

Abstract
The design, synthesis, docking, and biological evaluation of novel potent HDAC3 and HDAC8 isoxazole- and pyrazole-based diazide probes suitable for Binding Ensemble Profiling with Photoaffinity Labeling (BEProFL) experiments in cells is described. Both the isoxazole- and pyrazole-based probes exhibit low nanomolar inhibitory activity against HDAC3 and HDAC8, respectively. The pyrazole-based probe 3f appears to be one of the most active HDAC8 inhibitors reported in the literature with an IC50 of 17 nM. Our docking studies suggest that unlike the isoxazole-based ligands the pyrazole-based ligands are flexible enough to occupy the second binding site of HDAC8. Probes/inhibitors 2b, 3a, 3c, and 3f exerted the anti-proliferative and neuroprotective activities at micromolar concentrations through inhibition of nuclear HDACs, indicating that they are cell permeable and the presence of an azide or a diazide group does not interfere with the neuroprotection properties, or enhance cellular cytotoxicity, or affect cell permeability.
Keywords: histone deacetylases, HDAC, Binding Ensemble Profiling with Photoaffinity Labeling, BEProFL, diazide probes, pyrazoles, isoxazoles, antiproliferative activity, neuroprotection
Introduction
Histone deacetylases (HDACs)1 are well established enzymatic targets for multiple therapeutic applications.2 It is widely accepted that therapeutic application of HDAC inhibitors depends on their HDAC class and isoform selectivity profile,3–4 making isoform selective inhibitors an important issue in the design and development of novel HDAC-based therapeutics. One of the key challenges in HDAC inhibitor design is the limited amount of information regarding the binding modes (poses) available to the highly solvent exposed surface binding groups (SBG) of HDAC inhibitors. To address this problem we recently developed a Binding Ensemble Profiling with Photoaffinity Labeling (BEProFL) approach that utilizes a diazide probe to map experimentally the multiple binding poses of the SBGs of HDAC ligands.5 The method requires availability of the active and preferably isoform-selective HDAC probes containing an aromatic azide group, which can covalently cross-link to the protein of interest upon UV-irradiation, and an aliphatic azide group, which can react with a biotin or fluorescent tag for further purification or visualization. The design of the diazide-containing HDAC probes appears to be nontrivial because the extra bulk of the diazide group may affect the HDAC activity and selectivity of the parent HDAC inhibitors. For example, the diazide probe 1 (Figure 1), which was designed by adding a diazide portion to SAHA through a linker and used in our recent BEProFL approach, showed a 17-fold decrease in HDAC8 inhibitory activity compared to SAHA.5
Figure 1.

Structure of BEProFL probe 1 and general structure of novel isoxazole-based compounds 2 and pyrazole-based compounds 3.
Further extension of the BEProFL approach to cell-based studies would also require probes that are not highly cytotoxic, and, therefore, evaluation of antiproliferative properties of the probes in the cell-based studies needs to accompany the evaluation of the HDAC activity of the probes against recombinant proteins in vitro. Design of isoform-selective inhibitors within a class is particularly challenging because of the similarity between isoforms within the class. In this paper we decided to focus on diazide probes for the two class I isoforms HDAC8 and HDAC3, which inhibitors may be of particular value for the treatment of neuroblastoma and leukemia,6–7 and gastric, prostate, and colorectal cancer,8 respectively.
As a starting point for the design of HDAC3 diazide probes, we decided to use recently reported isoxazole-based inhibitors that are particularly active against HDAC3,9 whereas for the design of HDAC8 active probes we explored a novel 4-aminopyrazole-based scaffold. Herein, we report the design, synthesis, docking, and biological evaluation of active HDAC3 and HDAC8 diazide probes suitable for photolabeling experiments in cells.
Results and discussion
Probe design
Our preliminary docking studies showed that HDAC inhibitors with linear scaffolds can be accommodated by both HDAC3 and HDAC8 binding sites. The next step was to ensure that the highly lipophilic diazide portions of the probes necessary for the BEProFL are efficiently hidden in the ridges surrounding the binding site. The overall design of the probes is shown in Figure 2. The zinc binding group (ZBG) was chosen as in SAHA and many other similar hydroxamic acid-based ligands as it is known for its excellent Zinc (II) complexing properties. The flexible aliphatic chain in the linker allows the ligands to find the most appropriate place for their SBG. The next portion of the ligand – a five-membered heterocycle – was designed so that it can provide the necessary kink and facilitate the placement of the SBG containing the diazide next to the protein surface. The latter is necessary for both the binding and the efficient cross-linking of the diazide portion upon UV irradiation in BEProFL. Two heterocycles drew our attention based on the criteria described above and ease of synthesis – an isoxazole and a 4-aminopyrazole ring. The two substituents form an angle of ca 145° allowing the substituents in the isoxazole and the pyrazole rings to be positioned complementary to the pseudo-spherical shape of the protein. We envisioned that the heteroatoms in the ring systems, as being more hydrophilic, may be oriented toward the solvent, whereas the more hydrophobic portions of the heterocycles would be hidden in one of the grooves on the protein surface, thus facilitating the desired placement of the diazide portion of the probes next to the protein surface. In an opposite orientation of the five-membered heterocycles, the large portions of the SBG would not make any pronounced interactions with the protein surface. An amide group was chosen as connecting unit between the heterocyclic and linker portions of the ligands. On one hand, it provides the necessary versatility of the synthesis and, on the other hand, the amide NH is known to form hydrogen bond with the side chain of Asp101 at the gorge region of the binding site.10–11 Excellent potential of the design described above is supported by a series of highly HDAC3 active isoxazole-based HDAC inhibitors.9 Until recently,12 the activity of the published HDAC8 inhibitors was mostly in the high nanomolar to low micromolar range, leaving us no other options than to try a novel 4-aminopyrazole scaffold in an attempt to find high affinity probes for HDAC8.
Figure 2.

Design of isoxazole- and pyrazole-based diazide probes.
Synthesis
A practical and scalable synthetic route for diazide moiety 8 was furnished following the procedure described earlier for 8 by Hosoya et al.13 Several modifications were done to the original procedure to improve the yields, regioselectivity, and ease of handling. The synthesis of probe 8 is outlined in scheme 1. Dimethyl 5-aminoisophthalate (4) was treated with excess LiAlH4 in THF to give the corresponding amino-diol 5. We replaced the conventional diazotization conditions, NaNO2/AcOH–H2O/NaN3, with t-Bu-ONO and TMS-N3 in CH3CN14 for amine to azide transformation. In the former method, the polar nature of the resulting azido-diol 6 made the aqueous workup tedious, resulting in loss of the product 6 in the water fractions, particularly during large scale preparations. In our optimized conditions, the azido-diol 6 was obtained with a 90% yield by a direct chromatography purification of the reaction mixture after evaporation of the organic solvent. We also optimized the next step - the selective conversion of the azido-diol 6 to diazido-alcohol 7. The reported method for this conversion involves a two-step procedure – a selective bromination using TPP/CBr4 in DMF followed by a nucleophilic azide displacement reaction. However, we noted that, in our hands, the bromination step reproducibly yielded only 50% or less of the required mono-bromide along with a 10% of a dibromide and some starting material still unreacted after 3 h at 0 °C. Prolonged reaction conditions and/or elevated temperatures led to the formation of the undesired dibromide as the major product. Besides, it was difficult to recover the starting material from the byproduct of the reaction, TPPO, as both appeared with the same Rf on TLC. The newly adopted one step conversion of alcohol to azide using bis(2,4-dichlorophenyl) chlorophosphate, DMAP, and NaN3 in DMF15 gave the diazido-alcohol 7 in 70% yield along with 10% of a triazide (based on conversion) and with a 10% starting material recovery. The optimized conditions also allowed us to avoid possible formation of an amine by-product as TPP is known to reduce the azides to amines via phosphazene formation. Treatment of 7 with equivalents of tosyl chloride in the presence of triethylamine in CH2Cl2 at 0 °C afforded the required diazido-tosylate 8 in 85% yield in 2 h. It is worth mentioning that an increase in temperature of the reaction from 0 °C to rt resulted in an unwanted by-product.
Scheme 1.

Synthesis of diazide moietiesa
a Reagents and conditions: (a) LiAlH4, THF, 0 °C–rt, 8 h; (b) t-Bu-ONO, TMS-N3, MeCN, 0 °C–rt, 2 h; (c) Bis(2,4-dichlorophenyl) chlorophosphate, DMAP, NaN3, DMF, rt, 4 h; (d) Ts-Cl, Et3N, 0 °C, 2 h; (e) LiBH4, THF, 50 °C, 48 h; (f) 2N NaOH, MeOH–THF (1:1), 1 h.
The synthesis of the diazido-acid 12 was initiated from a common precursor 4 following a synthetic route similar to that used to prepare diazido-tosylate 8 (scheme 1). After considerable experimentation using different metal hydride reducing agents such as LiAlH4, NaBH4, and LiBH4 the required regioselective reduction of 4 to amino-ester 9 was achieved using LiBH4 in THF at 50 °C for 48 h. Azido-ester 10 and diazido-ester 11 were prepared in high yields employing the same reaction conditions described for the synthesis of azido-diol 6 and diazido-alcohol 7, respectively. The subsequent basic hydrolysis of ester group of 11 produced the desired diazido-acid 12.
Having an efficient access to both the diazide moieties, we moved on to design and synthesis of the isoxazole-based HDAC inhibitors (Scheme 2). The ester 16 was prepared according to the procedure described earlier.9 1,3-Dipolar cycloaddition of ethyl chlorooximidoacetate to N-Boc alkyne 13 in the presence of Na2CO3 in THF–H2O afforded the 5-arylisoxazole-3-carboxylic acid ethylester 14 in 85% yield. This reaction was preferred over triethylamine-mediated cycloaddition reaction because the latter was reported earlier to give a low 40% yield for the same ethylester 14.9 Hydrolysis of 14 followed by an amide coupling reaction of the resulting acid 15 with methyl 7-aminoheptanoate gave the key intermediate ester 16. Compound 16 was treated with TFA in CH2Cl2 resulting in formation of an aromatic amine 17 that was transformed to azide 18 using NaNO2/NaN3 in AcOH–H2O. The amide coupling reaction between amine 17 and acid 12 under the standard EDC/HOBt/DIPEA conditions was slow and gave only low yields (up to 15%) of 19 after prolonged reaction time. An alternative coupling reaction using DCC/HOBt/DMAP in CH2Cl2 resulted in 19 with a much better yield of 70%. Further treatment of the esters 18 and 19 with freshly prepared NH2OH in the presence of KOH in MeOH gave the desired hydroxamates 2a and 2b respectively.
Scheme 2.

Synthesis of 2a–2ba
aReagents and conditions:(a) ethyl chlorooximidoacetate, Na2CO3, THF–H2O (1:1), 0 °C–rt, 18 h; (b) 2N NaOH, MeOH–THF (1:1), 0 °C–rt, 1 h; (c) EDC, HOBt, methyl 7-aminoheptanoate hydrochloride, DIPEA, CH2Cl2, rt, 4 h; (d) TFA, CH2Cl2, rt, 4 h; (e) NaNO2, AcOH–H2O, 0 °C, 10 min, then NaN3, 0 °C–rt, 1 h; (f) 12, DCC, HOBt, DMAP, CH2Cl2, 0 °C–rt, 6 h; (g) NH2OH, KOH, MeOH, 0 °C–rt, 3 h.
