol. 2009;11:2574–84. doi: 10.1111/j.1462-2920.2009.01982.x. [DOI] [PubMed] [Google Scholar]
- 47.Qin J, Li R, Raes J, Arumugam M, Burgdorf KS, Manichanh C, et al. MetaHIT Consortium A human gut microbial gene catalogue established by metagenomic sequencing. Nature. 2010;464:59–65. doi: 10.1038/nature08821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wu GD, Chen J, Hoffmann C, Bittinger K, Chen Y-Y, Keilbaugh SA, et al. Linking long-term dietary patterns with gut microbial enterotypes. Science. 2011;334:105–8. doi: 10.1126/science.1208344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Louis P, Young P, Holtrop G, Flint HJ. Diversity of human colonic butyrate-producing bacteria revealed by analysis of the butyryl-CoA:acetate CoA-transferase gene. Environ Microbiol. 2010;12:304–14. doi: 10.1111/j.1462-2920.2009.02066.x. [DOI] [PubMed] [Google Scholar]
- 50.Arumugam M, Raes J, Pelletier E, Le Paslier D, Yamada T, Mende DR, et al. MetaHIT Consortium Enterotypes of the human gut microbiome. Nature. 2011;473:174–80. doi: 10.1038/nature09944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wang M, Ahrne S, Jeppsson B, Molin G. Comparison of bacterial diversity along the human intestinal tract by direct cloning and sequencing of 16S rRNA genes. FEMS Microbiol Ecol. 2005;54:219–31. doi: 10.1016/j.femsec.2005.03.012. [DOI] [PubMed] [Google Scholar]
- 52.Zoetendal EG, Raes J, van den Bogert B, Arumugam M, Booijink CCGM, Troost FJ, et al. The human small intestinal microbiota is driven by rapid uptake and conversion of simple carbohydrates. ISME J. 2012 doi: 10.1038/ismej.2011.212. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Salyers AA, Vercellotti JR, West SE, Wilkins TD. Fermentation of mucin and plant polysaccharides by strains of Bacteroides from the human colon. Appl Environ Microbiol. 1977;33:319–22. doi: 10.1128/aem.33.2.319-322.1977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Salyers AA, West SEH, Vercellotti JR, Wilkins TD. Fermentation of mucins and plant polysaccharides by anaerobic bacteria from the human colon. Appl Environ Microbiol. 1977;34:529–33. doi: 10.1128/aem.34.5.529-533.1977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Leitch ECM, Walker AW, Duncan SH, Holtrop G, Flint HJ. Selective colonization of insoluble substrates by human faecal bacteria. Environ Microbiol. 2007;9:667–79. doi: 10.1111/j.1462-2920.2006.01186.x. [DOI] [PubMed] [Google Scholar]
- 56.Cummings JH, Macfarlane GT. The control and consequences of bacterial fermentation in the human colon. J Appl Bacteriol. 1991;70:443–59. doi: 10.1111/j.1365-2672.1991.tb02739.x. [DOI] [PubMed] [Google Scholar]
- 57.Silvester KR, Englyst HN, Cummings JH. Ileal recovery of starch from whole diets containing resistant starch measured in vitro and fermentation of ileal effluent. Am J Clin Nutr. 1995;62:403–11. doi: 10.1093/ajcn/62.2.403. [DOI] [PubMed] [Google Scholar]
- 58.Macfarlane GT, Englyst HN. Starch utilization by the human large intestinal microflora. J Appl Bacteriol. 1986;60:195–201. doi: 10.1111/j.1365-2672.1986.tb01073.x. [DOI] [PubMed] [Google Scholar]
- 59.Nugent AP. Health properties of resistant starch. BNF Nutrition Bulletin. 2005;30:27–54. doi: 10.1111/j.1467-3010.2005.00481.x. [DOI] [Google Scholar]
- 60.Le Leu RK, Hu Y, Brown IL, Young GP. Effect of high amylose maize starches on colonic fermentation and apoptotic response to DNA-damage in the colon of rats. Nutr Metab (Lond) 2009;6:11. doi: 10.1186/1743-7075-6-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ze X, Duncan SH, Louis P, Flint HJ. Ruminococcus bromii is a keystone species for the degradation of resistant starch in the human colon. ISME J. 2012 doi: 10.1038/ismej.2012.4. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Slavin JL, Brauer PM, Marlett JA. Neutral detergent fiber, hemicellulose and cellulose digestibility in human subjects. J Nutr. 1981;111:287–97. doi: 10.1093/jn/111.2.287. [DOI] [PubMed] [Google Scholar]
- 63.Wedekind KJ, Mansfield HR, Montgomery L. Enumeration and isolation of cellulolytic and hemicellulolytic bacteria from human feces. Appl Environ Microbiol. 1988;54:1530–5. doi: 10.1128/aem.54.6.1530-1535.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Robert C, Bernalier-Donadille A. The cellulolytic microflora of the human colon: evidence of microcrystalline cellulose-degrading bacteria in methane-excreting subjects. FEMS Microbiol Ecol. 2003;46:81–9. doi: 10.1016/S0168-6496(03)00207-1. [DOI] [PubMed] [Google Scholar]
- 65.Chassard C, Delmas E, Robert C, Bernalier-Donadille A. The cellulose-degrading microbial community of the human gut varies according to the presence or absence of methanogens. FEMS Microbiol Ecol. 2010;74:205–13. doi: 10.1111/j.1574-6941.2010.00941.x. [DOI] [PubMed] [Google Scholar]
