

Background: Oxidized bases in mammalian genome are repaired via base excision repair (BER) process that utilizes both common repair and noncanonical proteins.
Results: hnRNP-U stimulates NEIL1-initiated BER via direct interaction and is required for enhancing oxidative stress-induced repair.
Conclusion: NEIL1's interaction with hnRNP-U is critical for regulating oxidized genome damage repair after oxidative stress.
Significance: The RNA-binding protein hnRNP-U plays a role in maintaining genomic integrity.
Keywords: Base Excision Repair, DNA Damage Response, DNA Enzymes, DNA Repair, Fluorescence, NEIL1, Noncanonical Proteins in Base Excision Repair, Oxidized Genome Damage, RNA-binding Proteins, hnRNP-U
Abstract
Repair of oxidized base lesions in the human genome, initiated by DNA glycosylases, occurs via the base excision repair pathway using conserved repair and some non-repair proteins. However, the functions of the latter noncanonical proteins in base excision repair are unclear. Here we elucidated the role of heterogeneous nuclear ribonucleoprotein-U (hnRNP-U), identified in the immunoprecipitate of human NEIL1, a major DNA glycosylase responsible for oxidized base repair. hnRNP-U directly interacts with NEIL1 in vitro via the NEIL1 common interacting C-terminal domain, which is dispensable for its enzymatic activity. Their in-cell association increases after oxidative stress. hnRNP-U stimulates the NEIL1 in vitro base excision activity for 5-hydroxyuracil in duplex, bubble, forked, or single-stranded DNA substrate, primarily by enhancing product release. Using eluates from FLAG-NEIL1 immunoprecipitates from human cells, we observed 3-fold enhancement in complete repair activity after oxidant treatment. The lack of such enhancement in hnRNP-U-depleted cells suggests its involvement in repairing enhanced base damage after oxidative stress. The NEIL1 disordered C-terminal region binds to hnRNP-U at equimolar ratio with high affinity (Kd = ∼54 nm). The interacting regions in hnRNP-U, mapped to both termini, suggest their proximity in the native protein; these are also disordered, based on PONDR (Predictor of Naturally Disordered Regions) prediction and circular dichroism spectra. Finally, depletion of hnRNP-U and NEIL1 epistatically sensitized human cells at low oxidative genome damage, suggesting that the hnRNP-U protection of cells after oxidative stress is largely due to enhancement of NEIL1-mediated repair.
Introduction
The genomes of aerobic organisms are constantly exposed to reactive oxygen species (ROS),2 which are formed endogenously, mostly as byproducts of cellular respiration, and also induced by environmental agents and during inflammatory responses (1–3). ROS induce oxidatively modified bases (e.g. 8-oxoguanine, dihydrouracil, thymine glycol, 5-hydroxyuracil (5-OHU), etc.), abasic (AP) sites, and DNA single-strand breaks; these have been implicated in the etiology of various diseases including cancer, degenerative neurological disorders, and arthritis, and in aging (4–9). If left unrepaired, most of these lesions are mutagenic and/or cytotoxic and are repaired via the evolutionarily conserved base excision/single-strand break repair (BER/SSBR) pathway. Repair of oxidized bases in the mammalian genome is initiated with their excision by one of five DNA glycosylases (DGs): OGG1, NTH1, and NEIL 1–3 (10, 11). Oxidized base-specific DGs have intrinsic AP lyase activity, as a result of which the AP site produced after damaged base excision is cleaved via β or βδ elimination to generate 3′-phospho α,β-unsaturated aldehyde and 3′-phosphate, respectively (10). NEIL1 and -2 belonging to the Nei family of DGs are distinct from OGG1/NTH1 in the Nth family because of their βδ-lyase activity and ability to excise base lesions from single-stranded DNA substrate in vitro (12). As we had shown earlier, the 3′-phosphate terminus generated by NEIL1 and -2 is removed by polynucleotide kinase/phosphatase in the second step of BER, whereas the 3′-phospho α,β-unsaturated aldehyde phosphate produced by OGG1/NTH1 is removed by AP-endonuclease (APE1) (12). Subsequent repair steps, which occur via the one-nucleotide gap filling (named SN-BER), are common for both families of DGs in most cells. Alternatively, long patch repair with incorporation of 2–8 nucleotides could occur at the lesion site in replicating cells where the replication enzymes are co-opted for the repair (13–15).
Recent evidence indicates the involvement of noncanonical proteins in BER, in particular, RNA-binding proteins that lack known repair-related activity (16–18). These were shown to modulate the repair activity of DGs via direct interaction. For example, NEIL2 interacts functionally with Y-box-binding protein (YB-1), and also with RNA binding activity (17), under conditions of oxidative stress. The immunoprecipitates (IPs) of both NEILs contain hnRNP-U. We previously characterized the involvement of NEIL2 in the preferential repair of transcribed genes, where NEIL2 interacts functionally with both RNA polymerase II and hnRNP-U (19). Unlike NEIL2, NEIL1 is up-regulated during the S-phase and interacts functionally with DNA replication proteins, including proliferating cell nuclear antigen (PCNA), replication protein A (RPA), FEN-1, and Werner helicase (WRN), etc. Based on these results, we have proposed that NEIL1 is preferentially involved in replication-coordinated BER (20–23). Because of the differential functions and mechanisms of NEIL1 versus NEIL2, we decided to investigate the specific role of hnRNP-U in NEIL1-mediated repair.
In addition to its role in mRNA processing and transport, hnRNP-U has some unique functions, as also observed for other members of the hnRNP family (24, 25). hnRNP-U was independently identified and named scaffold attachment factor (SAF)-A on the basis of its strong binding to nuclear scaffold/matrix with affinity for A-T-rich DNA (26). With a molecular mass of 90 kDa, it is the largest member of the abundant hnRNP family (24). Interestingly, hnRNP-U was also found to be associated with WT1 (Wilms tumor 1) protein and suggested to be a potential Wilms tumor gene (27). hnRNP-U interacts with MDM2, an E3 ubiquitin ligase involved in degradation of p53, a central player in DNA damage response and also in DNA repair (28). The hnRNP-U interaction with some proteins may be regulated via its phosphorylation by DNA-protein kinase, which is activated by double-strand breaks in the genome (29, 30). In this study, we have documented the physical and functional interaction of NEIL1 with hnRNP-U, which enhances cell survival after oxidative stress by increasing NEIL1-mediated oxidized base repair.
EXPERIMENTAL PROCEDURES
Oligonucleotide and Plasmid Substrates
A 51-mer oligonucleotide containing 5-OHU, a major oxidative deamination product of C in DNA, at position 26 from the 5′-end, the complementary oligonucleotide containing G opposite the lesion (Sigma), was used for generating duplex oligonucleotides and partial duplexes containing a bubble or fork structure (supplemental Table S1). To produce 5′-32P-labeled substrates, the single-stranded (ss) oligonucleotides were labeled at their 5′ termini with [γ-32P]ATP using T4-polynucleotide kinase prior to annealing. The labeled substrates were separated from unincorporated radioactivity by chromatography on Sephadex G25. The pUC19CPD plasmid substrate containing a single 5-OHU for complete repair assay was generated as described previously (31, 32).