The strategy for the synthesis of pyrazole inhibitors is presented in Scheme 3. It involves an amide coupling reaction between 4-aminopyrazole (21), derived from the corresponding commercially available 4-nitropyrazole (20) by hydrogenolysis, and monomethyl suberate to give the ester 22.16 Alkylation of 22 with 8 in presence of K2CO3 in refluxing acetone afforded ester 23. Compounds 24 and 25 were obtained by alkylating 22 with benzyl bromide or 4-nitrobenzyl bromide in the presence of NaH in DMF, respectively. Reduction of the nitro group of 25 using SnCl2·2H2O in refluxing methanol resulted in aniline 26, a key intermediate for compounds 27–29. Diazotization of amino group of aniline 26 followed by an azide displacement reaction with NaN3 gave the corresponding azido compound 27. Treatment of 26 with Boc anhydride and Et3N in CH2Cl2 furnished the carbamate 28. Ester 29 was obtained by an amide coupling reaction between 26 and diazido-acid 12. Treatment of the methyl esters 23–25 and 27–29 with KOH/NH2OH in MeOH gave the corresponding hydroxamates 3f and 3a–3e, respectively.
Scheme 3.

Synthesis of 3a–3fa
aReagents and conditions: (a) H2, Pd/C, MeOH, overnight; (b) monometyl suberate, EDC, HOBt, DIPEA, CH2Cl2, 0 °C–rt, 6 h; (c) 8, K2CO3, acetone, reflux, 6 h; (d) NH2OH, KOH, 0 °C–rt, 3 h; (e) BnBr or 4-nitrobenzyl bromide, NaH, DMF, 0 °C–rt, 4 h; (f) SnCl2·2H2O, MeOH, reflux, 2 h; (g) NaNO2, AcOH–H2O (9:1), 0 °C, 10 min, NaN3, 0 °C–rt, 1 h; (h) (Boc)2O, Et3N, CH2Cl2, rt, 4 h; (i) 12, EDC, HOBt, DIPEA, CH2Cl2, 0 °C–rt, 6 h.
Activity assays
The isoxazoles 2a–2b and pyrazoles 3a–3f were tested for inhibition of HDAC3 and HDAC8 isoforms and the procedure is identical to that published previously.5 The inhibition of HDAC8 was measured using the fluorescent acetylated HDAC substrate Fluor de Lys and commercially available recombinant human HDAC8, whereas the inhibition of HDAC3 was measured using the fluorescent HDAC substrate Boc-L-Lys(Ac)-AMC and commercial available recombinant human HDAC3/NCoR2. The HDAC3/8 activity profiles are summarized in Tables 1 and 2. All newly synthesized HDAC inhibitors/probes tested had IC50s ranging from 17 nM to 707 nM. As expected, the isoxazole-based probes 2a and 2b showed HDAC3 selectivity over HDAC8, with 2b being the most potent HDAC3 inhibitor with HDAC3 and HDAC8 IC50s of 45 nM and 651 nM, respectively, and an IC50 HDAC8/HDAC3 ratio above 14. Isoxazole-based probe 2a inhibited HDAC3 and HDAC8 with IC50s of 73 nM and 707 nM, respectively. Both probes 2a and 2b maintained the HDAC3/8 activity and selectivity comparable to that of SAHA.
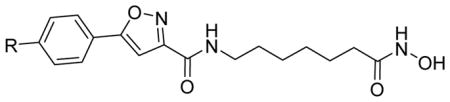
Table 1.
HDAC3 and HDAC8 isoform inhibitory activity (IC50, nM) of isoxazole-based compounds 2a and 2b.
| ||||
|---|---|---|---|---|
| Compd | Substituent R | IC50±SD (nM) | HDAC8/HDAC3a | |
| HDAC3a | HDAC8b | |||
| SAHA | 27 ± 1.0 | 440 ± 21 | 16.3 | |
| 1 | 1480 ± 220 | 7340 ± 22 | 4.96 | |
| 2a | N3 | 73 ± 15 | 707 ± 86 | 9.68 |
| 2b |
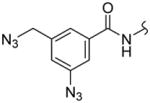
|
45 ± 1.3 | 651 ± 12 | 14.5 |
ratio of IC50 (nM)
Table 2.
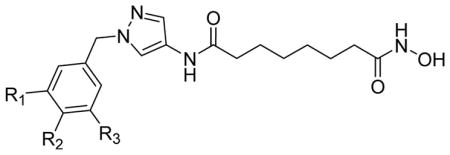
HDAC3 and HDAC8 isoform inhibitory activity (IC50, nM) of pyrazole-based compounds 3a–3f.
| ||||
|---|---|---|---|---|
| compd | Substituents R1, R2, R3 | IC50 ± SD (nM) | HDAC8/HDAC3a | |
| HDAC3 | HDAC8 | |||
| 3a | R1, R2, R3 = H | 44 ± 5.8 | 76 ± 5.0 | 1.7 |
| 3b | R2 = NO2, R1, R3 = H | 59 ± 1.0 | 82 ± 9.0 | 1.4 |
| 3c | R2 = N3 R1, R3 = H | 22 ± 1.3 | 28 ± 3.0 | 1.3 |
| 3d | R2 = NHBoc R1, R3 = H | 191 ± 18 | 147 ± 15 | 0.8 |
| 3e |
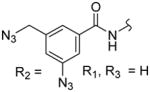
|
432 ± 52 | 487 ± 80 | 1.1 |
| 3f | R1 = CH2N3 R2 = H R3 = N3 | 128 ± 9.8 | 17 ± 3.0 | 0.13 |
ratio of IC50 (nM)
Among the pyrazole-based inhibitors, the simplest benzyl-substituted compound 3a inhibited HDAC3 and HDAC8 with IC50s of 44 nM and 76 nM, respectively. Introduction of a nitro group at 4-position of benzyl group of 3a resulted in compound 3b that showed slightly lower activity for both isoforms – 59 nM and 82 nM against HDAC3 and HDAC8, respectively, whereas the corresponding azido compound 3c exhibited a 2.0- and 2.7-fold better potency – 22 nM and 28 nM for HDAC3 and HDAC8, respectively. Overall, compounds 3a–3c exhibited an inhibitory activity against HDAC3 comparable to that of SAHA but a better double digit nanomolar activity against HDAC8. Introduction of a bulky Boc-protected amino group in 3d decreased the HDAC activity by about 10-fold, with an IC50 of 191 nM for HDAC3 and 147 nM for HDAC8 isoform. Replacement of the Boc group with a lipophilic aromatic diazide via a rigid amide bond in 3e further decreased the activity for both HDAC3 and HDAC8 to 432 nM and 487 nM, respectively. Comparison of the activity data of 3b–3c with 3d–3e clearly shows that the presence of the bulky substituent in the para position of the terminal phenyl ring of 3a leads to the lower activities for both HDAC3 and HDAC8 isoforms. This observation led us to the modification of 3- and 5-positions of the phenyl group of 3a and replacement of the phenyl group with a 3-azido-5-azidomethyl phenyl group resulting in 3f. To our surprise, compound 3f was 8-fold more active towards HDAC8 than for HDAC3, with IC50s equal to 17 nM and 128 nM, respectively.
Docking
To gain insights into the possible reasons for activity and selectivity of the two diazide probes 2b and 3f that would also facilitate the further BEProFL experiments, we docked these compounds to HDAC8 crystal structures available in the Protein Databank. Although we also docked these ligands to homology models of HDAC3 we decided not to discuss these results here as they may not represent the true binding poses. Indeed, the lack of any structural information on the HDAC3-NCoR2 complex, which formation is required for histone deacetylase activity of HDAC3, the likely malleability of the protein surface of HDAC3 (based on malleability of the surface of HDAC8), and the relative imprecision of homology models for interpretation of already theoretical docking poses, make these results highly hypothetical and very unreliable. The HDAC8 docking poses and MOE analysis of 2b and 3f are shown in Figures 3 and 4, respectively. The best scoring pose of both compounds are found with receptor structures based on a HDAC8 protein structure with an open second binding pocket. The best docking score for probe 2b was obtained for PDB:1T64, and in case of 3f – PDB:1VKG. The interactions between probe 2b and the binding site are consistent with those expected by design, i.e. ZBG interacts with the zinc atom, the amide NH between the linker and SBG forms hydrogen bond with the side chain COO− of Asp101, the heterocycle provides the important kink, the heteroatoms in the isoxazole point toward the solvent, whereas its opposite side points toward the protein, the amide group connecting the terminal phenyl ring forms hydrogen bond with Lys145, and the phenyl ring and the diazide portion slide along the surface. The SBG of 2b is linear and rigid and therefore has limited options in adjusting itself to the landscape of the protein surface and is mostly solvent exposed. Probe 3f, on the other hand, may have its SBG buried in a second binding pocket, leading to extensive non-polar interactions between the ligand and the protein and a π-cation charge transfer interactions between the terminal phenyl ring of 3f and Arg37. In comparison to probe 2b, probe 3f is much less solvent exposed (Fig. 4). The interpretation of these two poses agrees with the measured activities of the two compounds, as 3f, which is more active against HDAC8 than 2b, appears to find more favorable interactions with HDAC8, while the opposite is true for 2b. The binding of the SBG of the compounds in the pyrazole series to the secondary pocket is consistent with the SAR. Ligands with the relatively small substituents in the terminal phenyl ring 3a-3c and 3f show activity below 82 nM for HDAC8, with probe 3f peaking at 17 nM. As the size and rigidity of the substituent increases in ligands 3d and 3e their activity drops to 147 nM and 487 nM, respectively. Overall, availability of the second site for the binding of ligands is supported by X-ray crystallographic data, the upside-down pose of the probe 1 detected with the BEProFL approach,5 binding of the linkerless HDAC ligands,12 and the recent modeling studies by Wiest et al.17
Figure 3.

(A) 2b docked to HDAC8 PDB:1T64 (B) 3f docked to HDAC8 PDB:1VKG. Description of the grooves (G1–G3) and ridges (R1–R3) formed by the protein surface of HDAC8 is the same as in Ref. 5.
Figure 4.

MOE analysis of the interactions between HDAC8 receptors and ligands 2b (A) and 3f (B): (green) hydrophobic, (light-purple) polar, (blue ring) basic,(red ring) acidic, (purple ring) solvent exposed.
HDACs and MMPs activity profiling
We also conducted a preliminary HDAC profiling of 2b and 3f for the remaining class I HDACs 1 and 2, class II HDACs 4–7 and 9–10, and class IV HDAC 11 isoforms and a representative set of matrix metalloproteases (MMPs) 1, 3, 9, 14 (Table 3 and the Supplemental material). The IC50s of 2b and 3f were compared to those obtained for TSA for HDAC isoforms 1, 2, 4–7, and 9–11, SAHA – for class I HDACs, and galrdin (GM6001) – for MMPs. Neither of the two compounds exhibits a pronounced level of activity against the MMPs. When compared to activity of SAHA and TSA against class I, II, and IV HDACs, probe 3f demonstrated superior selectivity for HDAC8 (Table 3). Specifically, IC50s of 3f for all HDAC isoforms tested was between 3.11- and 119-fold higher than those of SAHA or TSA for the same HDAC isoforms except for HDAC8, for which the ratio of IC50s was 0.0386. A somewhat similar but less pronounced trend was observed for 2b. When compared to SAHA or TSA, 2b was relatively less potent against all HDAC isoforms, with only 1.67, 1.48, and 1.88-fold difference for HDAC3, HDAC8, and HDAC9, respectively. Within class I HDACs 2b and 3f were clearly more selective for HDAC3 and HDAC8, respectively, compared to SAHA.
Table 3.
