

Abstract
The transporter associated with antigen processing (TAP) comprises two subunits, TAP1 and TAP2, each containing a hydrophobic membrane-spanning region (MSR) and a nucleotide binding domain (NBD). The TAP1/TAP2 complex is required for peptide translocation across the endoplasmic reticulum membrane. To understand the role of each structural unit of the TAP1/TAP2 complex, we generated two chimeras containing TAP1 MSR and TAP2 NBD (T1MT2C) or TAP2 MSR and TAP1 NBD (T2MT1C). We show that TAP1/T2MT1C, TAP2/T1MT2C, and T1MT2C/T2MT1C complexes bind peptide with an affinity comparable to wild-type complexes. By contrast, TAP1/T1MT2C and TAP2/T2MT1C complexes, although observed, are impaired for peptide binding. Thus, the MSRs of both TAP1 and TAP2 are required for binding peptide. However, neither NBD contains unique determinants required for peptide binding. The NBD-switched complexes, T1MT2C/T2MT1C, TAP1/T2MT1C, and TAP2/T1MT2C, all translocate peptides, but with progressively reduced efficiencies relative to the TAP1/TAP2 complex. These results indicate that both nucleotide binding sites are catalytically active and support an alternating catalytic sites model for the TAP transport cycle, similar to that proposed for P-glycoprotein. The enhanced translocation efficiency of TAP1/T2MT1C relative to TAP2/T1MT2C complexes correlates with enhanced binding of the TAP1 NBD-containing constructs to ATP-agarose beads. Preferential ATP interaction with TAP1, if occurring in vivo, might polarize the transport cycle such that ATP binding to TAP1 initiates the cycle. However, our observations that TAP complexes containing two identical TAP NBDs can mediate translocation indicate that distinct properties of the nucleotide binding site per se are not essential for the TAP catalytic cycle.
The transporter associated with antigen processing (TAP) plays a key role in major histocompatibility complex (MHC) class I assembly and antigen presentation. The transporter functions in peptide transport from the cytosol into the endoplasmic reticulum, where a dynamic assembly of multiple proteins facilitate the assembly of peptides with newly synthesized MHC class I molecules (1, 2). Subsequently, MHC class I-peptide complexes exit the endoplasmic reticulum and are transported to the cell surface where the complexes are available for recognition by cytotoxic T lymphocytes. The structural organization of the TAP1/TAP2 complex [two nucleotide binding domains (NBDs) and two membrane-spanning regions (MSRs)] is characteristic of the ATP binding cassette family of transmembrane transporters (3). Early studies showed that TAP complexes contained a binding site for peptides and that the peptide binding site comprised elements of both TAP1 and TAP2 (4, 5). Further cross-linking experiments with radiolabeled peptides suggested that regions of the MSRs of TAP1 and TAP2, just N terminal to the NBD, form the peptide binding site (6). Neither TAP1 alone nor TAP2 alone is capable of binding peptides (4). The role of the NBD and nucleotides in peptide binding is controversial. It was first reported that the presence or absence of nucleotides had no effect on peptide binding to TAP complexes (5). More recent reports described impaired peptide binding to mutant TAP complexes in which nucleotide binding was impaired (7). We examined the effects of nucleotides on peptide binding to wild-type TAP complexes or a mutant TAP1(K544M)/TAP2 complex in which nucleotide binding to TAP1 was impaired (8). We showed that, at room temperature, peptide binding affinities and peptide dissociation kinetics were very similar for the TAP1(K544M)/TAP2 mutant complex as for the wild-type complex, both in the presence and absence of nucleotides. These observations indicated a lack of correlation between nucleotide binding to TAP1 and peptide binding to TAP1/TAP2 complexes (8). However, the role of nucleotide binding to the TAP2 subunit for peptide interactions with the TAP complex needs further investigation.
By contrast to peptide binding, it is well established that peptide translocation by TAP complexes is strictly ATP-dependent (4, 9). The two NBDs power the transport of peptides via the hydrolysis of ATP. Nonhydrolyzable ATP analogs do not allow substrate transport across microsomal membranes (10). Impairment in nucleotide interactions with either TAP1 or TAP2 NBDs impairs peptide translocation, indicating a catalytic coupling between the NBDs of TAP1 and TAP2 (8, 11). We noted functional differences between identical Walker A lysine mutations in TAP1 or TAP2, which completely abrogated peptide translocation with TAP2 mutant complexes, but permitted a low level of translocation with TAP1 mutant complexes (8). Other reports have described similar observations (11). These studies, taken together with reports that suggest reduced interactions of nucleotides with TAP2 NBD compared with TAP1 NBD (8, 11–15), raised the question of whether functional distinctions between TAP1 and TAP2 NBDs are important for coordinating the TAP transport cycle. Alternatively, the described differences might be a trivial consequence of structural differences between TAP1 and TAP2 NBDs, given that the structures are nonidentical (60% sequence identity), and therefore chemically distinct. To further understand the role of TAP1 and TAP2 NBDs in peptide binding and transport, we generated human TAP1 and TAP2 chimeras in which the NBDs were exchanged. In the studies described here, we characterize the abilities of different chimera/wild-type combinations to bind and translocate peptides, which allow us to draw mechanistic inferences about the peptide-interaction domains of TAP and the TAP transport cycle.
