

Background: The role of the 56-kDa gene 4 helicase-primase in T7 DNA replication is unknown.
Results: Individual hexamers of the 56-, 63-, or a mixture of the two have identical helicase activity. A mixture oligomerizes more efficiently than the 63-kDa protein.
Conclusion: An equimolar mixture is optimal for coordinated DNA synthesis.
Significance: The 56-kDa gp4 optimizes the ratio of primase to helicase.
Keywords: Bacteriophage, DNA, DNA Helicase, DNA Replication, Single Molecule Biophysics, Bacteriophage T7, DNA Replisome, Gene 4 Protein, Heterohexamer, Oligomerization
Abstract
Bacteriophage T7 expresses two forms of gene 4 protein (gp4). The 63-kDa full-length gp4 contains both the helicase and primase domains. T7 phage also express a 56-kDa truncated gp4 lacking the zinc binding domain of the primase; the protein has helicase activity but no DNA-dependent primase activity. Although T7 phage grow better when both forms are present, the role of the 56-kDa gp4 is unknown. The two molecular weight forms oligomerize by virtue of the helicase domain to form heterohexamers. The 56-kDa gp4 and any mixture of 56- and 63-kDa gp4 show higher helicase activity in DNA unwinding and strand-displacement DNA synthesis than that observed for the 63-kDa gp4. However, single-molecule measurements show that heterohexamers have helicase activity similar to the 63-kDa gp4 hexamers. In oligomerization assays the 56-kDa gp4 and any mixture of the 56- and 63-kDa gp4 oligomerize to form more hexamers than does the 63-kDa gp4. The zinc binding domain of the 63-kDa gp4 interferes with hexamer formation, an inhibition that is relieved by the insertion of the 56-kDa species. Compared with the 63-kDa gp4, heterohexamers synthesize a reduced amount of oligoribonucleotides, mediated predominately by the 63-kDa subunits via a cis mode. During coordinated DNA synthesis 7% of the tetraribonucleotides synthesized are used as primers by both heterohexamers and hexamers of the 63-kDa gp4. Overall, an equimolar mixture of the two forms of gp4 shows the highest rate of DNA synthesis during coordinated DNA synthesis.
Introduction
Bacteriophage T7 has a simple but efficient DNA replication system (1) consisting of gene 5 DNA polymerase (gp5), the processivity factor Escherichia coli thioredoxin (trx),2 gene 4 helicase-primase (gp4), and gene 2.5 ssDNA-binding protein (gp2.5) (Fig. 1A). gp5 forms a high affinity complex with trx (gp5/trx) to increase the processivity of nucleotide polymerization (2, 3). The helicase domain of the hexameric gp4 encircles the lagging-strand DNA and unwinds dsDNA (4, 5) in a reaction fueled by the hydrolysis of dTTP (1). The primase domain in the N-terminal half of gp4 recognizes specific sequences on the lagging-strand template at which it catalyzes the synthesis of tetraribonucleotides that are used as primers for the initiation of the synthesis of Okazaki fragments. Interactions between the lagging-strand gp5/trx and the C-terminal helicase domain of gp4 result in the formation of a replication loop containing the nascent Okazaki fragments. The replication loops have been observed by electron microscopy using a minicircle replication system (6), and their formation has also been observed using single-molecular techniques (7). gp2.5 coats the ssDNA to remove secondary structures and also physically interacts with both gp4 and gp5/trx, interactions essential for coordination of leading- and lagging-strand DNA synthesis (8).
FIGURE 1.

Model of the replisome of bacteriophage T7 and the organization of gp4. A, the T7 replisome consists of DNA polymerase (gp5), the processivity factor E. coli thioredoxin (trx), the hexameric helicase-primase (gp4), and the ssDNA-binding protein (gp2.5). The helicase unwinds dsDNA to generate two ssDNA templates for both leading- and lagging-strand DNA synthesis by gp5/trx. The primase synthesizes oligoribonucleotides for use as primers for the lagging-strand DNA polymerase to initiate the synthesis of Okazaki fragments. gp2.5 coats the lagging-strand DNA template. B, the 63-kDa gp4 has a C-terminal helicase domain (residues 272–566) with an acidic, flexible C-terminal tail (550–566) and an N-terminal primase domain (1–245). The primase domain has an N-terminal ZBD (1–56) and an RNA RPD (68–245). The 56-kDa gp4 (64–566) lacks the ZBD but retains the RPD and helicase domains.
T7 phage encodes two forms of gp4, a 63- and a 56-kDa form. The two molecular mass forms are present in equal amounts in phage-infected cells (9). The full-length 63-kDa gp4 consists of the C-terminal helicase domain (residues 272–566) and the N-terminal primase domain (residues 1–245) (Fig. 1B). The primase domain contains a zinc binding domain (ZBD, residues 1–56) and an RNA-polymerization domain (RPD, residues 68–245). The truncated 56-kDa gp4 (64–566) lacking the N-terminal 63 residues of the 63-kDa protein are also produced from an internal ribosome-binding site and a start codon located in the gene 4. Consequently the 56-kDa gp4 lacks the ZBD but contains the RPD of the primase and the helicase domain (10, 11). The 56-kDa gp4 cannot catalyze template-directed RNA synthesis, although it does form hexamers and exhibits normal helicase activity (10, 11). The 63-kDa gp4 alone is sufficient to support the growth of T7Δ4 lacking gene 4. However, the rate of DNA synthesis in vivo and the efficiency of plating of the phage are reduced in the absence of the 56-kDa gp4 (9). The 56-kDa gp4, lacking primase activity, cannot support the growth of phage T7Δ4.
The presence of helicase and primase domains within the same polypeptide is unique for T7 phage. In other prokaryotic systems such as Escherichia coli and bacteriophage T4, separate genes encode the two proteins. Nonetheless, the primase and helicase in the other systems have interacting domains, and the two proteins function together at the replication fork (1, 12, 13). Rather than having interacting motifs at the C terminus of the primase and the N terminus of the helicase, the two domains in gp4 are covalently connected via a linker of 26 residues (residues 246–271). This linker, together with N-terminal helices in the helicase domain, is also essential for the oligomerization of the protein into a functional hexamer (14, 15). A truncated gp4 (residues 241–566) containing the linker can oligomerize, whereas gp4 (residues 272–566) lacking this linker cannot (14). An alteration at position 257 (gp4-A257V) increases the oligomerization, whereas substitution at position 263 (gp4-D263N) reduces oligomerization compared with wild-type gp4 (15).
The association of helicase with primase provides distinct advantages. The T7 primase binds ssDNA weakly (Kd ∼ 10–150 μm) (16), whereas the hexameric helicase surrounds the DNA and binds tightly (Kd ∼ 10 nm) thus stabilizing the associated primase (17). The translocation of the helicase along ssDNA also enables the primase to search the primase recognition sites. The presence of the primase within the hexameric structure formed by the helicase domain may provide a mechanism for the coordination of leading- and lagging-strand synthesis.
Several lines of evidence clearly demonstrate the rather rapid and efficient equilibrium of gp4 monomers in solution with those in hexamers. gp4-E343Q with glutamic acid 343 replaced by glutamine loses the ability to hydrolyze dTTP and to unwind DNA (18). However, gp4-E343Q binds DNA tighter than does the wild-type gp4 in the presence of β,γ-methylene dTTP, which mimics dTTP. The titration of wild-type gp4 by gp4-E343Q results in an inhibition of DNA-independent hydrolysis of dTTP in a linear manner indicative of a progressive replacement of wild-type gp4 subunits by gp4-E343Q. However, insertion of a single subunit of gp4-E343Q into a hexamer of wild-type gp4 leads to a total inhibition of ssDNA-dependent dTTP hydrolysis. These results indicate that gp4 and gp4-E343Q can exchange freely within a hexamer (18). Hydrogen bonding between Asn-468 in one subunit and Arg-493 in an adjacent subunit of the gp4 hexamer is critical for helicase to bind DNA (17). Neither gp4-N468R nor gp4-R493N binds ssDNA, but a mixture of the two proteins binds DNA with optimal binding observed at an equimolar ratio of the two proteins. The results indicate that heterohexamers are readily formed, and hydrogen bonding is restored between these two residues in adjacent subunits (17). The C-terminal tail of gp4 interacts with the trx-binding domain of gp5/trx, increasing the processivity of leading-strand DNA synthesis from 5 to 17 kb (19). Mixing gp4-CΔ17 lacking the C-terminal tail with wild-type gp4 reduces the processivity to 5 kb at a ratio of gp4-CΔ17 to wild-type gp4 of 1:5. The results indicate that replacement of a single wild-type gp4 with gp4-CΔ17 is sufficient to prevent the exchange of gp5/trx at the replication fork (19). Finally, the 56-kDa gp4 cannot catalyze oligoribonucleotide synthesis due to the absence of the ZBD. An altered 63-kDa gp4 in which lysine-122 is replaced with alanine in the active site of the RPD (gp4-K122A) cannot catalyze the formation of phosphodiester bonds and thus also is unable to synthesize oligoribonucleotides (20). However, mixing the two proteins results in the template-directed synthesis of oligoribonucleotides (20). In this “trans” synthesis of oligoribonucleotides the ZBD of the gp4-K122A must contact the RPD of an adjacent 56-kDa gp4 subunit, indicating the formation of heterohexamer between the 63- and 56-kDa gp4. Therefore, the 56-kDa gp4 likewise freely interacts with the 63-kDa gp4, and thus, heterohexamers of gp4 should exist in vivo.
A flexible linker of 11 residues (residues 57–67) connects the ZBD and RPD. Both domains are essential for DNA-dependent oligoribonucleotide synthesis. Although there is no structure available on the association of the ZBD with the RPD, such an interaction would appear to be certain, an interaction enabled by the flexible linker connecting the two. In a crystal structure of the T7 primase domain, two primase domains form a dimer where the ZBD of one primase contacts the RPD of the other (21). A crystal structure of the truncated 56-kDa gp4 is also compatible with the ZBD of one subunit either contacting the RPD on the same polypeptide (cis mode) or on an adjacent subunit (trans mode) (22). Studies on the length of the linker between the ZBD and RPD show shorter linkers are defective in the synthesis of the initial dimer but are 2-fold more active in their extension (23).
Why does the presence of the 56-kDa gp4 in phage-infected cells enhance DNA replication and phage growth? In the present study we have examined the effect of 56-kDa gp4 on oligomerization and on both primase and helicase activity. We have also examined cis and trans mode in oligoribonucleotide synthesis by the heterohexamer and the ability of the heterohexamer to coordinate leading- and lagging-strand DNA synthesis.
EXPERIMENTAL PROCEDURES
Materials
Oligonucleotides were purchased from Integrated DNA Technologies. M13 ssDNA, λ DNA, and T4 polynucleotide kinase were purchased from New England Biolabs. The 63-kDa gp4, 56-kDa gp4, 63-kDa gp4-D237A, 56-kDa gp4-D237A, gp5/trx, and gp2.5 were overproduced and purified as described (2, 14, 20, 24, 25). Minicircular DNA was prepared as described (26).
Methods
dTTP Hydrolysis
DNA-dependent hydrolysis of dTTP (27) by a mixture of the 63- and 56-kDa gp4 was determined by incubation of 200 nm gp4 (monomeric concentration), 10 nm M13 ssDNA or 1 μm 60-mer ssDNA, and 0.3 mm [α-32P]dTTP (0.1 μCi) in 40 mm Tris-HCl (pH 7.5), 10 mm MgCl2, 10 mm DTT, and 50 mm potassium glutamate at 37 °C for 20 min. After termination of the reaction by the addition of EDTA to a final concentration of 25 mm, the reaction mixture was spotted onto a polyethyleneimine cellulose TLC plate. The TLC plate was developed with a solution containing 1 m formic acid and 0.8 m LiCl. After autoradiography of the TLC plate, the amount of [α-32P] dTDP formed in the reaction was measured using a Fuji BAS 1000 Bioimaging analyzer.
