

Abstract
The enantiomers and the cis isomers of two previously studied 4,5-disubstituted oxazolidinones have been synthesized and their binding to the T-box riboswitch antiterminator model RNA investigated in detail. Characterization of ligand affinities and binding site localization indicate that there is little stereospecific discrimination for binding antiterminator RNA alone. This binding similarity between enantiomers is likely due to surface binding, which accommodates ligand conformations that result in comparable ligand-antiterminator contacts. These results have significant implications for T-box antiterminator-targeted drug discovery and, in general, for targeting other medicinally relevant RNA that do not present deep binding pockets.
Introduction
Designing medicinal agents that specifically bind and disrupt the function of RNA is an evolving field of study1-6 whose importance will only grow as our knowledge of the role of noncoding RNAs continues to expand. Noncoding RNAs, as the name implies, do not encode a protein product, but instead are often critical regulators of gene expression.7,8 One medicinally relevant example of a noncoding RNA is the T-box transcription antitermination riboswitch.9 The T-box riboswitch is located in the 5′-untranslated mRNA region (5′-UTR) of many important Gram-positive bacterial genes and controls transcription of the protein-encoding region of the mRNA.10 We recently reported lead oxazolidinone compounds that act as molecular effectors of T-box riboswitch function.11,12 As part of a comprehensive effort to design and develop potential medicinal agents for disrupting the T-box riboswitch,11-15 we wished to investigate the details of the binding interaction between these lead oxazolidinones and their target, the T-box antiterminator RNA structural element.
During transcription of genes containing the T-box riboswitch, the 5′-UTR is transcribed before the protein-encoding region of the mRNA and the riboswitch responds to the presence or absence of non-aminoacylated (uncharged) cognate tRNA.9 The anticodon of the tRNA base pairs with a complementary specifier sequence located in Stem I near the beginning of the 5′-UTR while the tRNA acceptor end nucleotides base pair with four nucleotides in the bulge of the antiterminator (Figure 1a). The base pairing between the tRNA acceptor end nucleotides and the antiterminator leads to stabilization of the antiterminator and precludes the formation of an alternate terminator structural element.9 Without the extra stabilization provided by the antiterminator-tRNA base pairing, the terminator forms preferentially and transcription is terminated. While there are other important RNA elements within the T-box 5′-UTR, the interaction between the tRNA acceptor end and the antiterminator is critical for switching between antitermination of transcription (the entire gene is transcribed) and termination of transcription (transcription is terminated before reaching the protein encoding sequences).9 This critical role, the high degree of sequence conservation in the antiterminator and its prevalence in many pathogenic bacteria,10,16 make it an intriguing RNA target for drug discovery.
Figure 1.
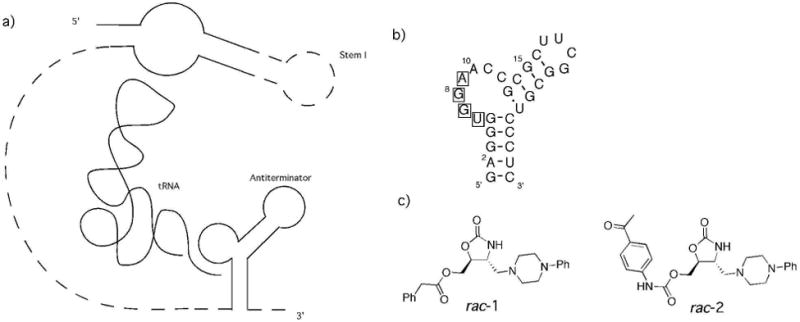
a) Schematic of T-box riboswitch binding tRNA b) antiterminator model RNA AM1A. Nucleotides that base pair with acceptor end of tRNA noted with boxes. c) Structure of racemic trans-4,5-disubstituted oxazolidinones rac-1 and rac-2.
While there have been a variety of studies related to targeting RNA with small molecules, few have involved an in-depth analysis of the RNA-ligand interactions.1-6 We previously reported 4,5-oxazolidinones as potential ligands for targeting T-box riboswitch antiterminator model RNA AM1A (Figure 1b).12,17 Further studies identified lead compounds that significantly altered the function of the riboswitch.11 One compound inhibited the riboswitch (rac-1, Figure 1c) while another acted as an agonist and promoted tRNA-independent antitermination (rac-2, Figure 1c). Preliminary characterization of the binding sites indicated that the two molecules were likely binding in different (but somewhat overlapping) regions of the antiterminator RNA.11 In this paper we report the enantiopure synthesis of these lead compounds as well as their cis analogs and the characterization of their RNA binding modes to the model antiterminator RNA, AM1A (Figure 1). These studies better elucidate the specific ligand-RNA interactions that may be critical for developing a medicinal agent targeted to the T-box antiterminator.
Results and Discussion
Chemistry
For the synthesis of both enantiomers of the lead compounds rac-1 and rac-2, we used a method previously developed in our laboratories18-20 and reported for the preparation of these racemic lead compounds (Scheme 1).11 The key step in this method is the intramolecular acylnitrene-mediated aziridination that provides the bicyclic aziridine 4 with very good diastereoselectivity.19 The bicyclic aziridine 4 can be further converted to the desired final compounds in three simple steps: nucleophilic aziridine ring opening, tritylether deprotection and reaction with phenylacetyl chloride or 4-acetylphenyl isocyanate. The azidoformate precursor 3 is readily accessible from 3-butene-1,2-diol (5, Scheme 1). In order to apply this synthetic plan for the synthesis of our target enantiomers, it was necessary to first prepare both enantiomers of 5.
Scheme 1.

The required enantiomers of 5 (Scheme 2) were obtained by the known hydrolytic kinetic resolution of commercially available racemic butadiene monoepoxide 6 using the catalyst (R,R)-7 and its enantiomer (S,S)-7.21 Prior to the determination of the enantiomeric purity (er), both enantiomers of 5 were converted to their corresponding tritylether, (R)-8 and (S)-8, derivatives. As determined by NMR, using (S)-(-)-α-methoxy-α-trifluoromethyl phenylacetyl chloride22 as the chiral derivatizing agent, the R enantiomer was obtained in 99:1 er and the S enantiomer was obtained in 98:2 er.
Scheme 2a.

aReagents and conditions: (i) AcOH, H2O, 18h, 20%; (ii) TrCl, Et3N, DMAP, CH2Cl2, 24h, 64%.
With both enantiomers of 8 in hand we proceeded with the synthesis as shown in Scheme 3. The (R)-8 was converted to its corresponding azidoformate (R)-3 in very good yield, by using p-nitrophenyl chloroformate followed by sodium azide.18 Azidoformate (R)-3 was then subjected to the thermal intramolecular aziridination to provide the bicyclic aziridine (4S,5R)-4. Next, the aziridine ring opening with N-phenyl piperazine followed by the acidic cleavage of trityl group afforded the (4S,5R)-10 oxazolidinone in excellent yield. The (4R,5S)-10 enantiomer was prepared in the same manner and with similar yields starting from (S)-8.
Scheme 3a.

aReagents and conditions: (i) a) p-NO2PhOC(O)Cl, pyridine, CH2Cl2, 2h, b) NaN3, acetone:H2O, 4d, (R)-3 (80% two steps), (S)-3 (80% two steps); (ii) sealed tube, CH2Cl2, 109 °C, (4S,5R)-4 (55%), (4R,5S)-4 (55%); (iii) N-phenyl piperazine, CH2Cl2, 2h, (4S,5R)-9 (91%), (4R,5S)-9 (86%); (iv) HCl, EtOAc, 30 min, (4S,5R)-10 (93%), (4R,5S)-10 (92%).
The desired final compounds, (4S,5R)-1 and (4S,5R)-2 were prepared from (4S,5R)-10 (Scheme 4) by reaction with phenylacetyl chloride providing (4S,5R)-1 and 4-acetyl-phenyl isocyanate affording (4S,5R)-2 in good yields. Their enantiomers, (4R,5S)-1 and (4R,5S)-2 were obtained in the same manner starting from (4R,5S)-10.
Scheme 4a.