Methods
Generation of Chimeric Baculoviruses.
We previously described the construction of PCR2.1 vectors, PCR2.1-Tap1-his (containing a DNA sequence encoding a histidine-tagged version of human TAP1 flanked by BamHI and XhoI sites at the 5′ end, and BamHI and NotI sites at the 3′ end) and PCR2.1-Tap2 (containing the human TAP2 sequence flanked by BglII and XhoI sites at the 5′ end, and BglII and NotI sites at the 3′ end) (8). We found unique SapI restriction sites in the sequences corresponding to the Walker A motifs of the TAP1 and TAP2. These sites were used to generate a TAP chimera (T1MT2C) encoding residues 1–541 of TAP1 (T1M) and residues 507–686 of TAP2 (T2C), and a second chimera (T2MT1C) encoding residues 1–506 of TAP2 (T2M) and residues 542–748 of TAP1 (T1C). DNA sequences encoding T1M and T1C were generated by digesting PCR2.1-Tap1-his with NotI and BamHI and subsequently with SapI. DNA sequences encoding T2M and T2C were purified by digesting the PCR2.1-Tap2 vector with BglII and NotI and finally with SapI. For generating a vector encoding T1MT2C, TIM and T2C-encoding fragments were ligated into the baculovirus expression vector pVL1393 (PharMingen) digested with BamHI and NotI, whereas for generating a vector encoding T2MT1C, T2M and T1C-encoding fragments were ligated into pVL1392 (PharMingen) digested with BglII and NotI. All constructs were confirmed by DNA sequencing across the junctions. Recombinant baculoviruses encoding T1MT2C and T2MT1C were generated by using established protocols (Baculovirus Expression Vector System Manual, PharMingen).
Microsome Preparations and Immunoblotting Analyses.
For microsome preparations, Sf21 cells were infected with the appropriate baculovirus at a multiplicity of infection in the range of 5 to 20. Microsomal membrane preparations were carried out as described (8). For direct immunoblotting analysis of the microsomes to determine expression levels, equal amounts of proteins were separated on 12% SDS/PAGE gels and transferred to nitrocellulose membranes, and the membranes were incubated in 15 ml of buffer containing 5 μl anti-his ascites (Covance Scientific, Richmond, CA), 150 μl 148.3 hybridoma supernatant (16), 200 μl 435.3 hybridoma supernatant (17), 2.5 μl Pep3 antiserum (6), or 150 μl anti-TAP2 antiserum (18). Membranes were treated with secondary antibodies conjugated to alkaline phosphatase, and proteins were visualized by using the Bio-Rad alkaline phosphatase-conjugated substrate kit.
Complex Formation Between Chimeras and Wild-Type Subunits.
To assess complex formation, microsomes were lysed in 1% Triton X-100, incubated with 20 μl Protein A Sepharose beads in a preclearing step, then with 0.75 μl anti-his ascites or 200 μl 148.3, then protein A-Sepharose beads, and finally processed for SDS/PAGE analysis as described (8). Immunoblotting analyses were carried out as described above.
Nucleotide Binding Assays.
For assessing the binding of individual constructs or specified combinations to nucleotide-agarose beads, infected cells or microsomes were lysed in 1% Triton-X-100 and incubated for 2 h with 30 μl preswollen ATP-, ADP-, or AMP-conjugated agarose beads (Sigma) (8). The beads were washed, and proteins were eluted with 6× SDS/PAGE buffer. Immunoblotting analyses were carried out as described above.
Peptide Binding Assays.
Peptide binding assays were carried out by using fluorescence quenching assays with the fluorescein-labeled peptide RRYQKCTEL, as described (8, 19) (see supplemental Methods, which are published on the PNAS web site, www.pnas.org).
Peptide Translocation Assays.
Peptide translocation assays were performed as described (8, 16) by using the 125I-labled peptide RRYNASTEL (refer to supplemental Methods).
Results
TAP Chimeras: Antibody Recognition, Complex Formation, and Nucleotide Binding.
An equivalent exchange was made between TAP1 and TAP2 to construct the T2MT1C and T1MT2C chimeras based on sequence alignments (refer to Fig. 6, which is published as supplemental material). We previously described the construction of a baculovirus encoding a histidine-tagged version of wild-type TAP1 (TAP1-his) (8), and we also had obtained baculoviruses encoding untagged versions of wild-type TAP1 and TAP2 from the laboratory of Robert Tampé, Philipps-Universitat, Marburg, Germany (16). We examined the abilities of TAP1- and TAP2-directed antibodies obtained from various laboratories, as well as a commercial anti-his antibody, to recognize T1MT2C and T2MT1C (Fig. 1A). T1MT2C is recognized by the Pep3 antiserum [which was generated against a synthetic peptide in the TAP1 MSR (6)], as well as by anti-TAP2, [generated against a synthetic peptide sequence from the C terminus of TAP2 (18)] (Fig. 1A, lane 4). On the other hand, T2MT1C was recognized by 435.3 [which was raised against a 280-aa C-terminal fragment of TAP2 (17)], by 148.3 [which was raised against a C-terminal peptide of TAP1 (16)], as well as the anti-his antibody (Fig. 1A, lane 3). The peptide sequence against which Pep3 was generated is somewhat conserved in the TAP2 MSR. Cross-reactive recognition by Pep3 of TAP2 and T2MT1C is seen in immunoprecipitation assays that use high quantities of the antibody, but not in immunoblotting assays (Fig. 1A, pep3 blot, lanes 1 and 4, compared with lanes 2 and 3).