DNA Unwinding
The DNA substrate used for DNA unwinding consisted of a 5′-32P-labeled 75-mer ssDNA partially annealed (54 base pairs) to a 95-mer ssDNA (see Fig. 3A, inset (5)). The unwinding assay contained 50 nm gp4 (monomeric concentration), 50 nm DNA substrate, 5 mm dTTP, 40 mm Tris-HCl (pH 7.5), 10 mm MgCl2, 10 mm DTT, and 50 mm potassium glutamate. After incubation at 37 °C for 2 min, the reaction was stopped by the addition of a stop buffer to a final concentration of 0.4% SDS, 40 mm EDTA, 8% glycerol, and 0.1% bromphenol blue. The ssDNA product was separated from the dsDNA substrate on a 10% non-denaturing Tris borate-EDTA (TBE) gel. The unwinding activities were determined by measuring the intensity of each band (5).
FIGURE 3.

DNA unwinding and strand-displacement DNA synthesis. A, the DNA substrate used in the unwinding assay (inset) was composed of a 5′ 32P-labeled 75-mer ssDNA partially annealed to a 95-mer ssDNA (54-bp duplex region). The unwinding assay contained 50 nm gp4 (monomeric concentration), 50 nm DNA substrate, and 5 mm dTTP in 40 mm Tris-HCl (pH 7.5), 10 mm MgCl2, 10 mm DTT, and 50 mm potassium glutamate. gp4 consisted of a mixture of 56- and 63-kDa gp4 in molar ratios ranging from 6:0 to 0:6, respectively. After incubation at 37 °C for 2 min, the unwinding activity was determined as described under “Experimental Procedures.” Error bars represent the S.D. of two independent experiments. B, the reaction for strand-displacement DNA synthesis contained 10 nm M13 dsDNA containing a replication fork (inset), 0.6 mm concentrations each of dATP, dTTP, dCTP, and [3H]dGTP (60 cpm/pmol), 200 nm gp4 (monomeric concentration), and 40 nm gp5/trx in 40 mm Tris-HCl (pH 7.5), 10 mm MgCl2, 10 mm DTT, and 50 mm potassium glutamate. gp4 consisted of a mixture of 56- and 63-kDa gp4 in molar ratios ranging from 6:0 to 0:6, respectively. After incubation at 37 °C for 10 min, the amount of [3H]dGMP incorporated into DNA was measured as described under “Experimental Procedures.” Representative data from multiple experiments are shown.
Strand-displacement DNA Synthesis
M13 dsDNA (7.2 kb) bearing a replication fork (see Fig. 3B, inset, and Ref. 28) was used as the substrate for strand-displacement DNA synthesis. The assay contained 40 nm gp5/trx, 200 nm gp4 (monomeric concentration), 10 nm M13 dsDNA, 0.6 mm concentrations each of dATP, dTTP, dCTP, and [3H]dGTP (60 cpm/pmol), 40 mm Tris-HCl (pH 7.5), 10 mm MgCl2, 10 mm DTT, and 50 mm potassium glutamate. After incubation at 37 °C for 10 min, the reaction was terminated by the addition of EDTA to a final concentration at 40 mm, and the amount of [3H]dGMP incorporated into DNA was measured (28).
Single-molecular Measurement of Leading-strand DNA Synthesis
Phage λ dsDNA (48.5 kb) containing a replication fork was attached to the surface of a flow cell via the 5′ end of one strand through biotin-streptavidin interaction (see Fig. 4A). The 3′ end of the same strand was attached to a 2.8-μm paramagnetic bead (Dynal) through a digoxigenin-anti-digoxigenin interaction (29, 30). A 3-piconewton magnetic force was applied upward by positioning a permanent magnet above the flow cell. Beads were imaged with a digital CCD camera with a frequency of 2 times/s. The flow cell containing the tethered DNA was incubated with 20 nm gp5/trx and 120 nm gp4 (monomeric concentration, the 63- or 56-kDa gp4, or 1:1 mixture) in 40 mm Tris-HCl (pH 7.5), 10 mm DTT, 50 mm potassium glutamate, 600 μm each of dATP, dTTP, dCTP, dGTP for 15 min. After thorough washing the flow cell to remove any free proteins, DNA replication was initiated by flowing the same buffer containing additional 10 mm MgCl2 at 25 °C. Data were collected and analyzed as described previously (29, 30).
FIGURE 4.

Single-molecular measurement of leading-strand DNA synthesis. A, shown is a scheme for single molecule experiment. Phage λ dsDNA (48.5 kb) containing a replication fork was attached between the flow cell surface and a paramagnetic bead. The replication reaction contained 120 nm 63-kDa gp4 or 56-kDa gp4 or a 1:1 mixture (each at 60 nm) of the two forms of gp4, 20 nm gp5/trx in a buffer containing 600 μm concentrations each of four dNTP, 10 mm DTT, and 10 mm MgCl2. During DNA synthesis, conversion of dsDNA to ssDNA coil shortens the length of the DNA strand attached to the bead and flow cell surface resulting in movement of the bead against the flow direction. B, rate and processivity of leading-strand DNA synthesis mediated by gp5/trx and gp4 were obtained by fitting using Gaussian and exponential decay distributions, respectively. Thirty-three, 32, and 32 single events were used, respectively, for 63-kDa gp4, 56-kDa gp4, and gp4 mixture to determine the rate and processivity. S.E represent the accuracy in fitting of these distributions. Examples of trajectory for each case are shown.
Oligomerization
The assays for oligomerization of gp4 were performed as described previously (31) with minor changes. The reactions contained 1–2 μm gp4 (monomeric concentration) in 40 mm Tris-HCl (pH 7.5), 10 mm MgCl2, 10 mm DTT, and 50 mm potassium glutamate. The assays for oligomerization of gp4 in the presence of ssDNA contained 50–800 nm gp4 (monomeric concentration) or 2 μm concentrations of a mixture of 56- and 63-kDa gp4 (monomeric concentration), 2.5 mm non-hydrolyzable β,γ-methylene dTTP, and the indicated amounts of 60-mer ssDNA (radioactively labeled or non-labeled) in 40 mm Tris-HCl (pH 7.5), 10 mm MgCl2, 10 mm DTT, and 50 mm potassium glutamate. For all the assays, after incubation at 37 °C for 20 min, fresh glutaraldehyde was added to a final concentration of 0.033%. The mixture was then incubated at 37 °C for 5 min to cross-link and stabilize the protein oligomers and then analyzed on a 10% non-denaturing TBE gel using a running buffer of 0.25 × TBE at 4 °C (5, 31).
A protein mixture containing non-labeled ssDNA was stained with Coomassie Blue (32). The same amounts of a mixture of 56- and 63-kDa gp4 without cross-linking were analyzed on a Tris-HCl denaturing gel containing SDS and stained simultaneously. gp4 oligomers in the non-denaturing gel and gp4 monomers in the denaturing gel were identified, and their intensities were measured using Odyssey v3.0 (33). The percentage of gp4 monomer that was oligomerized to hexamer was calculated by dividing the intensity of the gp4 hexamer band in the non-denaturing gel by the sum of the intensities of corresponding two monomer bands in the denaturing gel.
The oligomerization of gp4 containing radioactively labeled ssDNA was carried out as described above except for the use of 32P-labeled ssDNA. After electrophoresis on the 10% non-denaturing TBE gel, the gel was subjected to autoradiography, and the individual bands corresponding to gp4 hexamer and dodecamer encircling labeled ssDNA were observed. The amount of hexamer encircling ssDNA was analyzed by comparing the intensity of the radioactively labeled ssDNA in each band with the standards using Fuji BAS 1000 Bioimaging analyzer. The amount was plotted against the hexameric gp4 concentrations and fitted to a hyperbola equation using GraphPad Prism (Version 4.0c) to determine the maximal binding amount (Bindmax) and the binding affinity of hexameric gp4 to ssDNA (KD),
where Bind is the amount of hexamer-ssDNA complex at each concentration of hexameric gp4, Bindmax is the maximal amount of hexamer-ssDNA complex, and KD is the binding affinity of hexameric gp4 to ssDNA.
Template-directed Oligoribonucleotide Synthesis
The primer synthesis assay contained 0.2 μm concentrations of a mixture of 56- and 63-kDa gp4 (monomeric concentration), 4 μm 60-mer ssDNA containing a primase recognition sequence (5′-TGGTC-3′), 1 mm ATP, 1 mm [α-32P]CTP (0.1 mCi/ml), 40 mm Tris-HCl (pH 7.5), 10 mm MgCl2, 10 mm DTT, and 50 mm potassium glutamate. After incubation at 37 °C for 30 min, the reactions were stopped by the addition of EDTA, and the products were separated on a 25% sequencing gel containing 3 m urea. After autoradiography of the gel, the amount of products was determined using Fuji BAS 1000 Bioimaging analyzer as described previously (34).
Examination of oligoribonucleotide synthesis in cis or trans mode was carried out and analyzed similar to the described above. The reactions contained 0.2 μm gp4 (monomeric concentration), 50 nm 110-mer ssDNA containing two primase recognition sequences 5′-TGGTC-3′, 300 μm ATP, 300 μm [α-32P]CTP (0.1 mCi/ml), 1 mm dTTP, 40 mm Tris-HCl (pH 7.5), 10 mm MgCl2, 10 mm DTT, and 50 mm potassium glutamate. The gp4 preparations included 56-kDa gp4, 63-kDa gp4-D237A, 63-kDa gp4, an equimolar mixture of 56-kDa gp4-D237A and 63-kDa gp4, and an equimolar mixture of 63-kDa gp4-D237A and 56-kDa gp4. After incubation at 37 °C for 20 min, the products were separated, and the amount of ACCA was measured as described above.
Primase-dependent DNA Synthesis
The primase domain of gp4 synthesizes tetraribonucleotides and then transfers them to DNA polymerase for use as primers to initiate the synthesis of Okazaki fragments. The primase-dependent DNA synthesis reaction contained 20 nm gp5/trx, 200 nm gp4 mixture (monomeric concentration), 10 nm M13 ssDNA, 0.1 mm ATP and CTP or 0.1 mm ACCA, and 0.3 mm each of dATP, dCTP, dGTP, and [3H]dTTP (10 cpm/pmol) in 40 mm Tris-HCl (pH 7.5), 10 mm MgCl2, 10 mm DTT, and 50 mm potassium glutamate. After incubation at 37 °C for 10 min, the reactions were stopped by the addition of EDTA, and the amount of [3H]dTMP incorporated into DNA was measured as described (35).
Leading- and Lagging-strand DNA Synthesis Using Minicircular dsDNA
The minicircular DNA used in these assays consists of a dsDNA circle (70 bp) and a 5′-ssDNA tail (40 nt) (26). The lagging-strand template contains two primase recognition sites (5′-TGGTC-3′). The standard reaction (25 μl) contained 600 μm each of dATP, dCTP, dGTP, and dTTP, 300 μm each of ATP and CTP, 100 nm minicircular DNA, 2 μm gp2.5, and 100 mg/ml bovine serum albumin in 40 mm Tris-HCl (pH 7.5), 10 mm MgCl2, 10 mm DTT, and 50 mm potassium glutamate. To measure the leading- or lagging-strand DNA synthesis, [3H]dGTP or [3H]dCTP (60 cpm/pmol) was, respectively, added to identical but separate reactions. The 4 μm gp5/trx and 3 μm gp4 (monomeric concentration) were mixed and preincubated at 4 °C for 5 min followed by dilution with a buffer consisting of 40 mm Tris-HCl (pH 7.5) and 1 mm DTT to yield a final concentration of 80 nm gp5/trx and of 60 nm gp4 in the reaction. DNA synthesis was initiated by adding the proteins to the reaction mixture and then incubating at 30 °C. Aliquots of 4 μl were taken at the indicated times, and the reaction was stopped by the addition of EDTA. The amount of [3H]dGMP or [3H]dCMP incorporated into DNA was measured as described (26).