A preliminary screening for inhibitory activity (IC50, nM) of 2b and 3f against HDAC 1, 2, and 4–7, and 9–11 isoformsa and a representative set of metalloproteases (MMPs) 1, 3, 9, and 14.
| IC50 (nM) | IC50 ratios | ||||
|---|---|---|---|---|---|
| Target | Control inhibitorb | 2b | 3f | 2b/TSA 2b/SAHA | 3f/TSA 3f/SAHA |
| HDAC1c | 8.22 ± 1.45 (TSA), 22.37 ± 4.7 (SAHA) | 82.2 ± 1.22 | 99.1 ± 12.5 | 10.0 3.67 |
12.1 4.43 |
| HDAC2c | 68 ± 1.9 (TSA), 100.0 ± 0.25 (SAHA) | 780 ± 93 | 1000 ± 180 | 11.5 7.80 |
14.7 10.0 |
| HDAC3d | NDe 1.67 |
NDe 4.74 |
|||
| HDAC4f | 122 | 432 | 379 | 3.54 | 3.11 |
| HDAC5f | 8.08 | 932 | 892 | 115 | 110 |
| HDAC6f | 2.31 | 47.0 | 274 | 20.4 | 119 |
| HDAC7f | 139 | 2230 | 4510 | 16.0 | 32.5 |
| HDAC8d | NDe 1.48 |
NDe 0.0386 |
|||
| HDAC9f | 20.1 | 37.8 | 704 | 1.88 | 35.0 |
| HDAC10f | 11.3 | 776 | 140 | 68.7 | 12.4 |
| HDAC11f | 13.9 | 116 | 476 | 8.35 | 34.2 |
| MMP1f | 0.20 | 108000 | NAg | 540000 | ND |
| MMP3f | 7.62 | 12100 | NAg | 1590 | ND |
| MMP9f | 0.05 | 22900 | NAg | 458000 | ND |
| MMP14f | 0.41 | 9330 | NAg | 22800 | ND |
First number – TSA was used as a control inhibitor in HDAC 1, 2, 4–7, and 9–11 assays. Galardin (GM6001) was used as a control inhibitor in MMPs 1, 3, 9, and 14 assays. Second number – SAHA was used as an additional control inhibitor for class I isoforms.
IC50 values expressed as mean ± standard deviation of at least two independent experiments.
ND – IC50 for TSA were not determined for HDAC3 and HDAC8.
IC50 is a single measurement estimate provided by Reaction Biology (see supporting information).
NA – The remaining activity of the MMPs was above 70% at the highest tested concentration (10 mM).
Cell-based studies
In the addition to our primary goal – design of potent HDAC3 and HDAC8 BEProFL probes – our secondary objective was to demonstrate that the probes are compatible with cell-based experiments and have no effect on the integrity or the fitness of the cell. Specifically, it was important to show that the probes are cell membrane permeable, not overly cytotoxic, and the treatment of cells with them leads to physiological outcomes expected for HDAC inhibitors. Only compounds that meet those criteria can be used in future experiments as photoaffinity probes to assess the binding poses favored in the intracellular environment.
We explored the antiproliferative activity of the HDAC3 active diazide isoxazole probe 2b and the potent pyrazole inhibitor/probes 3a, 3c and 3f in three carcinoma cell lines of human origin. Our results show that the hepatocarcinoma HepG2 cells, cervical carcinoma Hela cells, and neuroblastoma SH-SY5Y cells are all sensitive to micromolar concentrations of the isoxazole and pyrazole HDAC inhibitors (Table 4). The antiproliferative action of the most HDAC8 active pyrazole inhibitor 3f (EC50–48 h = 24–36 μM) is in the same range compared to that obtained for the most HDAC3 active isoxazole inhibitor 2b (EC50–48 h = 11–32 μM) and SAHA (EC50–48 h = 8–26 μM). Also, the compounds 3a and 3c which inhibit HDAC3 and HDAC8 with comparable potencies (HDAC8/HDAC3 = 1.3–1.7, Tables 2–3), exert very comparable antiproliferative activity towards the three cell lines (EC50–48 h = 10–23 μM), indicating that difference in HDAC3/HDAC8 isoform activity observed in vitro has little impact on the value of EC50s. Comparison of the antiproliferative activity of azide 3c and diazides 3f and 2b with that of the non-azide inhibitor 3a suggests that the presence of an azide or a diazide group by itself does not enhance cell death and the antiproliferative activity observed is likely due to the inhibition of cellular HDACs of class I.
Table 4.
Antiproliferative activity (EC50) of 2b, 3a, 3c and 3f towards HepG2, Hela and SH-SY5Y carcinoma cell lines.
| Compd | HepG2a | Helaa | SH-SY5Ya | ||||||
|---|---|---|---|---|---|---|---|---|---|
| 24 h | 48 h | 72 h | 24 h | 48 h | 72 h | 24 h | 48 h | 72 h | |
| SAHA | >50 μM | 8.4± 1.8 | 3.8± 1.1 | 43.0± 2.3 | 14.2± 1.4 | 4.7± 1.2 | > 50 μM | 25.6± 1.5 | 17.4± 8.3 |
| 2b | >50 μM | 11.5± 2.5 | 4.9± 1.4 | 26.5± 1.1 | 10.7± 1.2 | 4.7± 1.1 | > 50 μM | 32.2± 1.1 | 23.0± 1.1 |
| 3a | >50 μM | 12.6± 1.6 | 5.5± 1.4 | 46.6± 1.2 | 10.6± 2.3 | 5.5± 1.3 | > 50 μM | 22.7± 1.1 | 23.2± 2.2 |
| 3c | >50 μM | 12.7± 1.3 | 5.9± 1.3 | 38.3± 1.1 | 9.9± 1.4 | 4.6± 1.3 | > 50 μM | 23.6± 1.4 | 23.1± 1.3 |
| 3f | >50 μM | 24.3± 1.5 | 13.4± 1.4 | 30.6± 3.3 | 23.7± 1.1 | 15.0± 1.1 | > 50 μM | 35.9± 1.1 | 20.8± 3.1 |
EC50 in μM concentration
The inhibitory effect of SAHA and 2b on cellular HDACs was assessed by monitoring the acetylation status of histone H4 in the HepG2 cell line using Western blot analysis. Both SAHA and 2b caused a marked increase in acetylation of histone H4 by 6 h with acetylation being sustained throughout 24 h post-treatment at low micromolar concentrations (Figure 5). These results suggest that 2b efficiently targets nuclear HDACs.
Figure 5.

Western blot detection of the acetylation of Histone H4 following treatment with the diazide probe 2b in HepG2 cells.
In an effort to delineate pathways responsible for the cellular death observed in the presence of our HDAC inhibitors, we looked at the activation/cleavage of caspase 3. Inhibitor 2b and the control compound SAHA, induced cleavage of caspase 3 by 14 h with maximum activation at 24 h (Figure 6). Taken together, our results indicate that in HepG2 cells, the inhibitor 2b successfully inhibits nuclear HDACs within 6 h of treatment (Figure 5), induces caspase 3 cleavage within 14 h of treatment (Figure 6) and cell death within 24–48 h (Table 4) as efficiently as SAHA does. This is consistent with another study where the apoptotic effect of 10 μM SAHA appears after a 12–16 h lag-phase in HepG2 cells and reached a maximum at about 36 h.18
Figure 6.

The isoxazole HDAC inhibitor/probe 2b induces apoptosis in HepG2 cells independent of p53 acetylation: A. Western blotting to detect the activation of caspase 3 and the acetylation of p53 in the treated HepG2 cells; B. Histograms representing the acetylation of p53 protein in the cell extracts.
SAHA has been shown to induce programmed cell death in HepG2 cells through the acetylation of p5318 and the decrease of BCL-219 and we sought to investigate whether 2b was exerting its pro-apoptotic effect via the same mechanism. HepG2 cells were treated with SAHA or 2b and the acetylation of p53 was monitored (Figure 6). In our conditions, the level of acetylated p53 is already significant in non-treated cells and neither SAHA nor 2b induced additional time and dose-dependent hyperacetylation of p53 in total cell extracts. Similar results were obtained independently on human lung carcinoma cells.20 The data reported by Carlisi et al.18 actually show a modest two-fold increase in p53-acetylation and very faint bands for acetylated-Lysine320 p53 and acetylated-Lys373/382 p53 immunoprecipitated from nuclear extracts with the p53 DO1 antibody. Another study performed in neuroblastoma cells also shows a modest acetylation of p53 upon treatment with 3 mM valproic acid, a pan HDAC inhibitor.21 We do not exclude that SAHA or 2b acts directly on the acetylation level of p53 and its function in the nucleus but did not pursue further investigation, as this effect is very modest in our conditions.
The HDAC inhibitor SAHA is FDA approved for treating T-cell lymphoma,22 yet, in cell and mouse research studies, can be protective to stressed neuronal cells.23–24 We, thus, next investigated whether the diazide isoxazole 2b exerted a neuroprotective effect in a retinoic acid differentiated SH-SY5Y human neuroblastoma cell model treated with 30 (LY294002), a widely used inhibitor of the phosphatidylinositol 3-kinase PI3K/AKT survival pathway.25 The HDAC inhibitors display differential cytotoxic and neuroprotective effects when cells stressed with 100 μM of 30 (Figure 7). The two isoxazoles 2a and 2b and the pyrazole 3f protect cells from stress induced by compound 30 at low micromolar concentrations but only the isoxazole 2a is not overly cytotoxic to retinoic acid-differentiated neuroblastoma cells at the highest concentrations tested (Figure 7). The pyrazole 3f more than doubles the amount of cell survival at 0.5 and 1 μM concentrations, yet shows subtle toxicity at 20 μM. The compound 3a, which does not contain an azide, and the compound 3c, which contains a monoazide substituent, are both non-neuroprotective, yet both are not toxic to the cells indicating that the single azide has no cytotoxic properties in differentiated neuroblastoma cells.
Figure 7.

Differential neuroprotective and cytotoxic effects of HDAC inhibitors in unstressed (dark grey) or treated with compound 30 differentiated SH-SY5Y cells (light grey). **p < 0.01, ***p < 0.001 when compared to vehicle DMSO control (0 μM HDAC inhibitor).
We sought to ascertain the in situ effects of these inhibitors on nuclear HDACs by monitoring the acetylation status of histone H4 in retinoic acid-differentiated SH-SY5Y cells. Our results show that treatment with 1 μM of probes 2a, 2b and 3f induce hyperacetylation of histone H4 in differentiated SH-SY5Y cells (Figure 8). Furthermore, this effect is also observed in cells stressed with compound 30.26–28 These results indicate that the three probes 2a, 2b, and 3f efficiently target nuclear HDACs in normal and stress conditions in differentiated human neuroblastoma cells.
Figure 8.

Small molecule HDAC inhibitors 2a, 2b, and 3f induce accumulation of acetylated histone H4 in a differentiated SH-SY5Y cellular model.
Conclusions
Two types of scaffolds were explored for design of novel HDAC3 and HDAC8 BEProFL diazide probes. A total of eight inhibitors/probes were synthesized, tested for HDAC3 and HDAC8 activity, and docked to HDAC8. Probes 2b and 3f were profiled against class I, II, and IV HDAC isoforms and MMPs 1, 3, 9, and 14 and compared to a set of control inhibitors. Four novel active HDAC3 and HDAC8 probes/inhibitors 2b, 3a, 3c, and 3f were tested for their antiproliferative and neuroprotective activity and compared to that of SAHA. Both the isoxazole and pyrazole scaffolds were found to be excellent starting points for the design of potent HDAC3 and HDAC8 BEPRoFL probes, respectively. The synthetic route to the diazide intermediates 8 and 12 was optimized for gram scale, making the final probes/inhibitors readily accessible in few synthetic steps in a convergent manner. A facile route for the synthesis of the pyrazole-based inhibitors developed in this study can be extended to a wide range of other 4-aminopyrazole-based inhibitors.
