

Abstract
Until recently, a capacity for apoptosis and synthesis of nitric oxide (⋅NO) were viewed as exclusive to multicellular organisms. The existence of these processes in unicellular parasites was recently described, with their biological significance remaining to be elucidated. We have evaluated l-arginine metabolism in Trypanosoma cruzi in the context of human serum-induced apoptotic death. Apoptosis was evidenced by the induction of DNA fragmentation and the inhibition of [3H]thymidine incorporation, which were inhibited by the caspase inhibitor Ac-Asp-Glu-Val-aspartic acid aldehyde (DEVD-CHO). In T. cruzi exposed to death stimuli, supplementation with l-arginine inhibited DNA fragmentation, restored [3H]thymidine incorporation, and augmented parasite ⋅NO production. These effects were inhibited by the ⋅NO synthase inhibitor Nω-nitroarginine methyl ester (l-NAME). Exogenous ⋅NO limited DNA fragmentation but did not restore proliferation rates. Because l-arginine is also a substrate for arginine decarboxylase (ADC), and its product agmatine is a precursor for polyamine synthesis, we evaluated the contribution of polyamines to limiting apoptosis. Addition of agmatine, putrescine, and the polyamines spermine and spermidine to T. cruzi sustained parasite proliferation and inhibited DNA fragmentation. Also, the ADC inhibitor difluoromethylarginine inhibited l-arginine-dependent restoration of parasite replication rates, while the protection from DNA fragmentation persisted. In aggregate, these results indicate that T. cruzi epimastigotes can undergo programmed cell death that can be inhibited by l-arginine by means of (i) a ⋅NO synthase-dependent ⋅NO production that suppresses apoptosis and (ii) an ADC-dependent production of polyamines that support parasite proliferation.
Trypanosoma cruzi is a hemoflagellate unicellular eukaryote that undergoes morphological and biochemical changes during its life cycle. This life cycle includes passage through the insect vector (triatomid hematophage arthropod) and the vertebrate host, causing Chagas disease in humans, an infectious process that is widespread in Latin America. In the gut of the insect vector, proliferating extracellular epimastigotes replicate and differentiate into the nondividing infective form, metacyclic trypomastigotes. Once in the bloodstream, trypomastigotes infect nucleated cells (macrophages, heart muscle cells) and differentiate into the proliferative intracellular amastigotes that emerge as nondividing trypomastigotes. It has been recently shown that, in response to different extracellular stimuli, epimastigotes of T. cruzi die by programmed cell death with features of apoptosis (1).
Apoptosis is characterized by typical morphological changes including nuclear DNA condensation and fragmentation, cell shrinkage, and, ultimately, formation of apoptotic bodies. The biochemical mechanisms leading to apoptosis depend on the cellular lineage and the death stimuli (2). The execution phase of apoptosis is typically triggered by the activation of a family of cysteine proteinases, known as caspases, thought to represent a major regulatory step in the apoptotic pathway (3). Among them, caspase 3 participates in the activation of the nuclear endonuclease, leading to DNA fragmentation, a hallmark of apoptosis (4). Apoptosis, first postulated to be an exclusive process of multicellular organisms, is now appreciated to occur in unicellular eukaryotic organisms such as T. cruzi (1), Trypanosoma brucei rhodesiense (5), and T. brucei brucei (6). The biological significance of this mode of cell death in unicellular organisms is unknown at present.
Another intriguing event in the biology of T. cruzi has been the discovery of a calcium-dependent nitric oxide (⋅NO) synthase (NOS), which can be modulated by glutamate, possibly through the N-methyl-d-aspartate (NMDA)-activated ion channel (7). The resultant calcium influx leads to a rise in cGMP, ascribable to ⋅NO-dependent activation of soluble guanylate cyclase (8). In mammalian cells, it has been postulated that ⋅NO can mediate the regulation of caspase activity by the S-nitrosylation of critical thiol groups present in the active site (9, 10), leading to inhibition of apoptotic death (11, 12). Alternatively, ⋅NO may exert an antiapoptotic role by influencing mitochondrial electrochemical gradients and by cGMP-dependent mechanisms, such as up-regulation of Bcl-2 expression (13–15).
As a unicellular eukaryote with a high replication rate, T. cruzi depends on polyamine production and/or uptake from the extracellular medium for propagation (16). Previous studies failed to detect ornithine decarboxylase activity in T. cruzi. Their polyamine biosynthesis is dependent on the decarboxylation of l-arginine by arginine decarboxylase (ADC), yielding agmatine, which can be further metabolized to putrescine, spermidine, and spermine (17–19). Moreover, studies with the inhibitor of ADC difluoromethylarginine (DFMA) reveal that l-arginine-derived polyamines are essential for parasite proliferation and infection in mammalian cells (20).
The utilization of l-arginine by T. cruzi for the production of bioactive ⋅NO and/or polyamines can be modulated by external conditions that could ultimately affect cellular physiology. This study reveals the contribution of these l-arginine-derived metabolites to the modulation of apoptosis of a unicellular eukaryote.
Materials and Methods
Materials.
α-Difluoromethyl-dl-ornithine and Ac-Asp-Glu-Val-aspartic acid aldehyde (DEVD-CHO) were from Bachem. 1-Hydroxy-2-oxo-3,3-bis(2-aminoethyl)-1-triazene (NOC-18) was from Dojindo, Kamimashiki Kumamoto, Japan. DFMA was from Aventis Pharmaceuticals, Bridgewater, NJ. MK-801 maleate was from Tocris Cookson, St. Louis. Diethylenetriamine 2,2′-(hydroxynitrosohydrazono)bisethanamine (DETA NONOate) was from Cayman Chemicals, Ann Arbor, MI. Apoptosis detection system-fluorescein was from Promega. Agmatine sulfate was from Aldrich. p-Nitroaniline (p-NA) was from Fluka and benzyloxycarbonyl-Asp-Glu-Val-Asp-p-NA (zDEVD-p-NA) was from Alexis, San Diego. l-[2,3-3H]Arginine (38.5 Ci/mmol; 1 Ci = 37 GBq), [5,6-3H]uridine (45 Ci/mmol), [3H]thymidine (1 mCi/mmol), Nω-nitro-l-arginine methyl ester (l-NAME), diphenylamine, AG50WX-8 Dowex resin, and all other substrates and reagents were from Sigma. Fresh human serum (FHS) was obtained from healthy volunteers. Heat-inactivated serum was obtained by exposure at 56°C for 40 min (21) and used in all control experiments.