- 66.Bétian HG, Linehan BA, Bryant MP, Holdeman LV. Isolation of a cellulolytic Bacteroides sp. from human feces. Appl Environ Microbiol. 1977;33:1009–10. doi: 10.1128/aem.33.4.1009-1010.1977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Oufir LE, Barry JL, Flourié B, Cherbut C, Cloarec D, Bornet F, et al. Relationships between transit time in man and in vitro fermentation of dietary fiber by fecal bacteria. Eur J Clin Nutr. 2000;54:603–9. doi: 10.1038/sj.ejcn.1600687. [DOI] [PubMed] [Google Scholar]
- 68.Gibson GR, Roberfroid MB. Dietary modulation of the human colonic microbiota: introducing the concept of prebiotics. J Nutr. 1995;125:1401–12. doi: 10.1093/jn/125.6.1401. [DOI] [PubMed] [Google Scholar]
- 69.Van Loo J. The specificity of the interaction with intestinal bacterial fermentation by prebiotics determines their physiological efficacy. Nutr Res Rev. 2004;17:89–98. doi: 10.1079/NRR200377. [DOI] [PubMed] [Google Scholar]
- 70.Macfarlane GT, Steed H, Macfarlane S. Bacterial metabolism and health-related effects of galacto-oligosaccharides and other prebiotics. J Appl Microbiol. 2008;104:305–44. doi: 10.1111/j.1365-2672.2007.03520.x. [DOI] [PubMed] [Google Scholar]
- 71.Roberfroid MB. Introducing inulin-type fructans. Br J Nutr. 2005;93(Suppl 1):S13–25. doi: 10.1079/BJN20041350. [DOI] [PubMed] [Google Scholar]
- 72.Warchol M, Perrin S, Grill JP, Schneider F. Characterization of a purified beta-fructofuranosidase from Bifidobacterium infantis ATCC 15697. Lett Appl Microbiol. 2002;35:462–7. doi: 10.1046/j.1472-765X.2002.01224.x. [DOI] [PubMed] [Google Scholar]
- 73.Bartosch S, Woodmansey EJ, Paterson JCM, McMurdo MET, Macfarlane GT. Microbiological effects of consuming a synbiotic containing Bifidobacterium bifidum, Bifidobacterium lactis, and oligofructose in elderly persons, determined by real-time polymerase chain reaction and counting of viable bacteria. Clin Infect Dis. 2005;40:28–37. doi: 10.1086/426027. [DOI] [PubMed] [Google Scholar]
- 74.Ramirez-Farias C, Slezak K, Fuller Z, Duncan A, Holtrop G, Louis P. Effect of inulin on the human gut microbiota: stimulation of Bifidobacterium adolescentis and Faecalibacterium prausnitzii. Br J Nutr. 2009;101:541–50. doi: 10.1017/S0007114508019880. [DOI] [PubMed] [Google Scholar]
- 75.Harmsen HJM, Wildeboer-Veloo ACM, Raangs GC, Wagendorp AA, Klijn N, Bindels JG, et al. Analysis of intestinal flora development in breast-fed and formula-fed infants by using molecular identification and detection methods. J Pediatr Gastroenterol Nutr. 2000;30:61–7. doi: 10.1097/00005176-200001000-00019. [DOI] [PubMed] [Google Scholar]
- 76.German JB, Freeman SL, Lebrilla CB, Mills DA. Human milk oligosaccharides: evolution, structures and bioselectivity as substrates for intestinal bacteria. Nestle Nutr Workshop Ser Pediatr Program. 2008;62:205–18, discussion 218-22. doi: 10.1159/000146322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Derrien M, Collado MC, Ben-Amor K, Salminen S, de Vos WM. The Mucin degrader Akkermansia muciniphila is an abundant resident of the human intestinal tract. Appl Environ Microbiol. 2008;74:1646–8. doi: 10.1128/AEM.01226-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.van Passel MWJ, Kant R, Zoetendal EG, Plugge CM, Derrien M, Malfatti SA, et al. The genome of Akkermansia muciniphila, a dedicated intestinal mucin degrader, and its use in exploring intestinal metagenomes. PLoS One. 2011;6:e16876. doi: 10.1371/journal.pone.0016876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.McCarthy RE, Kotarski SF, Salyers AA. Location and characteristics of enzymes involved in the breakdown of polygalacturonic acid by Bacteroides thetaiotaomicron. J Bacteriol. 1985;161:493–9. doi: 10.1128/jb.161.2.493-499.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Bayliss CE, Houston AP. Characterization of plant polysaccharide- and mucin-fermenting anaerobic bacteria from human feces. Appl Environ Microbiol. 1984;48:626–32. doi: 10.1128/aem.48.3.626-632.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Martens EC, Lowe EC, Chiang H, Pudlo NA, Wu M, McNulty NP, et al. Recognition and degradation of plant cell wall polysaccharides by two human gut symbionts. PLoS Biol. 2011;9:e1001221. doi: 10.1371/journal.pbio.1001221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Chassard C, Goumy V, Leclerc M, Del’homme C, Bernalier-Donadille A. Characterization of the xylan-degrading microbial community from human faeces. FEMS Microbiol Ecol. 2007;61:121–31. doi: 10.1111/j.1574-6941.2007.00314.x. [DOI] [PubMed] [Google Scholar]
- 83.Hayashi H, Abe T, Sakamoto M, Ohara H, Ikemura T, Sakka K, et al. Direct cloning of genes encoding novel xylanases from the human gut. Can J Microbiol. 2005;51:251–9. doi: 10.1139/w04-136. [DOI] [PubMed] [Google Scholar]
