

Background: Conotoxins can be utilized to investigate enzyme-assisted folding of disulfide-rich peptides.
Results: Various ER-resident cone snail enzymes act in concert to accelerate the oxidative folding of conotoxins and modulate their conformation by reconfiguring disulfide connectivities.
Conclusion: The folding of conotoxins is a tightly regulated multienzyme-assisted process.
Significance: Modulation of the conformation of conotoxins increases their molecular and functional diversity.
Keywords: Chaperone Chaperonin, Disulfide, Prolyl Isomerase, Protein Folding, Toxins, Conotoxins, Disulfide-rich Peptides, Foldases, Oxidative Folding
Abstract
The oxidative folding of large polypeptides has been investigated in detail; however, comparatively little is known about the enzyme-assisted folding of small, disulfide-containing peptide substrates. To investigate the concerted effect of multiple enzymes on the folding of small disulfide-rich peptides, we sequenced and expressed protein-disulfide isomerase (PDI), peptidyl-prolyl cis-trans isomerase, and immunoglobulin-binding protein (BiP) from Conus venom glands. Conus PDI was shown to catalyze the oxidation and reduction of disulfide bonds in two conotoxins, α-GI and α-ImI. Oxidative folding rates were further increased in the presence of Conus PPI with the maximum effect observed in the presence of both enzymes. In contrast, Conus BiP was only observed to assist folding in the presence of microsomes, suggesting that additional co-factors were involved. The identification of a complex between BiP, PDI, and nascent conotoxins further suggests that the folding and assembly of conotoxins is a highly regulated multienzyme-assisted process. Unexpectedly, all three enzymes contributed to the folding of the ribbon isomer of α-ImI. Here, we identify this alternative disulfide-linked species in the venom of Conus imperialis, providing the first evidence for the existence of a “non-native” peptide isomer in the venom of cone snails. Thus, ER-resident enzymes act in concert to accelerate the oxidative folding of conotoxins and modulate their conformation and function by reconfiguring disulfide connectivities. This study has evaluated the role of a number of ER-resident enzymes in the folding of conotoxins, providing novel insights into the enzyme-guided assembly of these small, disulfide-rich peptides.
Introduction
During synthesis on the ribosome, the nascent polypeptide chain enters the crowded environment of the ER,2 where it engages in a variety of intra- and intermolecular interactions. The newly synthesized protein associates with a number of ER-resident chaperones and foldases that accelerate rate-limiting steps along the folding pathway. The three enzymes that have repeatedly been implicated in the folding of protein substrates in the ER are protein-disulfide isomerase (PDI), peptidyl-prolyl cis-trans isomerase (PPI), and immunoglobulin-binding protein (BiP).
Polypeptides containing one or more disulfides in their native state are likely to become substrates for PDI, the enzyme that catalyzes the oxidation, isomerization, and reduction of disulfide bonds. PDI contains four thioredoxin-like domains, two of which have the catalytic CXXC motif (1). The thiol-disulfide redox state of the catalytic sites determines the direction of the reaction. Conversion of the disulfide site to a dithiol leads to oxidation of a substrate protein, whereas the alteration from a dithiol to a disulfide results in reduction of the substrate (2). Isomerization occurs either directly or through cycles of reduction and reoxidation (3). To date, 19 members of the PDI family have been identified in humans that significantly differ in their primary amino acid sequence, domain organization, catalytic activity, or mass (4, 5).
In addition to disulfide bond formation, the cis-trans isomerization of peptidyl-prolyl bonds can impede folding of proline-containing proteins. Peptide bonds to proline are synthesized in the trans conformation. Although the majority of these bonds remain in trans, some peptidyl-prolyl bonds need to undergo trans-cis isomerization. The enzyme that catalyzes this otherwise slow reaction is PPI, a ubiquitous protein that is present in almost all cellular compartments (6). In mammalian cells, a number of ER-resident PPIs have been identified, including PPI B. Interestingly, in addition to accelerating folding rates via isomerization of peptide bonds to proline, PPI was also shown to improve the efficiency of PDI-mediated folding of ribonuclease T1 by providing partially folded protein chains with the correct proline isomers (7). Similarly, oxidative folding rates of maurotoxin, a disulfide-rich peptide from scorpion venom, were highest in the presence of PDI and FKBP-12, a PPI isoform located in the cytosol (8).
Another enzyme known to cooperate with PDI in the folding of disulfide-containing proteins is BiP, a member of the heat shock protein 70 (Hsp70) family. BiP binds to unfolded and partially misfolded proteins upon their entry into the lumen of the ER and limits protein misfolding and aggregation. It is also a key enzyme in the retrograde transport of misfolded proteins from the ER to the proteasome (9). In recent years, a number of studies have addressed the potential cooperative folding of disulfide-containing proteins by PDI and BiP. Investigations into the folding of antibody chains revealed that both enzymes cooperatively act in the refolding of Fab fragments in vitro (10). It has been suggested that BiP binds the unfolded polypeptide chains and keeps them in a conformation in which the cysteine residues are accessible for PDI (10). Immunoprecipitation experiments further revealed that PDIA6, a member of the PDI family, forms a non-covalent complex with BiP and shows specificity toward BiP client proteins in human fibroblast cells (11). Subsequently, the in vitro interaction between another PDI family member, PDIA3, and BiP was shown to improve folding rates of ribonuclease B and α-lactalbumin (12).
Together, these findings indicate that members of the PDI family can recruit or are themselves recruited by other ER-resident enzymes and chaperones, such as BiP and/or PPI B, to cooperatively act in the oxidative folding of at least some client substrates. Recently, an ER-resident complex comprising PDI, PPI B, and BiP was identified in human hepatoma and mouse lymphoma cells (13), reflecting tightly regulated compartmentalization of protein folding in the ER.
Although our understanding of enzyme-guided folding of proteins is constantly improving, comparatively little is known about the folding and assembly of small, disulfide-rich peptides. Disulfide-rich peptides are widely distributed in the plant and animal kingdoms, where they serve diverse functions. Examples include antimicrobial peptides, such as the defensins; various protease inhibitors; hormones, including insulin; and a wide range of neurotoxins found in animal venoms (Table 1). Predatory marine snails of the genus Conus synthesize a great diversity of disulfide-rich peptides that often carry additional post-translational modifications, most commonly proline hydroxylations, γ-carboxylations, and C-terminal amidations (14). The diversity of peptides generated by cone snails is astonishing. Each of the 500–700 species of cone snail is estimated to synthesize hundreds of different peptides with distinct disulfide connectivities (15). With their vast structural diversity, conotoxins can therefore be regarded as model disulfide-rich peptides for understanding general mechanisms of peptide folding, including the formation of correct disulfide bonds.
TABLE 1.
Diversity of disulfide-rich peptides and enzymes implicated in their biosynthesis
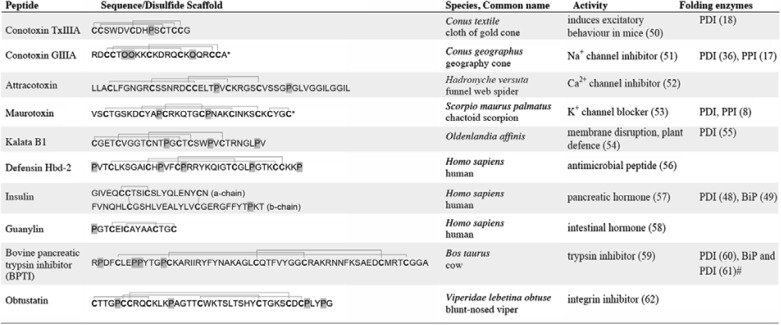
* C-terminal amidation, O = hydroxyproline, prolines and hydroxyprolines are highlighted in gray, cysteine residues are shown in boldface type.
# Findings were based on kinetic modeling.
The concerted effect of PDI, PPI, and BiP on the folding of these and other disulfide-rich peptides has not been systematically explored. Although PDI and PPI have been identified in the venom gland of Conus (16) and are known to play a role in the in vitro folding of conotoxins (17, 18), their combined effect has not been investigated. We recently sequenced Conus BiP from the venom gland of Conus novaehollandiae and demonstrated it to be highly expressed in the venom glandular cells (16). Whether BiP plays a functional role in the folding of conotoxins was not addressed in our earlier study. To investigate multienzyme-assisted folding of conotoxins, we cloned and expressed PDI, PPI B, and BiP from C. novaehollandiae and tested their potential in assisting in the oxidative folding of α-conotoxins GI and ImI. Here, we show that all three enzymes play a role in the oxidative folding of these two peptides and, in the case of PDI, also in the unfolding of disulfide bonds under reducing conditions. Importantly, a direct interaction between PDI and conotoxin α-GI was confirmed by affinity purification. We further reveal the presence of a complex between PDI and BiP, suggesting a concerted action of these enzymes, similar to what has been observed for the folding of larger proteins. Our findings indicate that the proper assembly of conotoxins relies upon the action of a number of ER-resident enzymes, including PDI, PPI B, and BiP. Importantly, these enzymes appear to modulate the disulfide isomers of conotoxins, a process that not only generates additional conformational diversity but also has the potential to enhance the functional diversity of these molecules.
EXPERIMENTAL PROCEDURES
Depending on the experimental requirements, a number of different cone snail species were utilized in this study. Conus PPI B and BiP were previously sequenced from the venom gland of C. novaehollandiae. Consequently, C. novaehollandiae was selected for molecular sequencing of PDI. Conotoxin α-ImI was originally isolated from the venom of Conus imperialis. Investigations into the presence of different isoforms of α-ImI were carried out on the venom of this species. Investigations of protein complexes were performed on the venom gland of Conus victoriae. This species has a particularly large venom gland, allowing the isolation of sufficient material. Conotoxin α-GI was originally isolated from the venom of Conus geographus. Co-immunoprecipitation (co-IP) experiments using the biotin-labeled propeptide of α-GI were performed on venom glands from this species.
Specimen Collection and Tissue Preparation
Specimens of C. novaehollandiae and C. victoriae were collected from Broome, Western Australia. Live animals were shipped to Melbourne, Australia, and processed within 24 h of collection. Venom ducts were extracted, snap-frozen and stored at −80 °C. Specimens of C. imperialis and C. geographus were collected from Marinduque Island, the Philippines. Venom ducts were prepared as described above. Prior to freezing, venom was extruded from isolated venom ducts of C. imperialis, air-dried, and stored at −80 °C.
Venom Extraction and Analysis
Twenty mg of dried C. imperialis venom was reconstituted in 1 ml of 40% acetonitrile (ACN), 0.1% trifluoroacetic acid (TFA) and homogenized using a Dounce tissue grinder. Insoluble material was pelleted by centrifugation at 12,000 × g for 10 min. The supernatant was collected, dried by vacuum centrifugation, and stored at −80 °C until processing. Four mg of crude venom was separated by reversed-phase high performance liquid chromatography (RP-HPLC) on a semipreparative C18 column (5-μm particle size, 10 × 250 mm, Vydac-Grace) using a linear gradient from 5 to 100% buffer B (90% ACN, 0.1% TFA) over 80 min. Peaks were manually collected and further analyzed by electrospray ionization tandem mass spectrometry (ESI-MS/MS) and matrix-assisted laser desorption ionization mass spectrometry (MALDI-MS). For ESI-MS/MS, samples were analyzed using a high resolution linear ion trap mass spectrometer (LTQ XL Orbitrap, Thermo Scientific). For MALDI-MS, peptides were spotted onto matrix-coated MALDI plates and analyzed using a MALDI-time of flight (TOF) mass spectrometer in positive reflector mode (Voyager, AB SCIEX).