Despite the presence of relatively bulky and lipophilic diazide moieties necessary for BEProFL, both the isoxazole- and the pyrazole-based probes exhibit excellent low double-digit nanomolar inhibitory activity against HDAC3 and HDAC8. Having IC50 of 17 nM against HDAC8, the meta-diazide pyrazole-based probe 3f not only shows 8-fold selectivity for HDAC8 vs HDAC3 but is also one of the most active HDAC8 inhibitors reported to date. The rest of the novel pyrazole-based HDAC inhibitors also displayed excellent low nanomolar activity against HDAC3 and HDAC8, though they were also lacking isoform selectivity. Both probes 2b and 3f showed no pronounced activity against MMPs and were generally not HDAC class selective based on the comparison of IC50 values. Probe 3f was more selective for HDAC8 compared to TSA and SAHA. Within class I HDACs, 2b and 3f were respectively HDAC3 and HDAC8 isoform-selective compared to SAHA. Our docking studies suggest that unlike the isoxazole-based ligands the pyrazole-based ligands may occupy the second binding site of HDAC8. This theoretical observation is consistent with our previous BEPRoFL studies as well as published X-ray data and molecular modeling studies of the HDAC8 linkerless ligands.
Comparison of anti-proliferative and neuroprotective properties and investigation of the pathways typically triggered by HDAC inhibitors showed that the presence of an azide or a diazide group does not interfere with the neuroprotection and does not enhance cellular cytotoxicity exerted through inhibition of intracellular HDACs. Taken together, cell-based studies show that the isoxazole- and pyrazole-based inhibitors and azide and diazide probes are able to freely enter the cell nucleus and trigger mechanisms known to respond to HDAC inhibition such as accumulation of acetylated H4 within 6 h, activation of caspase 3 within 14 h and cell death of otherwise highly proliferative human cancer cell lines within 24–48 h of treatment at low micromolar concentrations. Thus, the toxicity of these HDAC inhibitors/probes can be managed allowing their use at sub-micromolar concentrations and 0–6 h treatment in BEProFL experiments to identify the binding poses of the inhibitors in living cells. Unlike SAHA, probe 2a, 2b and 3f did not exert cytotoxic effects in neuronal-like cells and even protected these cells from chemically-induced apoptosis, making them interesting candidates for further thorough mechanistic studies in normal and stress conditions. Our future plans include BEProFL experiments with recombinant HDAC3/NCoR2 and HDAC8 proteins to improve our understanding of the binding poses of the probes and generate more data for further refinement of the docking approaches for HDACs and solvent exposed binding sites in general, extension of the BEPRoFL approach to experiments in cells, completion of the HDAC isoform profiling, and extensive investigation of SAR of the promising pyrazole-based scaffold identified in this study.
Experimental Section
Chemistry materials and methods
All the reagents and solvents were obtained from commercial sources and used without further purification. 1H NMR and 13C NMR spectra were recorded on Bruker spectrometers at 300/400 MHz and 75/100 MHz, respectively. Chemical shifts were reported on δ scale in ppm with solvent indicated as the internal reference. Coupling constants were reported in Hz and the standard abbreviations indicating multiplicity were used as follows: s = singlet, bs = broad singlet, d = doublet, t = triplet, q = quartet, and m = multiplet. Mass spectrometry experiment was carried out on Agilent 1100-MSD instrument. The purity of all the final compounds was confirmed to be ≥95% as it was analyzed by Shimadzu LCMS-2020 with UV detector at 214 and 254 nm using Kinetex C18 column (100 × 3.0 mm, 2.6 μ) eluting with mixtures of water–acetonitrile. TLC was performed with Merck 60F254 silica gel plates. Chromatography purification was performed on Biotage-Isolera four instrument using pre-filled KP-Sil (normal phase) and KP-C18-HS (reverse phase) SNAP cartridges with UV detection at 254 and 280 nm. Hexanes–ethyl acetate (normal phase) and H2O–MeOH (reverse phase) solvent systems were used as eluent for chromatography unless it is mentioned otherwise.
(3-Amino-5-hydroxymethylphenyl)methanol (5)
To a solution of dimethyl 5-aminoisophthalate 4 (10 g, 47.8 mmol) in dry THF (200 mL) at 0 °C was added LiAlH4 (4.9 g, 143 mmol) portionwise. The mixture was stirred for 8 h at rt, cooled to 0 °C, and quenched by slow addition of aqueous KOH solution (20%, 20 mL) followed by ethyl acetate (200 mL). The mixture was filtered through a pad of Celite and rinsed with ethyl acetate. The filtrate was dried over Na2SO4, filtered, and concentrated under reduced pressure. The resulting crude residue was purified by flash chromatography (100:0 → 90:10, EtOAc:MeOH) to give 5 as a pale yellow solid (6.6 g, 90%). 1H NMR (400 MHz, methanol-d4): δ 6.70 (s, 1H), 6.65 (s, 2H), 4.50 (s, 4H). 13C NMR (100 MHz, methanol-d4): δ 147.0, 141.9 (2C), 114.8, 112.4 (2C), 63.6 (2C).
(3-Azido-5-hydroxymethylphenyl)methanol (6)
To an ice cooled solution of amino-diol 5 (6.5 g, 42.4 mmol) in dry acetonitrile was added t-BuONO (5.3 g, 60 mmol) followed by TMS-N3 (6.2 mL, 50 mmol) dropwise. The resulting mixture was stirred for 2 h at rt, and concentrated under reduced pressure. The crude product was purified by flash chromatography (30:70 → 0:100, hexanes:EtOAc) to give azido-diol 6 as orange yellow solid (6.9 g, 91%). 1H NMR (400 MHz, methanol-d4): δ 7.13 (s, 1H), 6.97 (s, 2H), 4.60 (s, 4H). 13C NMR (100 MHz, methanol-d4): δ 143.4 (2C), 139.8, 120.9, 115.1 (2C), 62.7 (2C).
(3-Azido-5-azidomethylphenyl)methanol (7)
This reaction was carried out on 1–2 g scale repeatedly as per the requirement of the diazido-alcohol 7. To a stirred solution of azido-diol 6 (2 g, 11.2 mmol) in anhydrous DMF (20 mL) were added at rt NaN3 (2.9 g, 45 mmol) and DMAP (1.7 g, 13.4 mmol) followed by bis(2,4-dichlorophenyl)chlorophosphate (5 g, 12.3 mmol) and the stirring was continued for 4 h. Brine (40 mL) was added to the reaction mixture and extracted with ethyl acetate (3 × 50 mL). The organic fractions were dried over Na2SO4, filtered, and concentrated under reduced pressure. The crude residue was purified by flash chromatography (70:30 → 50:50, hexanes:EtOAc) to afford the desired diazido-alcohol 7 (1.5 g, 75%) as yellow oil along with triazide (0.22 g, 10%), based on 88% conversion and 12% of starting material recovery. 1H NMR (400 MHz, CDCl3): δ 7.08 (s, 1H), 7.02 (s, 1H), 6.89 (s, 1H), 4.70 (s, 2H), 4.35 (s, 2H), 2.15 (bs, 1H). 13C NMR (100 MHz, CDCl3): δ 143.2, 140.6, 137.3, 122.3, 117.3, 116.6, 64.0, 53.9.
Toluene-4-sulfonic acid 3-azido-5-azidomethylbenzyl ester (8)
A solution of 7 (1.5 g, 7.4 mmol) and triethylamine (2.6 mL, 18.4 mmol) in CH2Cl2 was treated with TsCl (1.6 g, 18.4 mmol) at 0 °C. The reaction was monitored by TLC while maintaining the temperature at 0 °C (otherwise undesired products were obtained). The reaction was quenched with water (20 mL) and extracted with CH2Cl2 (2 × 20 mL). The combined organic layers were washed with brine, dried over Na2SO4, filtered, and concentrated under reduced pressure. The residual oil was immediately purified by flash chromatography (90:10 → 70:30, hexanes:EtOAc) to give the diazido-tosylate 8 as pale yellow oil (2.25 g, 85%). 1H NMR (400 MHz, CDCl3): δ 7.80 (d, J = 8.0 Hz, 2H), 7.35 (d, J = 8.0 Hz, 2H), 6.98 (s, 1H), 6.92 (s, 1H), 6.83 (s, 1H), 5.05 (s, 2H), 4.32 (s, 2H), 2.46 (s, 3H). 13C NMR (100 MHz, CDCl3): δ 145.1, 141.1, 138.1, 136.0, 133.0, 129.9 (2C), 127.9 (2C), 124.0, 118.9, 118.4, 70.6, 53.9, 21.6.
3-Amino-5-hydroxymethylbenzoic acid methyl ester (9)
To a solution of dimethyl 5-aminoisophthalate 4 (5 g, 24.0 mmol) in dry THF at 0 °C was added portionwise LiBH4 (1.1 g, 50.0 mmol). The reaction mixture was heated at 50 °C for 48 h and quenched by adding MeOH at 0 °C. The solvent was evaporated and the crude solid was purified by flash chromatography (30:70 → 0:100, hexanes:EtOAc) to give amino-ester 9 as pale yellow solid (3.05 g, 71%). 1H NMR (400 MHz, methanol-d4): δ 7.33 (s, 1H), 7.26 (s, 1H), 6.94 (s, 1H), 4.55 (s, 2H), 3.87 (s, 3H). 13C NMR (100 MHz, CDCl3): δ 167.7, 147.9, 142.7, 130.6, 117.8, 117.0, 114.4, 63.5, 51.2.
3-Azido-5-hydroxymethylbenzoic acid methyl ester (10)
To an ice cooled solution of amino-ester 9 (3 g, 16.6 mmol) in dry acetonitrile (40 mL) was added dropwise t-BuONO (2.4 g, 23.2 mmol) followed by TMS-N3 (2.3 g, 20.0 mmol). The resulting mixture was stirred at rt for 1 h, concentrated, and the crude product was purified by flash chromatography (70:30 → 50:50, hexanes:EtOAc) to give azido-ester 10 as pale yellow solid (3.1 g, 91%). 1H NMR (400 MHz, CDCl3): δ 7.72 (s, 1H), 7.54 (s, 1H), 7.18 (s, 1H), 4.69 (s, 2H), 3.91 (s,3H), 2.87 (bs, 1H). 13C NMR (100 MHz, CDCl3): δ 166.2, 143.4, 140.4, 131.8, 124.0, 118.8, 64.0, 52.4.
3-Azido-5-azidomethylbenzoic acid methyl ester (11)
To a stirring solution of azido-alcohol 10 (3 g, 14.5 mmol) in anhydrous DMF (15 mL) were added at rt NaN3 (4.7 g, 72.4 mmol) and DMAP (2.6 g, 21.3 mmol) followed by bis(2,4-dichlorophenyl)chlorophosphate (7.07 g, 17.4 momol) and the stirring was continued for 4 h. Brine (40 mL) was added to the reaction mixture and extracted with ethyl acetate (3 × 50 mL). The organic fractions were dried over Na2SO4, filtered, and concentrated under reduced pressure. The crude residue was purified by flash chromatography (90:10 → 70:30, hexanes:EtOAc) to afford diazido-ester 11 as pale yellow solid (3.1 g, 92%). 1H NMR (400 MHz, CDCl3): δ 7.75 (s, 1H), 7.65 (s, 1H), 7.14 (s, 1H), 4.40 (s, 2H), 3.93 (s, 3H). 13C NMR (100 MHz, CDCl3): δ 165.7, 141.2, 137.9, 132.4, 125.3, 122.6, 119.6, 53.8, 52.5.