- 84.Robert C, Chassard C, Lawson PA, Bernalier-Donadille A. Bacteroides cellulosilyticus sp. nov., a cellulolytic bacterium from the human gut microbial community. Int J Syst Evol Microbiol. 2007;57:1516–20. doi: 10.1099/ijs.0.64998-0. [DOI] [PubMed] [Google Scholar]
- 85.Gherardini FC, Salyers AA. Characterization of an outer membrane mannanase from Bacteroides ovatus. J Bacteriol. 1987;169:2031–7. doi: 10.1128/jb.169.5.2031-2037.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Weaver J, Whitehead TR, Cotta MA, Valentine PC, Salyers AA. Genetic analysis of a locus on the Bacteroides ovatus chromosome which contains xylan utilization genes. Appl Environ Microbiol. 1992;58:2764–70. doi: 10.1128/aem.58.9.2764-2770.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Mirande C, Kadlecikova E, Matulova M, Capek P, Bernalier-Donadille A, Forano E, et al. Dietary fibre degradation and fermentation by two xylanolytic bacteria Bacteroides xylanisolvens XB1A and Roseburia intestinalis XB6B4 from the human intestine. J Appl Microbiol. 2010;109:451–60. doi: 10.1111/j.1365-2672.2010.04671.x. [DOI] [PubMed] [Google Scholar]
- 88.Mirande C, Mosoni P, Béra-Maillet C, Bernalier-Donadille A, Forano E. Characterization of Xyn10A, a highly active xylanase from the human gut bacterium Bacteroides xylanisolvens XB1A. Appl Microbiol Biotechnol. 2010;87:2097–105. doi: 10.1007/s00253-010-2694-0. [DOI] [PubMed] [Google Scholar]
- 89.Anderson KL, Salyers AA. Biochemical evidence that starch breakdown by Bacteroides thetaiotaomicron involves outer membrane starch-binding sites and periplasmic starch-degrading enzymes. J Bacteriol. 1989;171:3192–8. doi: 10.1128/jb.171.6.3192-3198.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Reeves AR, Wang GR, Salyers AA. Characterization of four outer membrane proteins that play a role in utilization of starch by Bacteroides thetaiotaomicron. J Bacteriol. 1997;179:643–9. doi: 10.1128/jb.179.3.643-649.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Martens EC, Koropatkin NM, Smith TJ, Gordon JI. Complex glycan catabolism by the human gut microbiota: the Bacteroidetes Sus-like paradigm. J Biol Chem. 2009;284:24673–7. doi: 10.1074/jbc.R109.022848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Shipman JA, Berleman JE, Salyers AA. Characterization of four outer membrane proteins involved in binding starch to the cell surface of Bacteroides thetaiotaomicron. J Bacteriol. 2000;182:5365–72. doi: 10.1128/JB.182.19.5365-5372.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Shipman JA, Cho KH, Siegel HA, Salyers AA. Physiological characterization of SusG, an outer membrane protein essential for starch utilization by Bacteroides thetaiotaomicron. J Bacteriol. 1999;181:7206–11. doi: 10.1128/jb.181.23.7206-7211.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Tancula E, Feldhaus MJ, Bedzyk LA, Salyers AA. Location and characterization of genes involved in binding of starch to the surface of Bacteroides thetaiotaomicron. J Bacteriol. 1992;174:5609–16. doi: 10.1128/jb.174.17.5609-5616.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Koropatkin NM, Martens EC, Gordon JI, Smith TJ. Starch catabolism by a prominent human gut symbiont is directed by the recognition of amylose helices. Structure. 2008;16:1105–15. doi: 10.1016/j.str.2008.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Schauer K, Rodionov DA, de Reuse H. New substrates for TonB-dependent transport: do we only see the ‘tip of the iceberg’? Trends Biochem Sci. 2008;33:331–8. doi: 10.1016/j.tibs.2008.04.012. [DOI] [PubMed] [Google Scholar]
- 97.D’Elia JN, Salyers AA. Contribution of a neopullulanase, a pullulanase, and an α-glucosidase to growth of Bacteroides thetaiotaomicron on starch. J Bacteriol. 1996;178:7173–9. doi: 10.1128/jb.178.24.7173-7179.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Cho KH, Cho D, Wang GR, Salyers AA. New regulatory gene that contributes to control of Bacteroides thetaiotaomicron starch utilization genes. J Bacteriol. 2001;183:7198–205. doi: 10.1128/JB.183.24.7198-7205.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Martens EC, Chiang HC, Gordon JI. Mucosal glycan foraging enhances fitness and transmission of a saccharolytic human gut bacterial symbiont. Cell Host Microbe. 2008;4:447–57. doi: 10.1016/j.chom.2008.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Benjdia A, Martens EC, Gordon JI, Berteau O. Sulfatases and a radical S-adenosyl-L-methionine (AdoMet) enzyme are key for mucosal foraging and fitness of the prominent human gut symbiont, Bacteroides thetaiotaomicron. J Biol Chem. 2011;286:25973–82. doi: 10.1074/jbc.M111.228841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Sonnenburg ED, Zheng H, Joglekar P, Higginbottom SK, Firbank SJ, Bolam DN, et al. Specificity of polysaccharide use in intestinal bacteroides species determines diet-induced microbiota alterations. Cell. 2010;141:1241–52. doi: 10.1016/j.cell.2010.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Bjursell MK, Martens EC, Gordon JI. Functional genomic and metabolic studies of the adaptations of a prominent adult human gut symbiont, Bacteroides thetaiotaomicron, to the suckling period. J Biol Chem. 2006;281:36269–79. doi: 10.1074/jbc.M606509200. [DOI] [PubMed] [Google Scholar]