Preparation of Rat Liver Microsomal Proteins
Microsomal proteins were prepared as described previously (19). Briefly, 20 mg of frozen rat liver tissue was homogenized in 20 ml of ice-cold homogenization buffer (10 mm Tris, 150 mm NaCl, pH 7.5, 1× protease inhibitor mixture (Roche Applied Science)) using a Dounce tissue grinder. Cellular debris was removed by centrifugation at 12,000 × g for 15 min. Membranous material containing microsomes was pelleted by ultracentrifugation at 100,000 × g for 1 h. Following ultracentrifugation, pellets were washed twice with homogenization buffer. Closed vesicles were converted to open membranes to release microsomal proteins by incubating with ice-cold 0.1 m sodium carbonate buffer (pH 11) for 20 min. Membranous material was removed by ultracentrifugation as described above. The pH of the supernatant was adjusted to 7.5 with 2 n HCl solution. Microsomal protein preparations were examined by SDS-PAGE, and protein concentrations were determined using Bradford reagent (Sigma-Aldrich). Protein-bound and free sulfhydryl groups were determined spectrophotometrically using Ellman's reagent (5,5′-dithiobis(2-nitrobenzoic acid)) as described previously (20).
Molecular Sequencing
We previously obtained full-length sequences of PPI B and BiP from the venom gland of C. novaehollandiae (accession numbers GU046312 and HM627497, respectively). In order to sequence Conus PDI, frozen venom glands from C. novaehollandiae were ground under liquid nitrogen, and total RNA was extracted using TRIzol® reagent (Invitrogen) and DNase I treated with Turbo DNase (Ambion Inc). RNA extraction and DNase treatment were performed according to the manufacturers' instructions. cDNA was reverse transcribed from 1 μg of DNase-treated RNA using the Superscript VILO cDNA synthesis kit (Invitrogen) with a 1:1 mixture of oligo(dT)s and random hexamers. Primary reverse transcription PCR (RT-PCR) was performed in volumes of 50 μl containing 4 μl of cDNA (200 ng), 0.5 μl of Velocity Taq DNA polymerase (Bioline), 1× PCR buffer (Bioline), 200 μm each deoxynucleoside triphosphate (dNTPs; Invitrogen), and 0.2 μm PDI sense (CGA CCA TAT GGA TGA TAT CAA ACA GGA GGA A) and antisense oligonucleotide primers (CGC TCG AGC AGT TCA TCT CTT GGC AGA TC). PCR amplicons were analyzed by gel electrophoresis, cloned, and sequenced as described previously (17). The novel Conus PDI sequence was deposited in GenBankTM (National Center for Biotechnology Information, United States National Library of Medicine, Bethesda, MD). Nucleotide sequences were translated into the predicted amino acid residues, and the putative signal peptide was predicted using SignalP software (21).
Cloning, Expression, and Purification of Recombinant Enzymes
Conus PPI B was expressed and purified as described previously (17). Conus PDI and BiP lacking the N-terminal signal sequences were cloned into the pET22b+ and pET30c+ expression vector, respectively (Novagen). Briefly, transcripts were PCR-amplified from venom gland cDNA prepared as described above and ligated into pET22b+ using the NdeI (5′) and XhoI (3′) restriction sites and into pET30c+ with the endonucleases NdeI (5′) and HindIII (3′) (New England Biolabs). Sequences were verified by nucleotide sequencing. The constructs, containing a C-terminal His6 tag, were transformed into Escherichia coli (Rosetta strain, Novagen) by heat treatment. For expression of recombinant proteins, LB broth containing antibiotics (100 μg/ml ampicillin for pET22b+ and 50 μg/ml kanamycin for pET30c+) was inoculated with overnight cultures and incubated at 37 °C with shaking until the A600 spectrophotometric reading was 0.6. Expression was induced by adding 0.1 mm isopropyl-β-d-thiogalactopyranoside followed by incubation for 3 h at 25 °C with shaking. Bacteria were harvested and resuspended in native lysis buffer (50 mm NaH2PO4, 300 mm NaCl, and 10 mm imidazole, pH 8.0) containing 1× protease inhibitor mixture (EDTA-free; Roche Applied Science). Bacterial cells were lysed by probe tip sonication. Cellular debris and insoluble protein were pelleted by centrifugation at 15,000 × g for 20 min, and the supernatants were used for subsequent protein purifications.
Recombinant proteins were purified on a 1-ml immobilized metal affinity column under native conditions (Bio-Scale Mini Profinity IMAC cartridge, Bio-Rad). Purification was performed using the Bio-Rad Profinia protein purification system. Protein lysates were loaded onto the column at 0.5 ml/min, and nonspecifically bound proteins were removed with native buffer (300 mm KCl, 50 mm KH2PO4, pH 8.0) containing 5 mm imidazole at 2 ml/min for 6 min, followed by a second wash with native buffer containing 15 mm imidazole for 5 min. His-tagged proteins were eluted with 250 mm imidazole in native buffer at 1 ml/min for 4 min. Further purification and buffer exchange into 10 mm Tris-HCl, 150 mm NaCl, pH 8, was accomplished by size exclusion chromatography (Superdex 200, HiLoad 16/60, GE Healthcare) at 1 ml/min. The purified recombinant proteins were analyzed by SDS-PAGE and sequence-verified by in-gel digestion and analysis of proteotypic tryptic peptides as described previously (17). Protein concentrations were determined spectrophotometrically using the proteins' molar absorption coefficients (22).
Functional Enzyme Analyses
The insulin reduction assay was performed using a modification of the insulin turbidity assay described previously (23). Briefly, recombinant PDI (0.5 and 2.5 μm final concentration) was incubated with 2 mm dithiothreitol (DTT) for 15 min prior to adding the assay mixture (100 mm potassium phosphate, 2 mm EDTA, 2 mm DTT, and 0.85 mg/ml insulin, pH 6.5). The absorbance at 650 nm was measured for 1 h at 30 °C.
The peptidyl-prolyl cis-trans isomerase activity of recombinant PPI B was determined using the coupled chymotrypsin assay (24) with modifications as described previously (17). Briefly, 0.3 or 3 μm PPI B was incubated in 100 mm Tris-HCl, pH 8, at 25 °C for 2 min. Samples were transferred to 10 °C for 5 min before the addition of 30 μl of 600 mm chymotrypsin (Sigma-Aldrich). After 5 min at 10 °C, reactions were initialized by adding the substrate N-succinyl-Ala-Ala-Pro-Phe-p-nitroanilide at a final concentration of 78 μm (Bachem AG; stock made in trifluoroethanol in the presence of 0.45 m LiCl (Sigma-Aldrich)). The cis-trans isomerization was measured by following the absorbance at 390 nm for 3 min.
The ATPase activity of recombinant BiP was verified using a modification of the NADH-coupled photometric assay described previously (25). The assay relies on the hydrolysis of ATP to ADP by the ATPase activity of BiP. Upon ATP hydrolysis, phosphoenolpyruvate is converted to pyruvate by pyruvate kinase, followed by further conversion into lactate by lactate dehydrogenase. The latter reaction requires oxidation of NADH, resulting in a decrease in absorbance at 340 nm. Briefly, reaction buffer containing 30 mm Tris (pH 7.6), 30 mm KCl, 5 mm MgCl2, 0.15 mm NADH, 4 mm phosphoenolpyruvate, 25 μg/ml lactate dehydrogenase, and 25 μg/ml pyruvate kinase was incubated at 25 °C for 10 min. For control reactions, ADP was added to a final concentration of 10 mm, and NADH depletion was monitored at 340 nm. To determine the ATPase activity of BiP, ATP was added to a final concentration of 10 mm. After base-line stabilization, BiP was added at a final concentration of 2.5 and 5 μm, and the absorbance was monitored for 35 min at 25 °C.
Peptide Synthesis
Based on their well characterized folding properties, the two α-conotoxins GI and ImI were selected for oxidative folding studies. Conotoxin α-ImI was obtained from the peptide synthesis core facility at the University of Utah. Conotoxin α-GI was synthesized with an Apex 396 automated peptide synthesizer (AAPPTec) using a standard solid-phase Fmoc protocol. The peptide was constructed on preloaded Fmoc-Rink Amide MBHA resin (substitution, 0.4 mmol/g; Peptides International Inc.) on a 50-μmol scale. All amino acids were purchased form AAPPTec, and side-chain protection for the following amino acids was as follows: Glu, O-tert-butyl; Arg, 2,2,4,6,7-pentamethyldihydrobenzofuran-5-sulfonyl; Tyr and Ser, tert-butyl; Asn, Cys, and His, trityl. Coupling activation was achieved with 1 eq of 0.4 m benzotriazol-1-yl-oxytripyrrolidinophosphonium hexafluorophosphate and 2 eq of 2 m N,N-diisopropylethyl amine in N-methyl-2-pyrrolidone as the solvent. A 10-fold excess of each amino acid was used, and each coupling reaction was conducted for 60 min. The Fmoc deprotection reaction was carried out for 20 min with 20% piperidine in dimethylfluoride. Peptides were cleaved from the resin by treatment with reagent K (TFA/thioanisole/ethanedithiol/water/phenol (82.5:5:2.5:5:5 by volume)) for 3 h and 3.5 h for α-GI and α-ImI, respectively. The peptides were subsequently filtered, precipitated, and washed with cold methyl tert-butyl ether. Linear peptides were purified by RP-HPLC on a semipreparative C18 column (5-μm particle size, 10 × 250 mm, Vydac-Grace) using a linear gradient from 5 to 40% buffer B (90% ACN, 0.1% TFA) over 20 min. Buffer A was 0.1% TFA, water. Absorbance was monitored at 220 nm. Concentrations were determined spectrophotometrically using the peptides' molar absorption coefficient at 280 nm. For co-elution experiments, correctly folded α-GI and α-ImI were obtained from the peptide synthesis facility at the Salk Institute.
The biotin-labeled propeptide of α-conotoxin GI (pro-GI; biotin-FPSERASDGRDDTAKDEGSDMEKLVEKKECCNPACGRHYSC-NH2) was synthesized as described previously (26) with the exception that the peptide was C-terminally tagged with biotin followed by 6-aminohexanoic acid (GL Biochem) and not oxidized prior to purification. The biotin-labeled control peptide (biotin-YARFQSQTTLKQKT) was purchased from the peptide synthesis facility at Monash University.