Figure 1.

(A) Immunoblotting analyses of microsome preparations expressing the indicated constructs. Pep3 recognizes TAP1 and T1MT2C. 148.3 and anti-his recognize TAP1 and T2MT1C. 435.3 recognizes TAP2 and T2MT1C. Anti-TAP2 recognizes TAP2 and T1MT2C. (B) Complex formation between T1MT2C and T2MT1C. Microsomes expressing the indicated constructs were directly analyzed by immunoblotting assays with 148.3 and anti-TAP2 antiserum (Top and Middle) or were first detergent solubilized, immunoprecipitated with 148.3, and analyzed by immunoblotting with anti-TAP2 (Bottom). (C) Complex formation between TAP1 and T1MT2C and between TAP2 and T2MT1C. Microsomes expressing the indicated constructs were directly analyzed by immunoblotting assays with anti-his or anti-TAP2 (Top and Middle) or were first detergent solubilized, immunoprecipitated with anti-his, followed by immunoblotting analyses with anti-TAP2 (Bottom).
Complex formation between T1MT2C and T2MT1C could be directly ascertained by first immunoprecipitating microsome lysates with the T1C-specific antibody 148.3, followed by immunoblotting analysis with the T2C-specific anti-TAP2 antiserum. Fig. 1B shows that in such immunoprecipitation experiments wild-type TAP2 coprecipitates with TAP1 as expected, and likewise, T1MT2C coprecipitated with T2MT1C. Because of the lack of antibodies with appropriate specificities, it was difficult to directly establish the formation of TAP1/T2MT1C and TAP2/T1MT2C complexes. As described above, Pep3, the only antibody we have directed against T1M, cross-reacts with T2M when used at the high concentrations required for immunoprecipitation assays. Conversely, when the T2M-specific antibody 435.3 is used as the immunoprecipitating antibody, pep3 blots are not interpretable, owing to high backgrounds. Thus, we used peptide binding assays to make inferences about complex formation between TAP1 and T2MT1C and between TAP2 and T1MT2C (see below). Complex formation between TAP1-his and T1MT2C or TAP2 and T2MT1C could be directly ascertained by first immunoprecipitating microsome lysates with the anti-his antibody, followed by immunoblotting analysis with the T2C-specific anti-TAP2 antiserum. We observed that T1MT2C coprecipitated with TAP1-his and that TAP2 coprecipitated with T2MT1C (Fig. 1C), indicating that these combinations indeed form stable complexes.
To test whether the NBD exchange had an effect on nucleotide binding by the chimeric TAP constructs, cell lysates or lysates of microsome preparations were examined for binding to ATP-, ADP-, or AMP-agarose beads. In the first set of experiments, binding to nucleotide agarose beads was compared of TAP1-his relative to T2MT1C (Fig. 2A) and TAP2 relative to T1MT2C (Fig. 2B), by using immunoblotting analyses with the anti-his and anti-TAP2 antibodies, respectively. We observed that both TAP1-his and T2MT1C interacted strongly with ATP- and ADP-agarose beads, but not with AMP-agarose beads (Fig. 2A). Similarly, binding of TAP2 and T1MT2C to ATP- and ADP-agarose beads but not to AMP-agarose beads was detectable (Fig. 2B). The interactions of TAP2 and T1MT2C with ATP-agarose and ADP-agarose beads are more difficult to detect and give weaker signals compared with the corresponding interactions of TAP1 and T2MT1C. Many laboratories have recently reported that TAP2 associates with nucleotides more weakly than TAP1 (11, 15), consistent with our previous experiments that showed that relatively weak signals were obtained for the binding of wild-type TAP2 to nucleotide agarose beads, as well as the binding of 8-azido ATP to TAP2 NBD (8, 14). In the present studies, we could directly compare the binding to nucleotide agarose beads of TAP1 relative to T1MT2C and TAP2 relative to T2MT1C, because the expression levels of both sets of proteins could be normalized with the antibodies pep3 and 435.3, respectively. These experiments also would allow an investigation of whether the extent of nucleotide binding depended on the MSR to which the NBD was anchored. Fig. 2C (pep3 blot) shows that at comparable expression levels of TAP1 and T1MT2C (lanes 1 and 5), significantly higher amounts of TAP1 are bound to ATP- and ADP-agarose beads compared with T1MT2C. Likewise, Fig. 2D (435.3 blot) shows that at comparable expression levels of T2MT1C and TAP2 (lanes 1 and 5), significantly higher levels of T2MT1C are bound to ATP- and ADP-agarose beads compared with TAP2. These observations indicate that TAP2 NBD has reduced ability to bind to nucleotide relative to TAP1 NBD and that reduced binding by TAP2 NBD is an inherent property of the NBD, or a function of its stability, but not a function of its MSR context. As expected from these observations, the TAP1/T2MT1C combination interacted more strongly with both ATP- and ADP-agarose beads compared with the TAP2/T1MT2C combination (Fig. 2 E, pep3 blot, and F, 435.3 blot).