Expression and Purification of Recombinant Proteins
Recombinant wild type (WT) NEIL1, its truncated polypeptides, polynucleotide kinase/phosphatase, DNA polymerase β, and DNA ligase IIIα were purified to homogeneity from Escherichia coli as described previously (12, 32–34). His-tagged WT human hnRNP-U (the plasmid was a kind gift from Gourisankar Ghosh, University of California, San Diego) was expressed in BL-21 Codon-Plus E. coli (Stratagene) and purified from the bacterial extract by affinity chromatography on a Ni-NTA-agarose column (Qiagen). It was further purified to homogeneity by elution from Q-Sepharose (Amersham Biosciences) using 100–750 mm NaCl gradient. The most purified fraction was eluted at 400 mm NaCl and stored after dialysis in PBS (pH 7.4) containing 450 mm NaCl, 50% glycerol, and 1 mm DTT. The hnRNP-U domains containing 1–100, 1–188, 1–235, 236–645, and 646–808 residues were cloned for GST fusion between the BamHI/EcoRI restriction sites in the pGEX-2T E. coli expression plasmid. The GST-tagged polypeptides were also expressed in E. coli, purified by glutathione-Sepharose affinity chromatography (GE Healthcare), and then eluted using 20 mm reduced glutathione. The GST fusion peptides were dialyzed and further purified by cation exchange chromatography.
Cell Culture and Co-immunoprecipitation (co-IP) Assays
The human embryonic kidney cell line HEK293 was grown in DMEM containing 10% FBS, 100 units/ml penicillin, 100 μg/ml streptomycin, and 2 mm glutamine, at 37 °C, 5% CO2, using reagents from Invitrogen. For co-IP experiments, we generated stable transfectants of HEK293 cells expressing FLAG-NEIL1 or empty-FLAG as follows. Mammalian expression plasmids for C-terminally FLAG-tagged NEIL1 were previously described (33, 35). Log-phase HEK293 cells (in 6-cm plates) were transfected with 1 μg of NEIL1-FLAG or the empty vector plasmid (19). After 24 h, the cells were trypsinized and plated in fresh medium containing Zeocin (100 μg/ml) to select clones carrying stably integrated plasmid DNA. After 10 days, Zeocin-resistant clones were individually expanded and screened for expression of FLAG-NEIL1; only the clones with ectopic NEIL1 expression comparable with that of endogenous NEIL1 were used in this study. For IP analysis, the cells were lysed in lysis buffer (20 mm Tris-HCl, pH 7.5, 150 mm NaCl, 1 mm EDTA, 1% Triton X-100, and protease inhibitor mixture (Roche Applied Science)) at 48 h after transfection. After digesting an aliquot with 500 units/ml each of DNase I and RNase A (Ambion) at 37 °C for 1 h followed by centrifugation as described previously (22, 23), the lysates were immunoprecipitated for 3 h at 4 °C with FLAG M2 antibody (Ab)-bound agarose beads (Sigma). The beads were collected by centrifugation and washed three times with cold Tris-buffered saline (TBS), and the FLAG immunocomplex was eluted from the beads by adding SDS loading buffer and separated by SDS-PAGE followed by immunoblotting with the appropriate antibodies.
To analyze the cell cycle dependence of the NEIL1 interaction with hnRNP-U, HEK293 cells were synchronized by double thymidine block (36). Briefly, at ∼30% confluency, the cells were treated with 2 mm thymidine in DMEM for 18 h and then transferred to fresh DMEM for 6 h. This was followed by a second thymidine treatment for 17 h. The cells were finally washed twice with PBS, allowed to grow in DMEM, and harvested at 0, 1.5, 3, 4.5, and 6 h. The cells were fixed in 70% prechilled (−20 °C) ethanol, stained with the staining solution (containing 100 μg/ml RNase A, 40 μg/ml propidium iodide in PBS), and analyzed by flow cytometry.
In Vitro Pulldown Assay
GST pulldown assays were performed as described previously (20). Briefly, the GST fusion hnRNP-U polypeptides (20 pmol) were bound to glutathione-Sepharose beads (20 μl) and then mixed with full-length NEIL1 or its deletion polypeptides (10 pmol) in 0.5 ml of TBS buffer and incubated with constant rocking for 4 h at 4 °C. After extensive washing, the bound proteins were separated by SDS-PAGE for immunoblotting analysis.
Far Western Analysis
The proteins (40 pmol) transferred to the nitrocellulose membrane after SDS-PAGE were denatured in situ with 6 m guanidine-HCl and renatured by successive incubation with serially diluted guanidine-HCl in PBS + 1 mm DTT (23). The membrane was then incubated with hnRNP-U or NEIL1 (10 pmol/ml) in PBS supplemented with 0.5% nonfat dried milk, 0.05% Tween 20, 10 mm trimethylamine N-oxide, and 1 mm DTT for 3 h followed by washing. Stable binding of interacting protein was confirmed by immunoblotting.
Fluorescence Spectroscopy
Interaction of hnRNP-U with NEIL1 C-terminal peptide (residues 312–349 lacking aromatic residues) was monitored by the change in intrinsic tryptophan fluorescence of hnRNP-U (λex = 295, λem = 300–450 nm) in an LS50 spectrofluorometer (PerkinElmer Life Sciences) after incubation in 10 mm PBS, pH 7.5, 5% glycerol at 25 °C for 5 min. The binding constant KD was calculated by plotting ΔF (change in hnRNP-U fluorescence at 345 nm) versus ligand concentration according to the equation ΔF = ΔFmax × [ligand]/KD + [ligand] (22, 23, 31).
Analysis of NEIL1 Activity
The strand cleavage of substrate DNA by NEIL1 after lesion base excision was analyzed after incubation of 5′-32P-labeled, 5-OHU-containing oligonucleotide substrates at 37 °C for 15 min in a 10-μl reaction mixture containing 40 mm HEPES-KOH, pH 7.5, 50 mm KCl, 1 mm MgCl2, 100 μg/ml bovine serum albumin (BSA), and 5% glycerol. The reaction was stopped with the formamide dye mix (80% formamide, 20 mm NaOH, 20 mm EDTA, 0.05% bromphenol blue, and 0.05% xylene cyanol). The products were separated by denaturing gel electrophoresis in 20% polyacrylamide containing 8 m urea in 1× Tris borate-EDTA buffer, pH 8.4 (22). The radioactivity was quantitated in a PhosphorImager using the ImageQuant software (Amersham Biosciences).
For kinetics studies, the fork substrate (2.5–120 nm) was incubated with 20 nm NEIL1 or NEIL plus hnRNP-U (10 nm) for 4 min at 37 °C after initiating the reaction by adding NEIL1. Km, Vmax, and kcat values were calculated from the linear range of the reaction by regression analysis using SigmaPlot (22).
Complete Repair Assay
NEIL1-initiated repair was assayed in reconstituted in vitro system using a 5-OHU-containing duplex oligonucleotide substrate (22) or 5-OHU-containing plasmid substrate (31). Briefly, after individual protein levels in the reaction mixture were optimized for maximum product formation, the repair reaction (20 μl) containing 50 fmol each of NEIL1, polynucleotide kinase/phosphatase, and DNA polymerase β, 100 fmol of DNA IIIα, 1 mmol of ATP, 10 μmol of [α-32P]dCTP, and unlabeled dNTPs (25 mmol) was incubated for 30 min at 37 °C. Appropriate controls lacking various components were included. For repair using the FLAG-NEIL1 IP, lysates of HEK293 cells stably expressing FLAG-NEIL1, untreated or treated with glucose oxidase (GO) (50 ng/ml) were incubated with beads attached with FLAG Ab and normalized for the FLAG level as before (31). Appropriately washed beads were then mixed with the repair reaction mixture containing the plasmid substrate, 1 mm ATP, and [α-32P]dATP and incubated with intermittent mixing for 30 min at 37 °C. The products were then analyzed in a PhosphorImager after separation in a denaturing gel (22).
In Situ Proximity Ligation Assay (PLA)
HEK293 cells grown overnight in 16-well chamber slides were fixed with 4% paraformaldehyde and then permeabilized with 0.2% Tween 20 followed by incubation with a primary Ab for NEIL1 (rabbit (33)) or hnRNP-U (mouse monoclonal; Santa Cruz Biotechnology). The PLA assay was performed using the Duolink PLA kit (Olink Bioscience catalogue number LNK-92101-KI01, Uppsala, Sweden) per the manufacturer's instructions. The nuclei were counterstained with DAPI, and the PLA signals were visualized in a fluorescence microscope (Olympus) at 200× magnification.