Parasites.
T. cruzi epimastigotes (Tulahuen-2 strain) were cultured at 28°C, pH 7.3, in BHI medium (which contains, in g/liter, brain–heart infusion 33, tryptose 3, hemin 0.02, KCl 0.4, and Na2HPO4 4), supplemented with complement-inactivated 10% fetal bovine serum, glucose (0.3 g/liter), streptomycin sulfate (0.2 g/liter), and penicillin (200,000 units/liter). Midexponential-phase parasites (5 days) were collected by centrifugation at 750 × g and washed three times in Krebs–Henseleit buffer (15 mM NaHCO3/5 mM KCl/120 mM NaCl/0.7 mM Na2HPO4/1.5 mM NaH2PO4, pH 7.3) containing 10 mM glucose.
Determination of ⋅NO Production and NOS Activity.
⋅NO production was assayed by detecting nitrite accumulation in reaction systems by using the Griess reagent (22). Briefly, 3 × 108 cells per ml were incubated in Krebs–Henseleit buffer for 10 h in various experimental conditions. After incubation, epimastigotes were lysed by three freeze–thaw cycles and centrifuged at 13,000 × g for 15 min, and the supernatant was used for quantification of nitrite as previously described (22). In experiments including 10% FHS as the death stimulus (see below), epimastigotes were first removed by centrifugation at 13,000 × g and the supernatant was discarded. Cells were resuspended in 0.5 ml of Krebs–Henseleit buffer and processed as above.
NOS activity was evaluated in vivo by following the conversion of l-[2,3-3H]arginine to l-citrulline as previously (7). The authenticity of the radiolabeled l-citrulline was validated by TLC analysis as previously described (7).
For the detection of ⋅NO fluxes from NOC-18 and DETA NONOate, oxidation of oxyhemoglobin to methemoglobin was measured at 577 nm (ɛ577 = 11 mM−1⋅cm−1) for 24 h in the presence of 10% FHS (23).
l-[2,3-3H]Arginine Transport.
T. cruzi epimastigotes (1 × 107 cells per ml) were incubated for 1 min in Krebs–Henseleit buffer, pH 7.3, containing 50 nM l-[2,3-3H]arginine (38.5 Ci/mmol) in the presence of increasing concentrations of l-arginine (0–10 mM) or l-NAME (10 mM). After incubation, epimastigotes were sedimented by centrifugation at 13,000 × g for 5 min and washed three times in Krebs–Henseleit buffer. Cells were then solubilized in 0.5 ml of TET buffer (10 mM Tris⋅HCl/1 mM EDTA, pH 8.0, containing 5% Triton X-100) and mixed with 5 ml of scintillation fluid, and their radioactivity was measured.
DNA Agarose Gel Electrophoresis.
Parasites (3 × 108 cells per ml) were incubated in Krebs–Henseleit buffer, pH 7.4, containing 10 mM glucose at 28°C in the presence or absence of various additives for 18–24 h. Cells were collected by centrifugation at 13,000 × g for 5 min. Pelleted cells were resuspended in 0.5 ml of TET buffer and allowed to sit on ice for 15 min. Intact chromatin (pellet) was separated from DNA fragments (supernatant) by centrifugation for 15 min at 13,000 × g at 4°C. The DNA fragments were precipitated by 5 μl of 1 M MgCl2, 100 μl of 5 M NaCl, and 1 ml of isopropyl alcohol for 12 h at −20°C, then centrifuged for 15 min at 13,000 × g, and the pellet was air dried. The DNA was then resuspended in 50 μl of TE buffer (10 mM Tris⋅HCl/1 mM EDTA, pH 8.0) and incubated at 37°C for 1 h with 100 μg/ml RNase followed by treatment for 1 h at 60°C with 100 μg/ml proteinase A. Samples were supplemented with loading solution [0.25% bromophenol blue/30% (vol/vol) glycerol] and electrophoretically separated on a 2% agarose gel containing 1 μg/ml ethidium bromide for 1 h at 80 V. Pictures were taken by UV transilumination. Epimastigote DNA fragmentation was explored in the presence or absence of 10% FHS. Photographs of the DNA gels were scanned and relative DNA fragmentation was determined by densitometric techniques using Scion Image (Scion, Frederick, MD). Results are expressed as percent DNA fragmentation with respect to conditions considered to be 100% fragmentation.
Quantitation of DNA Fragmentation.
DNA fragmentation was measured with diphenylamine reagent as previously (24). Intact chromatin and DNA fragments were obtained as above. Pellets (intact chromatin) were resuspended in 0.5 ml of TE buffer, precipitated with 10% trichloroacetic acid at 4°C, and sedimented at 13,000 × g for 10 min, and the supernatant was removed. Samples (intact chromatin and DNA fragments) were boiled for 15 min after addition of 5% trichloroacetic acid. DNA content was quantitated by using the diphenylamine reagent. DNA fragmentation was calculated as the percentage of DNA recovered in the supernatants with respect to the total DNA (pellet + supernatant).
Caspase-Like Activity.
Caspase-like activity was measured in cell extracts by using the chromogenic substrate zDEVD-p-NA (11). T. cruzi epimastigotes were exposed to 10% FHS for 6 h at 28°C, washed, and lysed. Then, 2 mg of parasite protein was preincubated in buffer (20 mM Pipes/100 mM NaCl/1 mM EDTA/0.1% Chaps/10% sucrose/5 mM DTT, pH 7.2) for 1 h at room temperature. In some experiments the inhibitor DEVD-CHO was present in the preincubation mixture at 200 μM. Then, 400 μM zDEVD-p-NA was added and changes in absorbance at 380 nm (ɛ = 5,000 M−1⋅cm−1) were followed for 2 h at 37°C.
[3H]Thymidine Incorporation.
[3H]Thymidine incorporation reflected changes in parasite proliferation rates. Epimastigotes (3 × 108 cells per ml) were incubated in Krebs–Henseleit buffer, pH 7.3, at 28°C under various experimental conditions. After 1 h, an aliquot was plated at a density of 1 × 106 cells per ml in 96-well plates with BHI medium (final volume 200 μl), and 1 μCi of [3H]thymidine was added for 18 h at 28°C. After incubations, cells were harvested over fiberglass filters and washed several times with distilled water. The filters were cut, mixed with 3 ml of scintillation fluid, and assayed for radioactivity. Results were expressed as percentage of [3H]thymidine incorporation with respect to the control.