Figure 2.

Nucleotide binding by chimeric and wild-type TAP constructs. (A) Cells expressing the indicated TAP constructs were lysed, immunoprecipitated with anti-his (lanes 1 and 5), or incubated with ATP (lanes 2 and 6), ADP (lanes 3 and 7) or AMP-agarose (lanes 4 and 8) beads, followed by immunoblotting analyses with anti-his. (B–F) Same procedure as in A was followed except that the microsome preparations rather than cell lysates were used to compare expression levels. Immunoblotting analysis was carried out with (B) anti-TAP2, (C and E) Pep3, and (D and F) 435.3.
Requirement of Different Structural Units of TAP for Peptide Binding.
Using the two TAP chimeras and the wild-type TAP subunits, we could generate five different combinations of chimeric TAP complexes to investigate the functions of the different structural units in peptide binding and translocation. These were TAP1/T2MT1C (two TAP1 NBDs), TAP2/T1MT2C (two TAP2 NBDs), T1MT2C/T2MT1C (double chimera), TAP1/T1MT2C (two TAP1 MSRs), and TAP2/T2MT1C (two TAP2 MSRs). For peptide binding experiments, we used a fluorescence quenching-based peptide binding assay done at room temperature in the absence of exogenous ATP (8, 19). For the wild type as well as for the T1MT2C/T2MT1C, TAP1/T2MT1C, and TAP2/T1MT2C complexes, increased fluorescence quenching was observed as microsomes were added to increasing concentrations of peptide (refer to Fig. 7, which is published as supplemental material). The steady-state fluorescence quenching was determined from an exponential fit of the quenching data and are plotted as a function of peptide concentration (Fig. 3). Each experiment summarized in Fig. 3 included a negative control with microsomes prepared from uninfected cells. The amplitudes of the fluorescence quenching signals obtained with microsomes containing wild type or any of the chimeric TAP complexes shown in Fig. 3 was consistently high compared with signals obtained (if any) for microsomes prepared from uninfected cells. The apparent binding constants (KD) were calculated from the steady-state fluorescence quenching vs. peptide concentration plots.
Figure 3.

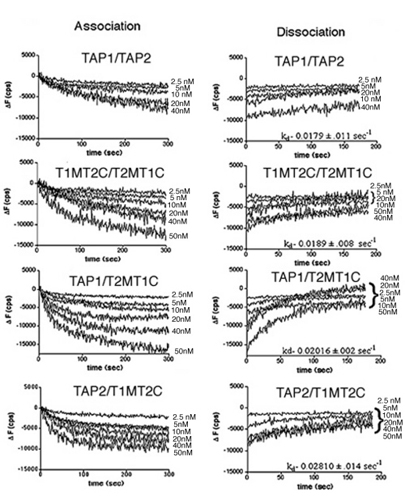
Effect of NBD switching on peptide binding. Microsomal vesicles expressing the indicated TAP complexes or microsomes prepared from uninfected Sf21 cells were assayed for their ability to bind fluorescein-labeled peptides RRYQKCTEL by using fluorescence quenching assays. At each peptide concentration, the steady-state fluorescence quenching was determined and plotted as a function of peptide concentration, and the KD values were estimated. The average of data points from at least two separate experiments with the same microsome preparation is indicated on each plot. At each peptide concentration, signals obtained for microsomes prepared from uninfected cells (negative control) also are indicated on each plot (not curve-fitted). KD values indicated are the average of four (for TAP1/TAP2), four (for T1MT2C/T2MT1C), eight (for TAP1/T2MT1C), and five (for TAP2/T1MT2C) independent binding experiments.
After stabilization of fluorescence signals, unlabeled peptide was added at a concentration of ≈30 μM, and the fluorescence recovery was monitored over several seconds as bound fluorescent peptide dissociated. As for wild-type TAP complexes, fluorescence recovery also was observed for the T1MT2C/T2MT1C, TAP1/T2MT1C, and TAP2/T1MT2C combinations, and the magnitude of the recovery was in proportion to the magnitude of the quenching (refer to Fig. 7). The dissociation rate constants, kd, estimated by fitting the fluorescence recovery signals to a monoexponential function, also are indicated in Fig. 3. The calculated binding constants and dissociation rate constants for peptide interactions with wild-type TAP1/TAP2 are very similar to the values derived for each of the chimeric complexes indicated and are also in agreement with previously reported values (8, 19). Taken together, these observations indicate that functional interactions occur between TAP1 and T2MT1C, TAP2 and T1MT2C, and T1MT2C and T2MT1C.