- 103.Xu J, Mahowald MA, Ley RE, Lozupone CA, Hamady M, Martens EC, et al. Evolution of symbiotic bacteria in the distal human intestine. PLoS Biol. 2007;5:e156. doi: 10.1371/journal.pbio.0050156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Martens EC, Roth R, Heuser JE, Gordon JI. Coordinate regulation of glycan degradation and polysaccharide capsule biosynthesis by a prominent human gut symbiont. J Biol Chem. 2009;284:18445–57. doi: 10.1074/jbc.M109.008094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.McBride MJ, Xie G, Martens EC, Lapidus A, Henrissat B, Rhodes RG, et al. Novel features of the polysaccharide-digesting gliding bacterium Flavobacterium johnsoniae as revealed by genome sequence analysis. Appl Environ Microbiol. 2009;75:6864–75. doi: 10.1128/AEM.01495-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Koebnik R. TonB-dependent trans-envelope signalling: the exception or the rule? Trends Microbiol. 2005;13:343–7. doi: 10.1016/j.tim.2005.06.005. [DOI] [PubMed] [Google Scholar]
- 107.Sonnenburg ED, Sonnenburg JL, Manchester JK, Hansen EE, Chiang HC, Gordon JI. A hybrid two-component system protein of a prominent human gut symbiont couples glycan sensing in vivo to carbohydrate metabolism. Proc Natl Acad Sci U S A. 2006;103:8834–9. doi: 10.1073/pnas.0603249103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Hehemann JH, Correc G, Barbeyron T, Helbert W, Czjzek M, Michel G. Transfer of carbohydrate-active enzymes from marine bacteria to Japanese gut microbiota. Nature. 2010;464:908–12. doi: 10.1038/nature08937. [DOI] [PubMed] [Google Scholar]
- 109.Lozupone CA, Hamady M, Cantarel BL, Coutinho PM, Henrissat B, Gordon JI, et al. The convergence of carbohydrate active gene repertoires in human gut microbes. Proc Natl Acad Sci U S A. 2008;105:15076–81. doi: 10.1073/pnas.0807339105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Bottacini F, Medini D, Pavesi A, Turroni F, Foroni E, Riley D, et al. Comparative genomics of the genus Bifidobacterium. Microbiology. 2010;156:3243–54. doi: 10.1099/mic.0.039545-0. [DOI] [PubMed] [Google Scholar]
- 111.Schell MA, Karmirantzou M, Snel B, Vilanova D, Berger B, Pessi G, et al. The genome sequence of Bifidobacterium longum reflects its adaptation to the human gastrointestinal tract. Proc Natl Acad Sci U S A. 2002;99:14422–7. doi: 10.1073/pnas.212527599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Sela DA, Chapman J, Adeuya A, Kim JH, Chen F, Whitehead TR, et al. The genome sequence of Bifidobacterium longum subsp. infantis reveals adaptations for milk utilization within the infant microbiome. Proc Natl Acad Sci U S A. 2008;105:18964–9. doi: 10.1073/pnas.0809584105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Zivkovic AM, German JB, Lebrilla CB, Mills DA. Human milk glycobiome and its impact on the infant gastrointestinal microbiota. Proc Natl Acad Sci U S A. 2011;108(Suppl 1):4653–8. doi: 10.1073/pnas.1000083107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Crittenden R, Laitila A, Forssell P, Mättö J, Saarela M, Mattila-Sandholm T, et al. Adhesion of bifidobacteria to granular starch and its implications in probiotic technologies. Appl Environ Microbiol. 2001;67:3469–75. doi: 10.1128/AEM.67.8.3469-3475.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Ryan SM, Fitzgerald GF, van Sinderen D. Screening for and identification of starch-, amylopectin-, and pullulan-degrading activities in bifidobacterial strains. Appl Environ Microbiol. 2006;72:5289–96. doi: 10.1128/AEM.00257-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.O’Connell Motherway M, Fitzgerald GF, Neirynck S, Ryan S, Steidler L, van Sinderen D. Characterization of ApuB, an extracellular type II amylopullulanase from Bifidobacterium breve UCC2003. Appl Environ Microbiol. 2008;74:6271–9. doi: 10.1128/AEM.01169-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Tannock GW, Munro K, Bibiloni R, Simon MA, Hargreaves P, Gopal P, et al. Impact of consumption of oligosaccharide-containing biscuits on the fecal microbiota of humans. Appl Environ Microbiol. 2004;70:2129–36. doi: 10.1128/AEM.70.4.2129-2136.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Costabile A, Kolida S, Klinder A, Gietl E, Bäuerlein M, Frohberg C, et al. A double-blind, placebo-controlled, cross-over study to establish the bifidogenic effect of a very-long-chain inulin extracted from globe artichoke (Cynara scolymus) in healthy human subjects. Br J Nutr. 2010;104:1007–17. doi: 10.1017/S0007114510001571. [DOI] [PubMed] [Google Scholar]