Synthesis of Oligoribonucleotides and Okazaki Fragments Using Minicircular DNA
The assays for the synthesis of oligoribonucleotides and Okazaki fragments were performed under similar conditions to the leading- and lagging-strand DNA synthesis. The assays contained 80 nm gp5/trx, 60 nm 63-kDa gp4, or the mixture of 30 nm 56-kDa gp4 and 30 nm 63-kDa gp4, 100 nm minicircular dsDNA, 2 μm gp2.5, 0.3 mm ATP, 0.3 mm [α-32P]CTP (0.1 mCi/ml), 0.6 mm concentrations each of dATP, dCTP, dGTP, and dTTP in 40 mm Tris-HCl (pH 7.5), 10 mm MgCl2, 10 mm DTT, and 50 mm potassium glutamate. After incubation at 30 °C for 4 min, the reactions were stopped by the addition of EDTA, and the products were separated on a 25% sequencing gel containing 3 m urea. Each Okazaki fragment contained a radioactive pppACCA at the 5′-terminus. After autoradiography of the gel, the Okazaki fragments were stacked in the well at the top of gel in confirmation of previous results (36). All the oligoribonucleotides (AC, ACC, ACCA) and Okazaki fragments, which all contained [α-32P]CMP, were quantified by comparing the intensity of [α-32P]CMP in each band to the standards. The percentage of oligoribonucleotides used for the synthesis of Okazaki fragments was calculated.
RESULTS
In this study we have addressed the role of the 56-kDa gp4 in T7 DNA replication. The overall approach is to examine the formation and function of heterohexamers containing various ratios of two molecular weight forms of gp4. Because gp4 provides both helicase and primase activities at the replication fork, we examined both of these reactions catalyzed by hexamers of 63-kDa gp4, 56-kDa gp4, and by heterohexamers containing varying ratios of the 56- and 63-kDa gp4.
Helicase Functions of gp4DNA-dependent Hydrolysis of dTTP
Gene 4 helicase translocates unidirectionally 5′ to 3′ on the ssDNA to which it is bound, fueled by the energy of hydrolysis of dTTP (27). Each of the six subunits of the hexameric helicase hydrolyzes dTTP randomly in the absence of ssDNA, albeit at a very slow rate. Upon binding to ssDNA, the rate of hydrolysis of dTTP is increased by ∼50-fold (18). In the experiment shown in Fig. 2, various ratios of the two molecular weight forms of gp4 were examined for DNA-dependent hydrolysis of dTTP. In the presence of dTTP the hexamer of 63-kDa gp4 translocates on ssDNA at a rate of 132 bases per second at 18 °C (37). DNA-dependent hydrolysis catalyzed by the 56-kDa gp4 is slightly higher than that catalyzed by the 63-kDa gp4 when measured on either M13 ssDNA (Fig. 2A) or a 60-mer oligonucleotide (Fig. 2B). Heterohexamers of gp4 had intermediate levels of dTTP hydrolysis that increased as the amount of 56-kDa gp4 in the hexamer increased (Fig. 2).
FIGURE 2.
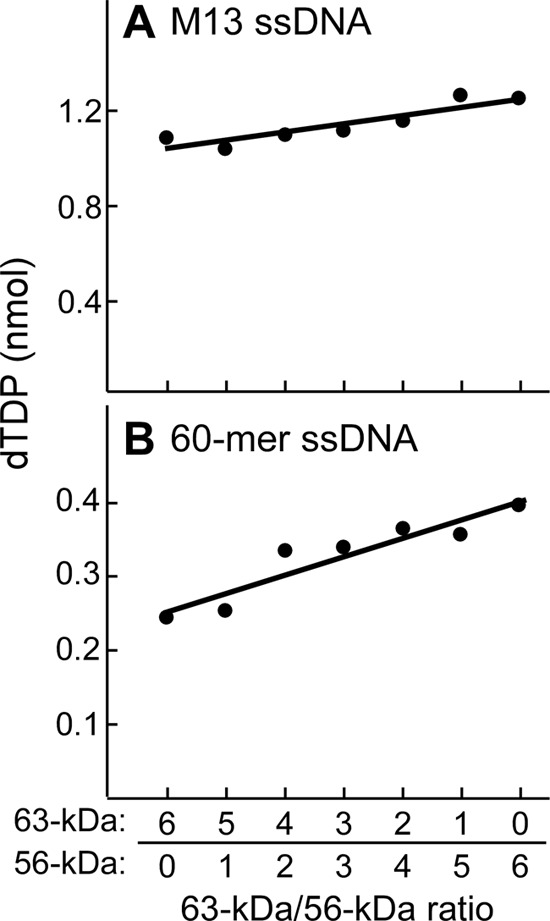
DNA-dependent dTTP hydrolysis. dTTP hydrolysis activity was measured by incubation of 200 nm gp4 (monomeric concentration) and 10 nm M13 ssDNA (A) or 1 μm 60-mer ssDNA (B) and 0.3 mm [α-32P]dTTP (0.1 μCi) in a buffer containing 40 mm Tris-HCl (pH 7.5), 10 mm MgCl2, 10 mm DTT, and 50 mm potassium glutamate at 37 °C for 20 min. gp4 consisted of a mixture of 56- and 63-kDa gp5 in molar ratios ranging from 6:0 to 0:6. The reaction products were separated by TLC, and dTTP hydrolysis was determined by measuring the amount of [α-32P]dTDP produced from the reaction. The product amount was fit to a linear line against the ratios of the 63- to 56-kDa gp4. Representative data from multiple experiments are shown.
DNA Unwinding and Strand-displacement DNA Synthesis
Unwinding of dsDNA by gp4 was measured using a DNA construct resembling a replication fork (Fig. 3A, inset). The assay measures the release of a radioactively labeled oligonucleotide partially annealed to a complementary ssDNA. In the unwinding assays (Fig. 3A), the total molar amount of the 63- and 56-kDa gp4 was kept constant, but their ratios were varied from 6:0 to 0:6, respectively. The 56-kDa gp4 had a 4-fold higher activity than that observed with the 63-kDa gp4 (Fig. 3A). All mixtures of the two proteins displayed higher activity than that observed with the 63-kDa gp4 alone.
T7 gp5/trx cannot polymerize nucleotides through duplex DNA and thus requires the helicase activity of gp4 to unwind the DNA to expose an ssDNA template. To measure the strand-displacement synthesis mediated by gp5/trx and gp4, we have used circular M13 dsDNA containing a 5′-ssDNA tail onto which the helicase can load (Fig. 3B, inset). Strand-displacement synthesis mimics leading-strand DNA synthesis. Again, the 56-kDa gp4 yielded a 4-fold higher activity than that observed with the 63-kDa gp4; any mixture of the two proteins also exhibited higher activity than the 63-kDa gp4 alone. To exclude the possibilities that these results were due to some difference in the proteins arising from purification, we have repeated these experiments using several different preparations of the 56- and 63-kDa gp4. Results essentially identical to those described here were obtained. We have also varied the protein and DNA concentrations as well as the temperature. Similar results were obtained (supplemental Fig. S1).
If both molecular weight forms of gp4 mediate unwinding and strand-displacement synthesis independently, the activity expected from a mixture of the two proteins would be the sum of the two individual activities, as represented by the dashed lines in Fig. 3. However, the results reveal a nonlinear increase in activity in which the activity is greater than that predicted by a mixture of the two proteins. These results clearly show that heterohexamers are formed as only hexamers bind to DNA and provide for DNA unwinding. Thus, these results are in agreement with earlier studies described in the Introduction that have revealed the assembly of heterohexamers (17–20).
Compared with the 63-kDa gp4, the 56-kDa gp4 and any mixture of the two proteins shows higher activities in unwinding and strand-displacement DNA synthesis. There are two possible reasons for the higher activities of gp4 containing the 56-kDa gp4. (i) A single 56-kDa gp4 hexamer or a single heterohexamer containing both forms of gp4 subunits has an intrinsic activity higher than a single 63-kDa gp4 hexamer. This possibility can be examined by determination of the helicase activity of a single gp4 hexamer in leading-strand DNA synthesis with gp5/trx by single-molecular measurement. (ii) The 56-kDa gp4 or the mixture of the two forms of gp4 can oligomerize to more functional hexamers than the 63-kDa gp4. This possibility can be addressed by measuring the oligomerization of each gp4 mixture.
Single-molecular Measurement of Leading-strand DNA Synthesis
We have used single-molecular measurement (29, 30) to examine whether a single 56-kDa gp4 hexamer or any single heterohexamer containing both gp4 subunits has higher activity and longer processivity in leading-strand DNA synthesis than a single 63-kDa gp4 hexamer (Fig. 4A). Bacteriophage λ duplex DNA containing a replication fork was attached between the flow cell surface and a bead. A constant flow yielded a 3-piconewton force on the DNA molecule, a force that does not influence the protein-DNA interactions (29). The 56-kDa gp4, 63-kDa gp4, or a 1:1 mixture of the two forms together with gp5/trx was flowed over the DNA in a buffer containing the four dNTP without Mg2+ to assemble gp4 and gp5/trx onto the replication fork. After removal of the free proteins by continuing the flow of buffer, DNA synthesis was initiated by flowing buffer containing the four dNTPs and Mg2+. Leading-strand DNA synthesis progressively converted dsDNA to ssDNA. The resulting ssDNA attached to the flow cell surface was less extended than dsDNA, resulting in the movement of the bead toward the flow source (Fig. 4A). The distance the bead moved reflects the number of nucleotides in the ssDNA and, hence, the amount of DNA synthesis (29).
Leading-strand DNA synthesis mediated by gp5/trx and the 63-kDa gp4 hexamer proceeds at a rate of 110 ± 10 nt/s with a processivity of 14 ± 2 kb (Fig. 4B), values similar to previous results (19). The 56-kDa gp4 hexamer and gp5/trx proceed at a rate of 80 ± 7 nt/s with a processivity of 11 ± 1 kb. gp5/trx and a 1:1 mixture of the 56- and 63-kDa gp4 mediates leading-strand DNA synthesis at a rate of 87 ± 5 nt/s with a processivity of 11 ± 2 kb. The three rates of DNA synthesis and the processivity are similar, values that reflect the activity of a single hexamer. The results show that all three forms of gp4 have similar helicase activity in leading-strand DNA synthesis.
Oligomerization of 56- and 63-kDa gp4
Unwinding of DNA and strand-displacement synthesis mediated by gp4 is dependent on the oligomerization of gp4 to form the functional hexamer. The single-molecular measurements presented above show heterohexamers have the same activity as hexamers of either molecular weight form alone. Therefore, the higher activity of leading-strand DNA synthesis observed in the ensemble experiments for the 56-kDa gp4 or for the mixture of 56- and 63-kDa gp4 may reflect their ability to oligomerize more efficiently than the 63-kDa gp4.