Oxidative Folding Studies
Oxidative folding reactions were carried out at room temperature in the presence of 0.1 m Tris-HCl (pH 7.5), 1 mm MgCl2, 10 mm ATP, 0.1 mm GSSG, 0.1 mm GSH, 3 μm PPI B and/or 1 μm PDI and/or 2 μm BiP. Because the exact concentrations of PDI, PPI B, and BiP in the ER are not known, the enzyme/substrate ratios were chosen to be in agreement with previous studies (7, 8). Folding was initiated by adding the linear peptide at a final concentration of 20 μm. Aliquots were taken at various time points, and reactions were quenched by acidification with formic acid to a final concentration of 8%. Folding reactions were analyzed by RP-HPLC on a C18 column (5-μm particle size, 4.6 × 250 mm, Vydac-Grace) using the conditions described above. For samples spiked with rat liver microsomal proteins, separation was performed on a diphenyl column (5-μm particle size, 4.6 × 250 mm, Vydac-Grace) using the same conditions. Native peptides were distinguished from linear forms based on characteristic elution profiles (27–29), co-elution experiments, and mass spectrometric (MS) analyses of manually collected reversed-phase fractions (MALDI-TOF mass spectrometer, positive reflector mode, Voyager, AB SCIEX). Statistical analyses were performed using two-tailed Student's t tests with unequal variance.
Conotoxin Reductase Assay
For reductase assays, 1 μm PDI was preincubated in reductase buffer (0.1 m Tris-HCl, 1 mm MgCl2, 10 mm ATP, 5 mm GSH, pH 7.5) for 1 h at room temperature. Peptide reduction was initiated by adding the folded peptide at a final concentration of 20 μm. Quenching and analysis were performed as described above.
Blue Native PAGE
Blue Native (BN)-PAGE techniques were adapted from those described previously (30). Briefly, frozen venom glands from C. victoriae were homogenized in ice-cold buffer (20 mm Tris-HCl, 50 mm NaCl, 10% glycerol, 1 mm PMSF, pH 7.5, containing 1× protease inhibitor mixture) using a Dounce tissue grinder. Cellular debris was removed by centrifugation at 6,000 × g for 8 min at 4 °C. The supernatant was collected and ultracentrifuged at 100,000 × g for 1 h at 4 °C to separate soluble proteins from membranous material. Supernatants and pellets were collected, and proteins were solubilized by adding between 1 and 3% of digitonin or CHAPS followed by incubation for 20 min on ice. An aliquot of the soluble and membranous protein sample was mixed with 5× BN-PAGE loading buffer (5% Coomassie Brilliant Blue G-250, 500 mm (E)-aminocaproic acid in 100 mm BisTris, pH 7) and loaded onto NuPAGE Native 4–16% gels (Invitrogen) along with an in-house molecular weight marker (kindly provided by Srgjan Civciristov). Samples were separated in the presence of blue cathode buffer (50 mm Tricine, 15 mm BisTris, 0.02% Coomassie Brilliant Blue G-250, pH 7) and anode buffer (50 mm BisTris, pH 7) for 50 min at 150 V on ice. The blue cathode buffer was then replaced with cathode buffer without Coomassie dye, and electrophoresis was completed at 200 V. Gels were either stained with Coomassie Blue followed by in-gel tryptic digest or second dimension SDS-PAGE or further analyzed by immunoblotting.
For second dimension SDS-PAGE, gel bands were excised and soaked in 1% SDS and 1% mercaptoethanol for 2 h followed by two washes in water. Gel bands were placed into wells on 12% NuPAGE BisTris gels (Invitrogen). Gels were run for 20 min at 100 V followed by 40 min at 200 V. Following SDS-PAGE, gels were either stained with Coomassie Blue or analyzed by in-gel tryptic digest or immunoblotting.
Immunoblotting
Proteins were transferred from gels onto polyvinylidene fluoride (PVDF) membranes and blocked with 5% skim milk in phosphate-buffered saline, 0.05% Tween 20 (PBS-T) followed by immunoblotting. Primary antibodies were rabbit polyclonal anti-BiP antibody raised against a synthetic peptide sequence derived from human BiP (anti-GRP78 BiP antibody, ab21685, Abcam) and rabbit polyclonal anti-PDI antibody raised against recombinant rat liver PDI (courtesy of Prof. M. Hubbard); The secondary antibody was sheep anti-rabbit, HRP-conjugated (Abcam). Incubations were carried out at room temperature for 1 h. Antibody stocks were diluted 1:2,000 in 2.5% skim milk, PBS-T. Detection was performed with ECL reagent (PerkinElmer Life Sciences). For immunoblotting following BN-PAGE, gels were briefly soaked in transfer buffer containing 0.1% SDS prior to transfer onto PVDF membranes. After transfer, membranes were washed in methanol for 3 min to remove residual Coomassie dye. For reprobing, membranes were washed 5 min in PBS-T, followed by incubation in 62 mm Tris-HCl, 100 mm mercaptoethanol, 2% SDS, pH 6.8, for 30 min at 70 °C. Membranes were washed three times for 10 min each in PBS-T, and immunoblotting was performed as described above.
Co-immunoprecipitation and Affinity Pull-down Studies
Co-IP assays were performed using either a polyclonal anti-PDI or anti-BiP antibody or a biotin-labeled propeptide of the α-conotoxin GI. Briefly, venom glands were homogenized in 1% digitonin in Tris-buffered saline (TBS; 100 mm Tris, 150 mm NaCl, pH 7.5) containing 1× protease inhibitor mixture by Dounce homogenization followed by incubation on ice for 30 min. Cellular debris was removed by centrifugation at 12,000 × g for 20 min, and supernatants were precleared with either 70 μl of Protein A-Sepharose pre-equilibrated in lysis buffer, MabSelect, GE Healthcare) or 70 μl of streptavidin-agarose (Sigma-Aldrich) for 1 h at 4 °C with mixing. Precleared supernatants were incubated with 40 μl of anti-PDI- or anti-BiP-conjugated Protein A-Sepharose slurry or pro-GI-conjugated biotin/streptavidin-agarose for 1 h at 4 °C with mixing. Non-immunized rabbit serum (Sigma-Aldrich) and a random biotin-labeled control peptide (YARF) were used to establish nonspecific binding. Following incubation, beads were washed six times with 0.1% digitonin in TBS containing 1× protease inhibitor mixture. Bound proteins were eluted by incubating with Laemmli sample buffer with and without 100 mm DTT for 10 min at 100 °C. Proteins were analyzed by SDS-PAGE followed by in-gel tryptic digest or immunoblotting as described above.
In-gel Digestion and Protein Identification
In-gel digestion was performed as described previously (31) with minor modifications. Briefly, gel spots were excised, washed in 50% ACN, triethylammonium bicarbonate, and reduced with 20 mm DTT, followed by alkylation in 100 mm iodoacetamide. In-gel digestion was performed using sequencing grade trypsin (Sigma-Aldrich) at a final concentration of 10 μg/ml in 25 mm triethylammonium bicarbonate. Peptides extracted after overnight digestions were separated on a C18 reversed-phase column (ProteCol nanocolumn, pore size 300 Å, particle size 3 μm, dimensions 75 μm × 100 mm, SGE Analytical Sciences) and analyzed using a Hybrid Quadrupole-TOF LC-MS/MS mass spectrometer (QSTAR Elite, AB SCIEX). Acquired data were analyzed using Analyst QS software (version 2.0, AB SCIEX). MS/MS data were searched against an in-house database that contained all molluscan proteins submitted to UniProt (n = 47,252) using Protein Pilot software (version 3.0, AB SCIEX) with the following selections: iodoacetamide, trypsin gel-based identification, biological modifications, thorough ID. The false discovery rate cut-off was set to 5%. Peptides with confidence scores of ≥99 were selected for protein identifications.
RESULTS
cDNA Sequencing of PDI
Conus PDI was successfully sequenced from the venom gland of C. novaehollandiae. Mature C. novaehollandiae PDI is encoded by 1449 nucleotides translating to a putative protein of 54,682 Da (GenBankTM accession number JQ745296). The predicted protein is most homologous to PDI sequenced from Conus virgo (92% amino acid sequence identity, 97% homology, GenBankTM accession number ADZ76590) and shares 61% sequence identity (77% homology) with the human homologue (GenBankTM accession number NP_000909). C. novaehollandiae PDI exhibits the classical PDI domain organization with two catalytic a and a′ domains and the non-catalytic b and b′ domains. The active site motif is -CGHC- in both catalytic domains.
Characterization of Recombinant Proteins
All three enzymes were expressed without their N-terminal signal sequence and purified to ∼90% purity by metal affinity and size exclusion chromatography (Fig. 1). The identity of recombinant proteins was confirmed by in-gel tryptic digestion of SDS-polyacrylamide gel bands (data not shown). Functional integrity was validated by enzyme-specific activity assays (Fig. 1). To assess enzymatic activity of Conus PDI, the insulin turbidity assay was performed. In the absence of PDI, insulin was slowly reduced by DTT, as observed by an increase in absorbance at 650 nm (Fig. 1A). The addition of PDI led to a substantial increase of insulin reduction, confirming that the active site cysteines efficiently unfolded the disulfide bonds of insulin (Fig. 1A).
FIGURE 1.

Purity and integrity of recombinant Conus enzymes as determined by SDS-PAGE and enzyme-specific activity assays. The purity of recombinant enzymes was determined by SDS-PAGE (right panels). A total of 3 μg of recombinant protein was separated on 12% Tris-glycine gels under reducing conditions. Proteins were visualized by Coomassie staining. A, PDI activity was measured using the insulin reductase assay. The addition of PDI (0.5 and 2.5 μm) caused rapid reduction of insulin, as observed by an increase in absorbance at 650 nm. This effect was concentration-dependent. B, the cis-trans isomerase activity of PPI B was measured using the coupled chymotrypsin assay. The addition of PPI B (0.3 or 3 μm) resulted in an increase in the rate of cis-trans isomerization of the oligopeptide N-succinyl-Ala-Ala-Pro-Phe-p-nitroanilide, a substrate of chymotrypsin (mean ± S.D.). Enzymatic cleavage leads to liberation of p-nitroanilide and an increase in absorbance at 390 nm. This reaction was concentration-dependent. C, the ATPase activity of BiP was determined using the NADH-coupled photometric assay. Hydrolysis of ATP was followed by monitoring depletion of NADH at 340 nm. The addition of ADP led to a sudden decrease in absorbance, confirming the functionality of the assay. In the presence of BiP (2.5 and 5 μm), absorbance slowly decreased over the time course of the assay, whereas no effect was observed for the no enzyme control. This effect was concentration-dependent.
Enzymatic activity of PPI B was tested using the coupled chymotrypsin assay. In the absence of enzyme, the peptidyl-prolyl bond of the oligopeptide N-succinyl-Ala-Ala-Pro-Phe-p-nitroanilide slowly underwent cis to trans isomerization, resulting in proteolytic cleavage by chymotrypsin. Accumulation of liberated p-nitroanilide led to an increase in absorbance at 390 nm (Fig. 1B). This reaction was accelerated in the presence of Conus PPI B, confirming the cis-trans isomerization activity of the recombinant enzyme (Fig. 1B).