Clonogenic Survival Assay
Log phase HEK293 cultures were treated with NEIL1 siRNA (80 nm; targeting the 3′-UTR region of the NEIL1 gene; sense sequence, 5′-CCGUGAUGAUGUUUGUUUAUU-3′; antisense sequence, 5′-UAAACAAACAUCAUCACGGUU-3′, Sigma) or hnRNP-U siRNA (Dharmacon, catalogue number J-013501-05), individually or together for 48 h. Scrambled siRNA was used as a control. NEIL1 or hnRNP-U down-regulation was confirmed by immunoblotting of the cell extracts at 48 h after transfection. Cells were trypsinized and transferred to 60-mm dishes (400 cells/dish). Twelve hours after plating, the cells were treated with GO (0–200 ng/ml) for 15 min. After allowing the cells to grow in fresh medium for 10 days, the colonies were counted after staining with crystal violet for calculating the surviving fraction (37).
PONDR Modeling
Secondary structures of NEIL1 and hnRNP-U were predicted using PONDR (Predictor of Naturally Disordered Regions) modeling software (38, 39). The protein sequences were obtained from the National Center for Biotechnology Information (NCBI) database. The PONDR score of 0.5 and higher indicate a disordered structure.
Circular Dichroism (CD) Spectroscopy
The CD spectra (195–260 nm) were recorded for the hnRNP-U terminal polypeptides 1–100 and 651–806 (2 μm) in 25 mm Tris-HCl buffer (pH 7.4), 50 mm NaCl in a JASCO J-720 spectropolarimeter (31), using a 1-mm cuvette, and four repetitions of the CD spectra were used for averaging and then corrected for the contribution from the buffer alone. The secondary structures of the enzyme were calculated from the spectra using the K2d software (40).
RESULTS
In-cell Association of NEIL1 with hnRNP-U and Its Enhancement after Oxidative Stress
After initial identification of hnRNP-U as one of the NEIL1 interacting partners by mass spectrometry of proteins in NEIL1 IP from HEK293 cells (data not shown), we decided to confirm in-cell association of endogenous NEIL1 with hnRNP-U using the in situ PLA in HEK293 cells. In this analysis, two proteins are immunostained with distinct species-specific secondary Abs that are linked to complementary oligonucleotides. When two different Ab molecules bind in close proximity (<40 nm), the linked DNA can be linearly amplified via the rolling circle mechanism and visualized as distinct foci with a fluorescent probe. This assay was shown to be highly specific for physically interacting endogenous proteins in the cell (41–43). We detected a significant number of foci with NEIL1-hnRNP-U, whereas such signal was essentially absent when the primary Abs for NEIL1 or hnRNP-U were replaced with normal IgGs (Fig. 1A). Interestingly, the association was significantly enhanced (∼4-fold) in oxidatively stressed cells after treatment with 50 ng/ml GO for 30 min. We then tested for the presence of hnRNP-U in NEIL1 IP from HEK293 cells stably transfected with NEIL1-FLAG or empty-FLAG expression plasmid. The levels of ectopic NEIL in these lines are comparable with that of the endogenous enzyme, and the 30-min GO exposure does not change the level of expression of endogenous NEIL1 or hnRNP-U (data not shown). The presence of hnRNP-U in the NEIL1-FLAG IP, but not in the vector control IP, and the hnRNP-U-enhanced association with NEIL1 in GO-treated cells (Fig. 1B), confirm the PLA results. Because both hnRNP-U and NEIL1 bind to DNA, we considered the possibility that the interaction is indirect, mediated by their independent binding to DNA. That prior treatment of the cell lysates with DNase I + RNase A (500 units/ml each) before co-IP did not affect hnRNP-U level in the NEIL1 IP strongly supports direct interaction between these polypeptides (Fig. 1C), which was also confirmed by similar analysis in HCT116 cells (data not shown). To confirm the specificity of oxidative stress dependence of enhancement of NEIL1-hnRNP-U association, we performed PLA analysis in HEK293 cells after irradiation with UV-C (254 nm), which predominantly induces cyclobutane dimers and (6–4) photoproducts in cellular genomes (44). No difference in NEIL1-hnRNP-U association was observed with low UV dose (5 J/m2) in control versus irradiated cells (supplemental Fig. S1). A moderate increase in PLA signal observed at higher dose could be due to secondary generation of ROS (45). Taken together, these data demonstrate the stable association of NEIL1 with hnRNP-U in human cells, which is enhanced by oxidative stress.
FIGURE 1.

In-cell association of NEIL1 with hnRNP-U and its enhancement after oxidative stress. A, in situ PLA analysis (Duolink) for association of endogenous NEIL1 with hnRNP-U in HEK293 cells (left panel), indicated by red foci in nuclei (stained blue with DAPI). The association increased significantly in cells treated with GO (50 ng/ml for 30 min; right panel). Right column, PLA controls with NEIL1 Ab (rabbit) versus IgG (mouse) or hnRNP-U Ab (mouse) versus IgG (rabbit). Histogram (right) represents quantitation of PLA foci/cell for NEIL1 versus hnRNP-U in untreated versus GO-treated cells. B, co-IP analysis of hnRNP-U with FLAG-NEIL1 in extracts from stably FLAG-NEIL1-expressing HEK293 cells. Increased association in oxidatively stressed cells is similar to A. IPs of extracts from cells stably expressing empty vector as the control are shown. The Western blot band intensity was quantified and represented as histogram. Error bars indicate S.D. C, association between FLAG-NEIL1 and hnRNP-U unaffected by treatment of HEK293 cell extracts with DNase I/RNase A (500 units/ml each).
S-phase Activation of NEIL1 Contributes to Its Increased Association with hnRNP-U
Because NEIL1 may be unique among oxidized base-specific DGs in transcriptional activation during the S-phase (34), we tested the possibility of cell cycle-dependent modulation of the NEIL1-hnRNP-U complex. Using double thymidine block (36), we synchronized HEK293 cells (supplemental Fig. S2), confirmed enrichment of S-phase cells (∼65%) 3 h after release, and monitored the level of hnRNP-U and NEIL1 polypeptides. As expected, the NEIL1 level was significantly higher (∼5-fold) in S-phase enriched cells when compared with G1 cells, but the hnRNP-U level was not significantly different in the two cell populations. However, the amount of the hnRNP-U in NEIL1 in IP from the S-phase cells was significantly higher (∼4-fold) than in the IP isolated from G1 cells. The comparable increase in NEIL1 level and its association with hnRNP-U in S-phase cells suggest that the enhanced association of NEIL1 with hnRNP-U in the S-phase was due to the increased level of NEIL1. This also predicts enhanced NEIL1-mediated repair during the S-phase.
Direct Binding of NEIL1 to hnRNP-U and the Mapping of Their Interaction Domains
To further examine whether hnRNP-U interacts with NEIL1 directly or is mediated by other proteins, we examined in vitro interaction using far Western and GST pulldown analysis. Fig. 2A shows that hnRNP-U interacted with NEIL1 and its deletion polypeptides containing residues 1–349, 289–389, 289–349, 312–389, and 312–349, but not with truncated polypeptides 1–311 or 1–288 or with BSA used as a control. hnRNP-U binding to the NEIL1 C-terminal peptide 312–349 (minimum interaction interface) was further confirmed by GST pulldown assays using GST fusion of the NEIL1 C-terminal peptides (Fig. 2, B and C).
FIGURE 2.