- 119.Ryan SM, Fitzgerald GF, van Sinderen D. Transcriptional regulation and characterization of a novel beta-fructofuranosidase-encoding gene from Bifidobacterium breve UCC2003. Appl Environ Microbiol. 2005;71:3475–82. doi: 10.1128/AEM.71.7.3475-3482.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Janer C, Rohr LM, Peláez C, Laloi M, Cleusix V, Requena T, et al. Hydrolysis of oligofructoses by the recombinant β-fructofuranosidase from Bifidobacterium lactis. Syst Appl Microbiol. 2004;27:279–85. doi: 10.1078/0723-2020-00274. [DOI] [PubMed] [Google Scholar]
- 121.Ehrmann MA, Korakli M, Vogel RF. Identification of the gene for β-fructofuranosidase of Bifidobacterium lactis DSM10140(T) and characterization of the enzyme expressed in Escherichia coli. Curr Microbiol. 2003;46:391–7. doi: 10.1007/s00284-002-3908-1. [DOI] [PubMed] [Google Scholar]
- 122.Rossi M, Corradini C, Amaretti A, Nicolini M, Pompei A, Zanoni S, et al. Fermentation of fructooligosaccharides and inulin by bifidobacteria: a comparative study of pure and fecal cultures. Appl Environ Microbiol. 2005;71:6150–8. doi: 10.1128/AEM.71.10.6150-6158.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Falony G, Lazidou K, Verschaeren A, Weckx S, Maes D, De Vuyst L. In vitro kinetic analysis of fermentation of prebiotic inulin-type fructans by Bifidobacterium species reveals four different phenotypes. Appl Environ Microbiol. 2009;75:454–61. doi: 10.1128/AEM.01488-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Falony G, Calmeyn T, Leroy F, De Vuyst L. Coculture fermentations of Bifidobacterium species and Bacteroides thetaiotaomicron reveal a mechanistic insight into the prebiotic effect of inulin-type fructans. Appl Environ Microbiol. 2009;75:2312–9. doi: 10.1128/AEM.02649-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Davis LMG, Martínez I, Walter J, Hutkins R. A dose dependent impact of prebiotic galactooligosaccharides on the intestinal microbiota of healthy adults. Int J Food Microbiol. 2010;144:285–92. doi: 10.1016/j.ijfoodmicro.2010.10.007. [DOI] [PubMed] [Google Scholar]
- 126.Davis LMG, Martínez I, Walter J, Goin C, Hutkins RW. Barcoded pyrosequencing reveals that consumption of galactooligosaccharides results in a highly specific bifidogenic response in humans. PLoS One. 2011;6:e25200. doi: 10.1371/journal.pone.0025200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Goulas T, Goulas A, Tzortzis G, Gibson GR. Comparative analysis of four beta-galactosidases from Bifidobacterium bifidum NCIMB41171: purification and biochemical characterization. Appl Microbiol Biotechnol. 2009;82:1079–88. doi: 10.1007/s00253-008-1795-5. [DOI] [PubMed] [Google Scholar]
- 128.Van den Abbeele P, Gérard P, Rabot S, Bruneau A, El Aidy S, Derrien M, et al. Arabinoxylans and inulin differentially modulate the mucosal and luminal gut microbiota and mucin-degradation in humanized rats. Environ Microbiol. 2011;13:2667–80. doi: 10.1111/j.1462-2920.2011.02533.x. [DOI] [PubMed] [Google Scholar]
- 129.van den Broek LAM, Hinz SWA, Beldman G, Vincken J-P, Voragen AGJ. Bifidobacterium carbohydrases-their role in breakdown and synthesis of (potential) prebiotics. Mol Nutr Food Res. 2008;52:146–63. doi: 10.1002/mnfr.200700121. [DOI] [PubMed] [Google Scholar]
- 130.Barboza M, Sela DA, Pirim C, Locascio RG, Freeman SL, German JB, et al. Glycoprofiling bifidobacterial consumption of galacto-oligosaccharides by mass spectrometry reveals strain-specific, preferential consumption of glycans. Appl Environ Microbiol. 2009;75:7319–25. doi: 10.1128/AEM.00842-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.LoCascio RG, Ninonuevo MR, Freeman SL, Sela DA, Grimm R, Lebrilla CB, et al. Glycoprofiling of bifidobacterial consumption of human milk oligosaccharides demonstrates strain specific, preferential consumption of small chain glycans secreted in early human lactation. J Agric Food Chem. 2007;55:8914–9. doi: 10.1021/jf0710480. [DOI] [PubMed] [Google Scholar]
- 132.Garrido D, Kim JH, German JB, Raybould HE, Mills DA. Oligosaccharide binding proteins from Bifidobacterium longum subsp. infantis reveal a preference for host glycans. PLoS One. 2011;6:e17315. doi: 10.1371/journal.pone.0017315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Boesten R, Schuren F, Ben Amor K, Haarman M, Knol J, de Vos WM. Bifidobacterium population analysis in the infant gut by direct mapping of genomic hybridization patterns: potential for monitoring temporal development and effects of dietary regimens. Microb Biotechnol. 2011;4:417–27. doi: 10.1111/j.1751-7915.2010.00216.