To determine the ATPase activity of BiP, the NADH depletion assay was employed (25). In the absence of BiP, the addition of ADP led to a rapid depletion of NADH, verifying the functionality of the assay (Fig. 1C, black arrows). In the presence of BiP and ATP, NADH was depleted via BiP-catalyzed ATP hydrolysis over the time course of the experiment, whereas depletion was not observed in the no enzyme control (Fig. 1C).
Enzyme-assisted Oxidative Folding Studies
To investigate the role of Conus PDI, PPI B, and BiP in the folding of disulfide-rich peptides, the two α-conotoxins GI and ImI were tested as folding substrates (Fig. 2). Peptides were selected based on their well characterized in vitro oxidative folding properties (27–29). All α-conotoxins contain 4 cysteine residues that can potentially adopt three disulfide conformations (Fig. 2). The Cys1-3 and Cys2-4 disulfide pairing has been confirmed for a number of α-conotoxins and is generally referred to as the native, globular fold. Although the three-dimensional conformation and bioactivity of the other two isomers, the ribbon and the bead form (Cys1-4 plus Cys2-3 and Cys1-2 plus Cys3-4, respectively), have been studied in detail, these isoforms have not been found in the venom (28, 32).
FIGURE 2.
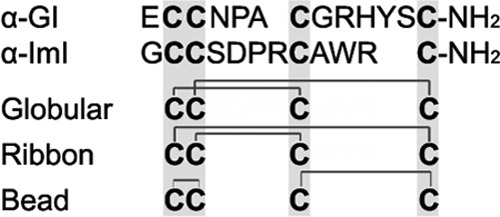
Amino acid sequences and possible disulfide connectivities of α-conotoxins used in this study. Cysteine residues are highlighted in gray. Both peptides are C-terminally amidated. Potential disulfide linkages of the three isomers, the globular, ribbon, and bead form, are depicted.
Conotoxins α-GI and α-ImI are 13 and 12 amino acids in length, respectively. Both peptides are C-terminally amidated and contain one proline. In α-GI, the proline is in position 5, and the Asn4–Pro5 bond most likely adopts a trans configuration (32). This peptide belongs to the 3/5 subfamily of α-conotoxins with 3 residues between the first and second cysteine and 5 amino acids between the third and fourth. While sharing the characteristic disulfide pattern with GI, conotoxin ImI belongs to the 4/3 subgroup of the α-conotoxins with 4 and 3 residues between the first and second set of cysteines, respectively (Fig. 2). ImI contains one proline in position 6, and the Asp5–Pro6 bond has a trans orientation (33).
All reversed-phase elution profiles of oxidative folding reactions concurred with those previously reported for these two peptides (27–29), and the molecular masses of the collected peaks agreed with the calculated masses as determined by MALDI-TOF MS analysis (data not shown). Identities of linear and native peptides were further confirmed by co-elution experiments using commercially purchased peptides as a reference (data not shown). Oxidative folding reactions were quenched by acidification. Previous studies have reported insufficient quenching by acidification of enzyme-assisted folding reactions in some cases (34). To ensure efficient termination of the reaction, replicate samples were analyzed in random order within 12 h following folding assays. No differences were observed between replicate samples run between 1 and 12 h following acidification, indicating efficient termination of the folding assay.
Effect of PDI on the Folding of α-GI and α-ImI
All oxidative folding reactions were carried out in 0.1 m Tris-HCl (pH 7.5), 1 mm MgCl2, 10 mm ATP, 0.1 mm GSH, and 0.1 mm GSSG. Under these conditions, steady state accumulation of native α-GI occurred after ∼64 min (Fig. 3A) with a final yield of 63.2% ± 2.0 (mean ± S.D.). The addition of PDI did not affect final folding yields but significantly increased folding rates (Fig. 3A). This effect was most apparent between 8 and 32 min of folding (Fig. 3A). The effect of PDI on the folding of α-GI was abolished when PDI was inactivated by denaturation for 20 min at 95 °C prior to the folding assay (data not shown).
FIGURE 3.

Analysis of PDI-assisted folding and reduction of α-GI and α-ImI. A–C, oxidative folding was carried out in the presence of 0.1 mm GSH, 0.1 mm GSSG, and 20 μm linear peptide with and without Conus 1 μm PDI. D–F, peptide reduction studies were performed in the presence of 5 mm GSH, 20 μm folded peptide with and without Conus PDI. Folding and reduction reactions were terminated by acid quenching and analyzed by reversed-phase chromatography (C18 column, Vydac-Grace). Relative abundances of fully folded globular α-GI (A and D), globular α-ImI (B and E), and ribbon α-ImI (C and F) were determined from three independent experiments (mean ± S.D. (error bars)).
Conotoxin α-ImI exhibited faster folding kinetics with steady state conditions reached after ∼16 min (Fig. 3, B and C). Unlike α-GI, this peptide folds into two distinct isomers in vitro, the globular and the ribbon form, with almost equal yields at steady state conditions (Fig. 3, B and C). Interestingly, PDI had little effect on the folding rates of globular α-ImI (Fig. 3B) but significantly increased the folding rates of the ribbon isomer (Fig. 3C).
In addition to its oxidation and isomerization activity, PDI is known to catalyze the reduction of disulfide bonds (4). To assess its reductase activity on α-GI and α-ImI, reductase assays were carried out in the presence of 5 mm GSH with and without PDI. The addition of PDI significantly accelerated unfolding rates of α-GI with a 6-fold decrease in the half-time of disappearance of the folded form when PDI was present (Fig. 3D). A similar effect, although less distinct, was observed for the ribbon isomer of α-ImI (Fig. 3F). The addition of PDI led to a 2-fold decrease in the half-time of the disappearance of this peptide. Unfolding kinetics of the globular form remained unchanged (Fig. 3E).
Effects of PDI and PPI B
To elucidate the effect of PDI in combination with PPI B, folding assays were carried out as described above in the presence of Conus PDI, PPI B, or both PDI and PPI B. The fastest folding rates for α-GI were observed in the presence of both PDI and PPI B, although both enzymes individually afforded a rate increase over the uncatalyzed reaction (Fig. 4). At 16 min, accumulation of the native peptide increased from 28.0 ± 0.33 to 32.0 ± 0.57% (p < 0.01) and 42.6 ± 1.35% (p < 0.01) in the presence of PPI B or PDI, respectively (Fig. 4 and Table 2). This further increased to 48.2 ± 0.25% in the presence of both enzymes.
FIGURE 4.

Analysis of PDI- and PPI-assisted folding of α-GI. Oxidative folding studies were performed in the presence of 0.1 mm GSH, 0.1 mm GSSG, and 20 μm linear peptide with and without Conus PDI and/or PPI. Folding reactions were acid-quenched at 0, 4, 8, 16, 32, and 64 min and analyzed by reversed-phase chromatography. A, relative abundance of the fully folded, globular peptide was determined by reversed-phase chromatography as shown in B and plotted against time points of folding. Plotted values are averages from three independent experiments (mean ± S.D. (error bars)). B, reversed-phase chromatograms of folding reactions quenched after 16 min. White circles denote the folded, globular peptide as determined by its characteristic elution profile (27, 29) and co-elution experiments.
TABLE 2.
Folding yields of globular α-GI and globular and ribbon α-ImI obtained at early and late time points of folding in the presence and absence of PDI and/or PPI B
Values represent mean ± S.D. calculated from three independent experiments.
| Peptide | Time point | Percentage of folded peptide |
|||
|---|---|---|---|---|---|
| No enzyme | PDI | PPI | PDI + PPI | ||
| min | % | % | % | % | |
| Globular α-GI | 16 | 28.0 ± 0.33 | 42.6 ± 1.35 | 32.0 ± 0.57 | 48.2 ± 0.25 |
| 64 (end point) | 63.2 ± 1.98 | 64.2 ± 0.18 | 62.3 ± 0.26 | 66.8 ± 1.94 | |
| Globular α-ImI | 4 | 35.4 ± 0.72 | 35.3 ± 0.93 | 38.8 ± 1.43 | 35.0 ± 0.47 |
| 16 (end point) | 43.5 ± 0.17 | 43.9 ± 0.19 | 40.3 ± 0.6 | 43.2 ± 1.94 | |
| Ribbon α-ImI | 4 | 11.5 ± 0.28 | 22.8 ± 1.18 | 15.6 ± 0.29 | 31.1 ± 0.18 |
| 16 (end point) | 45.6 ± 2.0 | 45.12 ± 0.19 | 44.2 ± 0.6 | 45.3 ± 1.94 | |
PDI and PPI B significantly increased the folding rates of the ribbon form of α-ImI (Fig. 5, B and C, and Table 2) but had little effect on the appearance of the globular form (Fig. 5A). Again, the fastest folding rates were achieved when both enzymes were present. At 4 min, accumulation of ribbon α-ImI increased from 11.5 ± 0.28% to 15.6 ± 0.29% (p < 0.001) and 22.8 ± 1.18% (p < 0.001) in the presence of PPI B and PDI, respectively (Fig. 5, B and C, and Table 2). This further increased to 31.1 ± 0.18% (p < 0.001) when both enzymes were present. The addition of PDI and PPI B significantly affected the folding rates of α-GI and ribbon α-ImI but did not alter their final folding yields (Figs. 4A and 5B and Table 2). Enzyme-mediated changes in folding rates were abolished when PDI and PPI B were heat-inactivated for 20 min at 95 °C prior to folding assays (data not shown).
FIGURE 5.

Analysis of PDI- and PPI-assisted folding of α-ImI. Oxidative folding studies were performed in the presence of 0.1 mm GSH, 0.1 mm GSSG, and 20 μm linear peptide with and without Conus PDI and/or PPI B. Folding reactions were acid-quenched at 0, 2, 4, 8, 16, and 32 min and analyzed by reversed-phase chromatography. Relative abundances of the fully folded globular (A) and fully folded ribbon peptide (B) were determined by reversed-phase chromatography as shown in C and plotted against time points of folding. Plotted values are averages from three independent experiments (mean ± S.D. (error bars)). C, reversed-phase chromatograms of folding reactions quenched after 4 min. White circles denote the folded globular peptide, and gray circles show the ribbon isomer, as determined by their characteristic elution profiles (28) and co-elution experiments.