Staining of Apoptotic Nuclei.
In situ staining of apoptotic nuclei was indicated by terminal deoxynucleotidyltransferase-mediated dUTP nick end labeling (TUNEL) with fluorescein (Promega). After incubations, epimastigotes were allowed to adhere to a poly(d-lysine)-precoated slide, air-dried, fixed with increasing concentrations of ethanol (30%, 50%, 70%, 95%, and 100%, vol/vol), and stored at −20°C until used. Apoptotic cells were visualized by fluorescence microscopy. Duplicate slides were stained with hematoxylin and eosin for visualization of morphological changes.
[5,6-3H]Uridine Release.
After 4 days in culture (BHI medium), epimastigotes were treated with 50 μCi of [5,6-3H]uridine and incubated for 48 h. After the incubation period, labeled parasites were collected by centrifugation and washed three times in Krebs–Henseleit buffer, pH 7.3, to eliminate unincorporated [5,6-3H]uridine. Parasites were resuspended at a density of 3 × 108 cells per ml and incubated for 24 h under various conditions. Aliquots (1 ml) were taken hourly, parasites were sedimented by centrifugation, the pellet (parasites) was resuspended in 500 μl of TET buffer, and supernatants and pellets were assayed for radioactivity. Results were expressed as percentage of [5,6-3H]uridine released (supernatant) with respect to total incorporation (pellet + supernatant).
Conversion of l-[2,3-3H]Arginine by Epimastigotes to Metabolic Intermediates.
Parasites (3 × 108 cells per ml) were incubated in the presence of 1 μCi of l-[2,3-3H]arginine (38.5 Ci/mmol) for 1 h at 28°C in Krebs–Henseleit buffer, pH 7.3. After incubation, 10% heat-inactivated human serum or FHS was added and the mixture was incubated for 18 h. Incubations were terminated by addition of 50 μl of 5% trichloroacetic acid and centrifuged at 13,000 × g for 30 min. A 5-μl sample of supernatant was spotted on a Whatman silica gel 150A TLC sheet and eluted with chloroform/methanol/8.6 M ammonium hydroxide/water (1:4:2:1, vol/vol). Agmatine and polyamines (putrescine, spermidine, and spermine) were identified by comigration with authentic standards. Identified bands were cut, scraped, mixed with 5 ml of scintillation fluid, and assayed for radioactivity.
Statistical Analysis.
Data were analyzed by a one-way analysis of variance (ANOVA) followed by a Student–Newman–Keuls test.
Results
l-Arginine Modulates Apoptotic Death in T. cruzi Epimastigotes.
Culture conditions that will induce apoptotic death in T. cruzi epimastigotes include culturing at 37°C or at low cellular densities, starvation, and addition of the antibiotic drug geneticin or FHS (1). Human serum-induced death of epimastigotes, dependent upon complement activation, was chosen to stimulate T. cruzi apoptosis (1). Because physiological concentrations of human serum result in massive T. cruzi death in less than 1 h (1, 21) whereas other pro-apoptotic stimuli induce apoptosis within several days (1), we used the less acute exposure to 10% FHS at 28°C. Under these conditions epimastigotes yielded an evident DNA laddering in agarose gel electrophoresis (Fig. 1A, lane 4) and positive TUNEL staining (Fig. 1Bb) with conservation of membrane integrity (Fig. 1C) after 18 h of incubation. Addition of 10% FHS for only 1 h was sufficient to activate epimastigote programmed cell death, since no difference in [3H]thymidine incorporation was observed when the death stimulus was removed from the incubation medium after 1 h (not shown). Nevertheless, long incubation periods (18–24 h) were required to visualize DNA fragmentation. Control experiments carried out with heat-inactivated human serum (10%) confirmed the thermally labile nature of the death stimulus (Fig. 1A, lane 3), as some complement components are heat sensitive (1, 21). Thus, using the cell death stimulus provided by 10% FHS, we evaluated the interplay between pro-apoptotic pathways and l-arginine metabolism in T. cruzi epimastigotes.
Figure 1.

Apoptotic death of epimastigotes exposed to 10% FHS. (A) DNA fragmentation analysis. Epimastigotes (3 × 108 cells per ml) were incubated for 18 h at 28°C in serum-free conditions (lane 2); 10% heat-inactivated human serum (lane 3); 10% FHS (lanes 4–8) in the presence of 10 mM d-arginine (lane 5); 10 mM l-arginine (lane 6); 10 mM l-arginine plus 10 mM l-NAME (lane 7); or 100 μM DEVD-CHO (lane 8). DNA marker was in lane 1. After the incubation period, DNA was extracted, subjected to electrophoresis on a 2% agarose gel, and visualized by staining with ethidium bromide. (B) TUNEL staining. (a) Control. (b) 10% FHS. (c) 10% FHS + 10 mM l-arginine. (d) 10% FHS + 10 mM l-arginine + 10 mM l-NAME. (C) Membrane permeability assays. Labeled epimastigotes were incubated at a density of 3 × 108 cells per ml in the various experimental conditions for 24 h at 28°C. Aliquots (1 ml) were taken every hour up to the end of the experiment. Parasites were collected by centrifugation. Supernatants and pellets were assayed for radioactivity. The results are expressed as percentage of [5,6-3H]uridine released (supernatant) with respect to the total incorporated (pellet + supernatant) in each condition. ○, Control; ●, 10% FHS; ▵, 10 mM l-arginine plus 10% FHS; ▿, 10 mM l-arginine plus 10 mM l-NAME plus 10% FHS; +, 50 mM H2O2.
l-Arginine (10 mM), but not d-arginine (10 mM), prevented DNA fragmentation, an effect that was abrogated by the NOS inhibitor l-NAME (10 mM) (Fig. 1A, lanes 6, 5, and 7, respectively). Apoptotic death was also evaluated by TUNEL staining and fluorescence microscopy. In the presence of the death stimulus and/or l-arginine (10 mM) plus l-NAME (10 mM) TUNEL staining of apoptotic cells was maximal (Fig. 1B, b and d, respectively), whereas in the presence of FHS plus l-arginine (10 mM) no significant increase in TUNEL staining was observed (Fig. 1B, c and a, respectively). To confirm that parasite membrane integrity was conserved in the context of putative apoptotic processes, membrane permeability was evaluated by [5,6-3H]uridine release. Incubation of T. cruzi epimastigotes with both 10% FHS and l-arginine (10 mM) showed no significant release of [5,6-3H]uridine, with respect to control conditions (10% heat-inactivated human serum). A high H2O2 concentration (50 mM) served as a positive control for necrotic death (Fig. 1C).