aReagents and conditions: (i) PhCH2COCl, Et3N, DMAP, CH2Cl2, 5h, (4S,5R)-1 (81%), (4R,5S)-1 (77%); (ii) 4-acetyl-phenyl isocyanate, Et3N, DMAP, CH2Cl2, 5h, (4S,5R)-2 (80%), (4R,5S)-2 (82%).
For the synthesis of cis isomers of rac-1 and rac-2 our synthetic plan started from commercially available 2-butyne-1,4-diol (11, Scheme 5). We first subjected compound 11 to a standard reduction with lithium aluminum hydride23 followed by the epoxidation of the resulting trans double bond using m-chloroperbenzoic acid in acetonitrile to provide the epoxy diol 1224. The next step in the synthesis was the nucleophilic ring opening of the epoxide 12 with sodium azide,25 which afforded the azido triol 1326 with the desired trans relative configuration. The differentiation between the three hydroxyl groups of 13 was accomplished by protecting the two adjacent groups as the isopropylidene acetal.27 None of the six-membered ring acetonide was observed.28 The free primary hydroxyl of acetonide 1429 was then tosylated and the resulting tosyloxy group was substituted with N-phenyl piperazine affording compound 16. The catalytic hydrogenation of azide 16 followed by the reaction of the resulting amine with phenyl chloroformate afforded the carbamate 17. The cleavage of the isopropylidene acetal with trifluoroacetic acid in water30 afforded only a low yield of the unprotected diol. A more suitable method for our substrate proved to be the use of an aqueous solution of oxalic acid in tetrahydrofuran that directly provided the cis-10 oxazolidinone after a basic work up.
Scheme 5a.

aReagents and conditions: (i) LiAlH4, THF, 18h, 67%; (ii) mCPBA, CH3CN, 4 °C, 4d, 90%; (iii) NaN3, NH4Cl, EtOH, 17h, 90%; (iv) p-TsOH, acetone, 12h, 67%; (v) TsCl, pyridine, CH2Cl2, 12h, 70%; (vi) N-phenyl piperazine, K2CO3, EtOH, 12h, 60%; (vii) H2, 10 mol % Pd/C, EtOAc, 97%; (viii) PhOCOCl, Et3N, DMAP, CH2Cl2, 15 min, 89%; (ix) oxalic acid, THF:H2O, 60 °C, 8h, 66%.
The final compounds, cis-1 and cis-2 (Scheme 6) were prepared from cis-10 in the same manner as their corresponding trans isomers. Treating cis-10 with phenylacetyl chloride afforded cis-1 while the reaction with 4-acetyl-phenyl isocyanate provided cis-2.
Scheme 6a.

aReagents and conditions: (i) PhCH2COCl, Et3N, DMAP, CH2Cl2, 5h, 36%; (ii) 4-acetyl-phenyl isocyanate, Et3N, DMAP, CH2Cl2, 5h, 50%.
Biological results
Binding Affinity
The binding affinity for each compound was determined using the previously described fluorescence resonance energy transfer (FRET) antiterminator RNA binding assay.11,13 The observed Kd values are summarized in Table 1.
Table 1.
| Compound | Kda (μM) | Emodelb (kcal/mol) |
|---|---|---|
| rac-1 | 13 ± 411 | -- |
| rac-2 | 0.9 ± 0.411 | -- |
| (4R,5S)-1 | 12 ± 5 | -101.0 |
| (4S,5R)-1 | 16 ± 5 | -98.9 |
| (4R,5S)-2 | 3 ±1 | -117.4 |
| (4S,5R)-2 | 1.6 ± 0.5 | -117.4 |
| cis-1 | NBc | -- |
| cis-2 | 1.8 ± 0.7 | -- |
Kd values determined using FRET-derived binding assay with 100 nM labeled AM1A RNA, see experimental section for details and supplementary material for the binding isotherms. All R2 values ≥ 0.8.
Glide calculated Emodel of most energetically favorable docked protonated ligand.
NB: No apparent binding was observed at 130 min (the standard incubation time for all other compounds), Kd > 100 μM. At 70 min incubation time, however, a binding isotherm was observed (Kd = 5.1 ± 2.2, R2 = 0.78). This apparent time-dependence will be investigated in future work.
For the racemic cis compounds, cis-1 did not bind AM1A while cis-2 bound with affinity similar to the corresponding racemic trans compound rac-2. The (4R,5S)-1/(4S,5R)-1 affinities for binding AM1A were approximately 7-fold weaker than those for (4R,5S)-2/(4S,5R)-2 consistent with previous binding studies of the corresponding racemic compounds.11 Interestingly, however, the enantiomeric pair (4R,5S)-1 and (4S,5R)-1 had similar RNA affinities. The (4R,5S)-2 and (4S,5R)-2 enantiomeric pair also had similar RNA affinities.
Ligand binding site
Ligand docking studies using the Glide module of FirstDiscovery 2.7 (Schrödinger) were conducted to begin to determine possible binding modes for the pairs of enantiomers. Glide performs flexible ligand docking to a rigid receptor by utilizing an OPLS-AA31 derived molecular-mechanics potential function, a grid-based docking method and a discretized version of the ChemScore empirical scoring function32 followed by energy minimization of the best-refined poses.33 The results of these docking studies were consistent with the binding studies. The most energetically favorable Emodel (an indicator of best docked structure) was similar between enantiomeric pairs (see Table 1). Between the two different structural series, however, the Emodel was ∼17% more stable for (4R,5S)-2 or (4S,5R)-2 with AM1A compared to (4R,5S)-1 or (4S,5R)-1 consistent with the ∼7-fold difference in the observed Kd values. In addition, the Glide predicted binding location was similar between enantiomeric pairs, but differed between the two structural series (Figure 2). The (4R,5S)-1/(4S,5R)-1 compounds bound along the major groove of helix A1 (G3-G5) extending up into the 5′ end of the bulge (U6-G8) while the (4R,5S)-2/(4S,5R)-2 compounds bound solely to the bulge region (U6-G8 and the major groove of the G5:C25 closing basepair). This difference in predicted binding location is consistent with the differing effects of rac-1 and rac-2 on T-box transcription antitermination.11
Figure 2.
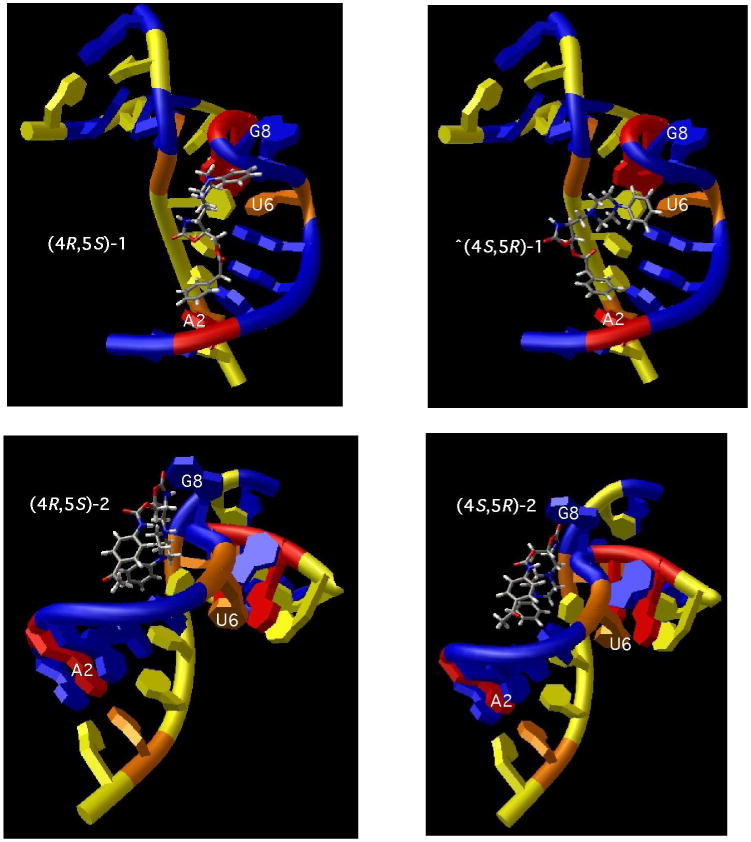
Lowest energy docked structure of monoprotonated oxazolidinones binding to AM1A.