The hnRNP-U direct interaction with NEIL1 and identification of the NEIL1 C-terminal residues (312–349) as the minimal interaction peptide. A, far Western analysis. Left panel, membrane-bound WT NEIL1 (lane 1) and its deletion polypeptides (lanes 2–8; Coomassie staining after SDS-PAGE) were incubated with hnRNP-U in solution and probed with hnRNP-U Ab (right panel). BSA (lane 9) and FEN-1 (lane 10) were used as negative and positive controls respectively (22). B, co-elution of hnRNP-U with the NEIL1 GST-tagged deletion polypeptides bound to glutathione-Sepharose beads. C, the NEIL1 C-terminal residues (312–349) were identified as minimal interaction residues for hnRNP-U as shown schematically.
The binding of hnRNP-U to the NEIL1 C-terminal peptide was further analyzed in-cell by PLA and IP studies. HEK293 cells stably expressing FLAG-NEIL1 or FLAG-NEIL1(1–311) were subjected to PLA using FLAG Ab (mouse; Sigma) and hnRNP-U Ab (rabbit; Santa Cruz Biotechnology). Interaction indicated by PLA signals was observed with FLAG-NEIL1/hnRNP-U, but not with FLAG-NEIL1(1–311)/hnRNP-U or IgG/hnRNP-U (Fig. 3A). Consistent with this, co-IP studies showed the presence of hnRNP-U in FLAG-NEIL1 but not in FLAG-NEIL1(1–311) IP (Fig. 3B).
FIGURE 3.

Specificity of NEIL1-hnRNP-U interaction. A, in situ PLA in HEK293 cells stably expressing FLAG-NEIL1 or FLAG-NEIL1(1–311) and probed with FLAG Ab (mouse) versus hnRNP-U Ab (rabbit). B, the lack of hnRNP-U interaction with NEIL1 deletion (1–311) polypeptide was confirmed by co-IP analysis.
We then characterized the regions in the hnRNP-U polypeptide required for NEIL1 interaction. GST pulldown analysis showed that the interaction interface of hnRNP-U is localized to two segments present within both the N-terminal (residues 1–100) and the C-terminal regions (residues 651–806; Fig. 4, A and B). PONDR analysis predicts that both these terminal segments in hnRNP-U are intrinsically disordered. This was further confirmed by CD spectroscopy of purified polypeptides (residues 1–100 and 651–806 from the termini of hnRNP-U (supplemental Fig. S3), which showed 94 and 82% random coil conformation, respectively (40).
FIGURE 4.

Mapping and correlation of NEIL1 interaction domains in hnRNP-U with predicted disordered regions. A, co-elution of NEIL1 with GST-tagged hnRNP-U polypeptide fragments bound to glutathione-Sepharose beads and analysis of eluted proteins by Western analysis with NEIL1 Ab or GST Ab. B, schematic representation of the hnRNP-U N- (residues 1–100) and C- (residues 651–806) terminal segments for NEIL1 binding. C, PONDR modeling-based prediction of disordered structure of the interaction segments in NEIL1 (C terminus) and hnRNP-U (N and C termini), which was confirmed by CD spectroscopy (see supplemental Fig. S3).
High Affinity Binding of hnRNP-U to NEIL1
Based on the change in intrinsic fluorescence of hnRNP-U in the presence of the NEIL1 C-terminal peptide (residues 312–349 lacking aromatic amino acids), we calculated the apparent dissociation constant (Kd) to be 54 nm for its binding to hnRNP-U (Fig. 5). Because our previous studies showed that this C-terminal peptide alone contributes to the NEIL1 interaction with all of its partners tested so far, and similar affinity was observed with the peptide versus full-length NEIL1 for FEN-1 (22), we are confident that the full-length NEIL1 interacts with hnRNP-U with comparable affinity. Further, a 1:1 molar stoichiometry for binding suggests simultaneous binding of the two terminal domains of hnRNP-U with NEIL1, implying the close proximity of the termini in the native conformation.
FIGURE 5.
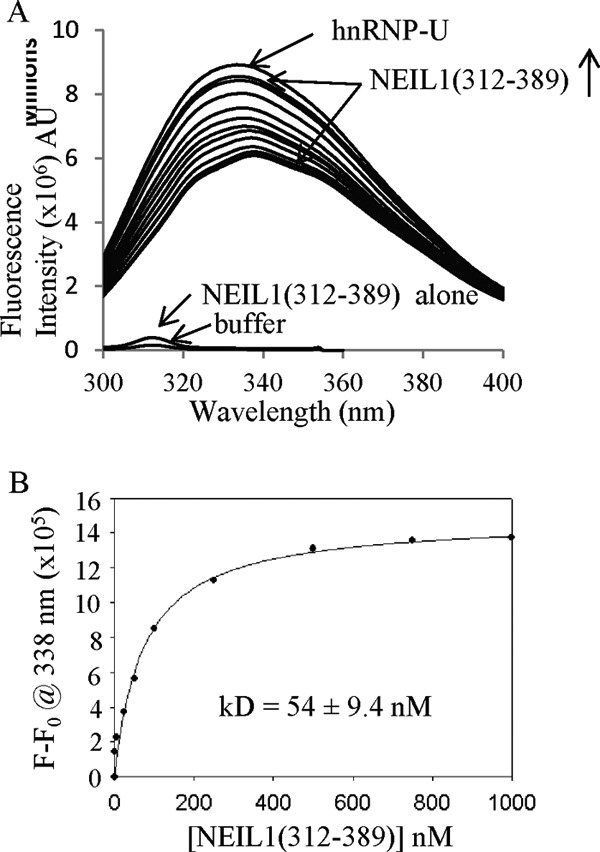
Intrinsic fluorescence of hnRNP-U bound to the NEIL1 interacting domain. A, change in the tryptophan fluorescence of hnRNP-U in the presence of increasing concentrations of the NEIL1 C-terminal peptide (residues 312–389). AU, arbitrary units. B, binding isotherm of NEIL1 with hnRNP-U monitored by fluorescence at 338 nm. The calculated affinity (kd) is shown.
hnRNP-U Increases NEIL1 Turnover
We investigated the effect of hnRNP-U on the NEIL1 excision of 5-OHU and strand cleavage for various oligonucleotide substrates. hnRNP-U stimulated the NEIL1 activity up to ∼4-fold in a concentration-dependent manner when the lesion was located in the single-stranded region or in close proximity to the primer-template junction of the partially duplex oligonucleotide (Fig. 6). The NEIL1 strand incision activity on a 5-OHU-containing duplex oligonucleotide substrate was stimulated ∼2-fold by hnRNP-U at 1:1 molar ratio (Fig. 6). Bubble and forked oligonucleotides showed similar levels of stimulation (∼2.5- and 2.0-fold, respectively, at a 1:1 molar ratio), whereas ssDNA showed the highest stimulation (∼3.5-fold). Furthermore, the lack of stimulation of C-terminal truncation mutant NEIL1(1–311), which does not bind to hnRNP-U, confirmed the requirement of their interaction for stimulation (supplemental Fig. S4). We further observed that the N- or C-terminal peptide of hnRNP-U alone also stimulated NEIL1, albeit to a lesser extent when compared with WT hnRNP-U (supplemental Fig. S5).
FIGURE 6.