The oligomerization of the 63- or 56-kDa gp4 was examined by the mobility of the proteins on a non-denaturing gel (Fig. 5A). Before electrophoresis, the oligomers were stabilized by cross-linking with glutaraldehyde (31). The 63-kDa gp4 oligomerized to approximately equal amounts of hexamers and heptamers as previously described (31). The 56-kDa gp4, however, formed hexamers exclusively. The amount of hexamer was ∼3-fold higher for the 56-kDa gp4 than that found for the 63-kDa gp4 (Fig. 5B). We previously showed that hexamers are the functional form of gp4 (31). The 63-kDa gp4 heptamer does not bind ssDNA; only hexamers bind to ssDNA (31). A higher molecular weight complex, dodecamer (two hexamers) encircling ssDNA, is also observed, although in much less amounts (31). In the experiment presented in Fig. 5C we examined the oligomerization of the 56- and 63-kDa gp4 in the presence of radioactively labeled ssDNA and non-hydrolyzable β,γ-methylene dTTP (38). After autoradiography of the gel containing the protein, two bands containing radioactively labeled DNA were observed (Fig. 5C). The major band migrating more rapidly corresponds to the hexamer containing ssDNA, whereas the slower migrating minor band represents the dodecamer bound to ssDNA, in agreement with previous results (31). The intensities of the hexamer encircling ssDNA were measured and plotted against the concentrations of hexameric gp4 and then fit to a hyperbola equation to determine the maximal binding amount and the binding affinity (Fig. 5D). The binding affinities of the 56- and 63-kDa gp4 to ssDNA are almost identical. Both molecular weight forms of gp4 have the same helicase domain and, therefore, would be expected to bind to ssDNA in a similar fashion. On the other hand, the 56-kDa gp4 formed at least 3-fold more hexamer-ssDNA complexes than did the 63-kDa gp4, most likely due to the more efficient oligomerization into hexamers in the absence of DNA (Fig. 5B).
FIGURE 5.

Oligomerization and DNA binding of gp4. A, oligomerization of gp4 in the absence of ssDNA is shown. Oligomerization reactions (31) contained various concentrations (1–2 μm, monomeric concentration) of 63- or 56-kDa gp4 in 40 mm Tris-HCl (pH 7.5), 10 mm MgCl2, 10 mm DTT, and 50 mm potassium glutamate. After incubation at 37 °C for 20 min, glutaraldehyde was added and incubated for 5 min to stabilize any oligomers present. The oligomeric forms of proteins were analyzed on non-denaturing TBE gels, and the proteins were stained as described under “Experimental Procedures.” The positions for hexamer and heptamer are indicated. B, the relative amount of hexamer formed from 56- or 63-kDa gp4 was measured and plotted against the total protein concentrations. C, oligomerization of gp4 in the presence of ssDNA is shown. Oligomerization reactions (31) contained varying concentrations (50–400 nm, monomeric concentration) of 63- or 56-kDa gp4, 50 nm 60-mer 5′ 32P-labeled ssDNA, and 2.5 mm β,γ-methylene dTTP in 40 mm Tris-HCl (pH 7.5), 10 mm MgCl2, 10 mm DTT, and 50 mm potassium glutamate. After incubation at 37 °C for 20 min, glutaraldehyde was added to cross-link and stabilize any oligomers present. The oligomeric proteins and ssDNA complex were analyzed on non-denaturing TBE gels, and the gel was subjected to autoradiography as described under “Experimental Procedures.” The positions of gp4-ssDNA complexes are indicated. D, the amount of hexamer-ssDNA complex was measured by comparing the intensity of the radioactively labeled ssDNA in each band with the standards, plotted against gp4 concentration, and fit to the hyperbola equation to determine maximal binding amount (Bindmax) and binding affinity (Kd).
Generally, the stability of a protein complex is affected by the ionic strength. We examined the formation of hexamers in the presence of ssDNA and β,γ-methylene dTTP at various concentrations of NaCl (0–1 M) (supplemental Fig. S2). The 56-kDa gp4 formed only hexamers at all concentrations of NaCl (supplemental Fig. S2A). The 63-kDa gp4 formed hexamers, but heptamers were also observed at higher NaCl concentrations. The 56-kDa gp4 formed more hexamer than did the 63-kDa gp4 at all concentrations of NaCl. The amounts of hexamer, heptamer, and dodecamer were quantified using Odyssey v3.0 (33) and plotted against the NaCl concentrations (supplemental Fig. S2B). The maximal amount of hexamers was observed at ∼0.4 m NaCl for both 56- and 63-kDa gp4, suggesting that both forms of gp4 are affected similarly by ionic strength.
Oligomerization of a Mixture of 56- and 63-kDa gp4
Both 56-kDa and 63-kDa gp4 contain the elements necessary for oligomerization. However, the difference in the oligomerization between the 63- and 56-kDa gp4 indicates that other factors also affect oligomerization. The obvious difference in the two molecular weight forms is the absence of the ZBD on the 56-kDa gp4, the form that more readily oligomerizes into the functional hexamer. What effect will the 56-kDa gp4 have on the formation of a heterohexamer containing both molecular weight forms? We have examined the oligomerization of a mixture of 56- and 63-kDa gp4 in the presence of ssDNA and β,γ-methylene dTTP (Fig. 6). The total molar amount of the two molecular weight forms of gp4 was kept constant, but their molar ratios were varied from 6:0 to 0:6. Consequently, the ratio of ZBD in one hexamer varied from 0 to 6. After mixing the two molecular weight forms, the mixtures were incubated with glutaraldehyde to cross-link and stabilize the oligomers that formed. The oligomerization state of the proteins was then analyzed on a non-denaturing TBE gel (Fig. 6A). The identical mixture of gp4 without cross-linking was also electrophoresed through a denaturing SDS gel (Fig. 6B). Proteins were stained with Coomassie Blue. The amount of hexameric gp4 derived from the intensity of the hexamer band in the non-denaturing gel (Fig. 6A) and the total amount of gp4 calculated from the sum of the intensities of the monomer bands in the denaturing gel (Fig. 6B) were quantified using Odyssey v3.0 (33) (Fig. 6, C and D), respectively. The percentage of gp4 that oligomerized to hexamer was calculated by dividing the amount of the hexameric gp4 by the corresponding total amount of gp4 (Fig. 6E). The percentage increased from ∼5 to 25% as the ratio of 56-kDa gp4 to 63-kDa gp4 went from 0:6 to 4:2. The percentage remained constant at 25% as the amount of 56-kDa gp4 in the hexamer was increased to 6:0.
FIGURE 6.

Oligomerization and DNA binding of gp4 mixture. A, the oligomerization assay for gp4 contained 2 μm gp4 (monomeric concentration), 2.5 mm β,γ-methylene dTTP, and 5 μm 60-mer ssDNA in 40 mm Tris-HCl (pH 7.5), 10 mm MgCl2, 10 mm DTT, and 50 mm potassium glutamate. gp4 consisted of a mixture of 56- and 63-kDa gp4 in molar ratios ranging from 6:0 to 0:6, respectively. After incubation at 37 °C for 20 min, the oligomers were cross-linked with glutaraldehyde and analyzed on a 10% non-denaturing TBE gel. The proteins were stained with Coomassie Blue. The positions for hexamer and dodecamer are indicated. B, the same amount of mixtures of 56- and 63-kDa gp4 without cross-linking was analyzed on a denaturing gel. The proteins were stained with Coomassie Blue. The positions for 63- and 56-kDa gp4 monomers are indicated. C, the intensity of the hexamer band in A was measured and plotted as a function of the ratios of 63–56-kDa gp4 in the mixture. D, the intensities of the monomer bands in B were measured and plotted as a function of 63–56-kDa gp4 in the mixture. The intensity of both monomers was added, and the total intensity is also plotted as a function of the ratios of 63–56-kDa gp4. E, the percentage of the gp4 that form hexamer was calculated by dividing the intensity of hexamer in C by the total intensities of the 63- and 56-kDa gp4 monomers in D. F, oligomerization of gp4 mixtures on ssDNA were performed as in A except for using 5′ 32P-labeled ssDNA instead of non-labeled ssDNA. After cross-linking and analysis on a non-denaturing TBE gel, the gel was subjected to autoradiography. The locations of the gp4 hexamer or dodecamer containing radioactively labeled ssDNA complex are indicated. G, the amount of hexamer encircling ssDNA was analyzed by comparing the intensity of the radioactively labeled ssDNA in each band with the standards. The percentage of gp4 that forms hexamers was calculated by dividing the amount of hexamer-labeled ssDNA complex by the total amount of gp4 in the mixture. The percentages are plotted as a function of the ratios of 63- to 56-kDa gp4. Representative data from multiple experiments are shown.
To examine the formation of gp4 hexamer encircling ssDNA, similar assays were carried out using radioactively labeled ssDNA instead of non-labeled ssDNA (Fig. 6F). Because only hexamer but not heptamer encircles ssDNA (31), hexamer-ssDNA and dodecamer-ssDNA complexes were formed. The amount of the hexamer-ssDNA complex was measured by comparing the amount of the radioactively labeled ssDNA in the band with standards. The percentage of gp4 that oligomerized to form the hexamer-ssDNA complex was then calculated by dividing the amount of the hexamer-ssDNA complex by the total amount of gp4 in the reaction and plotted against the ratios of the two molecular weight forms of gp4 (Fig. 6G). The percentage of hexamer formed increased from 4 to 15% as the amount of 56-kDa gp4 within the hexamer increased similar to hexamer formation in the absence of DNA (Fig. 6E).
Oligomerization of a mixture of 56- and 63-kDa gp4 in the absence of ssDNA and β,γ-methylene dTTP yielded a similar curve (supplemental Fig. S3). Oligomerization of a mixture of the two forms of gp4 in the presence of ssDNA and β,γ-methylene dTTP also showed a similar trend even at higher ionic strength (0.5 m NaCl) (supplemental Fig. S4). However, in the latter case more hexamer but less hexamer-ssDNA complex was formed compared with those at lower ion strength.
All the oligomerization assays show that the percentage of oligomers formed increases as the amount of 56-kDa gp4 subunits within the heterohexamer increases from a ratio of 0:6 to ∼4:2. However, a further increase in the ratio of 56-kDa gp4 to 63-kDa gp4 does not yield an increase in hexamer formation. These results do not match a proposed linear line connecting the percentages of the 63- and 56-kDa gp4. The lack of a liner relationship between the amount of hexamer formation and the ratio of the two molecular weight forms of gp4 suggests that the maximal oligomerization occurs at roughly equimolar amounts of the two forms of gp4. The oligomerization curves with respect to the ratio of 56- to 63-kDa are similar to the curves observed in DNA unwinding and strand-displacement DNA synthesis with different ratios of the two proteins (Fig. 3). We conclude that the various activities of strand-displacement DNA synthesis for the different mixtures of 56- and 63-kDa gp4 reflects the concentration of functional hexamers in solution with maximal hexamer formation occurring at equimolar amounts of the 56- and 63-kDa forms.
Primase Functions of gp4
Oligoribonucleotides are synthesized more efficiently by the primase within hexameric gp4 than by gp4 monomers (39). The enhanced activity arises from the high affinity of the hexamer to ssDNA and its ability to translocate along ssDNA to access primase recognition sites. The latter function is particularly important on long DNA molecules (1, 16, 17). As shown in the previous section, heterohexamers consisting of the 56- and 63-kDa gp4 are formed more readily than are hexamers of the 63-kDa gp4 alone. Consequently, more hexamers from a mixture of the two molecular weight forms of gp4 could be more efficient in the synthesis of oligoribonucleotides. A second consideration involving the primase activity is the relative use of the cis and trans mode of oligoribonucleotide synthesis by a heterohexamer. We have previously shown that the ZBD on one subunit of a hexamer can contact the RPD on an adjacent subunit for the template-directed synthesis of oligonucleotides (20). In the heterohexamer the 56-kDa gp4 subunits could function by supplying the RPD for a trans mode synthesis of oligoribonucleotides using the ZBD of an adjacent 63-kDa gp4. We have examined oligoribonucleotide synthesis catalyzed by heterohexamers of gp4 and measured the relative use of the cis and trans modes of synthesis.