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Turroni F, Bottacini F, Foroni E, Mulder I, Kim JH, Zomer A, et al. Genome analysis of Bifidobacterium bifidum PRL2010 reveals metabolic pathways for host-derived glycan foraging. Proc Natl Acad Sci U S A. 2010;107:19514–9. doi: 10.1073/pnas.1011100107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Barcenilla A, Pryde SE, Martin JC, Duncan SH, Stewart CS, Henderson C, et al. Phylogenetic relationships of butyrate-producing bacteria from the human gut. Appl Environ Microbiol. 2000;66:1654–61. doi: 10.1128/AEM.66.4.1654-1661.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Louis P, Flint HJ. Diversity, metabolism and microbial ecology of butyrate-producing bacteria from the human large intestine. FEMS Microbiol Lett. 2009;294:1–8. doi: 10.1111/j.1574-6968.2009.01514.x. [DOI] [PubMed] [Google Scholar]
- 137.Duncan SH, Louis P, Flint HJ. Lactate-utilizing bacteria, isolated from human feces, that produce butyrate as a major fermentation product. Appl Environ Microbiol. 2004;70:5810–7. doi: 10.1128/AEM.70.10.5810-5817.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Bernalier A, Willems A, Leclerc M, Rochet V, Collins MD. Ruminococcus hydrogenotrophicus sp. nov., a new H2/ CO2 –utilizing bacterium from human feces. Arch Microbiol. 1996;166:176–83. doi: 10.1007/s002030050373. [DOI] [PubMed] [Google Scholar]
- 139.Rey FE, Faith JJ, Bain J, Muehlbauer MJ, Stevens RD, Newgard CB, et al. Dissecting the in vivo metabolic potential of two human gut acetogens. J Biol Chem. 2010;285:22082–90. doi: 10.1074/jbc.M110.117713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Abell GCJ, Cooke CM, Bennett CN, Conlon MA, McOrist AL. Phylotypes related to Ruminococcus bromii are abundant in the large bowel of humans and increase in response to a diet high in resistant starch. FEMS Microbiol Ecol. 2008;66:505–15. doi: 10.1111/j.1574-6941.2008.00527.x. [DOI] [PubMed] [Google Scholar]
- 141.Martínez I, Kim J, Duffy PR, Schlegel VL, Walter J. Resistant starches types 2 and 4 have differential effects on the composition of the fecal microbiota in human subjects. PLoS One. 2010;5:e15046. doi: 10.1371/journal.pone.0015046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Kovatcheva-Datchary P, Egert M, Maathuis A, Rajilić-Stojanović M, de Graaf AA, Smidt H, et al. Linking phylogenetic identities of bacteria to starch fermentation in an in vitro model of the large intestine by RNA-based stable isotope probing. Environ Microbiol. 2009;11:914–26. doi: 10.1111/j.1462-2920.2008.01815.x. [DOI] [PubMed] [Google Scholar]
- 143.Lopez-Siles M, Khan TM, Duncan SH, Harmsen HJM, Garcia-Gil LJ, Flint HJ. Cultured representatives of two major phylogroups of human colonic Faecalibacterium prausnitzii can utilize pectin, uronic acids, and host-derived substrates for growth. Appl Environ Microbiol. 2012;78:420–8. doi: 10.1128/AEM.06858-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Aminov RI, Walker AW, Duncan SH, Harmsen HJM, Welling GW, Flint HJ. Molecular diversity, cultivation, and improved detection by fluorescent in situ hybridization of a dominant group of human gut bacteria related to Roseburia spp. or Eubacterium rectale. Appl Environ Microbiol. 2006;72:6371–6. doi: 10.1128/AEM.00701-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Duncan SH, Belenguer A, Holtrop G, Johnstone AM, Flint HJ, Lobley GE. Reduced dietary intake of carbohydrates by obese subjects results in decreased concentrations of butyrate and butyrate-producing bacteria in feces. Appl Environ Microbiol. 2007;73:1073–8. doi: 10.1128/AEM.02340-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Ramsay AG, Scott KP, Martin JC, Rincon MT, Flint HJ. Cell-associated alpha-amylases of butyrate-producing Firmicute bacteria from the human colon. Microbiology. 2006;152:3281–90. doi: 10.1099/mic.0.29233-0. [DOI] [PubMed] [Google Scholar]
- 147.Scott KP, Martin JC, Chassard C, Clerget M, Potrykus J, Campbell G, et al. Substrate-driven gene expression in Roseburia inulinivorans: importance of inducible enzymes in the utilization of inulin and starch. Proc Natl Acad Sci U S A. 2011;108(Suppl 1):4672–9. doi: 10.1073/pnas.1000091107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Chassard C, Delmas E, Robert C, Lawson PA, Bernalier-Donadille A. Ruminococcus champanellensis sp. nov., a cellulose-degrading bacterium from human gut microbiota. Int J Syst Evol Microbiol. 2012;62:138–43. doi: 10.1099/ijs.0.027375-0. [DOI] [PubMed] [Google Scholar]
- 149.Wolin MJ, Miller TL, Collins MD, Lawson PA. Formate-dependent growth and homoacetogenic fermentation by a bacterium from human feces: description of Bryantella formatexigens gen. nov., sp. nov. Appl Environ Microbiol. 2003;69:6321–6. doi: 10.1128/AEM.69.10.6321-6326.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Duncan SH, Hold GL, Barcenilla A, Stewart CS, Flint HJ. Roseburia intestinalis sp. nov., a novel saccharolytic, butyrate-producing bacterium from human faeces. Int J Syst Evol Microbiol. 2002;52:1615–20. doi: 10.1099/ijs.0.02143-0. [DOI] [PubMed] [Google Scholar]