By contrast to the results described in Fig. 3, fluorescence quenching was not observed upon addition of microsomes expressing the TAP1/T1MT2C and TAP2/T2MT1C combinations to fluorescent peptide (Fig. 4 C and D). The profiles seen for TAP1/T1MT2C and TAP2/T2MTIC combinations resembled the profiles seen for microsomal preparations from uninfected insect cells (cells that expressed neither TAP subunit, Fig. 4A), or microsomes expressing individual chimeras (data not shown). The amplitudes of fluorescence quenching signals were either negative values or low positive values over the concentration range of 2.5 to 80 nM, in several different experiments. Furthermore, in cases where low positive quenching signals were observed, fluorescence recovery was typically not observed upon addition of excess unlabeled peptide. Thus, the fluorescence quenching and recovery profiles for microsomes expressing the TAP1/T1MT2C and TAP2/T2MT1C combinations are distinct from those seen for wild-type TAP complexes (Fig. 4B) or the chimeric complexes described in Fig. 3. Lack of binding is not a consequence of low expression levels of either subunit in the TAP1/T1MT2C and TAP2/T2MT1C combinations (Fig. 4E).
Figure 4.

Requirement for TAP1 and TAP2 MSRs for peptide binding. (A–D) Equal amounts of microsomes expressing the indicated TAP constructs were assayed for binding to fluorescein-labeled RRYQKCTEL. (Left) The indicated concentrations of peptide were added to a constant amount of microsomes in buffer, and fluorescence quenching was recorded for 4–5 min at room temperature. (Right) Five minutes after microsome addition, unlabeled peptide was added and the fluorescence recovery was monitored over 3–4 min. Results shown are representative of at least three independent experiments using at least two separate preparations of microsomes. (E) Immunoblots of the various constructs, corresponding to the binding experiments illustrated in A–D indicate that expression levels of the chimeric TAP combinations were not limiting for binding.
Effect of NBD Switching on Peptide Translocation.
To further assess functionality of each complex, we performed translocation assays at 37°C in the presence of ATP, by using the 125I-labeled peptide RRYNASTEL. Peptide translocation efficiencies were quantitated by using an indirect assay that measures glycosylation of the peptide (10, 16). Microsomes expressing single TAP subunit or either chimera were used as negative controls. By varying viral clones as well as optimizing the multiplicity of infection, it was possible to obtain approximately equivalent expression of the wild-type and chimeric TAP constructs. Within an experiment, microsomes expressing the indicated TAP complexes or controls were tested in triplicate to minimize errors in individual readings, and the average was plotted. Most interestingly, the T2MT1C/T1MT2C complex gave a positive translocation signal (Fig. 5 and Table 1). Comparisons of the overall translocation signal [radioactivity associated with Con A-Sepharose beads in the presence of ATP (cpm(ATP))] in nine independent experiments consistently yielded signals several-fold greater than those observed for single infections with either chimera. However, the translocation efficiency for the double chimera is ≈56% relative to that observed for wild-type TAP [Table 1, compare average cpm(ATP)].
Figure 5.

Peptide translocation assays. Microsomal vesicles expressing the indicated TAP complexes were assayed for their ability to import 125I-labeled RRYNASTEL. Bar graphs represent the average of experiments done in triplicate. Translocation in the presence of ATP is indicated by solid bars and in the presence of Apyrase by open bars. (Insets) Immunoblots of various constructs used in translocation experiments, presented in the same sequence as the translocation experiments.
Table 1.
Microsomes expressing the indicated TAP constructs were assayed for their ability to translocate the radiolabeled model peptide RRYNASTEL
| Construct | N1 | N2 | cpm(ATP) | cpm(Apyrase) |
|---|---|---|---|---|
| TAP1/TAP2 | 8 | 4 | 16,736 ± 492 | 870 ± 190 |
| T1MT2C/T2MT1C | 9 | 5 | 9,479 ± 1,865 | 1,079 ± 249 |
| TAP1/T2MT1C | 4 | 3 | 7,825 ± 2,128 | 1,006 ± 163 |
| TAP2/T1MT2C | 7 | 4 | 4,300 ± 1,980 | 953 ± 217 |
| TAP2 | 6 | 3 | 2,589 ± 996 | 728 ± 195 |
| TAP1 | 3 | 2 | 1,118 ± 254 | 609 ± 290 |
| T1MT2C | 8 | 5 | 784 ± 407 | 534 ± 180 |
| T2MT1C | 4 | 2 | 319 ± 182 | 362 ± 146 |
The average radioactivities associated with Con-A-Sepharose in the presence of ATP [cpm(ATP)] or apyrase [cpm(Apyrase)] for N1 independent experiments each done in triplicate are shown. N2 represents the number of independent microsome preparations used to generate the data.
TAP complexes containing two TAP1 NBDs (TAP1/T2MT1C) complex also were capable of translocating peptide (Fig. 5 Upper). The translocation signals in four independent experiments were consistently significantly greater than those observed for the corresponding single infections with TAP1 or T2MT1C (Table 1). The translocation signal for the TAP1/T2MT1C combination was, on average, at 46% of the wild-type TAP signal and 83% relative to the double chimera. Backgrounds seen with the TAP2 single infection were, on average, higher than any of the other single subunit backgrounds (Table 1). In individual experiments, the translocation signal for the TAP2/T1MT2C combination was about 2- to 3-fold higher than the translocation signal for the TAP2 single infection, indicating that the TAP2/T1MT2C complexes were capable of translocating peptides with low efficiency, but were significantly impaired relative to wild type (Fig. 5 Lower). The translocation signal for the TAP2/T1MT2C combination was, on average, at 25% of the wild-type signal, 45% relative to the double chimera, and 54% relative to TAP1/T2MT1C complexes.