Oligoribonucleotide Synthesis by Heterohexamers of gp4
To examine the synthesis of oligoribonucleotides by heterohexamers of gp4, template-directed oligoribonucleotide synthesis was examined using mixtures of gp4 in which the ratio of 63- to 56-kDa gp4 ranged from 6:0 to 0:6 (Fig. 7A). The products pppAC, pppACC, and pppACCA were observed for all gp4 preparations except for the 56-kDa gp4. The amount of the functional pppACCA synthesized was measured and plotted against the ratios of the two molecular weight forms of gp4 (Fig. 7B). The amount of tetraribonucleotides decreased with the decrease in the ratio of the 63- to 56-kDa gp4 from 6:0 to 4:2 and then drastically decreased from 4:2 to 0:6. As anticipated, no synthesis was observed with 56-kDa gp4 exclusively.
FIGURE 7.
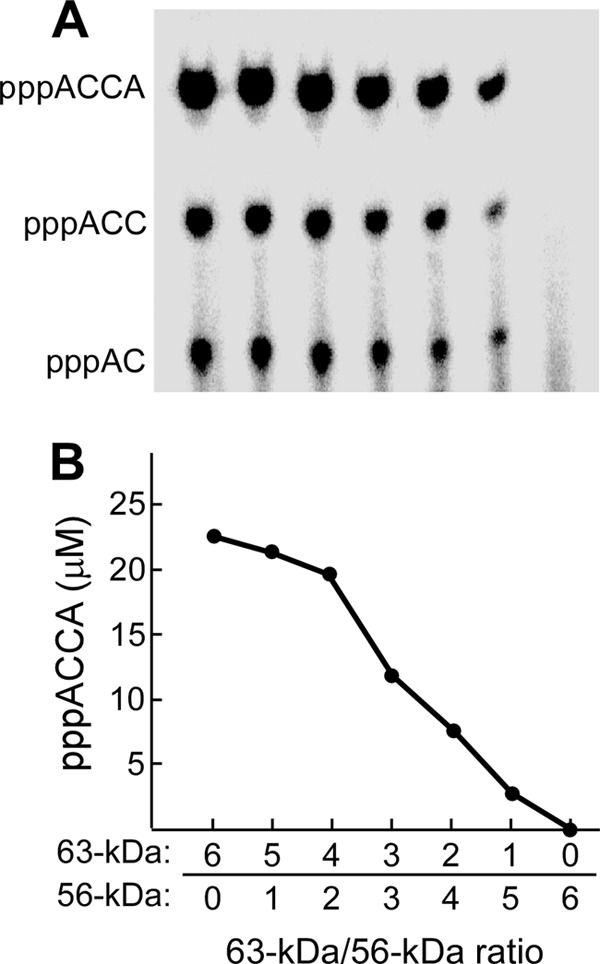
Oligoribonucleotide synthesis. A, oligoribonucleotide synthesis by gp4 mixture was measured in a reaction containing 4 μm 60-mer ssDNA with a primase recognition site, 5′-TGGTC-3′, 0.2 μm gp4 (monomeric concentration), 1 mm ATP, and 1 mm [α-32P]CTP (0.1 mCi/ml) in 40 mm Tris-HCl (pH 7.5), 10 mm MgCl2, 10 mm DTT, and 50 mm potassium glutamate. gp4 consisted of a mixture of 56- and 63-kDa gp4 in molar ratios ranging from 6:0 to 0:6, respectively. After incubation at 37 °C for 30 min, the products were separated as described under “Experimental Procedures.” B, the functional product (pppACCA) was measured and plotted against the ratios of the two molecular weight forms of gp4 in the reaction.
Cis and Trans Mode of Oligoribonucleotide Synthesis Catalyzed by Heterohexamers
Primer synthesis requires the interaction of the ZBD and RPD with the DNA template and NTPs. As described above, the ZBD and RPD from two adjacent gp4 subunits within a hexamer can synthesize oligoribonucleotides in a trans mode (20). The length of the linker between the ZBD and RPD affects oligoribonucleotide synthesis (23). These latter studies suggested that the de novo synthesis of diribonucleotides occurs in the trans mode but that their extension to the functional tetraribonucleotide occurs in the cis mode. In this study we have examined the two modes of oligoribonucleotide synthesis using heterohexamers containing equimolar amounts of the 56- and 63-kDa gp4. In a set in which primer synthesis occurs only in trans mode, equimolar amounts of the 63-kDa gp4-D237A and 56-kDa gp4 were used to prepare heterohexamers. The 63-kDa gp4-D237A has aspartate 237 replaced with alanine and, as a result, is unable to catalyze the synthesis of phosphodiester bonds (23). The 56-kDa gp4 is also unable to catalyze DNA-dependent oligoribonucleotide synthesis as it lacks a ZBD. Any oligoribonucleotide synthesis must, therefore, arise by an interaction of the ZBD of the 63-kDa gp4 with the RPD of an adjacent 56-kDa gp4 subunit within the heterohexamer. The cis mode was favored using a heterohexamer containing an equimolar amount of 56-kDa gp4-D237A and 63-kDa gp4. If the two molecular weight forms alternate within the hexamer, then synthesis is restricted to the cis mode. However, the only way to assure only the cis mode is to have one 63-kDa gp4 subunit within a hexamer. The mixtures of the various 56- and 63-kDa gp4, as expected, formed heterohexamers in the presence of ssDNA and β,γ-methylene dTTP (supplemental Fig. S5).
To compare the cis and trans mode of template-directed oligoribonucleotides synthesis by heterohexamers of gp4, we used a lower concentration of ssDNA (50 nm) (20). As expected, neither the 56-kDa gp4 nor the 63-kDa gp4-D237A alone catalyzed the synthesis of oligoribonucleotides (Fig. 8, lanes 1 and 2). In contrast, both the 63-kDa gp4 and the equimolar mixture of the 56-kDa gp4-D237A and 63-kDa gp4 catalyzed the synthesis of tetraribonucleotides (Fig. 8, lanes 3 and 4), but the latter mixture had approximately one-third that of the activity compared with the 63-kDa gp4. An equimolar mixture of the 56-kDa gp4 and the 63-kDa gp4-D237A did catalyze the synthesis of tetraribonucleotides but at only 2% that of the rate of the 63-kDa gp4 (Fig. 8, lanes 5 and 6). We conclude that the major contribution to oligoribonucleotide synthesis by a hexamer occurs in a cis mode.
FIGURE 8.
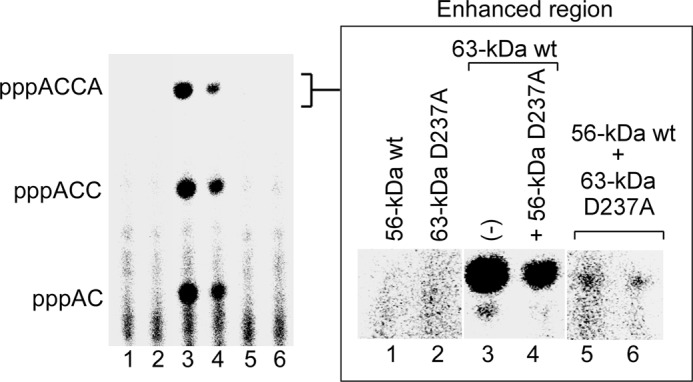
Cis and trans mode of oligoribonucleotide synthesis. Reactions for oligoribonucleotide synthesis contained 0.2 μm gp4 (monomeric concentration), 50 nm 110-mer ssDNA containing the primase recognition sequence 5′-TGGTC-3′, 300 μm ATP and [α-32P]CTP (0.1 mCi/ml), and 1 mm dTTP in 40 mm Tris-HCl (pH 7.5), 10 mm MgCl2, 10 mm DTT, and 50 mm potassium glutamate. The gp4 used in the reaction included the 56-kDa gp4 (lane 1), the 63-kDa gp4-D237A (lane 2), the 63-kDa gp4 (lane 3), an equimolar mixture of the 56-kDa gp4-D237A and 63-kDa gp4 (lane 4), and an equimolar mixture of 63-kDa gp4-D237A and 56-kDa gp4 (lanes 5 and 6). These 63-kDa gp4-D237A and 56-kDa in lanes 5 and 6 were prepared independently. After incubation at 37 °C for 20 min, the products were separated, and the amount of ACCA was measured. The enhanced region of pppACCA is shown to the right.
Primase-dependent DNA Synthesis
In the absence of exogenous DNA primer, DNA synthesis catalyzed by gp5/trx is dependent on the primase activity of gp4. The ability of heterohexamers of gp4 to mediate primase-dependent DNA synthesis is presented in Fig. 9A. T7 DNA primase can also bind and deliver preformed tetraribonucleotides complementary to a primase recognition site to gp5/trx (34, 40). The tetraribonucleotide 5′-ACCA-3′ was used as a primer to initiate DNA synthesis in the presence of gp4 (Fig. 9B). Both the de novo synthesis of primers and the utilization of the preformed tetraribonucleotides behaved similarly with the heterohexamers of gp4. The amount of DNA synthesis decreased with a decrease in the ratio of the 63- to 56-kDa gp4 from 6:0 to 3:3 and then drastically decreased from 3:3 to 0:6, with essentially no synthesis when only 56-kDa gp4 was present.
FIGURE 9.
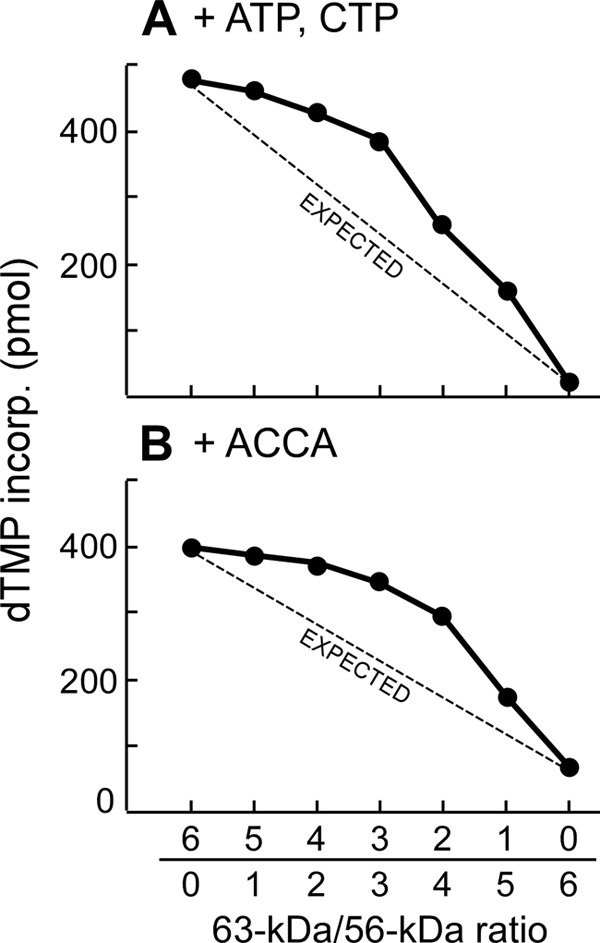
Primase-dependent DNA synthesis. A, the assay for primase-dependent DNA synthesis contained 0.2 μm gp4 (monomeric concentration), 20 nm gp5/trx, 10 nm M13 ssDNA, 0.1 mm ATP, and 0.1 mm CTP, and 0.3 mm concentrations each of dATP, dCTP, dGTP, and [3H]dTTP (10 cpm/pmol) in 40 mm Tris-HCl (pH 7.5), 10 mm MgCl2, 10 mm DTT, and 50 mm potassium glutamate. gp4 consisted of a mixture of 56- and 63-kDa gp4 in molar ratios ranging from 6:0 to 0:6, respectively. After incubation at 37 °C for 10 min, the amount of [3H]dTMP incorporated into DNA was measured as described under “Experimental Procedures.” B, the reactions were performed as in A except for using 0.1 mm ACCA instead of 0.1 mm ATP and CTP.