To explore whether parasite apoptotic death involved mediation by caspase-like proteolytic processing, the caspase-3 inhibitor DEVD-CHO was added (11, 25, 26). DEVD-CHO (100 μM) abolished the DNA fragmentation induced by FHS (Fig. 1A, lane 8). Moreover, caspase-like activity was directly detected in cell extracts of T. cruzi undergoing apoptosis. Caspase-like activity under 10% serum, 10% serum plus DEVD-CHO, and serum-free conditions was 0.205, 0.09, and 0.072 pmol of p-NA formed per min per mg of protein, respectively.
l-Arginine Restores [3H]Thymidine Incorporation in Serum-Treated T. cruzi.
Parasite [3H]thymidine incorporation was assayed in the presence or absence of death stimuli and during modulation of arginine metabolism (Fig. 2A). FHS (10%) inhibited [3H]thymidine incorporation by 50%. Coincubation with l-arginine (10 mM) completely restored parasite proliferation rates, whereas d-arginine (10 mM) had no effect. Moreover, the growth-restorative influence of l-arginine was abolished by l-NAME (10 mM). DNA fragmentation was quantitated by the diphenylamine method (Fig. 2B). In agreement with the cell proliferation-based observations, DNA fragmentation was prevented by l-arginine, an effect that was reversed by l-NAME.
Figure 2.

Effects of l-arginine on [3H]thymidine incorporation and DNA fragmentation. (A) Parasites (1 × 106 cells per ml) were incubated in the presence or absence of the various metabolites and inhibitors (10 mM l-arginine, 10 mM d-arginine, 10 mM l-NAME, or 100 μM DEVD-CHO) and pulsed with 1 μCi of [3H]thymidine for 18 h at 28°C. After the incubation period, parasites were harvested onto fiberglass filters and assayed for radioactivity. (B) Cells (3 × 108 per ml) were incubated in Krebs–Henseleit buffer for the quantification of DNA fragments by the diphenylamine method. * and ** denote statistical difference (P < 0.05) with respect to the control and 10% FHS conditions, respectively.
Serum-induced inhibition of [3H]thymidine incorporation was totally prevented by DEVD-CHO (100 μM) (Fig. 2A) and correlated well with the observed attenuation of DNA fragmentation (Fig. 2B). The IC50 for the protective effects of DEVD-CHO on serum-induced parasite death was ≈50 μM (not shown).
⋅NO Production in Serum-Treated Epimastigotes.
Parasite ⋅NO production in the presence or absence of
death stimuli was estimated by quantitation of NO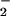 accumulation in the culture media (Table
1). Under basal culture conditions,
l-arginine (1 mM) caused a marginal stimulation of
NO
accumulation in the culture media (Table
1). Under basal culture conditions,
l-arginine (1 mM) caused a marginal stimulation of
NO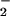 production, whereas the simultaneous presence of
l-glutamate (1 mM) stimulated NO
production, whereas the simultaneous presence of
l-glutamate (1 mM) stimulated NO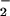 production by 60%. In the presence of l-arginine,
NO
production by 60%. In the presence of l-arginine,
NO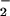 production as a function of
l-glutamate concentration yielded hyperbolic plots, with
≈12 μM glutamate required to obtain 50% of maximal
NO
production as a function of
l-glutamate concentration yielded hyperbolic plots, with
≈12 μM glutamate required to obtain 50% of maximal
NO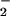 production and >100 μM for maximal
NO
production and >100 μM for maximal
NO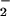 production (not shown). This increase was
inhibited by l-NAME (4 mM) and by the NMDA channel
inhibitor MK-801 (0.1 mM), as previously (upper part of Table 1) (7).
Furthermore, 1 mM d-arginine plus l-glutamate
failed to stimulate ⋅NO production (Table 1). Importantly, the
intracellular transport of
l-[2,3-3H]arginine was not affected
by the presence of l-NAME (not shown), as for mammalian
cells (27).
production (not shown). This increase was
inhibited by l-NAME (4 mM) and by the NMDA channel
inhibitor MK-801 (0.1 mM), as previously (upper part of Table 1) (7).
Furthermore, 1 mM d-arginine plus l-glutamate
failed to stimulate ⋅NO production (Table 1). Importantly, the
intracellular transport of
l-[2,3-3H]arginine was not affected
by the presence of l-NAME (not shown), as for mammalian
cells (27).
Table 1.
Nitric oxide formation by T. cruzi epimastigotes
| Compound (mM) | NO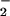 ,
nM⋅min−1⋅mg−1 ,
nM⋅min−1⋅mg−1
|
% change |
|---|---|---|
| In Krebs–Henseleit buffer | ||
| None | 1.94 ± 0.19a | 0 |
| l-Arginine (1) | 2.29 ± 0.28b | 18 |
| l-NAME (4) | 1.55 ± 0.18b | −20 |
| MK-801 (1) | 1.96 ± 0.33 | 1 |
| l-Arginine (1) + l-Glutamate (1) | 3.10 ± 0.05b | 60 |
| l-Arginine (1) + l-Glutamate (1) + l-NAME (4) | 1.84 ± 0.14 | −5 |
| l-Arginine (1) + l-Glutamate (1) + MK-801 (0.1) | 1.87 ± 0.1 | −3 |
| d-Arginine (1) + l-Glutamate (1) | 1.69 ± 0.14 | −13 |
| In the presence of 10% FHS | ||
| 10% heat-inactivated human serum | 2.32 ± 0.23 | 0 |
| 10% FHS | 2.36 ± 0.18c | 2 |
| + l-Glutamate (1) | 2.41 ± 0.13 | 4 |
| + l-Arginine (10) | 3.98 ± 0.37d | 72 |
| + l-Arginine (10) + l-Glutamate (1) | 3.92 ± 0.3d | 69 |
| + l-Arginine + l-NAME (10) | 2.10 ± 0.12 | −10 |
| + l-Arginine (10) + MK-801 (0.1) | 1.94 ± 0.26d | −16 |
⋅NO production was measured as
NO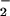 accumulation over a 10-h period. Results are
expressed as mean ± SD of at least three independent experiments.
accumulation over a 10-h period. Results are
expressed as mean ± SD of at least three independent experiments.
indicates statistical difference (P < 0.05) with respect to
;
indicates statistical difference (P < 0.05) with respect to
.