A lack of stereospecific ligand binding with relatively simple RNA structural motifs has been observed previously.34,35 Given that the known structure of AM1A36 presents multiple binding surfaces and few, if any, deep binding pockets, it was not a forgone conclusion that enantiomers would bind with widely differing affinities. Comparison of the energetically most favorable docked structures (Figure 2) indicates that each enantiomeric pair likely binds to a similar region in AM1A with comparable binding interactions (H-bonding, π-π stacking), but with different functional group partners within the antiterminator model RNA (Figure 3). With comparable electrostatics, the primary interaction differences between the two enantiomeric pairs are that the enantiomers of 2 form three hydrogen bonds with AM1A while the enantiomers of 1 form two hydrogen bonds along with differential amounts of π-π stacking. Futher, more detailed, structural studies are needed to best determine the role that these and other binding interactions may play in ligand binding specificity.
Figure 3.

Summary of H-bonding and π-stacking interactions between docked oxazolidinones and AM1A.
There is always the possibility, however, that the rigid receptor docking studies do not fully predict the structure of the RNA-ligand complex. It is conceivable that the ligand binds via an induced-fit and/or tertiary structure capture. In-line probing37 experiments were conducted to determine if there was a significant restructuring of the RNA upon ligand binding. In-line probing monitors ligand-induced changes in RNA flexibility. No dramatic secondary structural changes were detected (Figure 4). Instead, the observed ligand-induced changes in the in-line cleavage patterns were indicative of conformation/flexibility changes in the RNA without a significant reordering of secondary structure base pairing.
Figure 4.
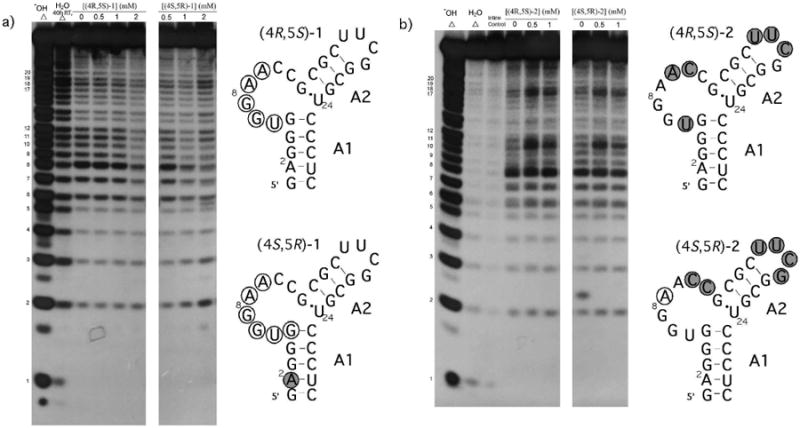
In-line probing of enantiomeric ligands binding to AM1A. a) (4R,5S)-1 and (4S,5R)-1 and b) (4R,5S)-2 and (4S,5R)-2. Significant changes are summarized on secondary structure of AM1A for increased (shaded circle) and decreased (open circle) in-line cleavage. Significant changes were identified by comparing the slopes of the ligand-induced relative band intensity changes (See Supplementary Material).
Each pair of enantiomers exhibited similar ligand-induced changes and the two structural series had very different ligand-induced changes. These observations are consistent with the binding and docking studies. For both (4R,5S)-1 and (4S,5R)-1 the ligand-induced cleavage changes observed were a slight increase along the 5′ side of the A1 helix (especially at nucleotide A2) and a more significant decrease in cleavage for bulge nucleotides U6-C11. These results indicate that ligand binding leads to reduced conformational flexibility for U6-C11 and increased flexibility at nucleotide A2. Both (4R,5S)-2 and (4S,5R)-2 resulted in an increase in cleavage at the 3′ end of the bulge, especially at C11 and also an increase in cleavage within the tetraloop (U17-G20). These results indicate that ligand binding leads to increased flexibility at C11 and in the tetraloop. Both effects could be due to the ligand binding to the 5′ end of the bulge (as predicted in the docking studies), resulting in a slight conformational change towards the 3′ end of the bulge. This change could induce a long-range topological strain that affects the tetraloop nucleotides.
These studies reveal that the lack of a deep binding pocket and redundancy of similar functional groups within the relatively simple structural motif of the antiterminator, can lead to a lack of stereospecific discrimination in ligand binding. In the context of the entire T-box riboswitch, there is a significant likelihood that a ligand functional group which did not interact with the antiterminator could be in a position to stereospecifically disrupt tRNA binding. Studies are currently in progress to investigate this possibility. The finding that the oxazolidinones likely bind AM1A via surface binding has significant implications for RNA drug discovery that is focused on targeting other medicinally relevant RNAs.
Experimental Section
General
Reagents and starting materials were obtained from Aldrich unless otherwise stated. All RNA experiments were conducted using RNase-free conditions and molecular biology grade reagents. Fluorescently labeled RNA, 3′-Fl-AM1A-(18)-Rhd was prepared as previously described.11,13 racemic butadiene monoepoxide 6 was purchased from Alfa Aesar. Both enantiomers of the (salen)Co(II) complex 7 are commercially available from Aldrich. CH2Cl2 and THF were dried using a SOLV-TEK solvent purification system. Acetone was dried over 4 Å molecular sieves and distilled immediately before use. Et3N was dried over calcium hydride and distilled immediately before use. Melting points were determined with a MEL-TEMP II melting point apparatus and are reported uncorrected. Specific rotations were measured on an AUTOPOL ® IV (Rudolph Research Analytical) polarimeter with a sodium (λ = 589 nm) lamp, and are reported as follows: [α]λT °C (c g/100 mL, solvent). The capillary GC analyses were performed on a Shimadzu GC-17A Gas Chromatograph employing the Rtx®-5 (15 m × 0.25 mm id × 0.25 μm df; Restek) column. The retention times are reported in minutes as follows: initial T °C to final T °C, rate, duration of run. The purity of tested compounds were determined via HPLC analysis on a Shimadzu LC-10AT machine equipped with a UV detector employing a Discovery-C8 (15 cm × 4.6 mm × 5 μm; Supelco) column eluting with MeOH in H2O at 1 mL/min flow rate (gradient started at 8 min with 50% MeOH/H2O ended after 5 min with 90% MeOH/H2O, run time 22 min). The compounds showed >95% purity unless otherwise stated. 1H NMR and 13C NMR spectra were recorded with a Brüker AVANCE (300 MHz) spectrometer. Chemical shifts are reported in ppm on the δ scale relative to deuterated chloroform as an internal standard. Data are reported as follows: chemical shift, multiplicity (s = singlet, d = doublet, t = triplet, q = quartet, m = multiplet), coupling constant in Hz, integration.
(R)-1-(Triphenylmethoxy)-2-[(azidocarbonyl)oxy]-3-butene ((R)-3).18
CAUTION: Azides are potentially explosive especially when heated. While we did not experience any problems, precaution should be taken when running reactions involving azides. p-Nitrophenyl chloroformate (2.4 g, 12 mmol) was added to a solution of allylic alcohol (R)-8 (2 g, 6 mmol) in CH2Cl2 (20 mL). The solution was cooled to 0 °C and pyridine (1.46 mL, 18 mmol) was added dropwise. The reaction mixture was allowed to warm to room temperature and stirred for 1 h. Then it was washed with saturated aqueous NaHCO3 solution (2 × 20 mL) and brine (2 × 20 mL). The organic layer was dried over MgSO4, filtered, concentrated to dryness and the residue was dissolved in acetone (22 mL). A solution of NaN3 (2.73 g, 42 mmol) in H2O (13 mL) was then added to this solution and the reaction mixture was stirred at room temperature for 72 h when was diluted with H2O and extracted with EtOAc (3 × 20 mL). The combined organic layers were washed with 10% K2CO3 aqueous solution (2 × 20 mL) and then dried over MgSO4, filtered and concentrated by rotary evaporation. The residue was purified by flash chromatography (5% EtOAc in hexanes) to provide 1.9 g (80%) product as colorless oil. Rf 0.36 (5% EtOAc in hexanes); 1H NMR (CDCl3, 300 MHz) δ 7.45-7.26 (m, 15H, Ar), 5.82 (ddd, J = 6.5, 10.6, 17.2 Hz, 1H, HC=CHH), 5.47-5.42 (m, 1H, OCH), 5.37 (d, J = 17.2 Hz, 1H, HC=CHH), 5.29 (d, J = 10.6 Hz, 1H, HC=CHH), 3.32 (dd, J = 7 .2, 10.2 Hz, 1H, TrOCHH), 3.23 (dd, J = 3.9, 10.2 Hz, 1H, TrOCHH); C NMR (CDCl3, 75 MHz) δ 156.9, 143.5, 132.1, 128.6, 127.9, 127.2, 119.4, 86.8, 78.5, 64.8; [α]D24 +21.1 (c 1.04, CHCl3). For (S)-3 [α]D24 -20.8 (c 1.0, CHCl3).