The Replication Fork
In earlier studies we used a synthetic minicircle to examine leading- and lagging-strand DNA synthesis with a reconstituted replisome (26). The minicircle, used in Fig. 10, consists of a dsDNA circle (70 bp) bearing a 5′-ssDNA tail (40 nt) onto which gp4 can assemble. The lagging-strand template contains the primase recognition site 5′-TGGTC-3′. Because the minicircle is chemically synthesized, we have dictated the base composition so that leading-strand synthesis can be monitored by the incorporation of radioactively labeled dGMP and lagging-strand synthesis by the incorporation of radioactively labeled dCMP (26). The leading strand is synthesized continuously to yield products >10 kb in length, and the lagging strand is synthesized discontinuously to yield Okazaki fragments with an average length of 2–3 kb (6). In these earlier studies the 63-kDa gp4 was used exclusively.
FIGURE 10.

Leading- and lagging-strand DNA synthesis mediated by gp5/trx and gp4 heterohexamers. Leading (blue)- and lagging (red)-strand synthesis were measured using the minicircle (26). The reaction contained 100 nm minicircle DNA, 80 nm gp5/trx, 60 nm gp4 (monomeric concentration), 100 mg/ml bovine serum albumin, 600 μm concentrations each of dATP, dCTP, dGTP, and dTTP, 300 μm concentrations each of ATP and CTP, and 2 μm gp2.5 in 40 mm Tris-HCl (pH 7.5), 10 mm MgCl2, 10 mm DTT, and 50 mm potassium glutamate. gp4 consisted of a mixture of 56- and 63-kDa gp4 in molar ratios ranging from 6:0 to 0:6 (panels A–G), respectively. After incubation at 30 °C for an indicated time, aliquots were removed, and the was reaction stopped by the addition of EDTA. The [3H]dGMP incorporated in the leading-strand DNA and the [3H] dCMP in the lagging-strand DNA were measured in identical but separate reactions as described under “Experimental Procedures.” Representative data from multiple experiments are shown. H, the rates and their S.D. of leading- and lagging-strand DNA synthesis were calculated based on the line slopes in (A–G) and plotted against the ratios of the two molecular weight forms of gp4.
Leading- and Lagging-strand DNA Synthesis
In the experiment shown in Fig. 10, the reactions contained minicircular dsDNA, hexameric gp4, gp5/trx, and gp2.5 at 100, 10, 80, and 2000 nm, respectively (6). gp4 consisted of a mixture of 56- and 63-kDa gp4 in molar ratios ranging from 6:0 to 0:6, respectively. Leading- and lagging-strand DNA synthesis were monitored for the first 5 min of the reaction during which the minicircle was replicated hundreds of times based on the product sizes of the leading-strand DNA synthesis (6). The amount of DNA synthesis on either strand was measured and plotted against the reaction time for each ratio of the two forms of gp4 (Fig. 10, A–G). The rates of DNA synthesis were calculated based on the slope of the line and were compared at each ratio of the two forms in the gp4 hexamer (Fig. 10H).
In the case of the 63-kDa gp4 alone, leading- and lagging-strand DNA synthesis were coordinated as judged by their similar rates of synthesis, in confirmation of previous results (6, 26) (Fig. 10A). The heterohexamers containing both the 63- and 56-kDa gp4 at ratios of 5:1 to 3:3 also showed coordinated DNA synthesis (Fig. 10, A–D). However, the rates of DNA synthesis gradually increased as the content of the 56-kDa gp4 increased. At ratios of 2:4 to 0:6, the rates of the lagging strand decreased significantly, and synthesis was no longer coordinated (Fig. 10, E–G). Not surprisingly, no lagging-strand synthesis was observed in the absence of 63-kDa gp4 (Fig. 10G). Overall, heterohexamers containing around equimolar amounts of the 56- and 63-kDa gp4 showed the highest rate of DNA synthesis with coordinated synthesis of the two strands (Fig. 10H).
Utilization of Oligoribonucleotides by gp5/trx during Coordinated Leading- and Lagging-strand DNA Synthesis
In the above experiments with the minicircle, it is remarkable that the initiation of Okazaki fragments occurs only every 2000–3000 thousand nucleotides as the replisome passes over the two primase recognition sites every 70 nucleotides. Therefore, the question arises as to whether oligoribonucleotides are synthesized in excess during passage of primase recognition sites, or perhaps oligoribonucleotides are only synthesized upon use by gp5/trx. We have used the minicircle replication system to address this question.
In the experiment shown in Fig. 11, we have used [α-32P]CTP to monitor the synthesis of oligoribonucleotides at the primase recognition sequence 5′-TGGTC-3′ present in the minicircle. At this site the functional tetraribonucleotide pppACCA is synthesized and thus will be radioactively labeled as a result of the incorporated [α-32P]CMP. Oligoribonucleotides synthesized but not used by gp5/trx can be detected as well as those located at the 5′-termini of Okazaki fragments. The products, both radioactively labeled oligoribonucleotides and radioactively labeled Okazaki fragments were separated on a 25% polyacrylamide gel (Fig. 11). The larger Okazaki fragments, as anticipated from earlier studies (36), remained in the well at the top of each lane.
FIGURE 11.
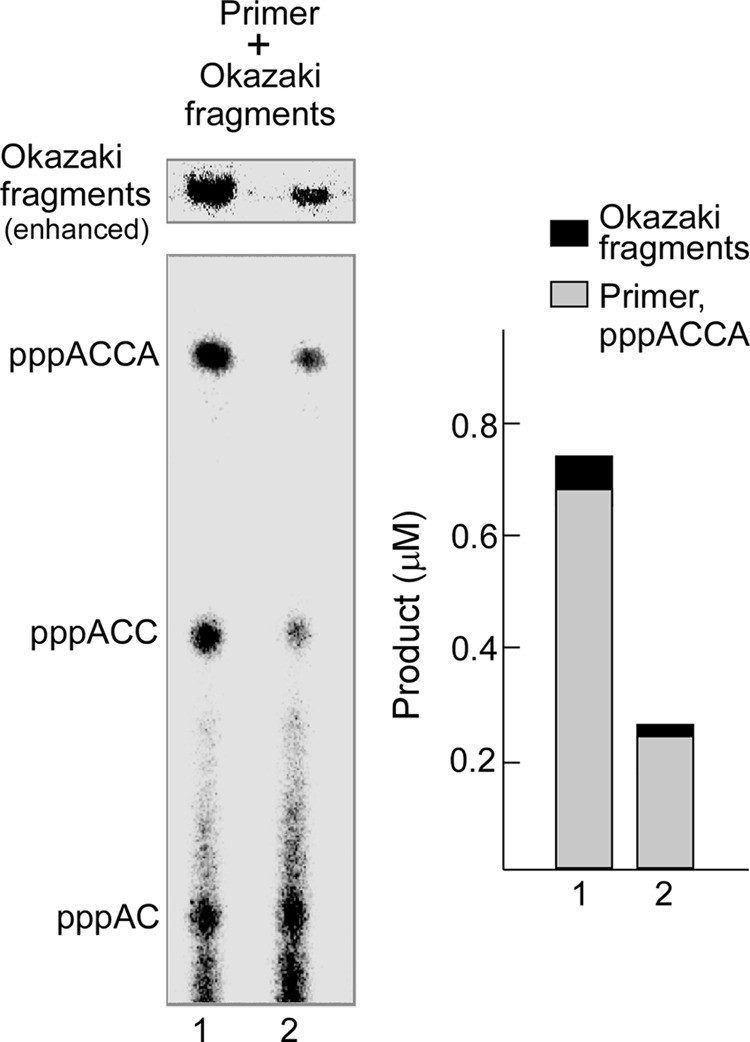
Utilization oligoribonucleotides by gp5/trx during coordinated leading- and lagging-strand DNA synthesis. Left, lane 1, the assay for synthesis of oligoribonucleotides and Okazaki fragments by 63-kDa gp4 and gp5/trx contained 80 nm gp5/trx, 60 nm 63-kDa gp4, 100 nm minicircle, 2 μm gp2.5, 0.3 mm ATP, 0.3 mm [α-32P]CTP (0.1 mCi/ml), 0.6 mm dATP, dCTP, dGTP, and dTTP in 40 mm Tris-HCl (pH 7.5), 10 mm MgCl2, 10 mm DTT, and 50 mm potassium glutamate. After incubation at 30 °C for 4 min, the products were separated as described under “Experimental Procedures.” Lane 2, synthesis of oligoribonucleotides and Okazaki fragments by 63- and 56-kDa gp4 is shown. The reaction was performed as in lane 1 except that 30 nm 63-kDa gp4 and 30 nm 56-kDa gp4 replaced 60 nm 63-kDa gp4. The oligoribonucleotides and Okazaki fragments are denoted to the right. The Okazaki fragment region was amplified. Right, the amount of tetraribonucleotides and Okazaki fragments was measured by comparing the intensity of [α-32P]CMP in each band to the standards. The percentage of the oligoribonucleotides used for the synthesis of Okazaki fragments was calculated.
Using the 63-kDa gp4 (lane 1), the amount of Okazaki fragments in the well at the top of gel was 0.056 μm, whereas the tetraribonucleotide pppACCA synthesized but not used by gp5/trx was 0.69 μm. Thus, only about 7.5% of the pppACCA was used by gp5/trx for the synthesis of Okazaki fragments. The same total molar amount of a 1:1 mixture of 63- and 56-kDa gp4 (lane 2) produced 0.018 μm pppACCA in Okazaki fragments and 0.24 μm free pppACCA. Both of these amounts were ∼3-fold less compared with those for the 63-kDa gp4 alone, whereas the percentage of the utilization of ACCA for Okazaki fragment synthesis remained approximately the same (6.9%). The percentages of pppACCA used for the synthesis of Okazaki fragments were similar and independent of the presence or absence of the 56-kDa gp4. The amount of Okazaki fragments synthesized is proportional to the amount of tetraribonucleotide synthesized, a value that is in turn dependent on the number of 63-kDa gp4 subunits in a hexamer. Compared with the 63-kDa gp4, an equimolar amount of 63- and 56-kDa gp4 is more efficient in lagging-strand DNA synthesis (Fig. 10) but produces fewer Okazaki fragments (Fig. 11). These results indicate that Okazaki fragments are longer than those produced by hexamers of the 63-kDa gp4. Alkaline agarose gel analysis of the length of Okazaki fragments confirmed these results (supplemental Fig. S6).