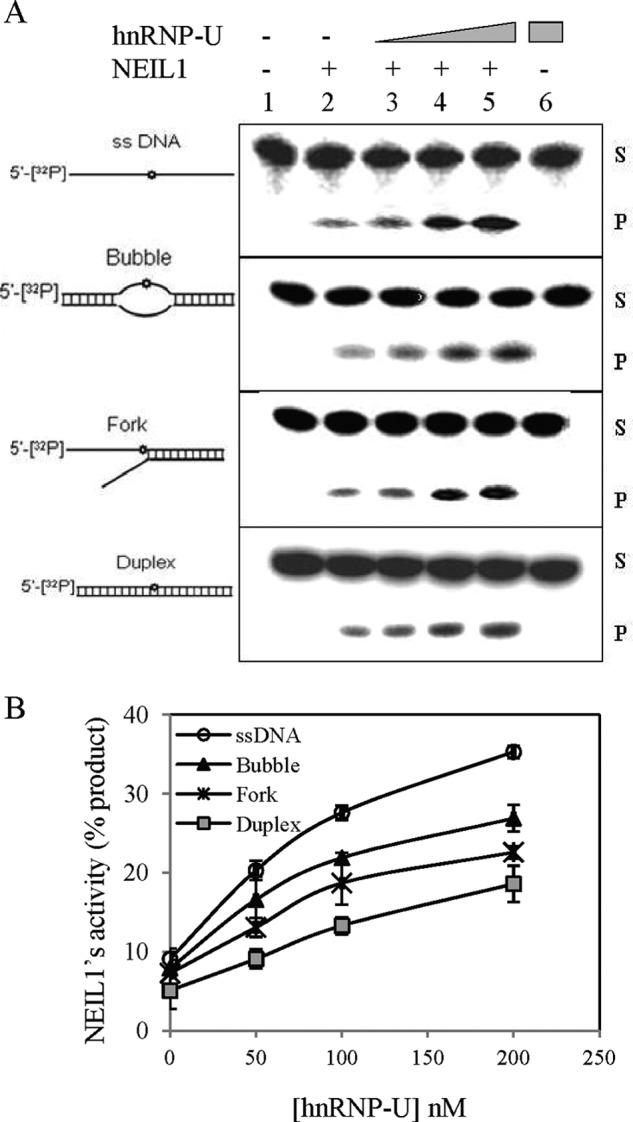
hnRNP-U stimulation of NEIL1 activity for excision of 5-OHU from various DNA substrates. A, a 5-OHU-containing 51-mer oligonucleotide (25 nm) was used alone (single-stranded) or annealed with complementary strands to generate duplex, bubble, or fork structures (supplemental Table S1) for use as NEIL1 substrate. Lanes 2–5, WT NEIL1 (1 nm; lane 2) alone or after preincubation of the DNA with various levels of hnRNP-U (0.1, 0.5, 1 nm; lanes 3–5). S and P indicate uncleaved substrate and cleaved product, respectively. Other details are described under “Experimental Procedures.” B, comparison of hnRNP-U stimulation with various repair substrates. Error bars indicate S.D.
We calculated the NEIL1 kinetic parameters using a forked substrate in the presence and absence of hnRNP-U (Table 1). The ∼2.5-fold decrease in Km indicated that hnRNP-U enhances the substrate affinity of NEIL1. However, ∼8-fold increase in kcat suggests that the hnRNP-U stimulatory effect is primarily due to its role in product release.
TABLE 1.
Kinetic parameters of NEIL1 glycosylase and its stimulation by hnRNP-U
| Reaction | Km | kcat | kcat/Km |
|---|---|---|---|
| nm | × 10−2/min | × 10−2/nm/min | |
| NEIL1 only | 15.9 ± 1.1 | 3.7 ± 0.9 | 0.23 |
| NEIL1 plus hnRNP-U | 6.9 ± 1.3 | 27.1 ± 3.4 | 3.9 |
hnRNP-U Is Required for Enhancement of NEIL1-mediated Repair under Oxidative Stress
We reconstituted NEIL1-initiated SN-BER in vitro using purified NEIL1, polynucleotide kinase/phosphatase, DNA polymerase β, and DNA ligase IIIα for repairing a 5-OHU lesion-containing duplex substrate (Fig. 7A). As expected, the presence of hnRNP-U stimulated the repair by 4-fold at 1:1 molar ratio (Fig. 7B).
FIGURE 7.
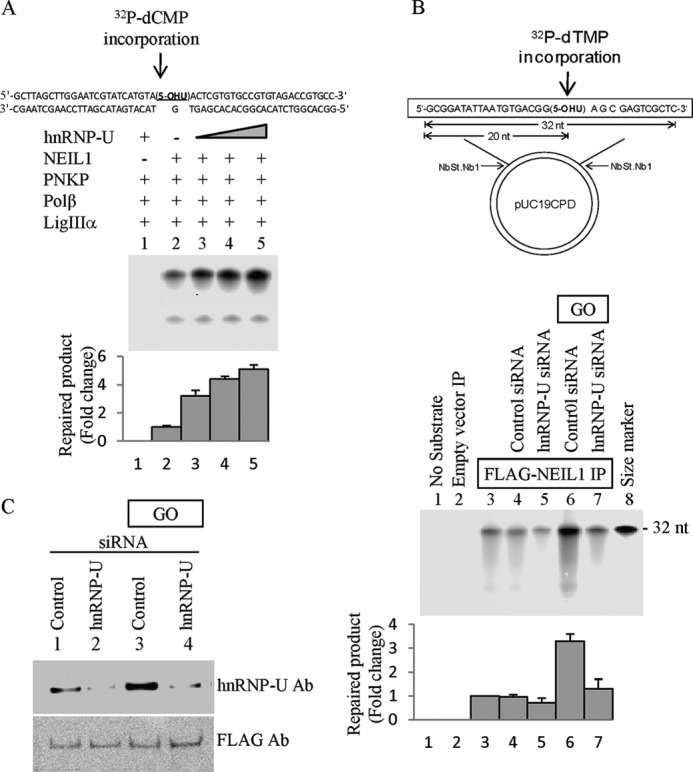
hnRNP-U enhances NEIL1-initiated repair. A, effect of hnRNP-U on NEIL1-initiated SN-BER in vitro. Repair of 5-OHU in DNA was analyzed using a 5-OHU-containing duplex oligonucleotide substrate (top) and purified proteins as indicated. SN-BER was measured by incorporation of [32P]dCMP in a reaction containing 50 fmol each of NEIL1, polynucleotide kinase/phosphatase (PNKP), and DNA polymerase β (Polβ) and 100 fmol of DNA ligase IIIα (LigIIIα). Increasing amounts (10, 25, and 50 fmol) of hnRNP-U were included in lanes 3–5. Error bars indicate S.D. B, repair of 5-OHU in a plasmid with proteins isolated from NEIL1 IP. FLAG-NEIL1 was immunoprecipitated from HEK293 cells, with or without pretreatment with treatment with GO (50 ng/ml, 30 min) or transfection with hnRNP-U siRNA (see Fig. 8B for the extent of depletion) or appropriate controls as indicated. Error bars indicate S.D. C, repair of a 5-OHU residue incorporated in a plasmid (top) was compared for various extracts normalized for the FLAG level using Western analysis.
We then analyzed repair of 5-OHU-containing plasmid with eluates from FLAG-NEIL1 IP isolated from HEK293 cells with or without prior GO treatment (50 ng/ml for 30 min). We normalized the FLAG level in the assay mixture for comparing repair activity (Fig. 7C). Complete repair by NEIL1 IP was reduced by 20% in hnRNP-U-depleted cells (siRNA-mediated, ∼75% down-regulation; Fig. 7B, lane 5). Enhancement of repair level by about 3-fold was observed in GO-treated cells when compared with the control cells (Fig. 7B, lower panel, lane 6). However, this enhancement was squelched in hnRNP-U-depleted cells (Fig. 7C). Taken together, these results strongly suggest the significant contribution of hnRNP-U to the enhancement of NEIL1-initiated repair due to oxidative stress, which should increase the level of oxidized bases.