As expected, TAP1/T1MT2C and TAP2/T2MT1C microsomes did not translocate peptides (data not shown), because these complexes were compromised at an earlier step in binding to the peptide (Fig. 4).
Discussion
To further understand the role of the MSRs and NBDs of TAP1 and TAP2 during peptide transport, we generated chimeras by exchanging residues C terminal to the Walker A serine (initiating at residues 542 and 507 of TAP1 and TAP2, respectively). Based on sequence alignments of TAP1 and TAP2 with other ATP binding cassette transporter NBDs (HisP and Malk) (3), we deduce that the MSR/NBD domain boundary in TAP complexes is about 43 residues N terminal to our switch site. Of these, the C-terminal 19 residues have 100% identity between TAP1 and TAP2, so that our switches in effect initiate at residues 523 of TAP1 (for T1MT2C) and 488 of TAP2 (for T2MT1C) (see Fig. 6). The nucleotide binding properties of each chimera resemble those of the parent TAP complex from which the NBD was derived. Thus, we believe that our chimeras are in effect switched in the entire NBD sequences.
No fluorescence quenching or recovery was obtained with microsomes expressing TAP1/T1MT2C or TAP2/T2MT1C complexes, indicating that although these complexes are formed (Fig. 1) both are nonfunctional (Fig. 4). By contrast, TAP1/T2MT1C, TAP2/T1MT2C, and T1MT2C/T2MT1C complexes bind peptides as do wild-type complexes (Fig. 3). These results indicate, as previously implicated (6), that the MSRs of both TAP1 and TAP2 are indispensable for forming a high-affinity peptide-binding site. In the absence of either, substrate binding to the TAP complex is impaired. On the other hand, peptide binding remains unaffected regardless of whether the NBD sequences are from TAP1, TAP2, or both (Fig. 3). These observations indicate that the NBDs do not contain any unique determinants required for peptide binding. These results are consistent with our recent observations of the lack of effect of nucleotides on peptide binding to TAP1/TAP2 complexes at room temperature, but differ from the reports of Knittler and colleagues (7, 11), who suggest that peptide binding by rat TAP complexes requires nucleotide binding (7, 11). We used a fluorescent peptide-based equilibrium binding assay, whereas Knittler and colleagues used photo cross-linking assays with radiolabeled peptides. Photo cross-linking experiments do not measure an equilibrium binding affinity; rather they measure reactivity of neighboring functional groups. It is possible that the photo cross-linking technique is more sensitive to small conformational changes that occur upon nucleotide interactions with TAP1 and TAP2. Alternatively, based on stability assessments of TAP (20), it is possible that rat TAP complexes expressed in mammalian cells cultured at 37°C are more rapidly inactivated in the absence of nucleotides than are human TAP complexes cultured in insect cells at 27°C.
Table 1 lists the chimeric complexes in the order of the relative translocation efficiencies, when efficiencies were compared after optimization of expressions levels to the extent possible. Wild-type TAP complexes are the most efficient, followed by T1MT2C/T2MT1C complexes, and TAP1/T2MT1C next. TAP2/T1MT2C complexes have the lowest efficiency, but are also capable of mediating translocation. The reduced translocation efficiency by all of the chimeric combinations relative to wild-type TAP1/TAP2 might arise due to folding constraints introduced into one or both components of the TAP chimeras. If such constraints exist, our data indicate that these do not interfere with nucleotide binding or peptide binding by these complexes.
It is now well established that in the TAP transport cycle, TAP1 and TAP2 do not independently catalyze peptide translocation. Mutations of Walker A residues of either TAP1 or TAP2 interfere with peptide translocation by TAP complexes (8, 11). These studies indicate that in the TAP transport cycle mutations that inactivate one of the catalytic sites also prevent catalysis at the second site. Our present observations that TAP complexes containing two TAP1 NBDs, and (to a lesser extent) two TAP2 NBDs, can mediate substrate translocation, suggests that both nucleotide binding sites (NBSs) are catalytically active for ATP hydrolysis. The alternating catalytic sites model first proposed by Senior et al. (reviewed in ref. 21) for P-glycoprotein is consistent with both sets of observations. By this model, in the first half-cycle, ATP hydrolysis at the first NBS drives the translocation of a substrate molecule across the membrane, but ATP hydrolysis at the second NBS is prohibited. In the second half-cycle, ATP hydrolysis at the first NBS would be conformationally disallowed, but ATP hydrolysis at the second NBS can drive the translocation of a second substrate molecule. Subsequent alternating catalysis at each NBS is essential for maintaining transport cycles. For some ATP binding cassette transporters such as the multidrug-resistance protein 1 (MRP-1), the observations of distinct functional properties of the NBD and NBD mutants, coupled with observations of significant sequence differences between the NBDs, have led to the postulation of a mechanism whereby one of the NBDs binds ATP with relatively high affinity and may function primarily as a regulatory site. It has been suggested that ATP binding and/or hydrolysis at this NBS would catalyze ATP exchange at the second subunit, with substrate translocation being driven primarily by ATP hydrolysis at the second NBS (ref. 22 and Roger Deeley, personal communication). Functional distinctions also have been proposed between cystic fibrosis transmembrane conductance regulator (CFTR) NBD1 and NBD2 (reviewed in ref. 21); however, more recent experiments with CFTR suggest that ATP binding and hydrolysis by either NBD can gate the CFTR channel (23). In considering functional equivalence vs. functional distinctions between TAP1 and TAP2 NBDs, it is noteworthy that the NBDs of TAP1 and TAP2 (60% sequence identity) are as closely related as are the N-terminal and C-terminal NBDs of P-glycoprotein (60–63% sequence identity), which appear to be functionally equivalent (24). By contrast to TAP and P-glycoprotein, the two NBDs of MRP-1 are considerably less similar to each other (34% sequence identity), consistent with the evidence for distinct functions of the two MRP NBDs.