Effects of PDI and BiP
To investigate the effect of PDI in combination with BiP, oxidative folding assays were carried out in the presence of Conus PDI, BiP, or both PDI and BiP. Conus BiP alone did not exhibit any apparent effect on the oxidative folding rates of α-GI and α-ImI under various enzyme concentrations and redox conditions tested. Although PDI maintained the ability to accelerate folding rates of α-GI and the ribbon isomer of α-ImI, no additional effect was observed when BiP was added (data not shown). To investigate whether recombinant BiP required additional co-factors for proper action, folding reactions were supplemented with rat microsomal extracts. Changes in the accumulation of folded peptides were determined at early time points of folding. Reactions were quenched at 4 and 8 min for α-ImI and α-GI, respectively. The addition of microsomal proteins alone led to an increase in the accumulation of folded α-GI (13.1 ± 0.19% versus 5.8 ± 0.16% without microsomes) and more rapid disappearance of the linear form after 8 min of folding (Fig. 6A), indicating the presence of foldases and/or chaperones in these preparations. This effect was only slightly reduced when microsomal proteins were heat-treated prior to folding (10.4 ± 0.07%), suggesting incomplete denaturing or renaturing of foldases and chaperones or the presence of non-protein co-factors. In the presence of microsomal proteins, the addition of PDI or BiP led to a significant increase in the accumulation of folded α-GI (28.7 ± 0.62 and 18.9 ± 0.3%, respectively (p < 0.001); Fig. 6A). Folding was most efficient when both enzymes were present in the same reaction (32.2 ± 0.27% (p < 0.05); Fig. 6A). For α-ImI, the addition of microsomal extracts alone had little effect on the accumulation of fully folded peptide but affected the disappearance of the linear form (Fig. 6B). However, the addition of PDI or BiP led to a significant increase in the accumulation of the folded ribbon isomer (20.1 ± 1.28 and 24.6 ± 0.75%, respectively, versus 10.9 ± 0.48% without enzyme (p < 0.01); Fig. 6B). This effect was further enhanced when both enzymes were present in the same reaction (30.1 ± 0.98% (p < 0.01); Fig. 6B). Interestingly, accumulation of the globular form was affected to a lesser extent. Although PDI and BiP individually increased the accumulation of the globular form (28.4 ± 0.89 and 28.8 ± 1.37%, respectively, versus 17.8 ± 0.60% without enzyme (p < 0.01); Fig. 6B), no additive effect was observed in the presence of both enzymes (28.5 ± 0.71%; Fig. 6B).
FIGURE 6.
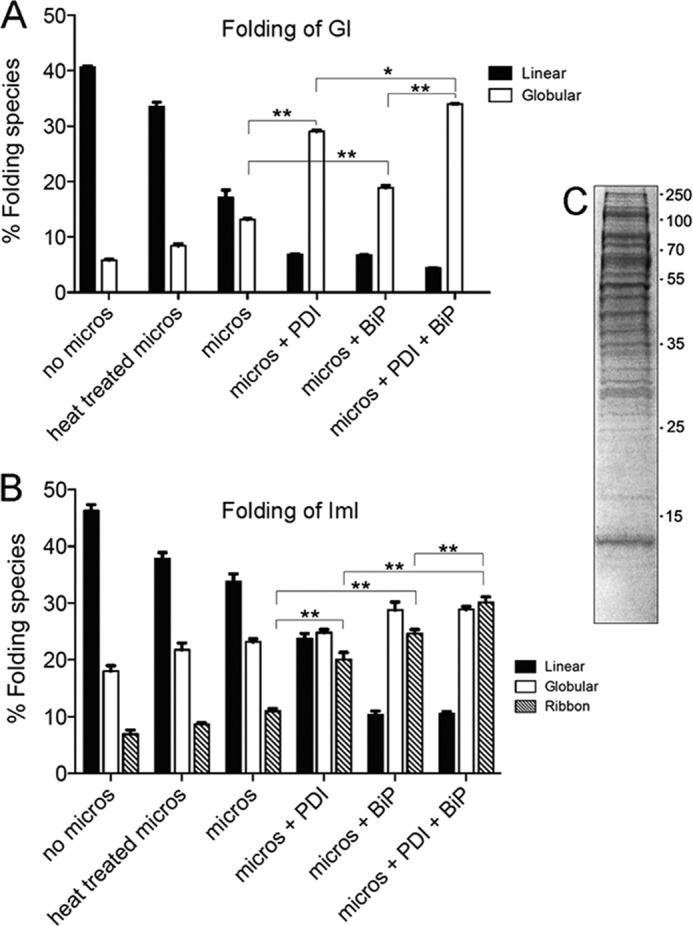
A and B, analysis of PDI and BiP-assisted folding of α-GI (A) and α-ImI (B) in the presence of rat liver microsomal proteins. Folding was carried out in the presence of 40 μg of microsomal proteins, 0.1 mm GSH, 0.1 mm GSSG, 1 mm MgCl2, 10 mm ATP, and 20 μm linear peptide with and without Conus PDI and/or BiP. Folding reactions were acid-quenched at 8 and 4 min for α-GI and α-ImI, respectively, and analyzed by reversed-phase chromatography. Plotted values are averages from three independent experiments (mean ± S.D. (error bars)). Statistical analysis was performed using two-tailed Student's t tests with unequal variance (*, p < 0.05; **, p < 0.001). C, microsomal proteins were analyzed by SDS-PAGE. A total of 20 μg was separated on a 12% Tris-glycine gel under reducing conditions.
Gel electrophoresis of microsomal extracts revealed a complex mixture of proteins characteristic of these preparations (Fig. 6C). To determine the concentration of protein-bound and free sulfhydryl groups that could potentially affect PDI-mediated thiol-disulfide exchange reactions, microsomal extracts were analyzed using Ellman's reagent. The concentration of protein-bound thiols was 67 ± 1.4 μm, and that of free sulfhydryl groups was below the detection limit of the assay (<15 μm).
In summary, all three recombinant enzymes affected the oxidative folding kinetics of α-GI and the ribbon isomer of α-ImI, a disulfide species believed to be absent from the venom. Folding rates of globular, so-called native α-ImI remained unchanged. These observations led us to subsequently investigate the presence of the ribbon isomer of α-ImI in the venom of C. imperialis.
Detection of α-ImI Isomers in the Venom of C. imperialis
To determine the presence of different isomers of α-ImI in the venom of C. imperialis, reversed-phase elution profiles of synthetic ribbon and globular α-ImI were compared with those of the crude venom. Using a linear gradient from 5 to 100% buffer B (90% ACN, 0.1% TFA) over 80 min, the synthetic ribbon and globular isomers eluted at 28.1 min (23.8% ACN) and 29.5 min (25.3% ACN), respectively (Fig. 7, A and B, yellow line). Analysis of the venom of C. imperialis revealed the presence of two corresponding peaks (Fig. 7, A and B, green line). Both peaks strongly increased in size upon the addition of synthetic peptides (Fig. 7, A and B, blue line), suggesting the presence of both isomers in the crude venom. The two corresponding venom fractions were collected and subjected to MALDI-MS analysis. Peptides with identical masses to folded α-ImI were identified in both peaks (m/z = 1351.5; Fig. 7, C and D), whereas no mass matches were observed in interjacent fractions (data not shown). To further verify the presence of the two isomers in the venom, venom fractions were subjected to ESI-MS/MS analysis. Comparison of the venom peptides with their co-eluting synthetic analogues revealed the presence of two characteristic fragment ions only present in the co-eluting synthetic and venom-derived globular isomer (m/z = 668.3 and 1099.3; data not shown). The absence of these peaks in both the synthetic and venom-derived co-eluting ribbon fractions strongly suggests a different disulfide connectivity of these two isobaric peptides. Collectively, reversed-phase chromatography and mass spectrometric analysis led to the identification of the ribbon isomer in the venom of C. imperialis and confirmed the presence of its globular form. To our knowledge, this represents the first evidence for the existence of a “non-native” peptide isoform in the venom of Conus.
FIGURE 7.

Analysis of α-ImI isomers in the venom of C. imperialis. A, overlay of reversed-phase chromatograms of crude C. imperialis venom (green; 4 mg) with the ribbon and globular isomer of ImI (orange; 2 nmol each) and crude venom spiked with 6 nmol of each ImI isomer (blue). Samples were separated on a semipreparative C18 column (5-μm particle size, 10 × 250 mm, Vydac-Grace) using a linear gradient from 5 to 100% buffer B (90% ACN, 0.1% TFA) over 80 min. B, enlargement of reversed-phase chromatogram depicted in A. Venom fractions co-eluting with ribbon (C) and globular ImI (D) were collected and subjected to MALDI-MS. Synthetic isomers were analyzed for comparison. MALDI-MS analysis shows the presence of peptides with the same mass/charge ratio as ImI in venom fractions that co-eluted with the ribbon (C) and globular (D) peptide (m/z 1351.5).
Investigation of Protein Associations in the Venom Glandular Cells
Our findings on enzyme-assisted folding and unfolding of conotoxins strongly indicate that their proper assembly is a multienzyme-assisted process. To determine whether PDI, PPI B, and BiP form a complex in the ER of the secretory cells of the venom gland, BN-PAGE and co-IP experiments were performed on the venom gland of C. victoriae. Soluble complexes were separated from membrane-associated ones by ultracentrifugation. Initial experiments showed that the best solubilization of protein complexes was achieved with 1% digitonin for soluble and 2% CHAPS for membrane preparations. Under these conditions, a complex with immunopositive staining for BiP and PDI was identified in both protein preparations (Fig. 8A, black arrows). To further verify the identity of BiP and PDI, bands were excised from BN-polyacrylamide gels (Fig. 8A, box Bi and Bii) and subjected to either in-gel tryptic digest combined with mass spectrometric identification or second dimension denaturing SDS-PAGE followed by immunoblotting. Both techniques confirmed the presence of PDI and BiP in the soluble and membrane protein preparations (Fig. 8, B and E). The soluble complex appeared smaller (200–300 kDa) and may have separated from the larger, membrane-bound complex (300–400 kDa) during protein preparation (Fig. 8A). Interestingly, besides the 55-kDa PDI protein, an additional PDI isoform of ∼120 kDa was observed in the soluble fraction (Fig. 8Bi). This 120-kDa protein is unlikely to represent a dimer of PDI because BN-polyacrylamide gel slices were incubated in denaturing buffer under strongly reducing conditions prior to second dimension electrophoresis (30 min in 2% SDS and 100 mm mercaptoethanol). A faint band of ∼120 kDa was also visible in the membrane fraction (Fig. 8Bii), with the main PDI isoform migrating at ∼90 kDa. In a previous study (16), immunoblotting of two-dimensional gels revealed the presence of larger molecular weight isoforms of PDI in the venom glands of C. victoriae and C. novaehollandiae. Whether the 90 kDa band represents a different PDI isoform remains to be determined because proteins excised from BN-PAGE can exhibit deviating migration patterns due to the presence of encasing BN gel matrices.3 Second dimension SDS-PAGE illustrated the presence of additional proteins of unknown identity, particularly for the membrane complex (Fig. 8, Bi and Bii). Bands corresponding to the size of PPI B (∼20 kDa) and Hsp40 (∼40 kDa) were observed. Unfortunately, due to low protein concentrations and/or a lack of sequence homology to published protein sequences, mass spectrometry could not resolve the identity of these species. Furthermore, immunoblotting using an anti-PPI B antibody was unsuccessful because commercial polyclonal antibodies raised against human PPI B did not cross-react with the Conus enzyme. Future studies using antibodies raised against Conus PPI B will determine whether PPI B forms part of the BiP-PDI complex.
FIGURE 8.