NEIL1 and hnRNP-U Protect HEK293 Cells from ROS Toxicity
The in vitro repair studies suggest the stimulatory role of hnRNP-U in repair of oxidized bases, particularly after oxidative stress, which may protect cells from ROS-induced toxicity. We tested this possibility by clonogenic survival assay of cells after GO treatment. NEIL1 deficiency significantly increased the sensitivity of cells to GO, implicating NEIL1 in repair of cytotoxic base lesions (Fig. 8). Moderate but definite increase in sensitivity to GO was also observed in hnRNP-U-deficient cells, which is consistent with the hnRNP-U stimulation of oxidized base damage repair (Fig. 7). However, ROS sensitivity was not significantly enhanced by combining hnRNP-U and NEIL1 deficiency, particularly at moderate GO level (Fig. 8), suggesting that the protection of cells after oxidative stress by hnRNP-U is primarily via activation of NEIL1-mediated repair.
FIGURE 8.
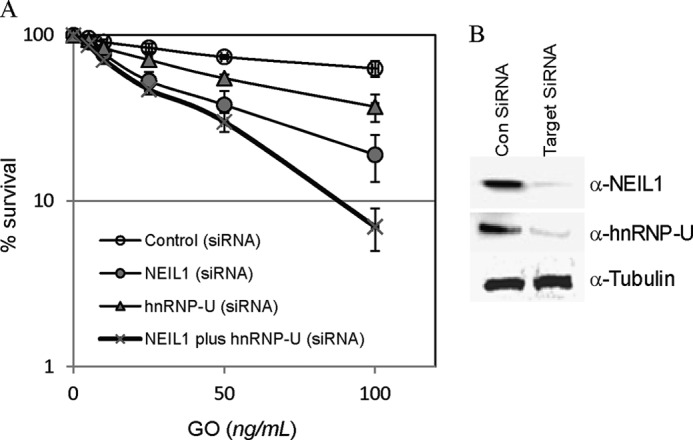
hnRNP-U and NEIL1 protect HEK293 cells from oxidative stress. A, survival of HEK293 cells transfected with control siRNA or siRNA for hnRNP-U, NEIL1, or hnRNP-U + NEIL1. At 48 h after transfection, 400 cells were plated into 60-mm dishes and treated with increasing concentration of GO (1–100 ng/ml medium) for 15 min. After changing to fresh medium, cells were maintained for 10 days before the colonies were stained with crystal violet. Other details are given under “Experimental Procedures.” Error bars indicate S.D. B, Western analysis of hnRNP-U and NEIL1 levels in HEK293 cells 48 h after transfection with corresponding siRNA.
DISCUSSION
Contrary to the common perception that the repair of oxidatively damaged bases in DNA via the BER pathway involves only a few conserved repair proteins, a more complex picture of BER is now emerging in that BER comprises several alternative subpathways that utilize distinct sets of repair enzymes and other proteins (16, 19, 21, 35, 46). We have been exploring the role of noncanonical proteins in oxidized base repair and have characterized the proteins present in immunoprecipitates of early BER enzymes including DGs. In addition to the BER proteins, we identified several additional proteins, including hnRNP-U, in the IPs of NEILs and APE1 (16, 19, 37, 47). Many of the non-repair proteins in such IPs are RNA-binding proteins that are usually abundant.3 To eliminate the possibility of their adventitious presence in the IPs, we examined their specific, binary interaction with NEIL1 and other DGs and mapped their interaction domains. Thus the studies described here provide evidence about the physiological relevance of the role of hnRNP-U in NEIL1-mediated repair. PLA results confirmed their association in the nucleus. In vitro studies show the hnRNP-U enhancement of NEIL1 activity via direct interaction. Definitive evidence for the role of hnRNP-U in oxidative damage repair in-cell was provided by the reduced survival of hnRNP-U-deficient cells caused by oxidative stress. Increased BER activity of the eluate from NEIL1 IP from ROS-treated cells was abrogated in hnRNP-U-depleted cells (Fig. 7). Because of the contribution of NEIL2 to oxidized base repair where hnRNP-U is also involved (19), it is likely that hnRNP-U provides cellular protection from ROS toxicity by activating both NEILs. The modest increase in ROS sensitivity due to hnRNP-U deficiency alone (Fig. 8) is consistent with its involvement in repair, primarily in oxidatively stressed cells. This is supported by the increase in its in-cell association with NEIL1 after GO treatment. Whether the enhanced affinity results from their covalent modifications remains to be tested. However, this may be logical because the DG is the rate-limiting enzyme in overall repair. Another common feature of the noncanonical proteins is their activation of DGs at the step of product release. A hallmark of DGs and other early BER enzymes is their product affinity, contrary to the notion of conventional enzyme kinetics. In fact, the affinity of OGG1 for the product AP site is higher than for the substrate 8-oxoG in DNA (48). It is possible that the noncanonical proteins such as hnRNP-U cause allosteric changes in the DGs to reduce their affinity for the cleaved DNA product (21, 22).
As mentioned earlier, hnRNP-U (and many other hnRNP family members) have additional functions in transcriptional regulation (24, 49), which may involve direct binding to sequence-specific or nonspecific DNA. Because of the presence of multiple domains and motifs in this large polypeptide, hnRNP-U interacts with diverse proteins (24). A recent study described the role of hnRNP-U-like proteins hnRNP-UL1 and -2 in DNA damage signaling and end resection at double-strand breaks, which may involve a distinct motif in these large polypeptides (50). Because of its nuclear scaffold attachment, hnRNP-U (and its homologues) may target the DNA repair site to the nuclear matrix where chromatin unfolding and refolding may occur. We have mapped the NEIL1-binding sequences to both the N termini and the C termini of hnRNP-U. The N-terminal region (∼160 residues) of hnRNP-U is rich in acidic (Asp and Glu) residues, and the C-terminal segment (∼112 amino acid residues) contains a cluster of RGG repeats. The function of the N terminus is not known, whereas the C-terminal RGG repeat motif is involved in RNA binding (24). Although the tertiary structures of hnRNP-U (and of other hnRNPs) have not been solved, the interaction mapping analysis strongly suggests close proximity of the hnRNP-U terminal segments in the native conformation (Figs. 4 and 5).
Our previous studies have shown that the disordered C-terminal extension of NEIL1 acts as the common interaction interface for diverse partners (12, 20–23, 39). The absence of this extension in E. coli Nei, the prokaryotic orthologue of NEIL1, underscores the physiological significance of protein-protein interactions in mammalian BER (10, 39), which may induce structure in the disordered region with multiple conformers. Interestingly, the NEIL1-hnRNP-U complex involves disorder-disorder interaction that would allow a large number of induced structures at the binding interface, which is common but poorly understood in mammalian cells (39).
In summary, these results together with past studies strongly suggest that BER for modified bases is tightly regulated in response to changes in cellular state and is facilitated by interaction with non-repair proteins such as hnRNP-U.
Acknowledgments
We thank Drs. D. W. Bolen and L. Holthauzen (Department of Biochemistry and Molecular Biology) for the use of the Biophysical Core Facility and Drs. M. Banadakoppa and C. Yallampalli (Department of Obstetrics and Gynecology) for fluorescence microscopy. We acknowledge the help of Dr. T. Wood of the NIEHS Center (National Institutes of Health), supported by Grant P30 ES06676 for large scale purification of pUC19CPD plasmid. We thank former Mitra laboratory members Drs. C. Theriot and A. Das for various help and Dr. D. Konkel for critically editing this manuscript.
This work was supported, in whole or in part, by National Institutes of Health Grants R01 CA81063 and P01 CA092584 (to S. M.), R01 NS073976 (to T. K. H.), and ES018948 (to I. B.) through the USPHS.

This article contains supplemental Table S1 and Figs. S1–S5.