3-Azido-5-azidomethylbenzoic acid (12)
An ice cooled solution of 11 (3 g, 12.9 mmol) in THF–MeOH (20 mL, 1:1, v/v) was treated with 2N NaOH (10 mL, 20.0 mmol) solution for 2 h. The reaction mixture was diluted with ethyl acetate (30 mL) and treated with 2N HCl solution (20 mL). The organic layer was dried over Na2SO4, filtered, and concentrated under reduced pressure. The crude residue was purified by flash chromatography (60:40 → 20:80, hexanes:EtOAc) to give diazido-acid 12 as white solid (2.5 g, 89%). 1H NMR (400 MHz, CDCl3): δ 7.83 (s, 1H), 7.73 (s, 1H), 7.22 (s, 1H), 4.45 (s, 2H). 13C NMR (100 MHz, CDCl3): δ 170.8, 141.5, 138.2, 131.5, 125.8, 123.5, 120.2, 53.8.
Method A
General amide coupling reaction using EDC/HOBt/DIPEA/CH2Cl2: To a stirred solution of an acid (1 eq.) in anhydrous CH2Cl2 was added EDC (1.1 eq.) followed by HOBt (1.05 eq.) at 0 °C. After 1 h, a solution of amine/amine hydrochloride (1 eq.) and DIEPA (3 eq.) in CH2Cl2 was added to the reaction mixture dropwise at 0 °C. The mixture was allowed to stir at rt for 4–6 h (monitored by TLC), treated with 2N HCl solution and extracted with CH2Cl2. The organic layer was washed with saturated aqueous NaHCO3 solution followed by brine. The organic extracts were dried over Na2SO4, filtered, and concentrated under reduced pressure. The crude product was further purified by flash chromatography.
5-(4-tert-Butoxycarbonylaminophenyl)isoxazole-3-carboxylic acid ethyl ester (14)
To a solution of alkyne 13 (3 g, 13.8 mmol) in THF–H2O (1:1, 60 mL) was added Na2CO3 (18 g, 170.0 mmol) in one portion at 0 °C. A solution of 2-chloro-2-hydroxyiminoacetic acid ethyl ester (5.3 g, 35.0 mmol) in THF (40 mL) was added slowly over 18 h using a syringe pump. The reaction mixture was diluted with ethyl acetate (50 mL) and washed with water followed by brine. The organic fractions were dried over Na2SO4, filtered, and concentrated under reduced pressure. The crude residue was purified by flash chromatography (90:10 → 70:30, hexanes:EtOAc) to give the isoxazole 14 as pale yellow solid (3.8 g, 83%). 1H NMR (400 MHz, CDCl3): δ 7.70 (d, J = 8.0 Hz, 2H), 7.51 (d, J = 8.0 Hz, 2H), 7.04 (s, 1H), 6.81 (s, 1H), 4.44 (q, J = 8.0, 16.0 Hz, 2H), 1.54 (s, 9H), 1.45 (t, J = 8.0, Hz, 3H). 13C NMR (100 MHz, CDCl3): δ 171.5, 160.1, 156.8, 152.5, 141.0, 126.8 (2C), 121.0, 118.4 (2C), 98.9, 81.0, 62.1, 28.2 (3C), 14.1.
7-{[5-(4-tert-Butoxycarbonylaminophenyl)isoxazole-3-carbonyl]amino}heptanoic acid methyl ester (16)
An ice cooled solution of 14 (3.0 g, 9.4 mmol) in THF–MeOH (20 mL, 1:1) was treated with 2N NaOH solution (7 mL, 14 mmol) and the reaction mixture was allowed to stir at rt for 1 h. The reaction was diluted with ethyl acetate (20 mL) and acidified with 2N HCl (20 mL). The organic layer was separated, dried over Na2SO4, filtered, and evaporated under reduced pressure. The crude acid (15) obtained as white solid was directly used for coupling reaction with methyl 7-aminoheptanoate hydrochloride following the method A to afford 16 as white solid (3 g, 72%). 1H NMR (400 MHz, CDCl): δ 7.72 (d, J = 8.4 Hz, 2H), 7.51 (d, J = 8.4 Hz, 2H), 6.90 (bs, 1H), 6.88 (s, 1H), 6.82 (bs, 1H), 3.67 (s, 3H), 3.46 (q, J = 6.8, 13.6 Hz, 2H), 2.32 (t, J= 7.2 Hz, 2H), 1.65 (m, 4H), 1.54 (s, 9H), 1.38 (m, 4H). 13C NMR (100 MHz, CDCl3): δ 174.1, 171.3, 159.2, 159.0, 152.3, 140.7, 126.8 (2C), 121.3, 118.4 (2C), 98.1, 81.1, 51.5, 39.4, 33.9, 29.2, 28.7, 28.3 (3C), 26.5, 24.7.
7-{[5-(4-Azidophenyl)isoxazole-3-carbonyl]amino}heptanoic acid methyl ester (18)
A solution of 16 (2 g, 4.5 mmol) in CH2Cl2 (20 mL) was treated with trifloroacetic acid (1 mL, 13.5 mmol) for 4 h at rt. The solvent was evaporated, and the residue was azeotroped with toluene (3 × 10 mL) under reduced pressure. The crude aniline 17 obtained as pale yellow solid was directly used in the next step without any further purification.
To a solution of 17 (0.5 g, 1.5 mml) in AcOH–H2O (9:1, v/v, 8 mL) at 0 °C was added NaNO2 (0.12 g, 1.74 mmol) and the mixture was stirred for 10 min. To this orange red colored solution was then added portionwise NaN3 (0.14 g, 2.17 mmol) at the same temperature and stirring was continued for additional 1 h. The reaction mixture was neutralized by adding saturated aqueous NaHCO3 solution and extracted with ethyl acetate (3 × 25 mL). The combined organic extracts were washed with brine, dried over Na2SO4, filtered, and concentrated under reduced pressure. The crude product was purified by flash chromatography (90:10 → 70:30, hexanes:EtOAc) to afford 18 (0.38 g, 71%). 1H NMR (400 MHz, CDCl3): δ 7.60 (d, J = 8.4 Hz, 2H), 7.10 (d, J = 8.4 Hz, 2H), 6.93 (bs, 1H), 6.91 (s, 1H), 3.65 (s, 3H), 3.45 (q, J = 6.6, 13.3 Hz, 2H), 2.30 (t, J = 7.4 Hz, 2H), 1.70-1.47 (m, 4H), 1.40–1.30 (m, 4H). 13C NMR (100 MHz, CDCl3): δ 174.9, 170.9, 159.3, 158.8, 127.47, 123.45, 119.69, 98.96, 51.48, 39.44, 33.92, 29.26, 28.72, 26.52, 24.76. MS (ESI) m/z 372.2 (M+H)+.
7-({5-[4-(3-Azido-5-azidomethylbenzoylamino)phenyl]isoxazole-3-carbonyl}amino)heptanoic acid methyl ester (19)
To a mixture of aniline 17 (500 mg, 1.45 mmol) and diazido-acid 12 (475 mg, 2.17 mmol) in anhydrous CH2Cl2 (10 mL) was added HOBt (315 mg, 2.32 mmol). After the solution was cooled to 0 °C in an ice bath, DCC (480 mg, 2.32 mmol) was added followed by DMAP (90 mg, 0.07 mmol). The reaction solution was stirred at rt for 6 h. The mixture was concentrated under reduced pressure and the crude residue obtained was purified by flash chromatography (95:05 → 80:20, hexanes:EtOAc) to afford 19 as white solid (550 mg, 70%). 1H NMR (400 MHz, CDCl3): δ 9.81 (s, 1H), 7.88 (d, J = 8.1 Hz, 2H), 7.70 (d, J = 8.1 Hz, 2H) 7.65 (s, 1H), 7.63 (s, 1H), 7.59 (s, 1H), 7.09 (m, 2H), 6.85 (s, 1H), 4.19 (s, 2H), 3.60 (s, 3H), 3.37 (m, 2H), 1.58–1.54 (m, 4H), 1.33 (br s, 4H). 13C NMR (100 MHz, CDCl3): δ 170.32, 169.87, 160.20, 158.79, 141.46, 140.20, 138.15, 137.71, 128.63, 1256.41, 122.83, 120.02, 118.64, 100.50, 53.29, 33.64, 29.84, 28.49, 26.45, 25.33; MS (ESI) m/z 546.1 (M+H)+.
7-(1H-Pyrazol-4-ylcarbamoyl)heptanoic acid methyl ester (22)
An amide coupling reaction between aminopyrazole 21 (1 g, 12.0 mmol) and monomethylsuberate (2.27 g, 12.0 mmol) following method A resulted in 22 as pale brown solid (2.1 g, 69%). 1H NMR (400 MHz, DMSO-d6): δ 12.50 (s, 1H), 9.82 (s, 1H), 7.95 (s, 1H), 7.81 (s, 1H), 3.54 (s, 3H), 2.28 (t, J = 7.4 Hz, 2H), 2.20 (t, J = 7.4 Hz, 2H), 1.55-1.50 (m, 4H), 1.29-1.24 (m, 4H). 13C NMR (100 MHz, DMSO-d6): δ 173.3, 168.8, 130.5, 121.7, 118.82, 51.6, 35.9, 33.8, 28.8, 25.5, 24.2. MS (ESI) m/z 254.2 (M+H)+.
7-[1-(3-Azido-5-azidomethylbenzyl)-1H-pyrazol-4-ylcarbamoyl]heptanoic acid methyl ester (23)
A mixture of 22 (200 mg, 0.79 mmol), 8 (300 mg, 0.84 mmol) and powdered K2CO3 (220 mg, 1.60 mmol) in acetone (15 mL) was refluxed for 6 h. The mixture was cooled to rt and concentrated leaving a residue which was dissolved in ethylacetate and washed with water followed by brine. The organic fractions were dried over Na2SO4, filtered, and concentrated under reduced pressure. The crude product was purified by flash chromatography (80:20 → 50:50, hexanes:EtOAc) to furnish 23 as pale yellow solid (260 mg, 75%). 1H NMR (400 MHz, CDCl3): δ 7.99 (s, 1H), 7.43 (s, 1H), 7.42 (s, 1H), 7.27(s, 1H), 6.91 (s, 1H), 6.81 (s, 1H), 5.30 (s, 2H), 4.31 (s, 2H), 2.36–2.30 (m, 4H), 1.72–1.64 (m, 4H), 1.34 (m, 4H). 13C NMR (100 MHz, CDCl3): δ 174.4, 170.5, 141.2, 139.1, 138.1, 130.6, 123.4, 121.9, 121.4, 118.0, 117.9, 55.5, 53.9, 51.5, 36.3, 33.9, 28.7, 28.6, 25.4, 24.6. MS (ESI) m/z 440.2 (M+H)+.
7-(1-Benzyl-1H-pyrazol-4-ylcarbamoyl)heptanoic acid methyl ester (24)
To a solution of 22 (200 mg, 0.79 mmol) in anhydrous DMF (5 mL) at 0 °C was added portionwise NaH (22 mg, 0.95 mmol). After 10 min, benzylbromide (150 mg, 0.88 mmol) was added to the reaction mixture at the same temperature. After 4 h stirring at rt, the reaction was quenched with ice water and extracted with ethyl acetate (3 × 10 mL). Organic layer was further washed with water followed by brine. The combined organic fractions were dried over Na2SO4, filtered, and concentrated under reduced pressure. The crude residue was purified by flash chromatography (75:25 → 50:50, hexanes:EtOAc) to afford 24 (190 mg, 70%). 1H NMR (400 MHz, CDCl3): δ 7.95 (s, 1H), 7.43 (s, 1H), 7.36–7.30 (m, 3H), 7.27–7.23 (m, 3H), 5.25 (s, 2H), 3.67 (s, 3H), 2.36–2.30 (m, 4H), 1.72–1.68 (m, 4H), 1.83 (m, 4H). MS (ESI) m/z 344.2 (M+H)+.