Investigations of protein-protein interactions in the venom gland of Conus. A, blue native polyacrylamide gel electrophoresis (BN-PAGE) of soluble and membrane-associated protein complexes isolated from the venom gland of C. victoriae. Gels were immunoblotted using anti-PDI- and anti-BiP-specific antibodies (AB). A band with immunopositive staining for PDI and BiP was identified in both preparations (black arrows). Corresponding bands were excised from BN-polyacrylamide gels and further analyzed by immunoblotting and second dimension SDS-PAGE under denaturing conditions (Bi and Bii) followed by in-gel tryptic digestion (see E for results). Both techniques confirmed the presence of PDI and BiP as part of the protein complex. Ci, co-IP of proteins in the venom gland of C. victoriae using an anti-BiP antibody. Immunoprecipitated proteins were separated by SDS-PAGE and analyzed by in-gel tryptic digestion (bands 1, 2, 3, and 4; see E for results) and immunoblotting using an anti-PDI-specific antibody. In-gel tryptic digestion identified BiP and PDI in BiP co-IP samples but not in rabbit serum controls. Immunoblotting further revealed the presence of PDI in BiP Co-IP samples (black arrow), confirming the association between BiP and PDI. Ciii, to rule out the possibility that PDI-positive staining was derived from the secondary antibody, membranes were stripped and reblotted using secondary antibody alone. No staining of the PDI band was observed for reblotted membranes. Di, affinity purification of PDI using the biotin-labeled propeptide of α-GI. Biotin-labeled pro-GI was incubated with venom gland protein lysates of C. geographus. Proteins interacting with pro-GI were pulled down using streptavidin beads and analyzed by SDS-PAGE, in-gel tryptic digestion (see E for results), and immunoblotting using an anti-PDI-specific antibody. In-gel tryptic digestion demonstrated that PDI was successfully affinity-purified using biotin-labeled pro-GI but not using a random biotin-labeled control peptide (YARF). Dii, immunoblotting using an anti-PDI antibody further confirmed the identity of the PDI band (white arrow). E, results of in-gel tryptic digestions. Tryptic peptides were analyzed by ESI-MS/MS on the QStar Elite Hybrid Q-TOF mass spectrometer (AB SCIEX). MS/MS data were searched against an in-house molluscan protein database using Protein Pilot software (version 3.0, AB SCIEX). Peptides with a confidence of ≥99 and a false discovery rate of ≤5 were accepted for protein identifications.
Because proteins that co-migrate on BN-polyacrylamide gels do not necessarily interact, co-IP experiments were performed to confirm the direct association between PDI and BiP. Initial IP approaches using the polyclonal anti-PDI antibody did not result in efficient immunoprecipitation of PDI, rendering this antibody unsuitable for co-IP experiments. However, the interaction between PDI and BiP was successfully confirmed using the anti-BiP-specific antibody (Fig. 8C). Co-immunoprecipitated proteins were separated by SDS-PAGE alongside whole lysates under reducing and non-reducing conditions (Fig. 8C). A band that was subsequently identified as Conus BiP was observed in co-IP samples, illustrating that the BiP antibody was suitable for IP experiments (Fig. 8Ci, bands 1 and 3). A very faint band corresponding to the molecular weight of PDI could also be observed in BiP co-IP lanes but not in non-immunized rabbit serum controls (Fig. 8Ci, bands 2 and 4). In-gel tryptic digestion unambiguously identified this protein as Conus PDI (Fig. 8E). A number of additional gel bands were observed in BiP co-IP lanes. However, these proteins could not be identified by in-gel tryptic digest. The identity of the PDI protein band was further confirmed by immunopositive staining of co-IP samples using the anti-PDI antibody (Fig. 8Cii, black arrow). Immunostaining of this band was also observed in whole lysates but not in the serum control (Fig. 8Cii). Both anti-PDI and anti-BiP were raised in rabbits, resulting in high background staining. To rule out the possibility that PDI staining resulted from the secondary anti-rabbit antibody, membranes were stripped and reblotted using secondary antibody only. PDI-associated staining could not be observed in BiP co-IP lanes. Together, in-gel tryptic digest and immunoblotting of BiP co-IP samples confirmed the association between PDI and BiP in the venom glandular cells of C. victoriae.
In Vitro Interaction between Conus PDI and Conotoxin α-GI
Several studies have proposed PDI-assisted folding of conotoxins (18, 27, 35–37). However, a direct interaction between PDI and conotoxins has never been demonstrated. To determine whether PDI directly interacts with a conotoxin in vitro, the biotin-labeled propeptide of α-GI was incubated with whole venom gland lysates of C. geographus and subsequently affinity-purified using streptavidin beads. Affinity-purified proteins were analyzed by SDS-PAGE under reducing conditions (Fig. 8Di). A number of proteins specifically associated with pro-GI but not with the random YARF control peptide (Fig. 8Di). Bands were excised and subjected to in-gel tryptic digest and mass spectrometric analysis. Due to a lack of Conus protein sequences represented in public databases, only a limited number of proteins could be identified. One of these proteins was Conus PDI (Fig. 8, Di (white arrow) and E). PDI was associated with pro-GI but not YARF (Fig. 8Di). Immunoblotting using anti-PDI antibody further confirmed that PDI was successfully affinity-purified with biotin-pro-GI but not biotin-YARF (Fig. 8Cii, white arrow).
DISCUSSION
The Conus venom gland can be regarded as one of the most sophisticated organs of disulfide-rich peptide biosynthesis. At any given time, hundreds of biologically diverse and structurally complex peptides are expressed, translated, folded, and secreted in the epithelial cells of this highly specialized organ. It is now well understood that the folding and assembly of larger polypeptides is guided by the action of a number of different foldases and chaperones. Enzymes known to cooperate in the folding of protein substrates include various members of the PDI family; heat shock proteins, such as Hsp90 and BiP; PPI-like enzymes; and lectins, such as calreticulin and calnexin (38). Although our understanding of these events is steadily improving, comparably little is known about the contribution of these enzymes to the assembly of small peptides, especially those comprising multiple disulfide bonds. Given the diversity of peptides generated by cone snails, we believe that conotoxins can serve as model peptides for understanding general mechanisms of peptide folding. In this study, we investigated the contribution of a number of enzymes to the folding of the two α-conotoxins GI and ImI. Our findings suggest that, similar to the assembly of larger polypeptide substrates, the proper folding of conotoxins is a multienzyme-assisted process.
Three enzymes were selected based on their well established roles in the folding of protein substrates and their high expression levels in the venom gland of Conus. We showed that all three enzymes, PDI, PPI B, and BiP, efficiently assist in the oxidative folding of α-GI and α-ImI. PDI represents one of the most abundant soluble proteins in the venom gland of several cone snail species (16, 37) and has previously been shown to improve in vitro folding rates of conotoxin substrates (18). We confirm this finding and further demonstrate the ability of PDI to efficiently unfold disulfide bonds in conotoxins under reducing conditions. The role of this enzyme in oxidative protein folding is well established. In contrast, in vivo PDI-mediated protein unfolding has only been demonstrated for cholera toxin. PDI catalyzes the reduction of the A subunit of this protein, which enables its retrograde transport into the cytosol (39). Whether the PDI-assisted unfolding of conotoxins observed in vitro here also plays a role in vivo remains to be determined.
PDI has been repeatedly implicated in the oxidative folding of conotoxins (18, 36), but a direct interaction has never been demonstrated. Here, we provide evidence for the direct interaction between PDI and the α-conotoxin GI by affinity purification, ultimately establishing the role of this enzyme in the folding of conotoxins. Furthermore, by investigating different folding isomers of α-ImI, we reveal a substrate selectivity of PDI for the ribbon isomer, which was previously believed to be absent from the venom (28, 32). Similar observations were made for the folding of the ribbon isomer in the presence of Conus PPI B. Both enzymes significantly accelerated the folding of ribbon α-ImI, a peptide isomer with otherwise slow cysteine oxidation rates. These observations suggest that both disulfide bond formation and isomerization of the peptide bond to proline are rate-limiting steps in the folding of this isomer. The three-dimensional structure of the ribbon isomer is unknown. However, PPI-assisted folding of ribbon α-ImI suggests that the Asp5–Pro6 bond has a cis orientation or transiently adopts this conformation during folding. For the globular form, the Asp5–Pro6 bond adopts a trans orientation (33) that most likely does not undergo transient trans-cis isomerization during folding. Previous studies have predominantly focused on the pharmacological properties of globular, so-called native conotoxin isomers. The few published studies on receptor selectivity of different α-conotoxin isomers all demonstrated a shift in target affinity upon disulfide rearrangement (40–42). For example, the ribbon isomer of α-Vc1.1 is a weaker antagonist of the bovine nicotinic receptor than the globular peptide (42). In contrast, the ribbon form of AuIB has significantly higher potency at rat nicotinic receptors than the globular peptide (40). These observations together with our findings of ribbon α-ImI in the venom of C. imperialis suggest that cone snails synthesize a variety of different disulfide isomers in order to extend their repertoire of biologically active neuropeptides. We show that whereas the globular isomer of α-ImI rapidly folds under oxidizing conditions, the in vitro folding of the ribbon form is guided by ER-resident foldases.
Folding studies on α-GI and α-ImI carried out in the presence of both enzymes showed an additive rather than a synergistic effect, suggesting that these two enzymes act independently in the folding of the two peptides. Consequently, PPI B is unlikely to present conotoxins containing the correct proline isomer to PDI as previously suggested for ribonuclease T1 (7). Investigations on the concerted folding by PDI and BiP resulted in similar observations. An additive rather than a synergistic effect was observed when PDI and BiP were simultaneously present in folding reactions supplemented with microsomal extracts. BiP does not directly contribute to disulfide-exchange reactions (38). Instead, it may have promoted faster folding by preventing aggregation and/or increasing the solubility of the toxins and/or of PDI (9, 38). The BiP-mediated increase in folding rates was abolished when microsomal proteins were omitted, strongly suggesting that this enzyme requires one or more ER-resident co-factors. Free thiols present in microsomal preparations are unlikely to have contributed to faster folding rates because their concentration was very low. Macromolecular crowding is another factor that is known to contribute to alterations in rates and equilibria of protein interactions. However, the total protein concentrations used in this study were too low to have caused crowding (43). It is now well understood that members of the Hsp70 family interact with a variety of Hsp40 co-chaperones. The major function of Hsp40 is to stimulate the otherwise weak ATPase activity of BiP by increasing the rate of hydrolysis of bound ATP (44). Microsomal extracts are likely to have contained ER-resident Hsp40 co-chaperones that could have contributed to BiP-assisted folding of conotoxins. Because endogenous ATP was not depleted from microsomal preparations, no conclusions can be drawn on the ATP dependence of this reaction.
Although PDI- and BiP-assisted folding did not appear to be synergistic for α-GI and α-ImI, this could be the case for conotoxins that contain very hydrophobic regions and more than two disulfide bonds. We acknowledge that more studies utilizing a wider range of conotoxin substrates are needed to elucidate this possibility in the future.