M. L. Hegde, P. M. Hegde, T. K. Hazra, I. Boldogh, and S. Mitra, unpublished observation.
- ROS
- reactive oxygen species
- Ab
- antibody
- BER
- base excision repair
- SN-BER
- single-nucleotide BER
- FEN-1
- flap endonuclease 1
- hnRNP-U
- heterogeneous nuclear ribonucleoprotein-U
- 5-OHU
- 5-hydroxyuracil
- DG
- DNA glycosylase
- GO
- glucose oxidase
- IP
- immunoprecipitate
- co-IP
- co-immunoprecipitation
- PLA
- proximity ligation assay
- APE1
- AP-endonuclease
- AP
- abasic
- NEIL
- Nei-like.
REFERENCES
- 1. Breen A. P., Murphy J. A. (1995) Reactions of oxyl radicals with DNA. Free Radic. Biol. Med. 18, 1033–1077 [DOI] [PubMed] [Google Scholar]
- 2. Cadet J. L., Brannock C. (1998) Free radicals and the pathobiology of brain dopamine systems. Neurochem. Int. 32, 117–131 [DOI] [PubMed] [Google Scholar]
- 3. Grisham M. B., Hernandez L. A., Granger D. N. (1986) Xanthine oxidase and neutrophil infiltration in intestinal ischemia. Am. J. Physiol. 251, G567–G574 [DOI] [PubMed] [Google Scholar]
- 4. Ames B. N., Shigenaga M. K., Hagen T. M. (1993) Oxidants, antioxidants, and the degenerative diseases of aging. Proc. Natl. Acad. Sci. U.S.A. 90, 7915–7922 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Bozner P., Grishko V., LeDoux S. P., Wilson G. L., Chyan Y. C., Pappolla M. A. (1997) The amyloid-β protein induces oxidative damage of mitochondrial DNA. J. Neuropathol. Exp. Neurol. 56, 1356–1362 [DOI] [PubMed] [Google Scholar]
- 6. DeWeese T. L., Shipman J. M., Larrier N. A., Buckley N. M., Kidd L. R., Groopman J. D., Cutler R. G., te Riele H., Nelson W. G. (1998) Mouse embryonic stem cells carrying one or two defective Msh2 alleles respond abnormally to oxidative stress inflicted by low-level radiation. Proc. Natl. Acad. Sci. U.S.A. 95, 11915–11920 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Meyer T. E., Liang H. Q., Buckley A. R., Buckley D. J., Gout P. W., Green E. H., Bode A. M. (1998) Changes in glutathione redox cycling and oxidative stress response in the malignant progression of NB2 lymphoma cells. Int. J. Cancer 77, 55–63 [DOI] [PubMed] [Google Scholar]
- 8. Mukherjee S. K., Adams J. D., Jr. (1997) The effects of aging and neurodegeneration on apoptosis-associated DNA fragmentation and the benefits of nicotinamide. Mol. Chem. Neuropathol. 32, 59–74 [DOI] [PubMed] [Google Scholar]
- 9. Multhaup G., Ruppert T., Schlicksupp A., Hesse L., Beher D., Masters C. L., Beyreuther K. (1997) Reactive oxygen species and Alzheimer disease. Biochem. Pharmacol. 54, 533–539 [DOI] [PubMed] [Google Scholar]
- 10. Hegde M. L., Hazra T. K., Mitra S. (2008) Early steps in the DNA base excision/single-strand interruption repair pathway in mammalian cells. Cell Res. 18, 27–47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Liu M., Bandaru V., Bond J. P., Jaruga P., Zhao X., Christov P. P., Burrows C. J., Rizzo C. J., Dizdaroglu M., Wallace S. S. (2010) The mouse orthologue of NEIL3 is a functional DNA glycosylase in vitro and in vivo. Proc. Natl. Acad. Sci. U.S.A. 107, 4925–4930 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Wiederhold L., Leppard J. B., Kedar P., Karimi-Busheri F., Rasouli-Nia A., Weinfeld M., Tomkinson A. E., Izumi T., Prasad R., Wilson S. H., Mitra S., Hazra T. K. (2004) AP endonuclease-independent DNA base excision repair in human cells. Mol. Cell 15, 209–220 [DOI] [PubMed] [Google Scholar]
- 13. Matsumoto Y. (2001) Molecular mechanism of PCNA-dependent base excision repair. Prog. Nucleic Acid Res. Mol. Biol. 68, 129–138 [DOI] [PubMed] [Google Scholar]
- 14. Klungland A., Lindahl T. (1997) Second pathway for completion of human DNA base excision-repair: reconstitution with purified proteins and requirement for DNase IV (FEN1). EMBO J. 16, 3341–3348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Garg P., Stith C. M., Sabouri N., Johansson E., Burgers P. M. (2004) Idling by DNA polymerase δ maintains a ligatable nick during lagging-strand DNA replication. Genes Dev. 18, 2764–2773 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Das S., Chattopadhyay R., Bhakat K. K., Boldogh I., Kohno K., Prasad R., Wilson S. H., Hazra T. K. (2007) Stimulation of NEIL2-mediated oxidized base excision repair via YB-1 interaction during oxidative stress. J. Biol. Chem. 282, 28474–28484 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Prasad R., Liu Y., Deterding L. J., Poltoratsky V. P., Kedar P. S., Horton J. K., Kanno S., Asagoshi K., Hou E. W., Khodyreva S. N., Lavrik O. I., Tomer K. B., Yasui A., Wilson S. H. (2007) HMGB1 is a cofactor in mammalian base excision repair. Mol. Cell 27, 829–841 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Marenstein D. R., Ocampo M. T., Chan M. K., Altamirano A., Basu A. K., Boorstein R. J., Cunningham R. P., Teebor G. W. (2001) Stimulation of human endonuclease III by Y box-binding protein 1 (DNA-binding protein B): interaction between a base excision repair enzyme and a transcription factor. J. Biol. Chem. 276, 21242–21249 [DOI] [PubMed] [Google Scholar]
- 19. Banerjee D., Mandal S. M., Das A., Hegde M. L., Das S., Bhakat K. K., Boldogh I., Sarkar P. S., Mitra S., Hazra T. K. (2011) Preferential repair of oxidized base damage in the transcribed genes of mammalian cells. J. Biol. Chem. 286, 6006–6016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Das A., Boldogh I., Lee J. W., Harrigan J. A., Hegde M. L., Piotrowski J., de Souza Pinto N., Ramos W., Greenberg M. M., Hazra T. K., Mitra S., Bohr V. A. (2007) The human Werner syndrome protein stimulates repair of oxidative DNA base damage by the DNA glycosylase NEIL1. J. Biol. Chem. 282, 26591–26602 [DOI] [PubMed] [Google Scholar]
- 21. Dou H., Theriot C. A., Das A., Hegde M. L., Matsumoto Y., Boldogh I., Hazra T. K., Bhakat K. K., Mitra S. (2008) Interaction of the human DNA glycosylase NEIL1 with proliferating cell nuclear antigen: the potential for replication-associated repair of oxidized bases in mammalian genomes. J. Biol. Chem. 283, 3130–3140 [DOI] [PubMed] [Google Scholar]
- 22. Hegde M. L., Theriot C. A., Das A., Hegde P. M., Guo Z., Gary R. K., Hazra T. K., Shen B., Mitra S. (2008) Physical and functional interaction between human oxidized base-specific DNA glycosylase NEIL1 and flap endonuclease 1. J. Biol. Chem. 283, 27028–27037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Theriot C. A., Hegde M. L., Hazra T. K., Mitra S. (2010) RPA physically interacts with the human DNA glycosylase NEIL1 to regulate excision of oxidative DNA base damage in primer-template structures. DNA Repair (Amst.) 9, 643–652 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kiledjian M., Dreyfuss G. (1992) Primary structure and binding activity of the hnRNP U protein: binding RNA through RGG box. EMBO J. 11, 2655–2664 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Xiao R., Tang P., Yang B., Huang J., Zhou Y., Shao C., Li H., Sun H., Zhang Y., Fu X. D. (2012) Nuclear matrix factor hnRNP U/SAF-A exerts a global control of alternative splicing by regulating U2 snRNP maturation. Mol. Cell 45, 656–668 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Romig H., Fackelmayer F. O., Renz A., Ramsperger U., Richter A. (1992) Characterization of SAF-A, a novel nuclear DNA-binding protein from HeLa cells with high affinity for nuclear matrix/scaffold attachment DNA elements. EMBO J. 11, 3431–3440 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Spraggon L., Dudnakova T., Slight J., Lustig-Yariv O., Cotterell J., Hastie N., Miles C. (2007) hnRNP-U directly interacts with WT1 and modulates WT1 transcriptional activation. Oncogene 26, 1484–1491 [DOI] [PubMed] [Google Scholar]