7-[1-(4-Nitrobenzyl)-1H-pyrazol-4-ylcarbamoyl]heptanoic acid methyl ester (25)
To a solution of 22 (1.6 g, 6.32 mmol) in anhydrous DMF (15 mL) at 0 °C was added portionwise NaH (175 mg, 7.6 mmol). After 10 min, 4-nitrobenzylbromide (1.5 g, 7.0 mmol) was added to the reaction mixture at the same temperature. After 4 h stirring at rt, the reaction was quenched with ice water and extracted with ethyl acetate (3 × 30 mL). Organic layer was further washed with water followed by brine. The combined organic fractions were dried over Na2SO4, filtered, and concentrated under reduced pressure. The crude residue was purified by flash chromatography (70:30 → 40:60, hexanes:EtOAc) to give 25 as pale yellow solid (1.62 g, 66%). 1H NMR (400 MHz, DMSO-d6): δ 8.17 (d, J = 8.3 Hz, 2H),8.05 (s, 1H), 7.43 (s, 1H), 7.34 (s, 1H), 7.32 (s, 1H), 5.34 (s, 2H), 3.66 (s, 3H), 2.33–3.29 (m, 4H), 1.71–1.64 (m, 4H), 1.23 (br s, 4H). 13C NMR (100 MHz, CDCl3): δ 174.3, 170.4, 147.6, 143.7, 130.8, 128.2 (2C), 124.0 (2C), 122.0, 121.7, 55.3, 51.5, 36.4, 33.9, 28.7, 28.6, 25.3, 24.6. MS (ESI) m/z 389.2 (M++H).
7-[1-(4-Aminobenzyl)-1H-pyrazol-4-ylcarbamoyl]heptanoic acid methyl ester (26)
A mixture of 25 (1.6 g, 4.1 mmol) and SnCl2·2H2O (2.8 g, 12.4 mmol) in methanol (20 mL) was heated at 70 °C for 2 h. The pH of the reaction mixture was adjusted to 7–8 by addition of saturated aqueous NaHCO3 solution. The resulting mixture was extracted with CHCl3 (3 × 20 mL). The organic phase was washed with brine, dried over Na2SO4, and concentrated under reduced pressure. The crude residue was purified by flash chromatography (100:00 → 90:10, CH2Cl2:MeOH) to give aniline 26 as orange yellow solid (1.4 g, 95%). 1H NMR (400 MHz, methanol-d4): δ 7.83 (s, 1H), 7.48 (s, 1H), 7.02 (d, J = 8.0 Hz, 2H), 6.68 (d, J = 8.0 Hz, 2H), 5.10 (s, 2H), 3.65 (s, 3H), 2.30 (m, 4H), 1.64 (m, 4H), 1.36 (s, 4H). 13C NMR (100 MHz, methanol-d4): δ 174.6, 171.7, 147.4, 129.9, 129.3, 128.7, 125.5, 121.7, 120.7, 115.1, 115.0, 55.4, 50.7, 35.7, 33.4, 28.5, 28.4, 25.3, 24.4.
7-[1-(4-Azidobenzyl)-1H-pyrazol-4-ylcarbamoyl]heptanoic acid methyl ester (27)
To a solution of 26 (300 mg, 0.84 mmol) in AcOH–H2O (9:1, v/v, 5 mL) at 0 °C was added NaNO2 (70 mg, 1.01 mmol) in one portion. The resulting orange red colored mixture was stirred at the same temperature for 10 min. To this was then added portionwise NaN3 (85 mg, 1.3 mmol) and stirring was continued for additional 1 h. The reaction mixture was neutralized by adding saturated aqueous NaHCO3 solution and extracted with ethyl acetate (3 × 20 mL). The combined organic fractions were washed with brine, dried over Na2SO4, filtered, and concentrated under reduced pressure. The crude residue was purified by flash chromatography (70:30 → 50:50, hexanes:EtOAc) to afford 27 (220 mg, 68%). 1H NMR (400 MHz, CDCl3): δ 8.18 (s, 1H), 8.08 (s, 1H), 7.42 (s, 1H), 7.18 (d, J = 8.4 Hz, 2H), 6.95 (d, J = 8.4 Hz, 2H), 5.17 (s, 2H), 3.65 (s, 3H), 2.29–2.19(m, 4H), 1.68–1.60 (m, 4H), 1.23 (br s, 4H). 13C NMR (100 MHz, CDCl3): δ 174.3, 170.6, 139.0, 133.0, 130.3, 129.3, 121.6, 121.3, 119.4, 55.7, 53.5, 36.4, 33.9, 28.7, 28.7, 24.7. MS (ESI) m/z 385.2 (M+H)+.
7-[1-(4-Azidobenzyl)-1H-pyrazol-4-ylcarbamoyl]heptanoic acid methyl ester (28)
To a stirred solution of 26 (200 mg, 0.56 mmol) and triethylamine (120 μL, 0.84 mmol) in CH2Cl2 (10 mL) was added Boc-anhydride (150 mg, 0.69 mmol) at rt. After 4 h, the mixture was concentrated and the crude residue was purified by flash chromatography (80:20 → 50:50, hexanes:EtOAc) to furnish 28 (210 mg, 82%). 1H NMR (400 MHz, CDCl3): δ 8.23 (s, 1H), 7.80 (s, 1H), 7.59 (s, 1H), 7.36 (d, J = 8.2 Hz, 2H), 7.20 (d, J = 8.2 Hz, 2H), 6.94 (s, 1H), 5.17 (s, 2H), 3.81 (s, 3H), 2.29–2.21 (m, 4H), 1.64–1.56 (m, 4H), 1.47 9 (s, 9H), 1.23 (br s, 4H). 13C NMR (100 MHz, CDCl3): δ 174.4, 70.6, 152.6, 138.4, 130.4, 130.4, 129.3, 128.0, 121.6, 121.1, 118.9, 55.5, 51.5, 36.4, 33.9, 29.7, 28.8, 28.3, 27.9, 25.4, 24.7; MS (ESI) m/z 459.3 (M+H)+.
7-{1-[4-(3-Azido-5-azidomethylbenzoylamino)benzyl]-1H-pyrazol-4-ylcarbamoyl}heptanoic acid methyl ester (29)
Compound 29 (190 mg, 61%) was obtained by a coupling reaction between acid 12 (122 mg, 0.56 mmol) and amine 26 (200 mg, 0.56 mmol) following method A. 1H NMR (400 MHz, CDCl3): δ 8.84 (s, 1H), 8.24 (s, 1H), 8.00 (s, 1H), 7.79 (s, 1H), 7.58 (s, 1H), 7.50 (d, J = 8.1 Hz, 2H), 7.40 (s, 1H), 7.34 (s, 1H), 7.06 (d, J = 8.2 Hz, 2H), 5.30 (s, 2H), 4.35 (s, 2H), 2.30–2.23 (m, 4H), 1.66–1.54 (m, 4H), 1.27 (br s, 4H). 13C NMR (CDCl3, 75 MHz): δ 174.4, 170.7, 164.7, 141.4, 138.1, 137.7, 137.0, 132.6, 130.6, 128.4, 123.2, 121.6, 121.4, 121.1, 121.1, 117.9, 55.8, 53.9, 36.6, 36.4, 33.9, 31.5, 28.8, 28.7, 25.4, 24.6, 65.3, 162.3, 160.7, 125.6. MS (ESI) m/z 560.2 (M+H)+.
General method for the preparation of hydroxamate
All the following hydroxamates were prepared from their corresponding methyl ester precursors on 100–200 mg scale with ca 60–70% yield.
To a solution of hydroxylamine hydrochloride (200 mmol) in MeOH at 0 °C was added portionwise powdered KOH (205 mmol). The solution was stirred for 1 h at rt after the addition was complete. The precipitate was filtered off and the filtrate containing NH2OH in MeOH was added dropwise to an ice cooled solution of methyl ester (1 mmol) in MeOH (5 mL). An additional amount of powdered KOH (10 mmol) was added to the reaction mixture and the white solution was stirred for 3 h at rt. MeOH was concentrated at rt under reduced pressure. The residual solid was treated with saturated aqueous NH4Cl solution (20 mL) and extracted with ethyl acetate (3 × 25 mL). The combined organic extracts were washed with brine, dried over Na2SO4, filtered, and concentrated under reduced pressure. The crude product was purified on Biotage using reverse phase C-18 column (90:10 → 10:90, H2O:MeOH). The purity of all hydroxamates was found to be ≥95% as it was analyzed by reversed-phase LCMS.
5-(4-Azidophenyl)isoxazole-3-carboxylic acid(6-hydroxycarbamoylhexyl)amide (2a)
1H NMR (400 MHz, DMSO-d6): δ 10.39 (s, 1H), 8.81 (br s, 1H), 8.66 (s, 1H), 7.92 (m, 2H), 7.56 (s, 1H), 7.36 (m, 2H), 3.24 (q, J = 6.5, 13.0 Hz, 2H), 1.94 (t, J = 7.2 Hz, 2H), 1.52–1.45 (m, 4H), 1.17 (br s, 4H). 13C NMR (100 MHz, DMSO-d6): δ 170.7, 169.9, 160.2, 158.9, 144.3, 143.3, 131.3, 129.8, 128.0, 126.8, 123.5, 120.5, 100.0, 32.7, 29.2, 28.6, 26.6, 25.5. MS (ESI) m/z 395.1 (M+Na)+.
5-[4-(3-Azido-5-azidomethylbenzoylamino)phenyl]isoxazole-3-carboxylic acid (6-hydroxycarbamoylhexyl)amide (2b)
1H NMR (400 MHz, DMSO-d6): δ 10.62 (s, 1H), 10.19 (s, 1H), 8.77 (m, 1H), 8.64 (s, 1H), 7.92 (m, 4H), 7.73 (s, 1H), 7.59 (s, 1H), 7.39 (s, 1H), 7.25 (s, 1H), 4.59 (s, 2H), 3.25 (q, J = 6.9, 13.3 Hz, 2H), 2.94 (t, J = 7.3 Hz, 2H), 1.92 (t, J = 7.1 Hz, 2H), 1.52–1.45 (m, 4H), 1.19 (br s, 4H). 13C NMR (100 MHz, DMSO-d6): δ 170.4, 169.3, 160.0, 158.9, 141.6, 140.7, 138.3, 137.1, 129.6, 126.8, 122.4, 120.9, 118.4, 99.5, 53.2, 32.7, 29.2, 28.7, 26.6, 25.5. MS (ESI) m/z 547.2 (M+H)+.
Octanedioic acid (1-benzyl-1H-pyrazol-4-yl)amide hydroxyamide (3a)
1H NMR (400 MHz, DMSO-d6): δ 10.35 (s, 1H), 10.24 (s, 1H), 9.92 (s, 1H), 7.97 (s. 1H), 7.41 (s, 1H), 7.35–7.28 (m, 3H), 7.23–7.20 (m, 2H), 5.26 (s, 2H), 2.23–2.17 (m, 2H), 1.95–1.91(m, 2H), 1.55–1.47(m, 4H), 1.24 (m, 4H). 13C NMR (100 MHz, methanol-d4): δ 171.8, 171.5, 136.3, 130.4, 128.6 (2C), 127.9, 127.4 (2C), 121.9, 121.4, 55.9, 35.9, 32.5, 28.4, 28.3, 25.3, 25.1. MS (ESI) m/z 345.2 (M+H)+.