However, our findings on the additive effect of ER-resident enzymes on the folding of α-conotoxins led us to investigate whether these events may occur in close spatial and/or temporal proximity. This would probably be corroborated by the existence of multienzyme complexes. Immunoprecipitation studies alongside with BN-PAGE experiments clearly identified a complex between PDI and BiP in the venom glandular cells of Conus. Because folding intermediates are highly susceptible to degradation, this multienzyme complex may decrease the time needed to generate conotoxins in their final three-dimensional structure. BN-PAGE followed by second dimension SDS-PAGE revealed that the PDI-BiP complex contained additional proteins, including those migrating at the same molecular weight as PPI B and Hsp40. Attempts to identify these proteins by mass spectrometry and immunoblotting failed due to low concentrations and/or lack of homology with published protein sequences. Future studies utilizing Conus-specific antibodies will determine whether proteins such as PPI B and Hsp40 form part of the BiP-PDI complex. A recent investigation into the existence of multiprotein complexes in mammalian cells identified a complex comprising, among other proteins, PDI, PPI B, BiP, and the Hsp40 protein ERdj3 (13). The authors suggested that a large fraction of ER-resident chaperones forms a network that can bind to unfolded protein substrates rather than existing as free proteins that assemble onto nascent polypeptide chains (13). Based on the contribution of PDI, PPI B, and BiP to conotoxin folding and the existence of a PDI-BiP complex in venom glandular cells, it may be hypothesized that this proposed ER network has been utilized by cone snails for toxin assembly for more than 55 million years and has remained conserved throughout evolution. Notably, high expression levels of PDI, BiP, and PPI were also found in the venom glands of snakes and wasps, animals that secrete disulfide-rich peptides as part of their venoms (45–47). The existence of multienzyme folding complexes in the venom glands of these and other venomous animals has not been investigated. However, studies on the oxidative folding of the scorpion toxin maurotoxin revealed most efficient folding in the presence of PDI and the cytosolic peptidyl-prolyl isomerase FKBP-12 (8). PDI has also been shown to accelerate the folding rates of insulin, an archetypal peptide hormone that contains multiple disulfide bonds (48). Interestingly, recent studies also implicated BiP in the folding and secretion of insulin (49). Based on these observations and our findings on the multienzyme-assisted folding of conotoxins, we hypothesize that small, disulfide-rich peptides abide by similar rules and experience folding environments comparable with those of larger polypeptide chains.
In conclusion, this study is the first to investigate the concerted contribution of a number of highly expressed, ER-resident Conus enzymes to the folding of conotoxins. All enzymes were shown to accelerate oxidative folding rates of two α-conotoxins in vitro, with the most efficient rates in the presence of PDI and PPI B or PDI, BiP, and microsomes. We were able to demonstrate multienzyme-assisted folding of the thermodynamically less favored ribbon isomer of α-ImI and reveal its presence in the venom of C. imperialis. We further provide the first evidence for the direct interaction between PDI and a nascent conotoxin, unequivocally establishing the role of this enzyme in the folding of cone snail peptides. Our subsequent investigations into the presence of multienzyme complexes led to the discovery of a novel Conus ER protein complex, strongly suggesting that conotoxins interact with a variety of enzymes to acquire their correct three-dimensional conformation. In effect, these enzymes form a molecular machine to process and fold these disulfide-rich toxins. Future studies addressing the full composition of this ER complex are likely to extend our understanding of the mechanisms underlying disulfide-rich peptide synthesis.
Acknowledgments
We thank Dr. Robyn Bradbury and John Ahern for specimen collection and maintenance, Dr. Yee-Foong Mok and Zachary Rosenes for assistance with the ATPase assay, Prof. Mike Hubbard for kindly providing the PDI antibody, Dr. Julita Imperial for advice on venom extraction, Dr. William Low for help with mass spectrometry, Srgjan Civciristov and Dr. Philippa Saunders for assistance with BN-PAGE, and Dr. Robert Schlaberg for reviewing the manuscript.
This work was supported, in whole or in part, by National Institutes of Health, NIGMS, Program Project GM48677. This work was also supported by Australian Research Council Grant DP110101331.
H. Safavi-Hemami, unpublished observations.
- ER
- endoplasmic reticulum
- PDI
- protein-disulfide isomerase
- PPI
- peptidyl-prolyl cis-trans isomerase
- BiP
- immunoglobulin-binding protein
- IP
- immunoprecipitation
- ACN
- acetonitrile
- RP-HPLC
- reversed-phase HPLC
- ESI
- electrospray ionization
- Fmoc
- N-(9-fluorenyl)methoxycarbonyl
- BN
- blue native
- BisTris
- 2-[bis(2-hydroxyethyl)amino]-2-(hydroxymethyl)propane-1,3-diol
- Tricine
- N-[2-hydroxy-1,1-bis(hydroxymethyl)ethyl]glycine.
REFERENCES
- 1. Edman J. C., Ellis L., Blacher R. W., Roth R. A., Rutter W. J. (1985) Sequence of protein-disulfide isomerase and implications of its relationship to thioredoxin. Nature 317, 267–270 [DOI] [PubMed] [Google Scholar]
- 2. Hawkins H. C., Freedman R. B. (1991) The reactivities and ionization properties of the active-site dithiol groups of mammalian protein-disulfide isomerase. Biochem. J. 275, 335–339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Schwaller M., Wilkinson B., Gilbert H. F. (2003) Reduction-reoxidation cycles contribute to catalysis of disulfide isomerization by protein-disulfide isomerase. J. Biol. Chem. 278, 7154–7159 [DOI] [PubMed] [Google Scholar]
- 4. Ellgaard L., Ruddock L. W. (2005) The human protein-disulfide isomerase family. Substrate interactions and functional properties. EMBO Rep. 6, 28–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Wilkinson B., Gilbert H. F. (2004) Protein disulfide isomerase. Biochim. Biophys. Acta 1699, 35–44 [DOI] [PubMed] [Google Scholar]
- 6. Galat A. (2003) Curr. Top. Med. Chem. 3, 1313–1347 [DOI] [PubMed] [Google Scholar]
- 7. Schönbrunner E. R., Schmid F. X. (1992) Peptidyl-prolyl cis-trans isomerase improves the efficiency of protein-disulfide isomerase as a catalyst of protein folding. Proc. Natl. Acad. Sci. 89, 4510–4513 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. di Luccio E., Azulay D. O., Regaya I., Fajloun Z., Sandoz G., Mansuelle P., Kharrat R., Fathallah M., Carrega L., Estève E., Rochat H., De Waard M., Sabatier J. M. (2001) Parameters affecting in vitro oxidation/folding of maurotoxin, a four-disulfide-bridged scorpion toxin. Biochemical Journal 358, 681–692 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Kleizen B., Braakman I. (2004) Protein folding and quality control in the endoplasmic reticulum. Curr. Opin. Cell Biol. 16, 343–349 [DOI] [PubMed] [Google Scholar]
- 10. Mayer M., Kies U., Kammermeier R., Buchner J. (2000) BiP and PDI cooperate in the oxidative folding of antibodies in vitro. J. Biol. Chem. 275, 29421–29425 [DOI] [PubMed] [Google Scholar]
- 11. Jessop C. E., Watkins R. H., Simmons J. J., Tasab M., Bulleid N. J. (2009) Protein disulphide isomerase family members show distinct substrate specificity: P5 is targeted to BiP client proteins. J. Cell. Sci. 122, 4287–4295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Okudo H., Kato H., Arakaki Y., Urade R. (2005) Cooperation of ER-60 and BiP in the oxidative refolding of denatured proteins in vitro. J. Biochem. 138, 773–780 [DOI] [PubMed] [Google Scholar]
- 13. Meunier L., Usherwood Y. K., Chung K. T., Hendershot L. M. (2002) A subset of chaperones and folding enzymes form multiprotein complexes in endoplasmic reticulum to bind nascent proteins. Mol. Biol. Cell 13, 4456–4469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Buczek O., Bulaj G., Olivera B. M. (2005) Conotoxins and the posttranslational modification of secreted gene products. Cell. Mol. Life Sci. 62, 3067–3079 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Davis J. M., Jones A., Lewis R. J. (2009) Remarkable inter- and intra-species complexity of conotoxins revealed by LC/MS. Peptides 30, 11222–11227 [DOI] [PubMed] [Google Scholar]
- 16. Safavi-Hemami H., Siero W. A., Gorasia D. G., Young N. D., Macmillan D., Williamson N. A., Purcell A. W. (2011) Specialization of the venom gland proteome in predatory cone snails reveals functional diversification of the conotoxin biosynthetic pathway. J. Proteome Res. 10, 3904–3919 [DOI] [PubMed] [Google Scholar]
- 17. Safavi-Hemami H., Bulaj G., Olivera B. M., Williamson N. A., Purcell A. W. (2010) Identification of Conus peptidylprolyl cis-trans isomerases (PPIases) and assessment of their role in the oxidative folding of conotoxins. J. Biol. Chem. 285, 12735–12746 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Wang Z. Q., Han Y. H., Shao X. X., Chi C. W., Guo Z. Y. (2007) Molecular cloning, expression, and characterization of protein-disulfide isomerase from Conus marmoreus. FEBS J. 274, 4778–4787 [DOI] [PubMed] [Google Scholar]
- 19. Fujiki Y., Hubbard A. L., Fowler S., Lazarow P. B. (1982) Isolation of intracellular membranes by means of sodium carbonate treatment. Application to endoplasmic reticulum. J. Cell Biol. 93, 97–102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Sedlak J., Lindsay R. H. (1968) Estimation of total, protein-bound, and nonprotein sulfhydryl groups in tissue with Ellman's reagent. Anal. Biochem. 25, 192–205 [DOI] [PubMed] [Google Scholar]
- 21. Emanuelsson O., Brunak S., von Heijne G., Nielsen H. (2007) Locating proteins in the cell using TargetP, SignalP, and related tools. Nat. Protoc. 2, 953–971 [DOI] [PubMed] [Google Scholar]