Our data indicate that the translocation efficiencies of TAP complexes containing two TAP2 NBDs are reduced relative to that observed for two TAP1 NBDs (Fig. 5), which correlates with reduced binding to nucleotides (both ATP and ADP) of TAP complexes containing two TAP2 NBDs (Fig. 2). It is a possibility that reduced nucleotide binding by TAP2 NBDs reflects reduced stability of TAP2 to in vitro purification procedures. Nevertheless, if the observed in vitro differences in the binding of TAP1 vs. TAP2 to nucleotide agarose beads extend to significant differences in the interactions of TAP1 and TAP2 with ATP in vivo at the millimolar concentrations of cytosolic ATP, then TAP1 rather than TAP2 would be expected to be initiator of the catalytic cycle. Once the cycle is initiated, by the alternating catalytic sites model described above, both TAP1 and TAP2 NBDs would have essentially equivalent functions. For P-glycoprotein, a more complex version of the alternating catalytic cycle model has recently been proposed in which ATP hydrolysis was suggested to be required at two distinct steps during a single turnover of the catalytic cycle (25). The first ATP hydrolysis event was suggested to drive extrusion of substrate and also induce an intermediate conformation with low affinity for substrate. The second ATP hydrolysis event was suggested to be a reset step for the reacquisition of a conformation with high affinity for substrate (25). During individual hydrolysis events, the two NBDs were suggested to be randomly recruited, such that neither the N-terminal NBS nor the C-terminal NBS were preferentially used at either step (24). If such a model were applicable to peptide transport by TAP complexes, it is possible that preferential ATP interaction with TAP1, if occurring in vivo, would polarize the transport cycle such that the cycle initiates at TAP1 NBS, resulting in apparently distinct functions for each NBD. However, our observation that TAP complexes containing two TAP1 NBDs are only slightly less efficient than the double chimera suggests that distinct properties of the NBSs per se are not essential for maintaining the TAP catalytic cycle. Thus, our data do not strongly support the possibility that distinct functions of TAP1 and TAP2 NBDs are important for TAP complex function. We predict that the NBDs of other ATP binding cassette transporters such as P-glycoprotein can functionally substitute for the NBDs of TAP, the major limitation being the ability of the heterologous NBDs to interact with TAP1 and TAP2 MSRs.
Supplementary Material
Acknowledgments
We thank Dr. Robert Tampé for the baculovirus constructs encoding TAP1 and TAP2 and the 148.3 antibody; Dr. F. Momburg for the Pep3 antiserum; Dr. M. J. Androlewicz for anti-TAP2 antiserum; the University of Michigan Biomedical Research core facilities for DNA sequencing, peptide synthesis, and purification; the Reproductive Sciences program at the University of Michigan for peptide iodination; the Cell Biology laboratories at the University of Michigan for use of computer resources; and the hybridoma core facility at the University of Michigan for maintenance of 435.3. We are grateful to Dr. R Neubig for helpful suggestions with fluorescence assays and for our use of the PTI fluorimeter, Dr. W. Dunnick for critical review of the manuscript, and Drs. M. Al-Shawi and J. Sheps for helpful discussions of P-glycoprotein mechanisms. We thank Drs. Apurva Sarin and K. Vijayaraghavan at the National Center for Biological Sciences, Bangalore, India for making possible the visiting studentship (to S.A.). This work was supported by National Institutes of Health Grant AI44115–03 (to M.R.).
Abbreviations
- TAP
transporter associated with antigen processing
- NBD
nucleotide binding domain
- MSR
membrane-spanning region
- NBS
nucleotide binding site
Note Added in Proof.
O. Daumke, P. Alberts, and M. Knittler have published an abstract describing the characterization of rat TAP1 and TAP2 chimeras with the NBD of TAP1 and the MSR of TAP2 and vice versa [FEBS Advanced Lecture Course: ATP Binding Cassette (ABC) Proteins: From Genetic Disease to Multidrug Resistance, Abstract Book Page 176]. Their observations were that rat TAP complexes containing identical NBDs had no transport activity.
Footnotes
This paper was submitted directly (Track II) to the PNAS office.