Octanedioic acid hydroxyamide [1-(4-nitrobenzyl)-1H-pyrazol-4-yl]amide (3b)
1H NMR (400 MHz, DMSO-d6): δ 10.32 (s, 1H), 9.93 (s, 1H), 8.99 (s, 1H), 8.20 (d, J = 8.6 Hz, 2H), 8.06 (s, 1H), 7.45 (s, 1H), 7.40 (d, J = 8.6 Hz, 2H), 5.44 (s, 2H), 2.21 (t, J = 7.3 Hz, 2H), 1.92 (t, J = 7.3 Hz, 2H), 1.53–1.45 (m, 4H), 1.26 (br s, 4H) ppm. 13C NMR (100 MHz, DMSO-d6): δ 170.7, 170.4, 146.7, 144.1, 130.4, 130.3, 127.5, 122.9, 121.5, 120.9, 53.7, 34.5, 31.7, 27.8, 27.7, 24.6, 24.5.MS (ESI) m/z 390.2 (M+H)+.
Octanedioic acid [1-(4-azidobenzyl)-1H-pyrazol-4-yl]amide hydroxyamide (3c)
1H NMR (400 MHz, DMSO-d6): δ 10.31 (s, 1H), 9.87 (s, 1H), 8.64 (s, 1H), 7.95 (s, 1H), 7.39 (s, 1H), 7.26 (d, J = 8.1 Hz, 2H), 7.10 (d, J = 8.1 Hz, 2H), 5.24 (s, 2H), 2.19 (t, J = 7.4 Hz, 2H), 1.92 (t, J = 7.2 Hz, 2H), 1.52–1.40 (m, 4H), 1.23 (br s, 4H). 13C NMR (100 MHz, DMSO-d6): δ 170.7, 170.4, 138.9, 133.1, 129.8, 128.5 (2C), 121.4, 120.4, 118.4 (2C), 54.1, 35.0, 31.7, 27.8, 27.7, 24.6, 24.5. MS (ESI) m/z 386.2 (M+H)+.
{4-[4-(7-Hydroxycarbamoylheptanoylamino)pyrazol-1-ylmethyl]phenyl}carbamic acid tert-butyl ester (3d)
1H NMR (400 MHz, DMSO-d6): δ 10.31 (s, 1H), 10.06 (s, 1H), 9.86 (s, 1H), 9.29 (s, 1H), 8.63 (s, 1H), 7.88 (s, 1H), 7.30 (d, J = 8.4 Hz, 2H), 7.13 (d, J = 8.4 Hz, 2H), 5.14 (s, 2H), 2.19 (t, J = 7.2 Hz, 2H), 1.92 (t, J = 7.1 Hz, 2H), 1.50–1.46 (m, 4H), 1.26 (br s, 4H). 13C NMR (100 MHz, DMSO-d6): δ 169.9, 169.6, 153.2, 139.5, 131.4, 128.7, 122.5, 120.5, 118.6, 79.5, 55.0, 35.9, 32.7, 28.8, 25.6, 25.5. MS (ESI) m/z 482.2 (M+Na)+.
Octanedioic acid {1-[4-(3-azido-5-azidomethylbenzoylamino)benzyl]-1H-pyrazol-4-yl}amide hydroxyamide (3e)
1H NMR (400 MHz, DMSO-d6): δ 10.38 (s, 1H), 10.34 (s, 1H), 9.89 (s, 1H), 7.93 (s, 1H), 7.74 (s, 1H), 7.70 (d, J = 8.2 Hz, 2H), 7.63 (s, 1H), 7.40 (s, 1H), 7.34 (s, 1H), 7.23 (d, J = 8.2 Hz, 2H), 5.22 (s, 2H), 4.58 (s, 2H), 2.21 (t, J = 7.2 Hz, 2H), 1.92 (t, J = 7.1 Hz, 2H), 1.55–1.45 (m, 4H), 1.23 (br s, 4H). 13C NMR (100 MHz, DMSO-d6): δ 171.0, 169.5, 164.6, 140.4, 138.6, 133.4, 128.6, 124.4, 122.4, 120.8, 118.3, 53.2, 49.1, 32.7, 28.8, 25.6, 25.5. MS (ESI) m/z 560.2 (M+H)+.
Octanedioic acid [1-(3-azido-5-azidomethylbenzyl)-1H-pyrazol-4-yl]amide hydroxyamide (3f)
1H NMR (400 MHz, DMSO-d6): δ 10.31 (bs, 1H), 9.89 (bs, 1H), 8.60 (s, 1H), 8.06 (s, 1H), 7.42 (s, 1H), 7.06 (s, 1H), 7.02 (s, 1H), 6.94 (s, 1H), 5.47 (s, 2H), 4.46 (s, 2H), 2.20 (t, J = 8.0 Hz, 2H), 1.93 (t, J = 8.0 Hz, 2H), 1.53–1.44 (m, 4H), 1.26 (m, 4H) ppm. 13C NMR (100 MHz, DMSO-d6): δ 170.1, 169.6, 147.3, 146.0, 132.0, 128.9, 124.2, 122.7, 121.4, 54.4, 49.1, 35.9, 32.7, 28.9, 28.8, 25.6, 25.5 ppm. MS (ESI) m/z 441.2 (M+H)+.
Modeling methods
Using Surflex-dock29 with the -pgeomx flag for more exhaustive docking, and otherwise default parameters, we docked all ligands to multiple crystal structures of HDAC8, choosing the best scoring as the final binding pose. For scoring we used a variation of a fragment based scoring approach that only considers interactions of the SBG and the linker, but not the ZBG. More details about the methodology are given elsewhere.30
Biological materials and methods
HDAC 3 and 8 activity assay
HDAC inhibition assay was performed in 96 well half-area microplate (Corning). 0.375U HDAC8 (BIOMOL) was diluted with HDAC8 assay buffer KI-311 (BIOMOL) and pre-incubated with 10 μL of inhibitor for 5 min. The assay mixture was treated with 25 μL of 10 μM HDAC substrate KI-178 (BIOMOL) for 25 min at rt. The reaction was quenched with 50 μL of 50 μg/mL trypsin and 4 μM trichostatin A in KI-143 (BIOMOL) for 30 min. The plate was read on a POLARstar OPTIMA (BMG labtech) microplate reader at excitation wavelength 360 nm and emission wavelength 460 nm.
10 ng HDAC3/NCoR2 (BPS Bioscience) was diluted with KI-311 (BIOMOL) and preincubated with 30 μL of inhibitor for 5 min then 10 μl, 125 μM HDAC fluorescent substrate Boc-L-Lys (Ac)-AMC (Chem-Impex) was added and the mixture incubated for 30 min under room temperature. The reaction was quenched with 50 μL of 1 mg/mL trypsin and 4 μM trichostatin A in KI-143 (BIOMOL) for 30 min. The plate was read on an OptimaPolar Starmicroplate reader (BMG labtech) at excitation wavelength 360 nm and emission wavelength 460 nm.
The IC50 values were determined using the GraphPad Prism 5 software (GraphPad Software Inc., La Jolla, CA).
The description of assays against HDAC 1, 2, 4–7, 9–11 and MMPs 1, 3, 9, 14 is given in the supporting information.
Cell Culture
HepG2 (ATCC) hepatocarcinoma cells and Hela (ATCC) cervical adenocarcinoma cells were grown in D7 medium containing DMEM supplemented with 7% FBS, 2 mM L-glutamine, 100 U/mL penicillin, 100 μg/mL streptomycin and 1 mM sodium pyruvate. SH-SY5Y (ATCC) neuroblastoma cells were grown in SH-SY5Y medium containing equal amounts of MEM/EBSS and Ham’s F12 supplemented with 10% FBS, 2 mM L-glutamine, 100 U/mL penicillin, 100 μg/mL streptomycin, 1% NEAA and 0.1% sodium bicarbonate. Cells were grown in 10 cm2 dishes to a density of 80% confluence and were differentiated in the SH-SY5Y media (1% FBS 2 mM L-glutamine, 100 U/mL penicillin, 100 μg/mL streptomycin, 1% NEAA, 0.1% sodium bicarbonate) supplemented with 10 μM retinoic acid (RA, Sigma) for 7–8 days. The differentiating medium was replaced every 2–3 days. Cells were maintained at 37 °C in a humidified atmosphere supplemented with 5% CO2.
Antiproliferation assays
Hepatocarcinoma HepG2 cells were seeded at 1.2 × 104 cells/well into 96 well plates in D7 medium. Hela cells were seeded at 0.2 × 104 cells/well into 96 well plates in D7 medium. Neuroblastoma SH-SY5Y cells were seeded at 0.9 × 104 cells/well into 96 well plates in 10% FBS SH-SY5Y medium. After 24 h, the medium (D7 prepared with phenol red free DMEM for HepG2 and Hela cells, 10% SH-SY5Y medium containing equal amounts of phenol red free DMEM and Ham’s F12 supplemented with 10% FBS, 2 mM L-glutamine, 100 U/mL penicillin, 100 μg/mL streptomycin, 1% NEAA and 0.1% sodium bicarbonate for the neuroblastoma cells) was changed to a medium containing either vehicle (DMSO) or 0.5–50 μM final concentrations of HDAC inhibitors. After 24, 48 or 72 h incubation, cell viability was measured using the XTT cell proliferation assay (Roche). Data from at least three independent experiments were analyzed using the GraphPad Prism 5.0 software and EC50s deduced from the best fits of the data using the non-linear regression with variable slope equations.
Western blot analysis in HepG2 cells
Hepatocarcinoma HepG2 cells were seeded at 2.7 × 105 cells/well into 6 well plates in D7 media. After 24 h the media was changed to fresh D7 media and the cells were incubated at 37 °C. After 1 h incubation, cells were treated with vehicle (DMSO) or 1, 5 or 10 μM 2b or SAHA. Cells were then incubated in the dark for 6 h, after which the cells were harvested and the cell pellets frozen and stored at −80 °C. Cells were lysed in ice cold 1% CHAPSO lysis buffer (50 mM Hepes pH 7.4, 150 mM NaCl, 1 mM EDTA, 1% CHAPSO, complete EDTA-free protease inhibitor cocktail from Roche) for 45 min at 4 °C. Clarified lysates were quantified by the method of Bradford31 and 40 μg of total protein per lysate was loaded on a Tris-glycine 12.5% acrylamide gel and submitted to SDS PAGE and electrophoretically transferred onto PVDF membrane (Millipore) using a Trans-blot® SD semi-dry electrophoretic transfer cell (Biorad). Membranes were blocked in Tris buffered saline solution containing 0.5% Tween 20 and 5% non-fat milk, then probed for total p53 (monoclonal antibody, Millipore), acetylated Lys373-p53 (Upstate), acetylated Lys8-Histone H4 (Upstate), cleaved caspase 3 Asp175 (Cell Signaling Technology) or GAPDH (Chemicon). Membranes were washed and subsequently incubated with the appropriate peroxidase conjugated secondary antibody (anti-Rabbit IgG or anti-Mouse IgG, GE). The detection was done using the SuperSignal West Pico Chemiluminescence Substrate (Pierce). Blots were analyzed on a G:Box gel documentation system and the images captured using the Gene Snap software (Syngene, Frederick, MD). Band intensities were quantified using the Image J freeware from the NIH web site (http://rsbweb.nih.gov/ij/).
Neuroprotection Assays
Differentiated SH-SY5Y cells were seeded at 2 × 104 cells/well into 96 well plates in 1% FBS media containing 5 μM retinoic acid. After 24 h, the medium was changed to 1% phenol red free SH-SY5Y media containing phenol red free DMEM and Ham’s F12 supplemented with 1% FBS, 2 mM L-glutamine, 100 U/mL penicillin, 100 μg/mL streptomycin, 1% NEAA and 0.1% sodium bicarbonate with 5 μM retinoic acid containing either vehicle (DMSO) or 0.5–50 μM final concentrations of HDAC inhibitors, in the absence (Figure 8, dark grey) or presence (Figure 8, light grey) of 100 μM of 30. After 24 h incubation, cell viability was measured using the XTT cell proliferation kit II (Roche Applied Science). Data are presented as the average percent survival of four replicates + standard deviation, relative to vehicle (100%). Statistical analysis was performed using one-way ANOVA with Dunnett’s secondary analysis.