In concert with exposure of T. cruzi to 10% FHS,
l-arginine (10 mM) increased
NO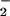 production by 72% with respect to controls
(lower part of Table 1). Addition of glutamate did not further enhance
NO
production by 72% with respect to controls
(lower part of Table 1). Addition of glutamate did not further enhance
NO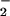 production, indicating that serum itself provides
one or more components able to promote calcium influx into T.
cruzi epimastigotes. The increment in NO
production, indicating that serum itself provides
one or more components able to promote calcium influx into T.
cruzi epimastigotes. The increment in NO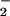 production in this condition was also completely inhibited by
l-NAME and MK-801 (Table 1). The presence of NOS
activity in T. cruzi epimastigotes was confirmed by
following the conversion of
l-[2,3-3H]arginine to
l-[3H]citrulline. In
Krebs–Henseleit buffer supplemented with
l-glutamate, citrulline production was 0.37
± 0.1 pmol of l-citrulline per h per
106 cells.
production in this condition was also completely inhibited by
l-NAME and MK-801 (Table 1). The presence of NOS
activity in T. cruzi epimastigotes was confirmed by
following the conversion of
l-[2,3-3H]arginine to
l-[3H]citrulline. In
Krebs–Henseleit buffer supplemented with
l-glutamate, citrulline production was 0.37
± 0.1 pmol of l-citrulline per h per
106 cells.
Exogenous ⋅NO Addition Inhibits Serum-Mediated DNA Fragmentation.
To confirm that the l-NAME-inhibitable,
l-arginine-mediated inhibition of DNA fragmentation was
mediated by parasite ⋅NO production, the influence of direct
cell exposure to ⋅NO was evaluated. Two different ⋅NO
donors (DETA NONOate and NOC-18) were selected to generate constant
fluxes of ⋅NO over long periods of time (30 h). Considering an
epimastigote volume of ≈3 × 10−14 liter
and an NO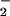 production of 0.40
pmol⋅h−1 per 106
cells under maximal stimulation conditions (10% FHS plus 10 mM
l-arginine, lower part of Table 1), the ⋅NO
concentration generated inside a single parasite was estimated to be
0.22 ± 0.1 μM ⋅NO per min. Taking this into account,
⋅NO donor concentrations were adjusted to generate fluxes
ranging from 0.01 to 0.25 μM ⋅NO per min. NOC-18 (5 mM, 0.245
μM ⋅NO per min) completely inhibited DNA fragmentation induced
by FHS during the 30-h incubation periods (Fig.
3 A and B, lane 8).
A dose-dependent effect of ⋅NO was observed at 18 h (Fig.
3A, lanes 4–8), with 1 mM NOC-18 generating 0.145 μM
⋅NO per min being sufficient to fully inhibit DNA fragmentation
induced by 18-h exposure to death stimuli (Fig. 3A, lane 7).
Similar results were obtained with DETA NONOate-derived ⋅NO (not
shown). Lower rates of ⋅NO generation (0.008–0.08 μM
⋅NO per min) had no effect on DNA fragmentation (Fig. 3
A and B, lanes 4 and 5). Additionally, no
detrimental effects were observed under control conditions with the
⋅NO donors at the highest concentrations tested (Fig. 3
A and B, lanes 9 and 10). While cell exposure to
exogenously generated ⋅NO inhibited DNA fragmentation, there was
no effect on [3H]thymidine incorporation, even
though l-arginine addition reestablished basal
cell proliferation rates (Fig. 3C). These results indicate
that other metabolites were also participating in the protective
effects yielded by l-arginine.
production of 0.40
pmol⋅h−1 per 106
cells under maximal stimulation conditions (10% FHS plus 10 mM
l-arginine, lower part of Table 1), the ⋅NO
concentration generated inside a single parasite was estimated to be
0.22 ± 0.1 μM ⋅NO per min. Taking this into account,
⋅NO donor concentrations were adjusted to generate fluxes
ranging from 0.01 to 0.25 μM ⋅NO per min. NOC-18 (5 mM, 0.245
μM ⋅NO per min) completely inhibited DNA fragmentation induced
by FHS during the 30-h incubation periods (Fig.
3 A and B, lane 8).
A dose-dependent effect of ⋅NO was observed at 18 h (Fig.
3A, lanes 4–8), with 1 mM NOC-18 generating 0.145 μM
⋅NO per min being sufficient to fully inhibit DNA fragmentation
induced by 18-h exposure to death stimuli (Fig. 3A, lane 7).
Similar results were obtained with DETA NONOate-derived ⋅NO (not
shown). Lower rates of ⋅NO generation (0.008–0.08 μM
⋅NO per min) had no effect on DNA fragmentation (Fig. 3
A and B, lanes 4 and 5). Additionally, no
detrimental effects were observed under control conditions with the
⋅NO donors at the highest concentrations tested (Fig. 3
A and B, lanes 9 and 10). While cell exposure to
exogenously generated ⋅NO inhibited DNA fragmentation, there was
no effect on [3H]thymidine incorporation, even
though l-arginine addition reestablished basal
cell proliferation rates (Fig. 3C). These results indicate
that other metabolites were also participating in the protective
effects yielded by l-arginine.
Figure 3.

Effects of ⋅NO donors on DNA fragmentation and [3H]thymidine incorporation. (A and B) DNA fragmentation in the presence of NOC-18 for 18 h (A) and 30 h (B). ⋅NO fluxes were as follows: 0.005 μM⋅min−1, 0.018 μM⋅min−1, 0.082 μM⋅min−1, 0.145 μM⋅min−1, and 0.245 μM⋅min−1, which correspond to 0.01 mM, 0.1 mM, 0.5 mM, 1 mM, and 5 mM NOC-18, respectively (lanes correspond to 1, control; 2, 10% inactivated serum; 3, 10% FHS; lanes 4–8 are 10% FHS plus 0.01, 0.1, 0.5, 1, and 5 mM NOC-18, respectively; lanes 9 and 10 correspond to 1 and 5 mM NOC-18 plus 10% heat-inactivated human serum.) (C) Effect of NOC-18 and serum on [3H]thymidine incorporation. Aliquots of cells from the experiments described in for A were pulsed with [3H]thymidine and assayed for radioactivity.