(4S,5R)-4-Trityloxymethyl-3-oxa-1-aza-bicyclo[3.1.0]hexan-2-one ((4S,5R)-4).18
CAUTION: Reactions carried out in pressure tubes are potentially explosive. While we did not experience any problems, the reactions should be carried out behind a protecting shield. A solution of the azidoformate (R)-3 (1.8 g, 4.5 mmol) in CH2Cl2 (70 mL) was placed in an Ace sealed tube (catalog no. 8648-79). The tube was cooled to –78 °C, evacuated, sealed and heated to 109 °C for 14 h. Then the tube was allowed to cool to room temperature and the solvent removed by rotary evaporation. The residue was washed with 5% EtOAc in hexanes and a precipitate was formed that was isolated by filtration to afford 0.93 g of (4S,5R)-4 (55%). Rf 0.24 (30% EtOAc in hexanes); 1H NMR (CDCl3, 300 MHz) δ 7.36-7.25 (m, 15H, Ar), 4.68 (t, J = 3.2 Hz, 1H, C(O)OCH), 3.56 (dd, J = 3.8, 10.5 Hz, 1H, TrOCHH), 3.25 (dd, J = 3.4, 10.5 Hz, 1H, TrOCHH), 3.05 (t, J = 4.1 Hz, 1H, NCH), 2.56 (d, J = 4.2 Hz, 1H, NCHH), 2.18 (d, J = 4.2, Hz, 1H, NCHH); [α]D24 -21.1 (c 1.04, CHCl3). For (4R,5S)-4 [α]D24 +20.9 (c 1.04, CHCl3).
(R)-1-(Triphenylmethoxy)-3-buten-2-ol ((R)-8).18
To a solution of (R)-521 (0.24 g, 2.72 mmol) in CH2Cl2 (5 mL) was added trityl chloride (0.909 g, 3.26 mmol) and DMAP (40 mg, 0.332 mmol) followed by Et3N (0.76 mL, 5.44 mmol). The reaction mixture was stirred under argon atmosphere at room temperature for 24 h. The reaction mixture was diluted with Et2O (20 mL) and washed with H2O (10 mL), cold 1M HCl (10 mL), saturated NaHCO3 (10 mL), H2O (10 mL) and brine. The organic phase was dried over MgSO4, filtered, and concentrated by rotary evaporation. The residue was purified by flash chromatography (10% EtOAc in hexanes) to provide 0.575 g (64%) of (R)-8 as colorless liquid. Rf 0.25 (10% EtOAc in hexanes); 1H NMR (CDCl3, 300 MHz) δ 7.45 (m, 15H, Ph), 5.82 (ddd, J = 5.6, 10.6, 17.2 Hz, 1H, CH=CH2), 5.35 (d, J = 17.2 Hz, 1H, =CHH), 5.20 (d, J = 10.6 Hz, 1H, =CHH), 4.27-4.32 (m, 1H, CHOH), 3.25 (dd, J = 3.8, 9.4 Hz, 1H, TrOCHH), 3.15 (dd, J = 7.4, 9.4 Hz, 1H, TrOCHH), 2.4 (d, J = 3.9 Hz, 1H, OH); 13C NMR (CDCl3, 75 MHz) δ 143.8, 137.0, 128.7, 127.9, 127.2, 116.4, 86.8, 72.1, 67.5; [α]D24 +27.8 (c 1.0, i-PrOH). For (S)-8 [α]D24 –28.0 (c 1.01, i-PrOH).
(4S,5R)-4-(4-Phenyl-piperazin-1-ylmethyl)-5-trityloxymethyl-oxazolidin-2-one ((4S,5R)-9)
To a solution of (4S,5R)-4 (0.78 g, 2.09 mmol) in CH2Cl2 (4 mL) was added freshly distilled N-phenyl-piperazine (0.35 mL, 2.3 mmol) neat. The reaction was stirred at room temperature until complete conversion shown by TLC (typically 2 h). The solvent was removed under reduced pressure and the residue was purified by flash chromatography (30% EtOAc in CH2Cl2) to provide 1.02 g (91%) of (4S,5R)-7 as white solid (mp 96.1-97.9). Rf 0.30 (30% EtOAc in CH2Cl2); 1H NMR (CDCl3, 300 MHz) δ 7.49 (d, 6H, Ar), 7.36-7.24 (m, 11H, Ar), 6.91 (d, 3H, Ar), 5.69 (s, 1H, NH), 4.28 (q, J = 4.5, 9.3 Hz, 1H, C(O)OCH), 3.87 (q, J = 6, 13.3 Hz, 1H, NHCH), 3.49 (dd, J = 4.5, 10.3 Hz, 1H, TrOCHH), 3.25 (dd, J = 4.2, 10.3 Hz, 1H, TrOCHH), 3.12-3.02 (m, 4H, PhNCH2), 2.64-2.42 (m, 6H, NCH2); 13C NMR (CDCl3, 75 MHz) δ 158.7, 151.1, 143.4, 129.1, 128.6, 128.0, 127.2, 119.9, 116.1, 86.9, 79.3, 63.8, 62.4, 53.4, 51.5, 49.0; Anal. Calcd for C34H35N3O3: C, 76.52; H, 6.61; N, 7.87. Found: C, 76.54; H, 6.21; N, 7.48; [α]D23 -32.1 (c 1.04, CHCl3). For (4R,5S)-9 [α]D23 +33.0 (c 1.02, CHCl3).
(4S,5R)-5-Hydroxymethyl-4-(4-phenyl-piperazin-1-ylmethyl)-oxazolidin-2-one ((4S,5R)-10)
To a solution of (4S,5R)-9 (0.69 g, 1.3 mmol) in EtOAc (5 mL) was added a solution of HCl in EtOAc (1.5M, 4.5 mL, 6.5 mmol). The mixture was allowed to stir at room temperature for 10 min when was diluted with EtOAc (20 mL) and H2O (30 mL) and the layers separated. Saturated aqueous NaHCO3 solution (10 mL) was added to the aqueous layer and extracted with EtOAc (4 × 10 mL). The combined organic layers were washed with H2O (10 mL), dried over MgSO4, filtered and concentrated by rotary evaporation to afford 0.36 g (93%) of (4S,5R)-10 as white solid (mp 140.5-141.9). No further purification was necessary. Rf = 0.16 (EtOAc); 1H NMR (CDCl3, 300 MHz) δ 7.31-7.26 (m, 2H, Ar), 6.94-6.87 (m, 3H, Ar), 5.98 (s, 1H, NH), 4.33 (q, J = 4.3, 10.3 Hz, 1H, C(O)OCH), 3.96 (q, J = 6.7, 13.3 Hz, 1H, NHCH), 3.87 (dd, J = 4.6, 12.0 Hz, 1H, HOCHH), 3.76 (dd, J = 4.0, 12.0 Hz, 1H, HOCHH), 3.21 (m, 4H, PhNCH2), 2.70 (m, 4H, NCH2), 2.66 (dd, J = 7.2, 12.5 Hz, 1H, NCHH), 2.53 (dd, J = 6.7, 12.5 Hz, 1H, NCHH); 13C NMR (CDCl3, 75 MHz) δ 157.3, 149.5, 127.7, 118.7, 114.8, 79.9, 61.3, 60.7, 52.3, 50.6, 47.7; HRMS: calcd for C15H21N3O3•Na+, 314.1475; found, 314.1476; [α]D28 -55.4 (c 1.05, CHCl3). For (4R,5S)-10 [α]D28 +54.5 (c 1.05, CHCl3).