Acetylation of H4 in differentiated SH-SY5Y cells
Differentiated SH-SY5Y cells were treated with either vehicle (DMSO) or 1 μM of probes 2b, 2a or 3f for 1 h followed by the addition of 100 μM of 30 and incubated for an additional 8 h treatment. Cells were lysed in 1% CHAPSO lysis buffer and analyzed by western blot as described above.
Supplementary Material
Acknowledgments
This study was funded by the National Cancer Institute/NIH Grant R01CA131970 (PAP and SYB) and, in part by the INSERM, the University Denis Diderot/Paris 7 and a Chaire of Excellence from the Agence Nationale pour la Recherche, France (SYB). Molecular modeling studies were in part performed with the UCSF Chimera package from the Resource for Biocomputing, Visualization, and Informatics at the University of California, San Francisco (supported by NIH P41 RR-01081), Open Eye Scientific Software, Santa Fe, NM, and Surflex package from Prof. Ajay N. Jain, San Mateo, CA (supported by NIH R01 GM070481). We thank Mr. Aditya Sudheer Vaidya and Mr. Hazem Abdelkarim for conducting HDAC1 and HDAC2 inhibitory assays for SAHA, TSA, 2b, and 3f.
Abbreviations
- HDAC
histone deacetylase
- BEProFL
(B)inding (E)nsemble (Pro)filing with (F)photoaffinity (L)abeling
- SBG
surface binding group
- ZBG
Zinc binding group
- SAHA
suberoylanilide hydroxamic acid
Footnotes
Supporting Information. Activity assay protocols for HDAC1, 2, 4–7, 9–11 and metallo proteases (MMPs)-1, 3, 9, 14. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.de Ruijter AJ, van Gennip AH, Caron HN, Kemp S, van Kuilenburg AB. Histone deacetylases (HDACs): characterization of the classical HDAC family. Biochem J. 2003;370:737–749. doi: 10.1042/BJ20021321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Acharya MR, Sparreboom A, Venitz J, Figg WD. Rational development of histone deacetylase inhibitors as anticancer agents: a review. Mol Pharmacol. 2005;68:917–932. doi: 10.1124/mol.105.014167. [DOI] [PubMed] [Google Scholar]
- 3.Korner M, Tibes U. Histone deacetylase inhibitors: a novel class of anti-cancer agents on its way to the market. Prog Med Chem. 2008;46:205–280. doi: 10.1016/S0079-6468(07)00005-7. [DOI] [PubMed] [Google Scholar]
- 4.Hahnen E, Hauke J, Trankle C, Eyupoglu IY, Wirth B, Blumcke I. Histone deacetylase inhibitors: possible implications for neurodegenerative disorders. Expert Opin Investig Drugs. 2008;17:169–184. doi: 10.1517/13543784.17.2.169. [DOI] [PubMed] [Google Scholar]
- 5.He B, Velaparthi S, Pieffet G, Pennington C, Mahesh A, Holzle DL, Brunsteiner M, van Breemen R, Blond SY, Petukhov PA. Binding ensemble profiling with photoaffinity labeling (BEProFL) approach: mapping the binding poses of HDAC8 inhibitors. J Med Chem. 2009;52:7003–7013. doi: 10.1021/jm9005077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Oehme I, Deubzer HE, Lodrini M, Milde T, Witt O. Targeting of HDAC8 and investigational inhibitors in neuroblastoma. Expert Opin Investig Drugs. 2009;18:1605–1617. doi: 10.1517/14728220903241658. [DOI] [PubMed] [Google Scholar]
- 7.Oehme I, Deubzer HE, Wegener D, Pickert D, Linke JP, Hero B, Kopp-Schneider A, Westermann F, Ulrich SM, von Deimling A, Fischer M, Witt O. Histone deacetylase 8 in neuroblastoma tumorigenesis. Clin Cancer Res. 2009;15:91–99. doi: 10.1158/1078-0432.CCR-08-0684. [DOI] [PubMed] [Google Scholar]
- 8.Witt O, Deubzer HE, Milde T, Oehme I. HDAC family: What are the cancer relevant targets? Cancer Lett. 2009;277:8–21. doi: 10.1016/j.canlet.2008.08.016. [DOI] [PubMed] [Google Scholar]
- 9.Kozikowski AP, Tapadar S, Luchini DN, Kim KH, Billadeau DD. Use of the Nitrile Oxide Cycloaddition (NOC) Reaction for Molecular Probe Generation: A New Class of Enzyme Selective Histone Deacetylase Inhibitors (HDACIs) Showing Picomolar Activity at HDAC6. J Med Chem. 2008;51:4370–4373. doi: 10.1021/jm8002894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Vannini A, Volpari C, Gallinari P, Jones P, Mattu M, Carfi A, De Francesco R, Steinkuhler C, Di Marco S. Substrate binding to histone deacetylases as shown by the crystal structure of the HDAC8-substrate complex. EMBO Rep. 2007;8:879–884. doi: 10.1038/sj.embor.7401047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dowling DP, Gantt SL, Gattis SG, Fierke CA, Christianson DW. Structural studies of human histone deacetylase 8 and its site-specific variants complexed with substrate and inhibitors. Biochemistry. 2008;47:13554–13563. doi: 10.1021/bi801610c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Balasubramanian S, Ramos J, Luo W, Sirisawad M, Verner E, Buggy JJ. A novel histone deacetylase 8 (HDAC8)-specific inhibitor PCI-34051 induces apoptosis in T-cell lymphomas. Leukemia. 2008;22:1026–1034. doi: 10.1038/leu.2008.9. [DOI] [PubMed] [Google Scholar]
- 13.Hosoya T, Hiramatsu T, Ikemoto T, Nakanishi M, Aoyama H, Hosoya A, Iwata T, Maruyama K, Endo M, Suzuki M. Novel bifunctional probe for radioisotope-free photoaffinity labeling: compact structure comprised of photospecific ligand ligation and detectable tag anchoring units. Org Biomol Chem. 2004;2:637–641. doi: 10.1039/b316221d. [DOI] [PubMed] [Google Scholar]
- 14.Barral K, Moorhouse AD, Moses JE. Efficient conversion of aromatic amines into azides: a one-pot synthesis of triazole linkages. Org Lett. 2007;9:1809–1811. doi: 10.1021/ol070527h. [DOI] [PubMed] [Google Scholar]
- 15.Yu C, Liu B, Hu L. A Simple One-Pot Procedure for the Direct Conversion of Alcohols to Azides via Phosphate Activation. Org Lett. 2000;2:1959–1961. doi: 10.1021/ol0060376. [DOI] [PubMed] [Google Scholar]
- 16.Suzuki T, Nagano Y, Kouketsu A, Matsuura A, Maruyama S, Kurotaki M, Nakagawa H, Miyata N. Novel inhibitors of human histone deacetylases: design, synthesis, enzyme inhibition, and cancer cell growth inhibition of SAHA-based non-hydroxamates. J Med Chem. 2005;48:1019–1032. doi: 10.1021/jm049207j. [DOI] [PubMed] [Google Scholar]
- 17.Estiu G, West N, Mazitschek R, Greenberg E, Bradner JE, Wiest O. On the inhibition of histone deacetylase 8. Bioorg Med Chem. 2010;18:4103–4110. doi: 10.1016/j.bmc.2010.03.080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Carlisi D, Vassallo B, Lauricella M, Emanuele S, D’Anneo A, Di Leonardo E, Di Fazio P, Vento R, Tesoriere G. Histone deacetylase inhibitors induce in human hepatoma HepG2 cells acetylation of p53 and histones in correlation with apoptotic effects. Int J Oncol. 2008;32:177–184. doi: 10.3892/ijo.32.1.177. [DOI] [PubMed] [Google Scholar]
- 19.Emanuele S, Lauricella M, Carlisi D, Vassallo B, D’Anneo A, Di Fazio P, Vento R, Tesoriere G. SAHA induces apoptosis in hepatoma cells and synergistically interacts with the proteasome inhibitor Bortezomib. Apoptosis. 2007;12:1327–1338. doi: 10.1007/s10495-007-0063-y. [DOI] [PubMed] [Google Scholar]
- 20.Picot CR, Blond SY. unpublished results. [Google Scholar]
- 21.Condorelli F, Gnemmi I, Vallario A, Genazzani AA, Canonico PL. Inhibitors of histone deacetylase (HDAC) restore the p53 pathway in neuroblastoma cells. Br J Pharmacol. 2008;153:657–668. doi: 10.1038/sj.bjp.0707608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mann BS, Johnson JR, Cohen MH, Justice R, Pazdur R. FDA Approval Summary: Vorinostat for Treatment of Advanced Primary Cutaneous T-Cell Lymphoma. Oncologist. 2007;12:1247–1252. doi: 10.1634/theoncologist.12-10-1247. [DOI] [PubMed] [Google Scholar]
- 23.Hockly E, Richon VM, Woodman B, Smith DL, Zhou X, Rosa E, Sathasivam K, Ghazi-Noori S, Mahal A, Lowden PA, Steffan JS, Marsh JL, Thompson LM, Lewis CM, Marks PA, Bates GP. Suberoylanilide hydroxamic acid, a histone deacetylase inhibitor, ameliorates motor deficits in a mouse model of Huntington’s disease. Proc Natl Acad Sci U S A. 2003;100:2041–2046. doi: 10.1073/pnas.0437870100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ryu H, Lee J, Olofsson BA, Mwidau A, Deodoglu A, Escudero M, Flemington E, Azizkhan-Clifford J, Ferrante RJ, Ratan RR. Histone deacetylase inhibitors prevent oxidative neuronal death independent of expanded polyglutamine repeats via an Sp1-dependent pathway. Proc Natl Acad Sci U S A. 2003;100:4281–4286. doi: 10.1073/pnas.0737363100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Vlahos CJ, Matter WF, Hui KY, Brown RF. A specific inhibitor of phosphatidylinositol 3-kinase, 2-(4-morpholinyl)-8-phenyl-4H-1-benzopyran-4-one (LY294002) J Biol Chem. 1994;269:5241–5248. [PubMed] [Google Scholar]
- 26.Li B, Desai SA, MacCorkle-Chosnek RA, Fan L, Spencer DM. A novel conditional Akt ‘survival switch’ reversibly protects cells from apoptosis. Gene Ther. 2002;9:233–244. doi: 10.1038/sj.gt.3301641. [DOI] [PubMed] [Google Scholar]
- 27.Mikami I, Zhang F, Hirata T, Okamoto J, Koizumi K, Shimizu K, Jablons D, He B. Inhibition of activated phosphatidylinositol 3-kinase/AKT pathway in malignant pleural mesothelioma leads to G1 cell cycle arrest. Oncol Rep. 2010;24:1677–1681. doi: 10.3892/or_00001033. [DOI] [PubMed] [Google Scholar]
- 28.Workman P, Clarke PA, Raynaud FI, van Montfort RL. Drugging the PI3 kinome: from chemical tools to drugs in the clinic. Cancer Res. 2010;70:2146–2157. doi: 10.1158/0008-5472.CAN-09-4355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jain AN. Surflex-Dock 2.1: Robust performance from ligand energetic modeling, ring flexibility, and knowledge-based search. J Comput-Aided Mol Des. 2007;21:281–306. doi: 10.1007/s10822-007-9114-2. [DOI] [PubMed] [Google Scholar]
- 30.Brunsteiner M, Petukhov PA. unpublished results. [Google Scholar]
- 31.Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 1976;72:248–254. doi: 10.1006/abio.1976.9999. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.