Role of l-Arginine Metabolites in Epimastigote Survival.
The polyamine precursors agmatine and putrescine, along with the polyamines spermidine and spermine, were used to evaluate the influence of these metabolites in 10% FHS-induced stimulation of apoptosis. Polyamines and their precursors agmatine and l-arginine were protective against loss of [3H]thymidine incorporation and DNA fragmentation (Table 2). l-Ornithine (10 mM) and the ornithine decarboxylase inhibitor α-difluoromethyl-dl-ornithine (10 mM) had no influence (data not shown). l-NAME addition inhibited the protective effects of only l-arginine (Table 2).
Table 2.
[3H]Thymidine incorporation and DNA fragmentation
| Compound (mM) | [3H]Thymidine incorporation, % | DNA fragmentation, % |
|---|---|---|
| None | 100 ± 10 | 15 ± 4 |
| 10% FHS | 47 ± 9a | 100 ± 20c |
| + l-Arginine (1) | 57 ± 8 | 99 ± 10 |
| + l-Arginine (5) | 70 ± 4b | 20 ± 4d |
| + l-Arginine (10) | 84 ± 6b | 18 ± 9d |
| + l-Arginine (10) + l-NAME (10) | 45 ± 7 | 115 ± 23 |
| + d-Arginine (10) | 50 ± 10 | 100 ± 14 |
| + Agmatine (10) | 100 ± 17b | 20 ± 3d |
| + Agmatine (10) + l-NAME (10) | 82 ± 3b | 17 ± 2d |
| + Putrescine (10) | 93 ± 8b | 16 ± 5d |
| + Putrescine (10) + l-NAME (10) | 82 ± 3b | 18 ± 6d |
| + Spermidine (10) | 88 ± 6b | 15 ± 10d |
| + Spermidine (10) + l-NAME (10) | 100 ± 10b | 17 ± 7d |
| + DFMA (10) | 60 ± 8 | 110 ± 10 |
| + DFMA (10) + l-Arginine (10) | 58 ± 7 | 15 ± 2d |
| + DFMA (10) + l-Arginine (10) + | ||
| l-NAME (10) | 60 ± 5 | 120 ± 15 |
[3H]Thymidine incorporation is expressed relative to nontreated epimastigotes (control condition) taken as 100%. Percent DNA fragmentation was estimated by digital analysis of the electrophoretic bands, taking as 100% the fragmentation observed with 10% FHS. Results are mean ± SD of at least three independent experiments.
and
indicate statistical significance (P < 0.05) compared with
and
, respectively.
To explore the influence of l-arginine as a biosynthetic precursor of the polyamine pathway, parasites were challenged with l-[2,3-3H]arginine. In the presence of FHS, radiolabeled polyamines (agmatine, putrescine, spermidine, and spermine) were detected by TLC, indicating that the l-arginine-dependent production of these metabolites (not shown). Furthermore, the ADC inhibitor DFMA [10 mM; Ki = 20 mM in T. cruzi (19)] prevented l-arginine (10 mM)-mediated recovery of [3H]thymidine incorporation but had no effect on DNA fragmentation. Importantly, the addition of l-NAME inhibited the protective effects of l-arginine on DNA fragmentation (Table 2). In aggregate, these results suggest the presence of two distinct but complementary l-arginine-dependent metabolic pathways contributing to the inhibition of serum-induced apoptotic death of epimastigotes.
Discussion
l-Arginine metabolism by both the ⋅NO and polyamine pathways is revealed to mediate protection of T. cruzi against apoptotic death induced by 10% FHS. External supplementation with l-arginine (but not d-arginine) was able to inhibit DNA fragmentation as seen by agarose gel electrophoresis, DNA-fragment quantitation, and TUNEL positive staining. Coincubation with the NOS inhibitor l-NAME completely inhibited the protective effect of l-arginine, suggesting that ⋅NO can serve as an antiapoptotic signaling molecule in T. cruzi. T. cruzi has a constitutive Ca2+/calmodulin-activatable NOS that can be regulated through l-glutamate-stimulated NMDA receptor (7). In agreement, our results show an MK-801-inhibitable l-glutamate stimulation of ⋅NO production by epimastigotes (7). Also, serum supplemented with l-arginine promoted parasite ⋅NO production (lower part of Table 1). While Ca2+ present in serum was sufficient for the activation of NOS, the concentration of glutamate in 10% serum [3–6 μM (28)] cannot account per se for maximal activation of parasite NOS activity. However, low concentrations of glutamate can be greatly potentiated by glycine and serine (7) also present in serum. In addition, other serum components also may be contributing to the MK-801-inhibitable activation of NOS.
Considering that intracellular rates of ⋅NO production by parasites was estimated to be 0.22 μM⋅min−1 during maximal stimulation, and that the protective actions of exogenously generated ⋅NO were observed within the same concentration range, it is concluded that parasite NOS is able to support the observed antiapoptotic actions. However, exogenous ⋅NO was unable to restore basal rates of parasite proliferation under these experimental conditions. Previous studies have shown that ⋅NO can inhibit ribonucleotide reductase, a critical enzyme in the synthesis of DNA precursors (29), and that ⋅NO produced by activated macrophages can be cytotoxic to T. cruzi (30, 31). Nevertheless, the ⋅NO fluxes used herein did not affect rates of parasite proliferation in the presence of heat-inactivated human serum (Fig. 3B), thus this is not a likely explanation. Thus, the contribution of l-arginine to the recovery of parasite replication rates is likely due to ⋅NO-independent pathways such as polyamine biosynthesis through ADC.
The polyamine biosynthetic pathway in T. cruzi differs from that in other trypanosomes in that ornithine decarboxylase activity is not present. There is support for the presence of ADC activity in epimastigotes of T. cruzi (17), and agmatine production by this parasite has been reported (17, 18, 32). More recently, significant DFMA-inhibitable ADC activity was detected in crude extracts of T. cruzi epimastigotes with a Km for l-arginine of 5 mM (19).