- 151.Rumney CJ, Duncan SH, Henderson C, Stewart CS. Isolation and characteristics of a wheatbran-degrading Butyrivibrio from human faeces. Lett Appl Microbiol. 1995;20:232–6. doi: 10.1111/j.1472-765X.1995.tb00435.x. [DOI] [PubMed] [Google Scholar]
- 152.Kleessen B, Hartmann L, Blaut M. Oligofructose and long-chain inulin: influence on the gut microbial ecology of rats associated with a human faecal flora. Br J Nutr. 2001;86:291–300. doi: 10.1079/BJN2001403. [DOI] [PubMed] [Google Scholar]
- 153.Manderson K, Pinart M, Tuohy KM, Grace WE, Hotchkiss AT, Widmer W, et al. In vitro determination of prebiotic properties of oligosaccharides derived from an orange juice manufacturing by-product stream. Appl Environ Microbiol. 2005;71:8383–9. doi: 10.1128/AEM.71.12.8383-8389.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Duncan SH, Scott KP, Ramsay AG, Harmsen HJM, Welling GW, Stewart CS, et al. Effects of alternative dietary substrates on competition between human colonic bacteria in an anaerobic fermentor system. Appl Environ Microbiol. 2003;69:1136–42. doi: 10.1128/AEM.69.2.1136-1142.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Walker AW, Duncan SH, McWilliam Leitch EC, Child MW, Flint HJ. pH and peptide supply can radically alter bacterial populations and short-chain fatty acid ratios within microbial communities from the human colon. Appl Environ Microbiol. 2005;71:3692–700. doi: 10.1128/AEM.71.7.3692-3700.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Scott KP, Duncan SH, Louis P, Flint HJ. Nutritional influences on the gut microbiota and the consequences for gastrointestinal health. Biochem Soc Trans. 2011;39:1073–8. doi: 10.1042/BST0391073. [DOI] [PubMed] [Google Scholar]
- 157.Hoskins LC. Mucin degradation in the human gastrointestinal tract and its significance to enteric microbial ecology. Eur J Gastroenterol Hepatol. 1993;5:205–13. doi: 10.1097/00042737-199304000-00004. [DOI] [Google Scholar]
- 158.Scott KP, Martin JC, Campbell G, Mayer C-D, Flint HJ. Whole-genome transcription profiling reveals genes up-regulated by growth on fucose in the human gut bacterium “Roseburia inulinivorans”. J Bacteriol. 2006;188:4340–9. doi: 10.1128/JB.00137-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Hooper LV, Xu J, Falk PG, Midtvedt T, Gordon JI. A molecular sensor that allows a gut commensal to control its nutrient foundation in a competitive ecosystem. Proc Natl Acad Sci U S A. 1999;96:9833–8. doi: 10.1073/pnas.96.17.9833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Duncan SH, Louis P, Thomson JM, Flint HJ. The role of pH in determining the species composition of the human colonic microbiota. Environ Microbiol. 2009;11:2112–22. doi: 10.1111/j.1462-2920.2009.01931.x. [DOI] [PubMed] [Google Scholar]
- 161.Brinkworth GD, Noakes M, Clifton PM, Bird AR. Comparative effects of very low-carbohydrate, high-fat and high-carbohydrate, low-fat weight-loss diets on bowel habit and faecal short-chain fatty acids and bacterial populations. Br J Nutr. 2009;101:1493–502. doi: 10.1017/S0007114508094658. [DOI] [PubMed] [Google Scholar]
- 162.Russell WR, Gratz SW, Duncan SH, Holtrop G, Ince J, Scobbie L, et al. High-protein, reduced-carbohydrate weight-loss diets promote metabolite profiles likely to be detrimental to colonic health. Am J Clin Nutr. 2011;93:1062–72. doi: 10.3945/ajcn.110.002188. [DOI] [PubMed] [Google Scholar]
- 163.De Preter V, Falony G, Windey K, Hamer HM, De Vuyst L, Verbeke K. The prebiotic, oligofructose-enriched inulin modulates the faecal metabolite profile: an in vitro analysis. Mol Nutr Food Res. 2010;54:1791–801. doi: 10.1002/mnfr.201000136. [DOI] [PubMed] [Google Scholar]
- 164.Scholz-Ahrens KE, Ade P, Marten B, Weber P, Timm W, Açil Y, et al. Prebiotics, probiotics, and synbiotics affect mineral absorption, bone mineral content, and bone structure. J Nutr. 2007;137(Suppl 2):838S–46S. doi: 10.1093/jn/137.3.838S. [DOI] [PubMed] [Google Scholar]
- 165.Smith KN, Queenan KM, Thomas W, Fulcher RG, Slavin JL. Physiological effects of concentrated barley beta-glucan in mildly hypercholesterolemic adults. J Am Coll Nutr. 2008;27:434–40. doi: 10.1080/07315724.2008.10719722. [DOI] [PubMed] [Google Scholar]
- 166.Lewis SJ, Heaton KW. Increasing butyrate concentration in the distal colon by accelerating intestinal transit. Gut. 1997;41:245–51. doi: 10.1136/gut.41.2.245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Lampe JW, Slavin JL, Melcher EA, Potter JD. Effects of cereal and vegetable fiber feeding on potential risk factors for colon cancer. Cancer Epidemiol Biomarkers Prev. 1992;1:207–11. [PubMed] [Google Scholar]