- 22. Pace C. N., Vajdos F., Fee L., Grimsley G., Gray T. (1995) How to measure and predict the molar absorption coefficient of a protein. Protein Sci. 4, 2411–2423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Lambert N., Freedman R. B. (1983) Kinetics and specificity of homogeneous protein-disulfide isomerase in protein disulfide isomerization and in thiol-protein-disulfide oxidoreduction. Biochem. J. 213, 235–243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Fischer G., Bang H., Mech C. (1984) [Determination of enzymatic catalysis for the cis-trans isomerization of peptide binding in proline-containing peptides]. Biomed. Biochim. Acta 43, 1101–1111 [PubMed] [Google Scholar]
- 25. Kreuzer K. N., Jongeneel C. V. (1983) Escherichia coli phage T4 topoisomerase. Methods Enzymol. 100, 144–160 [DOI] [PubMed] [Google Scholar]
- 26. Safavi-Hemami H., Siero W. A., Kuang Z., Williamson N. A., Karas J. A., Page L. R., MacMillan D., Callaghan B., Kompella S. N., Adams D. J., Norton R. S., Purcell A. W. (2011) Embryonic toxin expression in the cone snail Conus victoriae. Primed to kill or divergent function? J. Biol. Chem. 286, 22546–22557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Buczek O., Olivera B. M., Bulaj G. (2004) Propeptide does not act as an intramolecular chaperone but facilitates protein-disulfide isomerase-assisted folding of a conotoxin precursor. Biochemistry 43, 1093–1101 [DOI] [PubMed] [Google Scholar]
- 28. Kang T. S., Radić Z., Talley T. T., Jois S. D., Taylor P., Kini R. M. (2007) Protein folding determinants. Structural features determining alternative disulfide pairing in α- and χ/λ-conotoxins. Biochemistry 46, 3338–3355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Lopez-Vera E., Walewska A., Skalicky J. J., Olivera B. M., Bulaj G. (2008) Role of hydroxyprolines in the in vitro oxidative folding and biological activity of conotoxins. Biochemistry 47, 1741–1751 [DOI] [PubMed] [Google Scholar]
- 30. Wittig I., Braun H. P., Schägger H. (2006) Blue native PAGE. Nat. Protoc. 1, 418–428 [DOI] [PubMed] [Google Scholar]
- 31. Shevchenko A., Tomas H., Havlis J., Olsen J. V., Mann M. (2006) In-gel digestion for mass spectrometric characterization of proteins and proteomes. Nat. Protoc. 1, 2856–2860 [DOI] [PubMed] [Google Scholar]
- 32. Gehrmann J., Alewood P. F., Craik D. J. (1998) Structure determination of the three disulfide bond isomers of α-conotoxin GI. A model for the role of disulfide bonds in structural stability. J. Mol. Biol. 278, 401–415 [DOI] [PubMed] [Google Scholar]
- 33. Maslennikov I. V., Shenkarev Z. O., Zhmak M. N., Ivanov V. T., Methfessel C., Tsetlin V. I., Arseniev A. S. (1999) NMR spatial structure of α-conotoxin ImI reveals a common scaffold in snail and snake toxins recognizing neuronal nicotinic acetylcholine receptors. FEBS Lett. 444, 275–280 [DOI] [PubMed] [Google Scholar]
- 34. Hatahet F., Ruddock L. W. (2009) Protein-disulfide isomerase. A critical evaluation of its function in disulfide bond formation. Antioxid. Redox Signal. 11, 2807–2850 [DOI] [PubMed] [Google Scholar]
- 35. Bulaj G., Buczek O., Goodsell I., Jimenez E. C., Kranski J., Nielsen J. S., Garrett J. E., Olivera B. M. (2003) Efficient oxidative folding of conotoxins and the radiation of venomous cone snails. Proc. Natl. Acad. Sci. U.S.A. 100, 14562–14568 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Fuller E., Green B. R., Catlin P., Buczek O., Nielsen J. S., Olivera B. M., Bulaj G. (2005) Oxidative folding of conotoxins sharing an identical disulfide bridging framework. FEBS J. 272, 1727–1738 [DOI] [PubMed] [Google Scholar]
- 37. Gowd K. H., Krishnan K. S., Balaram P. (2007) Identification of Conus amadis disulfide isomerase. Minimum sequence length of peptide fragments necessary for protein annotation. Mol. Biosyst. 3, 554–566 [DOI] [PubMed] [Google Scholar]
- 38. Brodsky J. L., Skach W. R. (2011) Protein folding and quality control in the endoplasmic reticulum. Recent lessons from yeast and mammalian cell systems. Curr. Opin. Cell Biol. 23, 464–475 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Tsai B., Rodighiero C., Lencer W. I., Rapoport T. A. (2001) Protein-disulfide isomerase acts as a redox-dependent chaperone to unfold cholera toxin. Cell 104, 937–948 [DOI] [PubMed] [Google Scholar]
- 40. Dutton J. L., Bansal P. S., Hogg R. C., Adams D. J., Alewood P. F., Craik D. J. (2002) A new level of conotoxin diversity, a non-native disulfide bond connectivity in α-conotoxin AuIB reduces structural definition but increases biological activity. J. Biol. Chem. 277, 48849–48857 [DOI] [PubMed] [Google Scholar]
- 41. Nicke A., Samochocki M., Loughnan M. L., Bansal P. S., Maelicke A., Lewis R. J. (2003) α-Conotoxins EpI and AuIB switch subtype selectivity and activity in native versus recombinant nicotinic acetylcholine receptors. FEBS Lett. 554, 219–223 [DOI] [PubMed] [Google Scholar]
- 42. Townsend A., Livett B. G., Bingham J. P., Truong H. T., Karas J. A., O'Donnell P., Williamson N. A., Purcell A. W., Scanlon D. (2009) Mass spectral identification of Vc1.1 and differential distribution of conopeptides in the venom duct of Conus victoriae. Effect of post-translational modifications and disulfide isomerisation on bioactivity. Int. J. Pept. Res. Ther. 15, 195–203 [Google Scholar]
- 43. Ellis R. J. (2001) Macromolecular crowding. Obvious but underappreciated. Trends Biochem. Sci. 26, 597–604 [DOI] [PubMed] [Google Scholar]
- 44. Szabo A., Langer T., Schröder H., Flanagan J., Bukau B., Hartl F. U. (1994) The ATP hydrolysis-dependent reaction cycle of the Escherichia coli Hsp70 system DnaK, DnaJ, and GrpE. Proc. Natl. Acad. Sci. U.S.A. 91, 10345–10349 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Rioux V., Gerbod M. C., Bouet F., Ménez A., Galat A. (1998) Divergent and common groups of proteins in glands of venomous snakes. Electrophoresis 19, 788–796 [DOI] [PubMed] [Google Scholar]
- 46. Birrell G. W., Earl S. T., Wallis T. P., Masci P. P., de Jersey J., Gorman J. J., Lavin M. F. (2007) The diversity of bioactive proteins in Australian snake venoms. Mol. Cell. Proteomics 6, 973–986 [DOI] [PubMed] [Google Scholar]
- 47. Fernandes-Pedrosa Mde F., Junqueira-de-Azevedo I. D., Gonçalves-de-Andrade R. M., Kobashi L. S., Almeida D. D., Ho P. L., Tambourgi D. V. (2008) Transcriptome analysis of Loxosceles laeta (Araneae, Sicariidae) spider venomous gland using expressed sequence tags. BMC Genomics 9, 279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Winter J., Klappa P., Freedman R. B., Lilie H., Rudolph R. (2002) Catalytic activity and chaperone function of human protein-disulfide isomerase are required for the efficient refolding of proinsulin. J. Biol. Chem. 277, 310–317 [DOI] [PubMed] [Google Scholar]
- 49. Zhang L., Lai E., Teodoro T., Volchuk A. (2009) GRP78, but not protein-disulfide isomerase, partially reverses hyperglycemia-induced inhibition of insulin synthesis and secretion in pancreatic β-cells. J. Biol. Chem. 284, 5289–5298 [DOI] [PubMed] [Google Scholar]
- 50. Corpuz G. P., Jacobsen R. B., Jimenez E. C., Watkins M., Walker C., Colledge C., Garrett J. E., McDougal O., Li W., Gray W. R., Hillyard D. R., Rivier J., McIntosh J. M., Cruz L. J., Olivera B. M. (2005) Definition of the M-conotoxin superfamily. Characterization of novel peptides from molluscivorous Conus venoms. Biochemistry 44, 8176–8186 [DOI] [PubMed] [Google Scholar]
- 51. Cruz L. J., Kupryszewski G., LeCheminant G. W., Gray W. R., Olivera B. M., Rivier J. (1989) μ-Conotoxin GIIIA, a peptide ligand for muscle sodium channels. Chemical synthesis, radiolabeling, and receptor characterization. Biochemistry 28, 3437–3442 [DOI] [PubMed] [Google Scholar]
- 52. Wang X., Smith R., Fletcher J. I., Wilson H., Wood C. J., Howden M. E., King G. F. (1999) Structure-function studies of ω-atracotoxin, a potent antagonist of insect voltage-gated calcium channels. Eur. J. Biochem. 264, 488–494 [DOI] [PubMed] [Google Scholar]
- 53. Kharrat R., Mansuelle P., Sampieri F., Crest M., Oughideni R., Van Rietschoten J., Martin-Eauclaire M. F., Rochat H., El Ayeb M. (1997) Maurotoxin, a four-disulfide bridge toxin from Scorpio maurus venom. Purification, structure, and action on potassium channels. FEBS Lett. 406, 284–290 [DOI] [PubMed] [Google Scholar]
- 54. Huang Y. H., Colgrave M. L., Daly N. L., Keleshian A., Martinac B., Craik D. J. (2009) The biological activity of the prototypic cyclotide kalata b1 is modulated by the formation of multimeric pores. J. Biol. Chem. 284, 20699–20707 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Gruber C. W., Cemazar M., Clark R. J., Horibe T., Renda R. F., Anderson M. A., Craik D. J. (2007) A novel plant protein-disulfide isomerase involved in the oxidative folding of cystine knot defense proteins. J. Biol. Chem. 282, 20435–20446 [DOI] [PubMed] [Google Scholar]
- 56. Hamanaka Y., Nakashima M., Wada A., Ito M., Kurazono H., Hojo H., Nakahara Y., Kohno S., Hirayama T., Sekine I. (2001) Expression of human β-defensin 2 (hBD-2) in Helicobacter pylori-induced gastritis. Antibacterial effect of hBD-2 against Helicobacter pylori. Gut 49, 481–487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Murray I. (1971) Paulesco and the isolation of insulin. J. Hist. Med. Allied Sci. 26, 150–157 [DOI] [PubMed] [Google Scholar]
- 58. Forte L. R., Currie M. G. (1995) Guanylin. A peptide regulator of epithelial transport. FASEB J. 9, 643–650 [DOI] [PubMed] [Google Scholar]
- 59. Ascenzi P., Bocedi A., Bolognesi M., Spallarossa A., Coletta M., De Cristofaro R., Menegatti E. (2003) The bovine basic pancreatic trypsin inhibitor (Kunitz inhibitor). A milestone protein. Curr. Protein Pept. Sci. 4, 231–251 [DOI] [PubMed] [Google Scholar]
- 60. Ostermeier M., De Sutter K., Georgiou G. (1996) Eukaryotic protein-disulfide isomerase complements Escherichia coli dsbA mutants and increases the yield of a heterologous secreted protein with disulfide bonds. J. Biol. Chem. 271, 10616–10622 [DOI] [PubMed] [Google Scholar]
- 61. Gonzalez R., Andrews B. A., Asenjo J. A. (2002) Kinetic model of BiP- and PDI-mediated protein folding and assembly. J. Theor. Biol. 214, 529–537 [DOI] [PubMed] [Google Scholar]
- 62. Marcinkiewicz C., Weinreb P. H., Calvete J. J., Kisiel D. G., Mousa S. A., Tuszynski G. P., Lobb R. R. (2003) Obtustatin. A potent selective inhibitor of α1β1 integrin in vitro and angiogenesis in vivo. Cancer Res. 63, 2020–2023 [PubMed] [Google Scholar]