5-Hydroxymethyl-4-(4-phenyl-piperazin-1-ylmethyl)-oxazolidin-2-one (cis-10)
The acetonide 17 (1.1 g, 2.62 mmol) was placed in a flask followed by oxalic acid (1.65 g, 18.34 mmol), H2O (16.5 mL), THF (33 mL) and 12N HCl (0.2 mL) and the reaction mixture was stirred at 60 °C for 8 h. The reaction mixture was poured into a 5% aqueous K2CO3 and stirred for 30 minutes and then was extracted with CH2Cl2. The combined organic layers were dried over MgSO4, filtered and the solvent removed by rotary evaporation to provide 0.5 g (66%) of cis-10. No further purification was necessary. Rf 0.22 (95% EtOAc in hexanes); 1H NMR (CDCl3, 300 MHz) δ 7.27-7.22 (m, 2H, Ph), 6.90-6.84 (m, 3H, Ph), 5.73 (s, 1H, NH), 5.36 (br s, 1H, OH), 4.81 (ddd, J = 5, 7.7, 9 Hz, 1H, OCH), 4.16 (ddd, J = 4, 7.7, 11 Hz, 1H, NHCH), 3.90-3.77 (m, 2H, HOCH2), 3.25-3.12 (m, 4H, PhN(CH2)2), 2.90 (dd, J = 11, 13 Hz, 1H, NCHH), 2.83-2.76 (m, 2H, NCH2), 2.69-2.62 (m, 2H, NCH2), 2.42 (dd, J = 4, 13 Hz, 1H, NCHH); 13C NMR (CDCl3, 75 MHz) δ 158.7, 150.9, 129.4, 120.7, 116.6, 78.3, 59.3, 57.8, 53.7, 51.2, 49.2.
2-Azido-2-(2,2-dimethyl-[1,3]dioxolan-4-yl)-ethanol (13)
To a solution of epoxide 1224 (0.52 g, 5 mmol) in EtOH (8 mL) at room temperature were added successively NH4Cl (0.615 g, 11.5 mmol) and NaN3 (0.747 g, 11.5 mmol). The mixture was heated to reflux for 17 h. After cooling, the reaction mixture was diluted with EtOAc (20 mL) and then filtered. The filtrate was concentrated by rotary evaporation to provide 0.66 g (90%) of 13 as yellowish oil. Rf 0.3 (EtOAc); tR = 1.1 min (91 %) (40 °C to 100 °C at 10 °/min then to 250 °C at 25 °/min, 12 min); 1H NMR (D2O, 300 MHz) δ 3.93 (dd, J = 3, 11 Hz, 1H), 3.81-3.62 (m, 5H); 13C NMR (D2O, 75 MHz) δ 70.4, 64.2, 61.9, 60.5. Matched analytical data previously reported.26
2-Azido-2-(2,2-dimethyl-[1,3]dioxolan-4-yl)-ethanol (14)
To a solution of triol 13 (0.6 g, 4 mmol) in dry acetone (110 mL/mmol) was added p-toluenesulfonic acid (0.3 g, 1.6 mmol). The reaction mixture was stirred at room temperature for 18 h. K2CO3 (0.77 g, 5.6 mmol) was added to the reaction flask and stirred another 2 h. Then the reaction mixture was filtered and the filtrate was concentrated by rotary evaporation. The residue was purified by flash chromatography (20% EtOAc in hexanes) to provide 0.5 g (67%) of 14 as colorless oil. Rf 0.18 (20% EtOAc in hexanes); tR = 3.13 min (100 %) (75 °C to 300 °C at 25 °/min, 12 min); 1H NMR (CDCl3, 300 MHz) δ 4.14-4.04 (m, 2H, OCH and OCHH), 3.92 (dd, J = 5, 8 Hz, 1H, OCHH), 3.89-3.81 (m, 1H, HOCHH), 3.68 (dd, J = 6, 12 Hz, 1H, HOCHH), 3.63-3.57 (m, 1H, N3CH), 2.61 (t, 1H, OH), 1.44 (s, 3H, CH3), 1.34 (s, 3H, CH3); 13C NMR (CDCl3, 75 MHz) δ 109.9, 75.5, 66.6, 64.8, 62.7, 26.5, 25.2. Matched analytical data previously reported.29
2-Azido-2-(2,2-dimethyl-[1,3]dioxolan-4-yl)-ethyl-4-methylbenzenesulfonate (15)
To a solution of acetonide 14 (0.5 g, 2.67 mmol) in CH2Cl2 (5 mL) was added toluenesulfonyl chloride (0.61 g, 3.2 mmol). The mixture was cooled to 0 °C and pyridine (0.65 mL, 8.01 mmol) was added. The reaction mixture was allowed to warm up to room temperature and stirred overnight. Then it was diluted with EtOAc and washed with saturated aqueous NaHCO3 and brine. The organic phase was dried over MgSO4, filtered and the solvent removed by rotary evaporation. The residue was purified by flash chromatography (10% EtOAc in hexanes) to afford 0.64 g (70%) of 15 as colorless oil. Rf 0.35 (20% EtOAc in hexanes); tR = 7.57 min (100 %) (75 °C to 300 °C at 25 °/min, 10 min); 1H NMR (CDCl3, 300 MHz) δ 7.83 (d, J = 8 Hz, 2H, Ph), 7.38 (d, J = 8 Hz, 2H, Ph), 4.33 (dd, J = 3, 10.7 Hz, 1H, OCH), 4.08-4.02 (m, 2H, OCH2), 3.99-3.87 (m, 2H, TsOCH2), 3.71-3.65 (m, 1H, N3CH), 2.47 (s, 3H, PhCH3), 1.40 (s, 3H, CH3), 1.31 (s, 3H, CH3); 13C NMR (CDCl3, 75 MHz) δ 145.5, 132.6, 130.2, 128.2, 110.4, 74.4, 69.4, 66.7, 62.2, 26.7, 25.1, 21.9; HRMS: calcd for C14H19N3O5S•Na+, 364.0938; found, 364.0956.
1-Azido-1-(2,2-dimethyl-[1,3]dioxolan-4-yl)-2-(4-phenyl-piperazine-1-yl)-ethane (16)
To a solution of 15 (0.51 g, 1.5 mmol) in EtOH (5 mL) was added K2CO3 (0.41 g, 3 mmol) followed by N-phenyl-piperazine (0.25 mL, 1.65 mmol). The reaction mixture was heated to reflux overnight. After cooling to room temperature the reaction mixture was transferred in a separatory funnel, diluted with EtOAc and water. The aqueous layer was extracted with EtOAc (3 × 10 mL) and the combined organic layers were dried over MgSO4, filtered and the solvent removed by rotary evaporation. The residue was purified by means of flash chromatography (10% EtOAc in toluene) to provide 0.3 g (60%) of 16 as colorless oil. Rf 0.25 (10% EtOAc in toluene); tR = 8.09 min (100 %) (75 °C to 300 °C at 25 °/min, 10 min); IR 2100 cm-1 (s, N3); 1H NMR (CDCl3, 300 MHz) δ 7.31-7.26 (m, 2H, Ph), 6.96-6.85 (m, 3H, Ph), 4.11-4.04 (m, 2H, OCH2), 3.97-3.91 (m, 1H, OCH), 3.77-3.71 (m, 1H, N3CH), 3.23-3.18 (m, 4H, PhN(CH2)2), 2.79-2.72 (m, 2H, NCH2), 2.70-2.63 (m, 3H, NCH2 and NCHH), 2.56 (dd, J = 9, 13.3 Hz, 1H, NCHH), 1.49 (s, 3H, CH3), 1.38 (s, 3H, CH3); 13C NMR (CDCl3, 75 MHz) δ 151.4, 129.3, 119.9, 116.3, 109.9, 76.4, 66.5, 61.0, 59.7, 53.8, 49.4, 26.6, 25.4; HRMS: calcd for C17H25N5O2•Na+, 354.1900; found, 354.1899.