- 168.Pimentel M, Lin HC, Enayati P, van den Burg B, Lee H-R, Chen JH, et al. Methane, a gas produced by enteric bacteria, slows intestinal transit and augments small intestinal contractile activity. Am J Physiol Gastrointest Liver Physiol. 2006;290:G1089–95. doi: 10.1152/ajpgi.00574.2004. [DOI] [PubMed] [Google Scholar]
- 169.Hertog MGL, Hollman PCH, Katan MB, Kromhout D. Intake of potentially anticarcinogenic flavanoids and their determinants in adults in the Netherlands. Nutr Cancer- Int J. 1993;20:21–9. doi: 10.1080/01635589309514267. [DOI] [PubMed] [Google Scholar]
- 170.Roberfroid MB. Caloric value of inulin and oligofructose. J Nutr. 1999;129(Suppl):1436S–7S. doi: 10.1093/jn/129.7.1436S. [DOI] [PubMed] [Google Scholar]
- 171.Turnbaugh PJ, Ley RE, Mahowald MA, Magrini V, Mardis ER, Gordon JI. An obesity-associated gut microbiome with increased capacity for energy harvest. Nature. 2006;444:1027–31. doi: 10.1038/nature05414. [DOI] [PubMed] [Google Scholar]
- 172.Flint HJ. Obesity and the gut microbiota. J Clin Gastroenterol. 2011;45(Suppl):S128–32. doi: 10.1097/MCG.0b013e31821f44c4. [DOI] [PubMed] [Google Scholar]
- 173.Duncan SH, Lobley GE, Holtrop G, Ince J, Johnstone AM, Louis P, et al. Human colonic microbiota associated with diet, obesity and weight loss. Int J Obes (Lond) 2008;32:1720–4. doi: 10.1038/ijo.2008.155. [DOI] [PubMed] [Google Scholar]
- 174.Jumpertz R, Le DS, Turnbaugh PJ, Trinidad C, Bogardus C, Gordon JI, et al. Energy-balance studies reveal associations between gut microbes, caloric load, and nutrient absorption in humans. Am J Clin Nutr. 2011;94:58–65. doi: 10.3945/ajcn.110.010132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Bäckhed F, Ding H, Wang T, Hooper LV, Koh GY, Nagy A, et al. The gut microbiota as an environmental factor that regulates fat storage. Proc Natl Acad Sci U S A. 2004;101:15718–23. doi: 10.1073/pnas.0407076101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Fleissner CK, Huebel N, Abd El-Bary MM, Loh G, Klaus S, Blaut M. Absence of intestinal microbiota does not protect mice from diet-induced obesity. Br J Nutr. 2010;104:919–29. doi: 10.1017/S0007114510001303. [DOI] [PubMed] [Google Scholar]
- 177.Delzenne NM, Cani PD. Gut microbiota and the pathogenesis of insulin resistance. Curr Diab Rep. 2011;11:154–9. doi: 10.1007/s11892-011-0191-1. [DOI] [PubMed] [Google Scholar]
- 178.Cani PD, Delzenne NM. The gut microbiome as therapeutic target. Pharmacol Ther. 2011;130:202–12. doi: 10.1016/j.pharmthera.2011.01.012. [DOI] [PubMed] [Google Scholar]
- 179.Vrieze A, Holleman F, Zoetendal EG, de Vos WM, Hoekstra JB, Nieuwdorp M. The environment within: how gut microbiota may influence metabolism and body composition. Diabetologia. 2010;53:606–13. doi: 10.1007/s00125-010-1662-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Sleeth ML, Thompson EL, Ford HE, Zac-Varghese SE, Frost G. Free fatty acid receptor 2 and nutrient sensing: a proposed role for fibre, fermentable carbohydrates and short-chain fatty acids in appetite regulation. Nutr Res Rev. 2010;23:135–45. doi: 10.1017/S0954422410000089. [DOI] [PubMed] [Google Scholar]
- 181.Arora T, Sharma R, Frost G. Propionate. Anti-obesity and satiety enhancing factor? Appetite. 2011;56:511–5. doi: 10.1016/j.appet.2011.01.016. [DOI] [PubMed] [Google Scholar]
- 182.Canani RB, Costanzo MD, Leone L, Pedata M, Meli R, Calignano A. Potential beneficial effects of butyrate in intestinal and extraintestinal diseases. World J Gastroenterol. 2011;17:1519–28. doi: 10.3748/wjg.v17.i12.1519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Gao Z, Yin J, Zhang J, Ward RE, Martin RJ, Lefevre M, et al. Butyrate improves insulin sensitivity and increases energy expenditure in mice. Diabetes. 2009;58:1509–17. doi: 10.2337/db08-1637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Bienenstock J, Collins S. 99th Dahlem conference on infection, inflammation and chronic inflammatory disorders: psycho-neuroimmunology and the intestinal microbiota: clinical observations and basic mechanisms. Clin Exp Immunol. 2010;160:85–91. doi: 10.1111/j.1365-2249.2010.04124.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Bercik P, Denou E, Collins J, Jackson W, Lu J, Jury J, et al. The intestinal microbiota affect central levels of brain-derived neurotropic factor and behavior in mice. Gastroenterology. 2011;141:599–609, 609, e1-3. doi: 10.1053/j.gastro.2011.04.052. [DOI] [PubMed] [Google Scholar]
- 186.Jarchum I, Pamer EG. Regulation of innate and adaptive immunity by the commensal microbiota. Curr Opin Immunol. 2011;23:353–60. doi: 10.1016/j.coi.2011.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Muñoz-Tamayo R, Laroche B, Walter E, Doré J, Leclerc M. Mathematical modelling of carbohydrate degradation by human colonic microbiota. J Theor Biol. 2010;266:189–201. doi: 10.1016/j.jtbi.2010.05.040. [DOI] [PubMed] [Google Scholar]