DISCUSSION
Because the 63-kDa gp4 is sufficient to support the growth of bacteriophage T7 (9), what is the advantage for T7 to express the additional 56-kDa gp4? Over the years many hypotheses have been put forth (9, 11, 41, 42) only to be eliminated as additional biochemical and structural information became available. Here we show that the 56-kDa gp4 and any mixture of the 56- and 63-kDa gp4 is more active than the 63-kDa gp4 in the unwinding of DNA and in mediating strand-displacement synthesis together with gp5/trx. A single molecule analysis of individual hexamer of gp4 revealed that each of the single hexamer consisting of 56-kDa gp4, 63-kDa gp4, or heterohexamer containing both molecular weight forms mediates strand-displacement synthesis with gp5/trx at equal rates and processivity. The 56-kDa gp4 oligomerizes more efficiently than does the 63-kDa gp4 as do heterohexamers containing 56- and 63-kDa gp4. Clearly, the increased helicase activity observed with the 56-kDa gp4 and the heterohexamers of gp4 reflects the contribution of the 56-kDa gp4 to oligomerization.
The above conclusions are based on the assumption that mixing of the 56- and 63-kDa gp4 as well as genetically altered forms of gp4 results in a fairly rapid equilibrium between gp4 molecules in solution and those within the hexamer. This communication as well as several earlier studies supports this interpretation (17–20). The 56-kDa gp4 and the 63-kDa gp4-K122A form heterohexamer that catalyzes the DNA-dependent synthesis of oligoribonucleotides in a trans mode (20) shows that both molecular weight forms are present in the hexamer; no oligoribonucleotide synthesis occurs with either the 56-kDa gp4 or the 63-kDa gp4-K122A. Heterohexamers are also formed by gp4-N468R and gp4-R493N (17) and wild-type gp4 and gp4-CΔ17 (19). Particularly convincing is the titration of gp4-E343Q into a hexamer of wild-type gp4 where a single subunit of gp4-E343Q entering the hexamer inhibits DNA-dependent hydrolysis of dTTP, whereas the DNA-independent hydrolysis of dTTP is inhibited in a linear manner as the wild-type subunits are increasingly replaced by gp4-E343Q (18). In our present study, DNA unwinding, strand-displacement DNA synthesis, oligomerization of gp4 heterohexamers, and primer synthesis all exhibit experimental curves that support this exchange of subunits within the hexamer.
An interesting question is why the 56-kDa gp4 and heterohexamers containing 56-kDa gp4 are more prone to oligomerize as compared with the 63-kDa gp4. Both proteins contain all of the motifs located in the helicase domain and the linker connecting the helicase to the primase known to be important in oligomerization (14, 15). The obvious and only structural difference between the 56-kDa gp4 and the 63-kDa gp4 is the presence of the additional 7-kDa ZBD on the 63-kDa gp4. If the addition of each 63-kDa gp4 subunit to a hexamer contributes equally to the inhibition, then oligomerization should increase in a linear fashion corresponding to the increase in the content of 56-kDa gp4. However, the experimental data show that the extent of oligomerization remains constant when the ratio of the two forms lies between 2/4 to 0/6.
The simplest explanation for the decreased oligomerization with the 63-kDa gp4 is that the presence of ZBD structurally interferes with the assembly of the hexamer, an interference that increases as additional ZBDs are present in the hexamer. Replacing subunits of the 63-kDa gp4 with the 56-kDa gp4 reduces this steric hindrance and improves the oligomerization. When the number of subunits of 56-kDa gp4 in a hexamer is sufficient for optimal oligomerization (ratio ranged from 2:4 to 0:6), the addition of more 56-kDa gp4 does not further increase the efficiency of oligomerization. It suggests that steric interference is present if two ZBDs are adjacent in a hexamer so that the 63-kDa gp4 is prone to align alternatively with the 56-kDa gp4. In vivo, the equimolar amount of the 56- and 63-kDa gp4 should optimally oligomerize. Thus, although oligomerization of gp4 requires components of the helicase domain and the linker connecting it to the primase domain, the more distant N-terminal ZBD has a rather striking influence on hexamer formation. Previously, studies on the length of the linker between the ZBD and RPD led to a model in which the cis mode performs de novo oligoribonucleotide synthesis and primer extension, whereas the trans mode catalyzed only de novo oligoribonucleotide synthesis (23). In this study we find that oligoribonucleotide synthesis mainly arises from a cis mode with only minor contributions from the trans mode. Nonetheless, our results do not rule out a role of the trans mode of oligoribonucleotide synthesis during coordinated DNA synthesis. Although we examined oligoribonucleotide synthesis and Okazaki fragment synthesis in a coordinated system, the large amount of unused oligoribonucleotides makes it difficult to definitively identify the specific mode occurring at the replication fork.
The amount of Okazaki fragments synthesized is proportional to the amount of oligoribonucleotide synthesized, which in turn, is dependent on the number of 63-kDa gp4 subunits in a hexamer. The percentage of oligoribonucleotides synthesized was similar for the 63-kDa gp4 and heterohexamers of gp4, independent of the ratios of 63- and 56-kDa gp4 within the hexamer. On the basis of our results, we propose a model for oligoribonucleotide synthesis and utilization. A hexamer containing a high ratio of the 63-kDa gp4 more efficiently synthesizes oligoribonucleotides, most likely due to a higher probability that one of the 63-kDa gp4 within the hexamer will contact a primase recognition site. If the trans mode was preferred, the presence of 56-kDa gp4 within the hexamer should, if anything, increase oligoribonucleotide synthesis. During coordinated leading- and lagging-strand DNA synthesis a priming loop is formed upon oligoribonucleotide synthesis by the primase (43). However, due to weak binding of the T7 primase to its recognition site, as determined by surface plasmon resonance (44), priming loops are most likely quickly resolved, and the oligoribonucleotides are dissociated into solution. In this study we estimate that 93% of the oligoribonucleotides are present in solution and not used to initiate the synthesis of Okazaki fragments. Only when the primase-DNA complex at the replication fork interacts with gp5/trx, the complex is sufficiently stable for the initiation of synthesis of an Okazaki fragment.
In vivo the mixture of 56- and 63-kDa gp4 can support T7Δ4 growth 10-fold more efficiently than by only the 63-kDa gp4 (9). In vivo DNA synthesis is ∼5–6-fold more efficient in the presence of both proteins than that in the presence of only 63-kDa protein (9). In vitro strand-displacement DNA synthesis is increased 4–5-fold for the mixture of the two molecular weight forms of gp4 relative to the 63-kDa gp4 alone. However, coordinated DNA synthesis is increased about 50%. It is often difficult to compare directly the results obtained by different assays. For example, overexpression of gene 4 in T7Δ4 complementation assay leads to an underestimate of the defect in an altered gene 4 protein. However, as clearly demonstrated in previous as well as this study, the presence of 56-kDa gene 4 protein is advantageous to all gp4 functions.
In the replisome systems of E. coli or phage T4 (12) as well as in eukaryotic systems, the helicase and primase are encoded by two separate genes but require a physical association to function properly. In the replisome of E. coli, three DnaG primases are associated with a hexamer of DnaB helicase to form a functional complex (1, 13). The crystal structures have identified three DnaG primases bound to the hexameric DnaB helicase (45). DnaB helicase alone catalyzes the unwinding of duplex DNA with poor processivity. However, when associated with the other proteins in the replisome, DnaB displays a high processivity of unwinding DNA in the replisome (46). At the replication fork of phage T4, the gene 41 helicase also assembles as a hexamer but with up to six gene 61 primase monomers (1). The binding of the T4 primase to its cognate helicase stimulates helicase activity (47). It has been suggested that three or less primases form a stable complex with a helicase hexamer in T4 phage and Bacillus stearothermophilus (48, 49). Bacteriophage T7 expresses equal molar amounts of the 63- and 56-kDa gp4. At this ratio, heterohexamers are formed that consist of equimolar amounts of the 56- and 63-kDa gp4. Because the 56-kDa gp4 is devoid of DNA-directed primase activity, the hexamer consists of six helicase domains and three functional primase domains. Consequently, the primase to helicase ratio is the same as in these other systems where the two activities are not covalently linked. At this ratio, leading- and lagging-strand DNA synthesis are coordinated and faster than that obtained with the hexamers of the 63-kDa gp4.
In the replisome of bacterial B. stearothermophilus, one molecule of the helicase binding domain of DnaG primase binds to a dimer comprised of two N-terminal domains of DnaB helicase. Consequently, three DnaG primases are spaced around the DnaB helicase hexamer (45). In the T7 system the repulsion between two adjacent ZBD in a hexamer reduces hexamer stability. Thus the equimolar amount of 63-kDa gp4 and 56-kDa gp4 in the T7-infected cells is prone to heterohexamers formation with the two forms arranged alternatively. However, it is difficult to examine directly the alignment of the subunits within a heterohexamer by electron microscopy as the 7-kDa difference is near the limit of resolution. We have been reluctant to increase the size of the 63-kDa gp4 by the creation of fusion proteins or the addition of other bulky moieties as they may well interfere with hexamer formation on their own. We have attempted to identify the 56- and 63-kDa subunits using fluorescent probes, but again the resolution is difficult.
The reduction of the number of active primase subunits within the hexamer may have advantages in addition to the favored oligomerization. Leading-strand DNA synthesis in a coordinated T7 replication system pauses for 4–6 s upon the synthesis of an oligoribonucleotide as measured by single molecule techniques (29). How does oligoribonucleotide synthesis serve as a “molecular brake” for leading-strand synthesis? We had originally proposed that if oligoribonucleotide synthesis occurs in a trans mode, then contact of the ZBD of one subunit with the RPD of another subunit within the hexamer would prevent conformational changes that essentially move the DNA through the central core of the hexameric helicase (1). Despite this elegant design of a molecular brake, our current experiments do not support nor completely eliminate this model. Whatever the mechanism of the molecular brake, the reduction of the number of active primases within the replisome could eliminate unnecessary halting events during DNA synthesis. In addition it should be noted that the 56-kDa gp4 is much less stringent in its ability to transfer oligoribonucleotides of varying complexity to gp5/trx (40). Even E. coli total tRNA with the 3′ sequence 5′-ACCA-3′ can be delivered by the 56-kDa gp4 to initiate the lagging-strand DNA synthesis, whereas the 63-kDa gp4 cannot (40). Consequently, the 56-kDa gp4 provides a backup system in the event that de novo synthesis of oligoribonucleotides cannot occur. Recently, we also found that the 56-kDa gp4 takes a more important role in the initiation of DNA replication than does the 63-kDa gp4.
Acknowledgments
We greatly thank Drs. Ajit Satapathy and Joseph Lee for helpful discussion and Steve Moskowitz (Advanced Medical Graphics) for preparing the figures.
This work was supported, in whole or in part, by National Institutes of Health Public Health Service Grant GM 54937 (to C. C. R.).

This article contains supplemental Figs. S1–S6.
- trx
- thioredoxin
- gp5
- gene 5 DNA polymerase
- gp4
- gene 4 helicase-primase
- gp2.5
- gene 2.5 ssDNA-binding protein
- nt
- nucleotides
- ZBD
- zinc binding domain
- RPD
- RNA polymerase domain.