- 28. Davis M., Hatzubai A., Andersen J. S., Ben-Shushan E., Fisher G. Z., Yaron A., Bauskin A., Mercurio F., Mann M., Ben-Neriah Y. (2002) Pseudosubstrate regulation of the SCFβ-TrCP ubiquitin ligase by hnRNP-U. Genes Dev. 16, 439–451 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Britton S., Froment C., Frit P., Monsarrat B., Salles B., Calsou P. (2009) Cell nonhomologous end joining capacity controls SAF-A phosphorylation by DNA-PK in response to DNA double-strand breaks inducers. Cell Cycle 8, 3717–3722 [DOI] [PubMed] [Google Scholar]
- 30. Berglund F. M., Clarke P. R. (2009) hnRNP-U is a specific DNA-dependent protein kinase substrate phosphorylated in response to DNA double-strand breaks. Biochem. Biophys. Res. Commun. 381, 59–64 [DOI] [PubMed] [Google Scholar]
- 31. Hegde M. L., Hegde P. M., Holthauzen L. M., Hazra T. K., Rao K. S., Mitra S. (2010) Specific Inhibition of NEIL-initiated repair of oxidized base damage in human genome by copper and iron: potential etiological linkage to neurodegenerative diseases. J. Biol. Chem. 285, 28812–28825 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Das A., Wiederhold L., Leppard J. B., Kedar P., Prasad R., Wang H., Boldogh I., Karimi-Busheri F., Weinfeld M., Tomkinson A. E., Wilson S. H., Mitra S., Hazra T. K. (2006) NEIL2-initiated, APE-independent repair of oxidized bases in DNA: evidence for a repair complex in human cells. DNA Repair (Amst.) 5, 1439–1448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Hazra T. K., Izumi T., Boldogh I., Imhoff B., Kow Y. W., Jaruga P., Dizdaroglu M., Mitra S. (2002) Identification and characterization of a human DNA glycosylase for repair of modified bases in oxidatively damaged DNA. Proc. Natl. Acad. Sci. U.S.A. 99, 3523–3528 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Hazra T. K., Kow Y. W., Hatahet Z., Imhoff B., Boldogh I., Mokkapati S. K., Mitra S., Izumi T. (2002) Identification and characterization of a novel human DNA glycosylase for repair of cytosine-derived lesions. J. Biol. Chem. 277, 30417–30420 [DOI] [PubMed] [Google Scholar]
- 35. Guan X., Bai H., Shi G., Theriot C. A., Hazra T. K., Mitra S., Lu A. L. (2007) The human checkpoint sensor Rad9-Rad1-Hus1 interacts with and stimulates NEIL1 glycosylase. Nucleic Acids Res. 35, 2463–2472 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Whitfield M. L., Sherlock G., Saldanha A. J., Murray J. I., Ball C. A., Alexander K. E., Matese J. C., Perou C. M., Hurt M. M., Brown P. O., Botstein D. (2002) Identification of genes periodically expressed in the human cell cycle and their expression in tumors. Mol. Biol. Cell 13, 1977–2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Chattopadhyay R., Das S., Maiti A. K., Boldogh I., Xie J., Hazra T. K., Kohno K., Mitra S., Bhakat K. K. (2008) Regulatory role of human AP-endonuclease (APE1/Ref-1) in YB-1-mediated activation of the multidrug resistance gene MDR1. Mol. Cell. Biol. 28, 7066–7080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Romero P., Obradovic Z., Li X., Garner E. C., Brown C. J., Dunker A. K. (2001) Sequence complexity of disordered protein. Proteins 42, 38–48 [DOI] [PubMed] [Google Scholar]
- 39. Hegde M. L., Hazra T. K., Mitra S. (2010) Functions of disordered regions in mammalian early base excision repair proteins. Cell. Mol. Life Sci. 67, 3573–3587 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Yang J. T., Wu C. S., Martinez H. M. (1986) Calculation of protein conformation from circular dichroism. Methods Enzymol. 130, 208–269 [DOI] [PubMed] [Google Scholar]
- 41. Johansson H., Svensson F., Runnberg R., Simonsson T., Simonsson S. (2010) Phosphorylated nucleolin interacts with translationally controlled tumor protein during mitosis and with Oct4 during interphase in ES cells. PloS One 5, e13678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Fredriksson S., Gullberg M., Jarvius J., Olsson C., Pietras K., Gústafsdóttir S. M., Ostman A., Landegren U. (2002) Protein detection using proximity-dependent DNA ligation assays. Nat. Biotechnol. 20, 473–477 [DOI] [PubMed] [Google Scholar]
- 43. Mandal S. M., Hegde M. L., Chatterjee A., Hegde P. M., Szczesny B., Banerjee D., Boldogh I., Gao R., Falkenberg M., Gustafsson C. M., Sarkar P. S., Hazra T. K. (2012) Role of human DNA glycosylase Nei-like 2 (NEIL2) and single-strand break repair protein polynucleotide kinase 3′-phosphatase in maintenance of mitochondrial genome. J. Biol. Chem. 287, 2819–2829 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Douki T., Laporte G., Cadet J. (2003) Interstrand photoproducts are produced in high yield within A-DNA exposed to UVC radiation. Nucleic Acids Res. 31, 3134–3142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Cadet J., Sage E., Douki T. (2005) Ultraviolet radiation-mediated damage to cellular DNA. Mutat. Res. 571, 3–17 [DOI] [PubMed] [Google Scholar]
- 46. Otterlei M., Warbrick E., Nagelhus T. A., Haug T., Slupphaug G., Akbari M., Aas P. A., Steinsbekk K., Bakke O., Krokan H. E. (1999) Postreplicative base excision repair in replication foci. EMBO J. 18, 3834–3844 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Sengupta S., Mantha A. K., Mitra S., Bhakat K. K. (2011) Human AP endonuclease (APE1/Ref-1) and its acetylation regulate YB-1-p300 recruitment and RNA polymerase II loading in the drug-induced activation of multidrug resistance gene MDR1. Oncogene 30, 482–493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Hill J. W., Hazra T. K., Izumi T., Mitra S. (2001) Stimulation of human 8-oxoguanine-DNA glycosylase by AP-endonuclease: potential coordination of the initial steps in base excision repair. Nucleic Acids Res. 29, 430–438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Vizlin-Hodzic D., Runnberg R., Ryme J., Simonsson S., Simonsson T. (2011) SAF-A forms a complex with BRG1, and both components are required for RNA polymerase II-mediated transcription. PloS One 6, e28049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Polo S. E., Blackford A. N., Chapman J. R., Baskcomb L., Gravel S., Rusch A., Thomas A., Blundred R., Smith P., Kzhyshkowska J., Dobner T., Taylor A. M., Turnell A. S., Stewart G. S., Grand R. J., Jackson S. P. (2012) Regulation of DNA end resection by hnRNPU-like proteins promotes DNA double-strand break signaling and repair. Mol. Cell 45, 505–516 [DOI] [PMC free article] [PubMed] [Google Scholar]