N-[1-(2,2-Dimethyl-[1,3]dioxolan-4-yl)-2-(4-phenyl-piperazine-1-yl)-ethyl]-benzamide (17)
To a solution of 16 (0.7 g, 2.11 mmol) in EtOAc (4 mL) was added 10% Pd/C (w/w) (0.225 g, 0.211 mmol). The reaction flask was equipped with a three-way stopper with a H2 balloon attached. The flask was evacuated three times using and aspirator and then the reaction mixture was stirred under a hydrogen atmosphere for 24 h. The reaction mixture was then filtered through a short pad of celite. The solvent was removed by rotary evaporation and the residue (0.625 g, 97%) was dissolved in CH2Cl2 (3 mL). To this solution DMAP (25 mg, 0.211 mmol) was added followed by Et3N (1.7 mL, 12.6 mmol). The mixture was cooled to 0 °C and phenyl chloroformate (1 mL, 3.1 mmol) was added neat and the reaction mixture was allowed to warm to room temperature. After 15 minutes of stirring the reaction mixture was diluted with EtOAc and transferred in a separatory funnel. The reaction mixture was washed with H2O, saturated aqueous NaHCO3 and brine. The organic layer was dried over MgSO4, filtered and concentrated down. The solid residue was washed with hexanes and filtered to afford 0.774 g (89%) of 17. No further purification was necessary. Rf 0.3 (30% EtOAc in hexanes); tR = 8.3 min (100 %) (75 °C to 300 °C at 25 °/min, 10 min); 1H NMR (CDCl3, 300 MHz) δ 7.30-7.02 (m, 7H, Ph), 6.86-6.75 (m, 3H, Ph), 5.38 (d, J = 5 Hz, 1H, NH), 4.22-4.16 (m, 1H, OCH), 4.03 (dd, J = 6, 8.7 Hz, 1H, OCHH), 3.92 (dd, J = 6, 8.7 Hz, 1H, OCHH), 3.87-3.78 (m, 1H, NHCH), 3.13-3.10 (m, 4H, PhN(CH2)2), 2.69-2.54 (m, 6H, N(CH2)3), 1.38 (s, 3H, CH3), 1.29 (s, 3H, CH3); 13C NMR (CDCl3, 75 MHz) δ 154.8, 151.2, 150.9, 129.3, 129.1, 125.4, 121.6, 119.7, 116.0, 109.6, 76.8, 66.8, 57.7, 53.7, 51.2, 49.3, 29.7, 26.4, 25.1; HRMS: calcd for C24H31N3O4•Na+, 448.2207; found, 448.2199.
General method for the synthesis of enantiomers and cis isomers of 1 and 2
To a solution of oxazolidinone alcohol 10 (1 equiv) in CH2Cl2 (0.1 M) was added DMAP (0.12 equiv), Et3N (2 equiv) and phenylacetyl chloride or 4-acetylphenyl isocyanate (1.2 equiv). The mixture was stirred at room temperature for 5 h. The solvent was removed by rotary evaporation and the residue purified by flash chromatography.
(4S,5R)-2-oxo-4-(4-phenyl-piperazin-1-ylmethyl)-oxazolidin-5-ylmethyl phenyl acetate ((4S,5R)-1)
Compound (4S,5R)-1 was prepared from (4S,5R)-10 on a 0.22 mmol scale following the general procedure to afford 73 mg (81%) of product. Rf = 0.21 (50% EtOAc in hexanes); tR = 14.9 min (89 %); 1H NMR (CDCl3, 300 MHz) δ 7.30-7.17 (m, 7H, Ar), 6.85-6.78 (m, 3H, Ar), 5.34 (s, 1H, NH), 4.31 (q, J = 4.6, 9.1 Hz, 1H, C(O)OCH), 4.22 (d, J = 4.3 Hz, 2H, C(O)OCH2), 3.60 (s, 2H, PhCH2), 3.58-3.51 (m, 1H, HNCH), 3.07 (t, J = 4.8 Hz, 4H, PhNCH2), 2.56-2.41 (m, 5H, NCH2 and NCHH), 2.30 (dd, J = 5.5, 12.5 Hz, 1H, NCHH); 13C NMR (CDCl3, 75 MHz) δ 171.2, 158.0, 151.3, 133.7, 129.5, 129.4, 128.9, 127.5, 120.2, 116.4, 77.6, 64.4, 62.3, 53.7, 51.7, 49.4, 41.4; HRMS: calcd for C23H27N3O4•Na+, 432.1894; found, 432.1887; [α]D25 -51.7 (c 1.00, CHCl3). For (4R,5S)-1 [α]D25 +52.8 (c 1.04, CHCl3), tR = 15.0 min (87 %).
(4S,5R)-2-oxo-4-(4-phenyl-piperazin-1-ylmethyl)-oxazolidin-5-ylmethyl (4-acetyl-phenyl)- carbamate ((4S,5R)-2)
Compound (4S,5R)-2 was prepared from (4S,5R)-10 on a 0.41 mmol scale following the general procedure to provide 0.15 g (80%) of product as white solid (mp 102.3-103.9). Rf 0.3 (95% EtOAc in hexanes); 1H NMR (CDCl3, 300 MHz) δ 7.74 (d, J = 8.5 Hz, 2H, Ar), 7.29 (d, J = 8.5 Hz, 2H, Ar), 7.09-7.04 (m, 2H, Ar), 6.96 (s, 1H, NH), 6.72-6.65 (m, 3H, Ar), 5.26 (s, 1H, NH), 4.32 (q, J = 4.5, 8.0 Hz, 1H, C(O)OCH), 4.20 (m, 2H, C(O)OCH2), 3.66 (q, J = 5.6, 13.0 Hz, 1H, HNCH), 2.98 (t, J = 4.6 Hz, 4H, PhNCH2), 2.54-2.30 (m, 6H, NCH2), 2.37 (s, 3H, COCH3); 13C NMR (CDCl3, 75 MHz) δ 196.9, 158.4, 152.7, 151.3, 142.2, 132.9, 130.1, 129.4, 120.3, 118.1, 116.4, 78.3, 65.2, 62.4, 53.9, 51.7, 49.4, 26.5; Anal. Calcd for C24H28N4O5: C, 63.70; H, 6.24; N, 12.38. Found: C, 63.71; H, 6.23; N, 12.21; [α]D22 -41.7 (c 1.05, CHCl3). For (4R,5S)-2 [α]D22 +42.4 (c 1.02, CHCl3).
2-oxo-4-(4-phenyl-piperazin-1-ylmethyl)-oxazolidin-5-ylmethyl phenyl acetate (cis-1)
Compound cis-1 was prepared from cis-10 on a 0.34 mmol scale following the general procedure to provide 50 mg (36%) of product as colorless oil. Rf 0.21 (50% EtOAc in hexanes); tR = 3.4 min (88 %; isocratic 60% MeOH/H2O); 1H NMR (CDCl3, 300 MHz) δ 7.29-7.16 (m, 7H, Ar), 6.84-6.77 (m, 3H, Ar), 5.56 (s, 1H, NH), 4.73 (ddd, J = 4.5, 6.5, 8 Hz, 1H, C(O)OCH), 4.35 (dd, J = 4.5, 12 Hz, 1H, C(O)OCHH), 4.19 (dd, J = 6.5, 12 Hz, 1H, C(O)OCHH), 3.94 (ddd, J = 5, 8, 9.5 Hz, 1H, NHCH), 3.60 (s, 2H, PhCH2), 3.08-3.05 (m, 4H, PhN(CH2)), 2.58-2.51 (m, 2H, NCH2), 2.41-2.34 (m, 3H, NCH2 and NCHH), 2.25 (dd, J = 5, 12 Hz, 1H, NCHH); 13C NMR (CDCl3, 75 MHz) δ 171.1, 158.3, 151.2, 133.6, 129.5, 129.3, 128.9, 127.5, 120.2, 116.3, 75.4, 61.9, 57.5, 53.6, 51.2, 49.3, 41.4.