REFERENCES
- 1. Hamdan S. M., Richardson C. C. (2009) Motors, switches, and contacts in the replisome. Annu. Rev. Biochem. 78, 205–243 [DOI] [PubMed] [Google Scholar]
- 2. Tabor S., Huber H. E., Richardson C. C. (1987) Escherichia coli thioredoxin confers processivity on the DNA polymerase activity of the gene 5 protein of bacteriophage T7. J. Biol. Chem. 262, 16212–16223 [PubMed] [Google Scholar]
- 3. Wuite G. J., Smith S. B., Young M., Keller D., Bustamante C. (2000) Single-molecule studies of the effect of template tension on T7 DNA polymerase activity. Nature 404, 103–106 [DOI] [PubMed] [Google Scholar]
- 4. Ahnert P., Patel S. S. (1997) Asymmetric interactions of hexameric bacteriophage T7 DNA helicase with the 5′- and 3′-tails of the forked DNA substrate. J. Biol. Chem. 272, 32267–32273 [DOI] [PubMed] [Google Scholar]
- 5. Satapathy A. K., Kochaniak A. B., Mukherjee S., Crampton D. J., van Oijen A., Richardson C. C. (2010) Residues in the central beta-hairpin of the DNA helicase of bacteriophage T7 are important in DNA unwinding. Proc. Natl. Acad. Sci. U.S.A. 107, 6782–6787 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Lee J., Chastain P. D., 2nd, Griffith J. D., Richardson C. C. (2002) Lagging-strand synthesis in coordinated DNA synthesis by bacteriophage T7 replication proteins. J. Mol. Biol. 316, 19–34 [DOI] [PubMed] [Google Scholar]
- 7. Hamdan S. M., Loparo J. J., Takahashi M., Richardson C. C., van Oijen A. M. (2009) Dynamics of DNA replication loops reveal temporal control of lagging-strand synthesis. Nature 457, 336–339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Marintcheva B., Hamdan S. M., Lee S. J., Richardson C. C. (2006) Essential residues in the C terminus of the bacteriophage T7 gene 2.5 single-stranded DNA-binding protein. J. Biol. Chem. 281, 25831–25840 [DOI] [PubMed] [Google Scholar]
- 9. Mendelman L. V., Notarnicola S. M., Richardson C. C. (1992) Roles of bacteriophage T7 gene 4 proteins in providing primase and helicase functions in vivo. Proc. Natl. Acad. Sci. U.S.A. 89, 10638–10642 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Bernstein J. A., Richardson C. C. (1989) Characterization of the helicase and primase activities of the 63-kDa component of the bacteriophage T7 gene 4 Protein. J. Biol. Chem. 264, 13066–13073 [PubMed] [Google Scholar]
- 11. Bernstein J. A., Richardson C. C. (1988) A 7-kDa region of the bacteriophage-T7 gene 4 protein is required for primase but not for helicase activity. Proc. Natl. Acad. Sci. U.S.A. 85, 396–400 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Frick D. N., Richardson C. C. (2001) DNA primases. Annu. Rev. Biochem. 70, 39–80 [DOI] [PubMed] [Google Scholar]
- 13. O'Donnell M. (2006) Replisome architecture and dynamics in Escherichia coli. J. Biol. Chem. 281, 10653–10656 [DOI] [PubMed] [Google Scholar]
- 14. Guo S., Tabor S., Richardson C. C. (1999) The linker region between the helicase and primase domains of the bacteriophage T7 gene 4 protein is critical for hexamer formation. J. Biol. Chem. 274, 30303–30309 [DOI] [PubMed] [Google Scholar]
- 15. Lee S. J., Richardson C. C. (2004) The linker region between the helicase and primase domains of the gene 4 protein of bacteriophage T7 Role in helicase conformation and activity. J. Biol. Chem. 279, 23384–23393 [DOI] [PubMed] [Google Scholar]
- 16. Frick D. N., Richardson C. C. (1999) Interaction of bacteriophage T7 gene 4 primase with its template recognition site. J. Biol. Chem. 274, 35889–35898 [DOI] [PubMed] [Google Scholar]
- 17. Lee S. J., Qimron U., Richardson C. C. (2008) Communication between subunits critical to DNA binding by hexameric helicase of bacteriophage T7. Proc. Natl. Acad. Sci. U.S.A. 105, 8908–8913 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Crampton D. J., Mukherjee S., Richardson C. C. (2006) DNA-induced switch from independent to sequential dTTP hydrolysis in the bacteriophage T7 DNA helicase. Mol. Cell 21, 165–174 [DOI] [PubMed] [Google Scholar]
- 19. Hamdan S. M., Johnson D. E., Tanner N. A., Lee J. B., Qimron U., Tabor S., van Oijen A. M., Richardson C. C. (2007) Dynamic DNA helicase-DNA polymerase interactions assure processive replication fork movement. Mol. Cell 27, 539–549 [DOI] [PubMed] [Google Scholar]
- 20. Lee S. J., Richardson C. C. (2002) Interaction of adjacent primase domains within the hexameric gene 4 helicase-primase of bacteriophage T7. Proc. Natl. Acad. Sci. U.S.A. 99, 12703–12708 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kato M., Ito T., Wagner G., Richardson C. C., Ellenberger T. (2003) Modular architecture of the bacteriophage T7 primase couples RNA primer synthesis to DNA synthesis. Mol. Cell 11, 1349–1360 [DOI] [PubMed] [Google Scholar]
- 22. Toth E. A., Li Y., Sawaya M. R., Cheng Y., Ellenberger T. (2003) The crystal structure of the bifunctional primase-helicase of bacteriophage T7. Mol. Cell 12, 1113–1123 [DOI] [PubMed] [Google Scholar]
- 23. Qimron U., Lee S. J., Hamdan S. M., Richardson C. C. (2006) Primer initiation and extension by T7 DNA primase. EMBO J. 25, 2199–2208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Notarnicola S. M., Richardson C. C. (1993) The nucleotide binding site of the helicase-primase of bacteriophage T7 interaction of mutant and wild-type proteins. J. Biol. Chem. 268, 27198–27207 [PubMed] [Google Scholar]
- 25. Kim Y. T., Tabor S., Churchich J. E., Richardson C. C. (1992) Interactions of gene 2.5 protein and DNA-polymerase of bacteriophage T7. J. Biol. Chem. 267, 15032–15040 [PubMed] [Google Scholar]
- 26. Lee J., Chastain P. D., 2nd, Kusakabe T., Griffith J. D., Richardson C. C. (1998) Coordinated leading- and lagging-strand DNA synthesis on a minicircular template. Mol. Cell 1, 1001–1010 [DOI] [PubMed] [Google Scholar]
- 27. Zhu B., Lee S. J., Richardson C. C. (2009) An in trans interaction at the interface of the helicase and primase domains of the hexameric gene 4 protein of bacteriophage T7 modulates their activities. J. Biol. Chem. 284, 23842–23851 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Zhang H., Lee S. J., Zhu B., Tran N. Q., Tabor S., Richardson C. C. (2011) Helicase-DNA polymerase interaction is critical to initiate leading-strand DNA synthesis. Proc. Natl. Acad. Sci. U.S.A. 108, 9372–9377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Lee J. B., Hite R. K., Hamdan S. M., Xie X. S., Richardson C. C., van Oijen A. M. (2006) DNA primase acts as a molecular brake in DNA replication. Nature 439, 621–624 [DOI] [PubMed] [Google Scholar]
- 30. Akabayov B., Akabayov S. R., Lee S. J., Tabor S., Kulczyk A. W., Richardson C. C. (2010) Conformational dynamics of bacteriophage T7 DNA polymerase and its processivity factor, Escherichia coli thioredoxin. Proc. Natl. Acad. Sci. U.S.A. 107, 15033–15038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Crampton D. J., Ohi M., Qimron U., Walz T., Richardson C. C. (2006) Oligomeric states of bacteriophage T7 gene 4 primase/helicase. J. Mol. Biol. 360, 667–677 [DOI] [PubMed] [Google Scholar]
- 32. Notarnicola S. M., Park K., Griffith J. D., Richardson C. C. (1995) A domain of the gene-4 helicase-primase of bacteriophage-T7 required for the formation of an active hexamer. J. Biol. Chem. 270, 20215–20224 [DOI] [PubMed] [Google Scholar]
- 33. Bianchi M., Baulieu E. E. (2012) 3β-Methoxy-pregnenolone (MAP4343) as an innovative therapeutic approach for depressive disorders. Proc. Natl. Acad. Sci. U.S.A. 109, 1713–1718 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Lee S. J., Richardson C. C. (2005) Acidic residues in the nucleotide-binding site of the bacteriophage T7 DNA primase. J. Biol. Chem. 280, 26984–26991 [DOI] [PubMed] [Google Scholar]
- 35. Lee S. J., Chowdhury K., Tabor S., Richardson C. C. (2009) Rescue of bacteriophage T7 DNA polymerase of low processivity by suppressor mutations affecting gene 3 endonuclease. J. Virol. 83, 8418–8427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Yang J., Nelson S. W., Benkovic S. J. (2006) The control mechanism for lagging-strand polymerase recycling during bacteriophage T4 DNA replication. Mol. Cell 21, 153–164 [DOI] [PubMed] [Google Scholar]
- 37. Kim D. E., Narayan M., Patel S. S. (2002) T7 DNA helicase. A molecular motor that processively and unidirectionally translocates along single-stranded DNA. J. Mol. Biol. 321, 807–819 [DOI] [PubMed] [Google Scholar]
- 38. Satapathy A. K., Kulczyk A. W., Ghosh S., van Oijen A. M., Richardson C. C. (2011) Coupling dTTP hydrolysis with DNA unwinding by the DNA helicase of bacteriophage T7. J. Biol. Chem. 286, 34468–34478 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Frick D. N., Baradaran K., Richardson C. C. (1998) An N-terminal fragment of the gene 4 helicase/primase of bacteriophage T7 retains primase activity in the absence of helicase activity. Proc. Natl. Acad. Sci. U.S.A. 95, 7957–7962 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Zhu B., Lee S. J., Richardson C. C. (2010) Direct role for the RNA polymerase domain of T7 primase in primer delivery. Proc. Natl. Acad. Sci. U.S.A. 107, 9099–9104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Nakai H., Beauchamp B. B., Bernstein J., Huber H. E., Tabor S., Richardson C. C. (1988) DNA Replication and Mutagenesis, pp. 85–97, American Society for Microbiology, Washington, D. C [Google Scholar]
- 42. Mendelman L. V., Richardson C. C. (1991) Requirements for primer synthesis by bacteriophage T7 63-kDa gene 4 protein roles of template sequence and T7 56-kDa gene-4 protein. J. Biol. Chem. 266, 23240–23250 [PubMed] [Google Scholar]
- 43. Pandey M., Syed S., Donmez I., Patel G., Ha T., Patel S. S. (2009) Coordinating DNA replication by means of priming loop and differential synthesis rate. Nature 462, 940–943 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Lee S. J., Zhu B., Hamdan S. M., Richardson C. C. (2010) Mechanism of sequence-specific template binding by the DNA primase of bacteriophage T7. Nucleic Acids Res. 38, 4372–4383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Bailey S., Eliason W. K., Steitz T. A. (2007) Structure of hexameric DnaB helicase and its complex with a domain of DnaG primase. Science 318, 459–463 [DOI] [PubMed] [Google Scholar]
- 46. Tanner N. A., Hamdan S. M., Jergic S., Loscha K. V., Schaeffer P. M., Dixon N. E., van Oijen A. M. (2008) Single-molecule studies of fork dynamics in Escherichia coli DNA replication. Nat. Struct. Mol. Biol. 15, 170–176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Richardson R. W., Nossal N. G. (1989) Characterization of the bacteriophage T4 gene 41 DNA helicase. J. Biol. Chem. 264, 4725–4731 [PubMed] [Google Scholar]
- 48. Jing D. H., Dong F., Latham G. J., von Hippel P. H. (1999) Interactions of bacteriophage T4-coded primase (gp61) with the T4 replication helicase (gp41) and DNA in primosome formation. J. Biol. Chem. 274, 27287–27298 [DOI] [PubMed] [Google Scholar]
- 49. Bird L. E., Pan H., Soultanas P., Wigley D. B. (2000) Mapping protein-protein interactions within a stable complex of DNA primase and DnaB helicase from Bacillus stearothermophilus. Biochemistry 39, 171–182 [DOI] [PMC free article] [PubMed] [Google Scholar]