Our findings show that increasing concentrations of l-arginine, agmatine, putrescine, and spermine, but not ornithine, mediate a dose-dependent effect on [3H]thymidine incorporation (Table 2). Furthermore, l-[2,3-3H]arginine-dependent polyamine biosynthesis was observed when epimastigotes were exposed to FHS (not shown), implying involvement of ADC in the process. Indeed, DFMA had been previously used to demonstrate the role of the polyamine pathway on T. cruzi infectivity and proliferation (20, 32). DFMA inhibited T. cruzi proliferation when parasites were supplemented with l-arginine. Moreover, in the presence of l-arginine and DFMA, no DNA fragmentation was detected unless l-NAME was present, supporting both the role of parasite NOS in the protection of DNA and a role for l-arginine-derived polyamines in sustaining parasite proliferation rates in human-serum-induced apoptotic death. Unexpectedly, l-NAME also inhibited l-arginine-mediated [3H]thymidine incorporation under the death stimuli (Table 2). Because ⋅NO does not account for the recovery observed (Fig. 3C), we hypothesize that l-NAME, a structural analogue of l-arginine, may be inhibiting ADC activity, although this needs confirmation.
The fact that the caspase inhibitor DEVD-CHO protects epimastigotes from apoptotic death at concentrations previously used for mammalian cells (25, 26) and that a proteolytic activity on DEVD was detected in T. cruzi lysates of cells undergoing apoptosis, indicates the presence of a caspase-like activity in this trypanosomatid. This observation points to a possible enzymic step that can be down-regulated by ⋅NO by means of S-nitrosylation (10, 33).
Previous reports have shown that polyamines may play antiapoptotic roles. Polyamine depletion, observed in apoptosis, halts the cell cycle in G0/G1, with this checkpoint necessary to execute the apoptosis program (34). Thus, polyamine supplementation could influence cell cycle progression. Our results also show a polyamine-dependent apoptotic death protection when these compounds are present in high concentrations (Table 2). While the mechanism of action of polyamines is not well defined, possible explanations include parasite DNA and RNA stabilization and regulation of gene expression.
Human serum has long been considered lytic for T. cruzi epimastigotes (21). However, it has been more recently shown and proposed that physiological levels of serum complement at 37°C promote epimastigote apoptosis rather than necrosis (1). Massive death of epimastigotes, in response to human complement, has been postulated as an evolutionary process in which the nonadapted epimastigotes die by apoptosis, probably, for the maintenance of an immunological silent state of the host during the infection process (1). Also, phagocytosis of apoptotic T cells by host macrophages renders them more susceptible to T. cruzi infection (35) in a process that involves accelerated synthesis of polyamines by the macrophage, which in turn causes an augmented intracellular proliferation of T. cruzi. Thus, phagocytosis of apoptotic epimastigotes may serve as a mechanism to render the macrophages more susceptible to T. cruzi infection. On the other hand, activation of l-arginine metabolism in infective stages of the parasite may facilitate the evasion of the cellular immune response. This hypothesis remains to be tested.
In summary, the present work shows that T. cruzi epimastigotes can undergo programmed cell death that can be inhibited by l-arginine by means of (i) a NOS-dependent ⋅NO production that suppresses apoptosis and (ii) an ADC-dependent synthesis of polyamines that support parasite proliferation (Fig. 4). In addition, the data support the mediation of a caspase-like-dependent proteolytic processing during apoptosis of a unicellular eukaryote parasite.
Figure 4.

Proposed pathways of l-arginine metabolism in the modulation of programmed cell death in T. cruzi.
Acknowledgments
We thank Dr. Bruce A. Freeman for critical reading of the manuscript. This work was supported by grants from the Swedish Agency for Research Cooperation and the Howard Hughes Medical Institute. L.P. was supported by a fellowship from the Programa de Desarrollo de las Ciencias Básicas, Uruguay, and G.P. was partially supported by a fellowship from the Swedish Agency for Research Cooperation. R.R. is an International Research Scholar of the Howard Hughes Medical Institute.
Abbreviations
- NOC-18
1-hidroxy-2-oxo-3,3-bis(2-aminoethyl)-1-triazene
- ⋅NO
nitric oxide
- NOS
⋅NO synthase
- NMDA
N-methyl-d-aspartate
- ADC
arginine decarboxylase
- DFMA
difluoromethylarginine
- DETA NONOate
diethylenetriamine 2,2′-(hydroxynitrosohydrazono)bisethanamine
- p-NA
p-nitroaniline
- DEVD-CHO
Ac-Asp-Glu-Val-aspartic acid aldehyde
- zDEVD-p-NA
benzyloxycarbonyl-Asp-Glu-Val-Asp-p-NA
- l-NAME
Nω-nitro-l-arginine methyl ester
- FHS
fresh human serum
- TUNEL
terminal deoxynucleotidyltransferase-mediated dUTP nick end labeling
Footnotes
This paper was submitted directly (Track II) to the PNAS office.