2-oxo-4-(4-phenyl-piperazin-1-ylmethyl)-oxazolidin-5-ylmethyl (4-acetyl-phenyl)-carbamate (cis-2)
Compound cis-2 was prepared from cis-10 on a 0.68 mmol scale following the general procedure to provide 0.15 g (50%) of product as white solid (mp 102.5-104.0). Rf 0.23 (80% EtOAc in hexanes); 1H NMR (CD2Cl2, 300 MHz) δ 7.92 (dt, J = 8.8 Hz, 2H, Ar), 7.50 (dt, J = 8.8 Hz, 2H, Ar), 7.26-7.21 (m, 3H, Ar and NH), 6.92-6.89 (dd, 2H, Ar), 6.84 (t, 1H, Ar), 5.54 (s, 1H, NH), 4.90 (ddd, J = 3.5, 7.5, 11.5 Hz, 1H, C(O)OCH), 4.56 (dd, J = 3.5, 12 Hz, 1H, OCHH), 4.35 (dd, J = 7.5, 12.0 Hz, 1H, OCHH), 4.16 (ddd, J = 4.8, 10.0, 12.0 Hz, 1H, C(O)NHCH), 3.22-3.12 (m, 4H, PhNCH2), 2.77-2.70 (m, 2H, NCH2), 2.67-2.50 (m, 7H, C(O)CH3 and N(CH2)2); C NMR (CD2Cl2, 75 MHz) δ 197.0, 158.5, 152.9, 151.8, 142.6, 133.1, 130.2, 129.6, 120.2, 118.3, 116.5, 76.2, 63.2, 57.9, 51.5, 49.7, 30.3, 26.8.
General Method for In-line probing of RNA
AM1A was prepared via in vitro transcription and 32P-5′-end labeled as previously described.38 Labeled RNA (1 μL) was mixed with 5 μL of 2× in-line probing buffer (100 mM Tris-HCl, 200 mM KCl, 40 mM MgCl2, pH 8.3) and 1 μL of DMSO ligand stock solution. For the control experiment 1 μL of DMSO was added instead. After bringing the total volume to 10 μL with H2O the in-line probing experiments were incubated at room temperature (∼ 25 °C) for 40 hours. The resulting cleavage products were separated via 20 % denaturing polyacrylamide gel electrophoresis (19:1 acrylamide:bis-acrylamide) and the bands visualized via autoradiography. The relative normalized band intensities were determined using Nucleo Vision (NucleoTech) and plotted against ligand concentration to determine the slope for ligand-induced relative band intensity changes.
RNA Ligand Binding Assay
Ligand RNA affinities were determined using the previously described FRET-derived ligand binding assay and 100 nM 3′-Fl-AM1A-(18)-Rhd or 3′-Fl-C11U-(18)-Rhd.11,13 All RNAs were dialyzed and renatured prior to use. The FRET-labeled RNA was mixed with a series of different concentrations of the ligand (previously dissolved in DMSO) up to a final ligand concentration of 28 μM ((4S,5R)-2), 50 μM ((4R,5S)-2, cis-2) or 100 μM ((4R,5S)-1, (4S,5R)-1, cis-1) in 50 mM NaH2PO4, pH 6.5, 50 mM NaCl, 5 mM MgCl2 and 0.01 mM EDTA. Binding reactions were incubated for 130 min at 25 °C prior to analysis. Using a Molecular Devices FlexStation 96-well plat reader (Sunnyvale, CA), the FRET-labeled RNA was excited at 467 nm with a 515 nm cutoff filter, while the emission spectra were obtained over the range of 515-640 nm. The FRET was determined using the equation Qrel = |Q-Q0|/Q0 where Q is the fluorescence (F) ratio of F585nm / F525nm in the presence of ligand (or absence for Q0). Binding isotherms were constructed from duplicate Qrel data and the dissociation constant for each ligand determined using Graphpad Prism v.4. Single-site versus two-site binding models were compared and in all cases where binding was observed, the single-site binding was the preferred model.
Ligand Docking
Mono-protonated ligands were constructed in Spartan 04 (Wavefunction) and their energy minimized using molecular mechanics with MMFF.39 The subsequent structures were then exported to Macromodel (Schrödinger) and docked to the antiterminator model RNA structure 1N5336 using the Glide module of FirstDiscovery 2.7 (Schrödinger). Parameters were set such that the enclosing box encompassed the entire 1N53 structure (bulge region, A1 and A2 helices of AM1A) while the bounding box (for placing the ligand center) encompassed the bulge region and the first 1-2 adjacent base pairs for each helix. Images were rendered from the resulting structural files using PMV.40
Supplementary Material
Acknowledgments
We thank the NIH (GM073188) and Ohio University, through the BioMolecular Innovation & Technology (BMIT) project, for support of this work.
Abbreviations List
- UTR
untranslated region
Footnotes
Supporting information available: Binding isotherms, in-line probing of AM1A binding with the enantiomers, 1H and 19F NMR spectra for Mosher ester of 6 and 1H and 13C NMR spectra for all compounds. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Drysdale MJ. RNA as a drug target. Prog Med Chem. 2002;39:73–119. doi: 10.1016/s0079-6468(08)70069-9. [DOI] [PubMed] [Google Scholar]
- 2.Foloppe N, Matassova N, Aboul-Ela F. Towards the Discovery of Drug-Like RNA Ligands? Drug Discov Today. 2006;11:1019–1027. doi: 10.1016/j.drudis.2006.09.001. [DOI] [PubMed] [Google Scholar]
- 3.Gallego J, Varani G. Targeting RNA with Small-Molecule Drugs: Therapeutic Promise and Chemical Challenges. Acc Chem Res. 2001;34:836–843. doi: 10.1021/ar000118k. [DOI] [PubMed] [Google Scholar]
- 4.Thomas JR, Hergenrother PJ. Targeting RNA with Small Molecules. Chem Rev. 2008;108:1171–1224. doi: 10.1021/cr0681546. [DOI] [PubMed] [Google Scholar]
- 5.Tor Y. Targeting RNA with Small Molecules. Chembiochem. 2003;4:998–1007. doi: 10.1002/cbic.200300680. [DOI] [PubMed] [Google Scholar]
- 6.Zaman GJR, Michiels PJA, van Boeckel CAA. Targeting RNA: New Opportunities to Address Drugless Targets. Drug Discov Today. 2003;8:297–306. doi: 10.1016/s1359-6446(03)02624-2. [DOI] [PubMed] [Google Scholar]
- 7.Iorio MV, Piovan C, Croce CM. Interplay between microRNAs and the Epigenetic Machinery: An Intricate Network. Biochim Biophys Acta. 2010;1799:694–701. doi: 10.1016/j.bbagrm.2010.05.005. [DOI] [PubMed] [Google Scholar]
- 8.Smith AM, Fuchs RT, Grundy FJ, Henkin TM. Riboswitch RNAs Regulation of Gene Expression by Direct Monitoring of a Physiological Signal. RNA Biol. 2010;7:104–110. doi: 10.4161/rna.7.1.10757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Green NJ, Grundy FJ, Henkin TM. The T-box Mechanism: tRNA as a Regulatory Molecule. FEBS Lett. 2010;584:318–324. doi: 10.1016/j.febslet.2009.11.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gutierrez-Preciado A, Henkin TM, Grundy FJ, Yanofsky C, Merino E. Biochemical Features and Functional Implications of the RNA-Based T-Box Regulatory Mechanism. Microbiol Mol Biol Rev. 2009;73:36–61. doi: 10.1128/MMBR.00026-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Anupam R, Nayek A, Green NJ, Grundy FJ, Henkin TM, Means JA, Bergmeier SC, Hines JV. 4,5-Disubstituted Oxazolidinones: High Affinity Molecular Effectors of RNA Function. Bioorg Med Chem Lett. 2008;18:3541–3544. doi: 10.1016/j.bmcl.2008.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Means JA, Katz S, Nayek A, Anupam R, Hines JV, Bergmeier SC. Structure-Activity Studies of Oxazolidinone Analogs as RNA-Binding Agents. Bioorg Med Chem Lett. 2006;16:3600–3604. doi: 10.1016/j.bmcl.2006.03.068. [DOI] [PubMed] [Google Scholar]