References
- 1.Cresswell P, Bangia N, Dick T, Diedrich G. Immunol Rev. 1999;172:21–28. doi: 10.1111/j.1600-065x.1999.tb01353.x. [DOI] [PubMed] [Google Scholar]
- 2.Momburg F, Hammerling G J. Adv Immunol. 1998;68:191–256. doi: 10.1016/s0065-2776(08)60560-x. [DOI] [PubMed] [Google Scholar]
- 3.Holland I B, Blight M A. J Mol Biol. 1999;293:381–399. doi: 10.1006/jmbi.1999.2993. [DOI] [PubMed] [Google Scholar]
- 4.Androlewicz M J, Ortmann B, van Endert P M, Spies T, Cresswell P. Proc Natl Acad Sci USA. 1994;91:12716–12720. doi: 10.1073/pnas.91.26.12716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Androlewicz M J, Cresswell P. Immunity. 1994;1:7–14. doi: 10.1016/1074-7613(94)90004-3. [DOI] [PubMed] [Google Scholar]
- 6.Nijenhuis M, Hammerling G J. J Immunol. 1996;157:5467–5477. [PubMed] [Google Scholar]
- 7.Knittler M R, Alberts P, Deverson E V, Howard J C. Curr Biol. 1999;9:999–1008. doi: 10.1016/s0960-9822(99)80448-5. [DOI] [PubMed] [Google Scholar]
- 8.Lapinski P E, Neubig R R, Raghavan M. J Biol Chem. 2001;276:7526–7533. doi: 10.1074/jbc.M009448200. [DOI] [PubMed] [Google Scholar]
- 9.Shepherd J C, Schumacher T N, Ashton-Rickardt P G, Imaeda S, Ploegh H L, Janeway C A, Tonegawa S. Cell. 1993;74:577–584. doi: 10.1016/0092-8674(93)80058-m. [DOI] [PubMed] [Google Scholar]
- 10.Neefjes J J, Momburg F, Hammerling G J. Science. 1993;261:769–771. doi: 10.1126/science.8342042. [DOI] [PubMed] [Google Scholar]
- 11.Alberts P, Daumke O, Deverson E V, Howard J C, Knittler M R. Curr Biol. 2001;11:242–251. doi: 10.1016/s0960-9822(01)00073-2. [DOI] [PubMed] [Google Scholar]
- 12.Muller K M, Ebensperger C, Tampé R. J Biol Chem. 1994;269:14032–14037. [PubMed] [Google Scholar]
- 13.Russ G, Esquivel F, Yewdell J W, Cresswell P, Spies T, Bennink J R. J Biol Chem. 1995;270:21312–21318. doi: 10.1074/jbc.270.36.21312. [DOI] [PubMed] [Google Scholar]
- 14.Lapinski P E, Miller G G, Tampé R, Raghavan M. J Biol Chem. 2000;275:6831–6840. doi: 10.1074/jbc.275.10.6831. [DOI] [PubMed] [Google Scholar]
- 15.Hewitt E W, Gupta S S, Lehner P J. EMBO J. 2001;20:387–396. doi: 10.1093/emboj/20.3.387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Meyer T H, van Endert P M, Uebel S, Ehring B, Tampé R. FEBS Lett. 1994;351:443–447. doi: 10.1016/0014-5793(94)00908-2. [DOI] [PubMed] [Google Scholar]
- 17.van Endert P M, Tampé R, Meyer T H, Tisch R, Bach J F, McDevitt H O. Immunity. 1994;1:491–500. doi: 10.1016/1074-7613(94)90091-4. [DOI] [PubMed] [Google Scholar]
- 18.Androlewicz M J, Anderson K S, Cresswell P. Proc Natl Acad Sci USA. 1993;90:9130–9134. doi: 10.1073/pnas.90.19.9130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Neumann L, Tampé R. J Mol Biol. 1999;294:1203–1213. doi: 10.1006/jmbi.1999.3329. [DOI] [PubMed] [Google Scholar]
- 20.van Endert P M. J Biol Chem. 1999;274:14632–14638. doi: 10.1074/jbc.274.21.14632. [DOI] [PubMed] [Google Scholar]
- 21.Senior A E, Gadsby D C. Semin Cancer Biol. 1997;8:143–150. doi: 10.1006/scbi.1997.0065. [DOI] [PubMed] [Google Scholar]
- 22.Gao M, Cui H R, Loe D W, Grant C E, Almquist K C, Cole S P, Deeley R G. J Biol Chem. 2000;275:13098–13108. doi: 10.1074/jbc.275.17.13098. [DOI] [PubMed] [Google Scholar]
- 23.Ikuma M, Welsh M J. Proc Natl Acad Sci USA. 2000;97:8675–8680. doi: 10.1073/pnas.140220597. . (First Published July 4, 2000, 10.1073/pnas.140220597) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sauna Z E, Ambudkar S V. J Biol Chem. 2001;276:11653–11661. doi: 10.1074/jbc.M011294200. [DOI] [PubMed] [Google Scholar]
- 25.Sauna Z E, Ambudkar S V. Proc Natl Acad Sci USA. 2000;97:2515–2520. doi: 10.1073/pnas.97.6.2515. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}