References
- 1.Ameisen J, Idziorek T, Billaut-Mulot O, Loyens M, Tissier J P, Potentier A, Ouaissi A. Cell Death Differ. 1995;2:285–300. [PubMed] [Google Scholar]
- 2.Vaux D L, Strasser A. Proc Natl Acad Sci USA. 1996;93:2239–2244. doi: 10.1073/pnas.93.6.2239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Martin S J, Green D R, Cotter T G. Trends Biochem Sci. 1994;19:26–30. doi: 10.1016/0968-0004(94)90170-8. [DOI] [PubMed] [Google Scholar]
- 4.Janicke R U, Sprengart M L, Wati M R, Porter A G. J Biol Chem. 1998;273:9357–9360. doi: 10.1074/jbc.273.16.9357. [DOI] [PubMed] [Google Scholar]
- 5.Welburn S, Dale C, Ellis D, Beecroft B, Pearson T W. Cell Death Differ. 1996;3:229–236. [PubMed] [Google Scholar]
- 6.Ridgley E L, Xiong Z H, Ruben L. Biochem J. 1999;340:33–40. [PMC free article] [PubMed] [Google Scholar]
- 7.Paveto C, Pereira C, Espinosa J, Montagna A E, Farber M, Esteva M, Flawia M M, Torres H N. J Biol Chem. 1995;270:16576–16579. doi: 10.1074/jbc.270.28.16576. [DOI] [PubMed] [Google Scholar]
- 8.Pereira C, Paveto C, Espinosa J, Alonso G, Flawia M M, Torres H N. J Eukaryot Microbiol. 1997;44:155–156. doi: 10.1111/j.1550-7408.1997.tb05952.x. [DOI] [PubMed] [Google Scholar]
- 9.Melino G, Bernassola F, Knight R A, Corasaniti M T, Nistico G, Finazzi-Agro A. Nature (London) 1997;388:432–433. doi: 10.1038/41237. [DOI] [PubMed] [Google Scholar]
- 10.Rossig L, Fichtlscherer B, Breitschopf K, Haendeler J, Zeiher A M, Mulsch A, Dimmeler S. J Biol Chem. 1999;274:6823–6826. doi: 10.1074/jbc.274.11.6823. [DOI] [PubMed] [Google Scholar]
- 11.Kim Y M, Talanian R V, Billiar T R. J Biol Chem. 1997;272:31138–31148. doi: 10.1074/jbc.272.49.31138. [DOI] [PubMed] [Google Scholar]
- 12.Potter C L, Hanson P J. Gut. 2000;46:156–162. doi: 10.1136/gut.46.2.156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Genaro A M, Hortelano S, Alvarez A, Martinez C, Bosca L. J Clin Invest. 1995;95:1884–1890. doi: 10.1172/JCI117869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Brookes P S, Salinas E P, Darley-Usmar K, Eiserich J P, Freeman B A, Darley-Usmar V M, Anderson P G. J Biol Chem. 2000;275:20474–20479. doi: 10.1074/jbc.M001077200. [DOI] [PubMed] [Google Scholar]
- 15.Beltran B, Mathur A, Duchen M R, Erusalimsky J D, Moncada S. Proc Natl Acad Sci USA. 2000;97:14602–14607. doi: 10.1073/pnas.97.26.14602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gonzalez N S, Ceriani C, Algranati I D. Biochem Biophys Res Commun. 1992;188:120–128. doi: 10.1016/0006-291x(92)92358-5. [DOI] [PubMed] [Google Scholar]
- 17.Majumder S, Wirth J J, Bitonti A J, McCann P P, Kierszenbaum F. J Parasitol. 1992;78:371–374. [PubMed] [Google Scholar]
- 18.Schwarcz de Tarlovsky M N, Hernandez S M, Bedoya A M, Lammel E M, Isola E L. Biochem Mol Biol Int. 1993;30:547–558. [PubMed] [Google Scholar]
- 19.Hernandez S, Schwarcz de Tarlovsky S. Cell Mol Biol (Noisy-le-grand) 1999;45:383–391. [PubMed] [Google Scholar]
- 20.Yakubu M A, Basso B, Kierszenbaum F. J Parasitol. 1992;78:414–419. [PubMed] [Google Scholar]
- 21.Nogueira N, Bianco C, Cohn Z. J Exp Med. 1975;142:224–229. doi: 10.1084/jem.142.1.224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Grisham M B, Johnson G G, Lancaster J R., Jr Methods Enzymol. 1996;268:237–246. doi: 10.1016/s0076-6879(96)68026-4. [DOI] [PubMed] [Google Scholar]
- 23.Feelisch D S, Kubitzek D, Werringloer J. In: Methods in Nitric Oxide Research. Feelisch M, Stamler J S, editors. West Sussex, U.K.: Wiley; 1996. pp. 455–478. [Google Scholar]
- 24.Mebmer U K, Reed J C, Brune B. J Biol Chem. 1996;271:20192–20197. doi: 10.1074/jbc.271.33.20192. [DOI] [PubMed] [Google Scholar]
- 25.Li J, Bombeck C A, Yang S, Kim Y M, Billiar T R. J Biol Chem. 1999;274:17325–17333. doi: 10.1074/jbc.274.24.17325. [DOI] [PubMed] [Google Scholar]
- 26.Tanabe K, Nakanishi H, Maeda H, Nishioku T, Hashimoto K, Liou S Y, Akamine A, Yamamoto K. J Biol Chem. 1999;274:15725–15731. doi: 10.1074/jbc.274.22.15725. [DOI] [PubMed] [Google Scholar]
- 27.Wu G, Morris S M., Jr Biochem J. 1998;336:1–17. doi: 10.1042/bj3360001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Droge W, Gross A, Hack V, Kinscherf R, Schykowski M, Bockstette M, Mihm S, Galter D. Antioxidants in Disease Mechanisms and Therapy. San Diego: Academic; 1997. [DOI] [PubMed] [Google Scholar]
- 29.Lepoivre M, Flaman J-M, Henry I. J Biol Chem. 1992;267:22994–23000. [PubMed] [Google Scholar]
- 30.Muñoz-Fernández M A, Fernández M A, Fresno M. Eur J Immunol. 1992;22:301–307. doi: 10.1002/eji.1830220203. [DOI] [PubMed] [Google Scholar]
- 31.Muñoz-Fernández M A, Fernández M A, Fresno M. Immunol Lett. 1992;33:35–40. doi: 10.1016/0165-2478(92)90090-b. [DOI] [PubMed] [Google Scholar]
- 32.Kierszenbaum F, Wirth J J, McCann P P, Sjoerdsma A. Proc Natl Acad Sci USA. 1987;84:4278–4282. doi: 10.1073/pnas.84.12.4278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kim Y M, Chung H T, Kim S S, Han J A, Yoo Y M, Kim K M, Lee G H, Yun H Y, Green A, Li J, et al. J Neurosci. 1999;19:6740–6747. doi: 10.1523/JNEUROSCI.19-16-06740.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Dypbukt J M, Ankarcrona M, Burkitt M, Sjoholm A, Strom K, Orrenius S, Nicotera P. J Biol Chem. 1994;269:30553–30560. [PubMed] [Google Scholar]
- 35.Freire-de-Lima C G, Nascimento D O, Soares M B, Bozza P T, Castro-Faria-Neto H C, de Mello F G, DosReis G A, Lopes M F. Nature (London) 2000;403:199–203. doi: 10.1038/35003208. [DOI] [PubMed] [Google Scholar]