- 13.Means JA, Hines JV. Fluorescence Resonance Energy Transfer Studies of Aminoglycoside Binding to a T Box Antiterminator RNA. Bioorg Med Chem Lett. 2005;15:2169–2172. doi: 10.1016/j.bmcl.2005.02.007. [DOI] [PubMed] [Google Scholar]
- 14.Anupam R, Denapoli L, Muchenditsi A, Hines JV. Identification of Neomycin B-binding Site in T-box Antiterminator Model RNA. Bioorg Med Chem. 2008;16:4466–4470. doi: 10.1016/j.bmc.2008.02.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Acquaah-Harrison G, Zhou S, Hines JV, Bergmeier SC. Library of 1,4-Disubstituted 1,2,3-Triazole Analogs of Oxazolidinone RNA-Binding Agents. J Comb Chem. 2010;12:491–496. doi: 10.1021/cc100029y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Grundy FJ, Moir TR, Haldeman MT, Henkin TM. Sequence Requirements for Terminators and Antiterminators in the T-box Transcription Antitermination System: Disparity between Conservation and Functional Requirements. Nucl Acids Res. 2002;30:1646–1655. doi: 10.1093/nar/30.7.1646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Maciagiewicz I, Zhou S, Bergmeier SC, Hines JV. Structure activity studies of RNA-binding oxazolidinone derivatives. Bioorg Med Chem Letters. 2011:4524–4527. doi: 10.1016/j.bmcl.2011.05.130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bergmeier SC, Katz SJ. A Method for the Parallel Synthesis of Multiply Substituted Oxazolidinones. J Comb Chem. 2002;4:162–166. doi: 10.1021/cc010050f. [DOI] [PubMed] [Google Scholar]
- 19.Bergmeier SC, Stanchina DM. Synthesis of Vicinal Amino Alcohols via a Tandem Acylnitrene Aziridination-Aziridine Ring Opening. J Org Chem. 1997;62:4449–4456. doi: 10.1021/jo970473x. [DOI] [PubMed] [Google Scholar]
- 20.Bergmeier SC, Stanchina DM. Acylnitrene Route to Vicinal Amino Alcohols. Application to the Synthesis of (-)-Bestatin and Analogues. J Org Chem. 1999;64:2852–2859. doi: 10.1021/jo9823893. [DOI] [PubMed] [Google Scholar]
- 21.Schaus SE, Brandes BD, Larrow JF, Tokunaga M, Hansen KB, Gould AE, Furrow ME, Jacobsen EN. Highly Selective Hydrolytic Kinetic Resolution of Terminal Epoxides Catalyzed by Chiral (salen)Co-III Complexes. Practical Synthesis of Enantioenriched Terminal Epoxides and 1,2-Diols. J Am Chem Soc. 2002;124:1307–1315. doi: 10.1021/ja016737l. [DOI] [PubMed] [Google Scholar]
- 22.Dale JA, Dull DL, Mosher HS. α-Methoxy-α-Trifluoromethylphenylacetic Acid, a Versatile Reagent for Determination of Enantiomeric Composition of Alcohols and Amines. J Org Chem. 1969;34:2543–2549. [Google Scholar]
- 23.Rottger S, Waldmann H. Solid-Phase Synthesis of Decalin Scaffolds by Robinson Annulation with Immobilised Nazarov Reagents. Eur J Org Chem. 2006:2093–2099. [Google Scholar]
- 24.Nelson WL, Freeman DS, Vincenzi FF. Stereochemical Analogs of a Muscarinic, Ganglionic Stimulant .2. Cis and Trans Olefinic, Epoxide, and Cyclopropane Analogs Related to 4-[n-(3-Chlorophenyl)carbamoyloxy]-2-butynyltrimethylammonium Chloride (McN-A-343) J Med Chem. 1976;19:153–158. doi: 10.1021/jm00223a026. [DOI] [PubMed] [Google Scholar]
- 25.Katoh T, Itoh E, Yoshino T, Terashima S. Total Synthesis of an Enantiomeric Pair of FR900482 .1. Synthetic and End-Game Strategies. Tetrahedron. 1997;53:10229–10238. [Google Scholar]
- 26.Calderon F, Doyaguez EG, Fernandez-Mayoralas A. Synthesis of Azasugars through a Proline-Catalyzed Reaction. J Org Chem. 2006;71:6258–6261. doi: 10.1021/jo060568b. [DOI] [PubMed] [Google Scholar]
- 27.SanchezSancho F, Valverde S, Herradon B. Stereoselective Syntheses and Reactions of Chiral Oxygenated alpha,beta-Unsaturated-gamma- and delta-Lactones. Tetrahedron-Asymmetry. 1996;7:3209–3246. [Google Scholar]
- 28.Previous 13C NMR studies on acetonides indicated that the chemical shift of the acetalic carbon is located in the region of 107-110 ppm for a five-membered ring acetonide and 98.5-100.6 ppm for a six-membered ring acetonide. In the 13C NMR spectrum of our acetonide was only observed a signal at 109.0 ppm clearly indicating the desired five-membered ring acetonide.
- 29.Fadnavis NW, Sharfuddin M, Vadivel SK. Porcine Pancreatic Lipase Mediated Regio- and Stereoselective Hydrolysis: Chemoenzymatic Synthesis of (2S,3S)-2-Amino-3,4-dihydroxybutyric Acid. Tetrahedron-Asymmetry. 2001;12:691–693. [Google Scholar]
- 30.Nicolaou KC, Reddy KR, Skokotas G, Sato F, Xiao XY, Hwang CK. Total Synthesis of Hemibrevetoxin-b and (7a-α)-epi-Hemibrevetoxin-b. J Am Chem Soc. 1993;115:3558–3575. [Google Scholar]
- 31.Jorgensen WL, Maxwell DS, TiradoRives J. Development and Testing of the OPLS All-Atom Force Field on Conformational Energetics and Properties of Organic Liquids. J Am Chem Soc. 1996;118:11225–11236. [Google Scholar]
- 32.Eldridge MD, Murray CW, Auton TR, Paolini GV, Mee RP. Empirical Scoring Functions .1. The Development of a Fast Empirical Scoring Function to Estimate the Binding Affinity of Ligands in Receptor Complexes. J Comput-Aided Mol Des. 1997;11:425–445. doi: 10.1023/a:1007996124545. [DOI] [PubMed] [Google Scholar]
- 33.FirstDiscovery 2.7 Technical Notes. Schrodinger; 2003. [Google Scholar]
- 34.Litovchick A, Rando RR. Stereospecificity of Short Rev-derived Peptide Interactions with RRE IIB RNA. RNA. 2003;9:937–948. doi: 10.1261/rna.2172103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Paulsen RB, Seth PP, Swayze EE, Griffey RH, Skalicky JJ, Cheatham TEI, Davis DR. Inhibitor-induced structural change in the HCV IRES domain IIa RNA. Proc Natl Acad Sci. 2010;107:7263–7268. doi: 10.1073/pnas.0911896107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gerdeman MS, Henkin TM, Hines JV. Solution Structure of the Bacillus Subtilis T-box Antiterminator RNA: Seven Nucleotide Bulge Characterized by Stacking and Flexibility. J Mol Biol. 2003;326:189–201. doi: 10.1016/s0022-2836(02)01339-6. [DOI] [PubMed] [Google Scholar]
- 37.Regulski EE, Breaker RR. In-line Probing Analysis of Riboswitches. Methods Mol Biol. 2008;419:53–67. doi: 10.1007/978-1-59745-033-1_4. [DOI] [PubMed] [Google Scholar]
- 38.Gerdeman MS, Henkin TM, Hines JV. In Vitro Structure-Function Studies of the Bacillus Subtilis tyrS mRNA Antiterminator: Evidence for Factor-Independent tRNA Acceptor Stem Binding Specificity. Nucleic Acids Res. 2002;30:1065–1072. doi: 10.1093/nar/30.4.1065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hehre VJ, K PE. A Guide to Molecular Mechanics and Molecular Orbital Calculations in Spartan. Irvine, CA: Wavefunction, Inc.; 1997. [Google Scholar]
- 40.Sanner MF. Python: A Programming Language for Software Integration and Development. J Mol Graphics Mod. 1999;17:57–61. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.