

Abstract
Satellite tobacco mosaic virus (STMV) is an icosahedral T=1 single-stranded RNA virus with a genome containing 1058 nucleotides. X-ray crystallography revealed a structure containing 30 double-helical RNA segments, with each helix having nine base pairs and an unpaired nucleotide at the 3’ end of each strand. Based on this structure, Larson and McPherson proposed a model of 30 hairpin-loop elements occupying the edges of the icosahedron and connected by single-stranded regions. More recently, Schroeder et al. have combined the results of chemical probing with a novel helix searching algorithm to propose a specific secondary structure for the STMV genome, compatible with the Larson-McPherson model. Here we report an all-atom model of STMV, using the complete protein and RNA sequences and the Schroeder RNA secondary structure. As far as we know, this is the first all-atom model for the complete structure of any virus (100% of the atoms) using the natural genomic sequence.
Keywords: Virus structure, Molecular modeling, Viral assembly, RNA hairpin loop
1. Introduction
Satellite tobacco mosaic virus (STMV) is a T=1 icosahedral virus with a diameter of 17 nm (Mirkov et al., 1989). The genome of STMV is composed of a single-stranded RNA that has 1058 nucleotides. The 1.8 Å X-ray structure of this virus identified 30 RNA double helices, one on each edge of the icosahedron (Larson et al., 1993a; Larson et al., 1993b) (Figure 1). Each helix is composed of 9 base pairs, plus an unpaired nucleotide at the 3’ end of each strand. Altogether the visible nucleotides account for 57% of the entire genome (Larson et al., 1998). The RNA components closer to the center of the virus (and the linkers connecting these to the crystallographically visible RNA) are missing from the crystal structure, because this part of the RNA does not have icosahedral symmetry; when the viral particle crystallizes, it can enter the lattice in any of 60 orientations, and the RNA density is averaged among these orientations. RNA sequence information is also absent in the crystal structure for the same reason.
Figure 1.

The crystal structure of STMV reveals thirty RNA double helices, each centered on a crystallographic two-fold axis.
Larson and McPherson proposed that the RNA is organized in a series of 30 hairpin loops that are connected by single-stranded linkers. They presented a model in which the 30 hairpins are situated on the 30 two-fold axes of the icosahedron and are connected by the shortest path, suggesting both a structure for the mature virus and an efficient pathway for viral assembly; they then built a three-dimensional model with 30 identical hairpin loops arranged along the proposed path (Larson and McPherson, 2001). Since all RNA double helices appear identical in the icosahedrally averaged crystal structure, this model did not use the actual RNA sequence. Instead, each hairpin loop was composed of a stem of 9 A-U base pairs and a loop of 9 nucleotides. The stability of this model was subsequently demonstrated in molecular dynamics (MD) simulations (Freddolino et al., 2006). Recent atomic force microscopic images of STMV RNA shortly after release from the capsid are consistent with this linear arrangement of structural domains (Kuznetsov et al., 2010).
There is also an all-atom model for a larger icosahedral RNA virus, Pariacoto Virus (PaV) (T=3; 4322 nucleotides) (Devkota et al., 2009). This model also used an artificial sequence designed to match secondary structure constraints derived from the previously published crystal structure (Tang et al., 2001).
Neither the model for STMV (Larson and McPherson, 2001) nor the PaV model (Devkota et al., 2009) contains the actual genomic sequence, so neither model examined the possible three-dimensional organization of secondary structures that might be formed by a naturally existing viral genome. It is now possible to do so. Schroeder et al. recently proposed a secondary structure for STMV RNA, based on a combination of chemical probing and helix searching algorithms (Schroeder et al., 2011). Those authors emphasized that there are many possible secondary structures consistent with the Larson/McPherson hypothesis that the STMV genome contains 30 local hairpin loops connected by single-stranded linkers. They examined all hairpins that are consistent with this hypothesis and the chemical probing data, describing their final secondary structure model (Figure 2, Figure S1) as "the best representative structure of the ensemble" of possible structures (Schroeder et al., 2011).
Figure 2.

Secondary structure of STMV RNA determined by Schroeder et al. (Schroeder et al., 2011). Figure S1 (Supplementary Material) contains an expanded version of this structure that permits visualization of the sequence.
In this paper, we report an all-atom three-dimensional model of STMV. We demonstrate that STMV RNA adopting the proposed secondary structure (Schroeder et al., 2011) is able to cover all the edges of the icosahedron as originally proposed (Larson and McPherson, 2001). Further, we compare the predicted electron density map for the model with maps derived from the crystal structure, finding that the model’s RNA tertiary structure shares a strong similarity with that of the crystal structure. We also discuss the implications of our results for the process of viral assembly.
2. Materials and Methods
2.1. Building the all-atom model
The secondary structure in Figure 2 can be organized with hairpin loops around the icosahedron so that each edge is covered by one hairpin loop (Figure 3). We arranged the hairpin loops on neighboring edges according to the lengths of the connections between them: for linkers of less than 8 nucleotides, the neighboring hairpins must adopt the tail-to-tail conformation, while for linkers longer than 13 nucleotides, head-to-tail or head-to-head conformations can be used as necessary. We located the long connection between hairpin loop 1 and hairpin loop 2 in the interior of the virus, rather than closer to the globular domains of the capsid proteins. This arrangement is necessary, because this connection is too large to be accommodated right under the capsid. We emphasize that the arrangement shown in Figure 3 is just one of a very large number of possibilities.
Figure 3.

Implementation of the Larson-McPherson scheme (Larson and McPherson, 2001) for mapping the hairpin loops (red) of the secondary structure in Figure 2 onto the icosahedron, with single-stranded connections colored in blue. Each triangle represents one face of the icosahedron.
We generated all-atom models for each hairpin loop in Figure 2, using MC-sym (Parisien and Major, 2008). Each of these was minimized by NAMD (Phillips et al., 2005) (CHARMM force field, conjugate gradient, 300 steps), and then superposed onto the corresponding RNA stem in the crystal structure (PDB ID 1A34). The superposition was done by matching the positions of corresponding phosphorus atoms, using VMD (Humphrey et al., 1996). Single-stranded connections between successive stems were built with the conformation of one strand of an A-form RNA helix; each of these was linked to the appropriate hairpin loops using Sybyl-X (Tripos, St. Louis, Missouri). The one exception to this procedure was the very long connection between hairpin 1 and hairpin 2 (which reaches toward the center of the virus), for which a coarse-grained three-dimensional model was generated using NAST (Jonikas et al., 2009b), and then converted to all-atom structure by the program C2A (Jonikas et al., 2009a).
After completion of the entire RNA model, the structure was minimized using NAMD, as described in the previous paragraph. The positions of P and C1’ atoms in the double-helical stems of the crystal structure were used as restraints during minimization, assuring that the stems of the hairpin loops stayed aligned with those in the crystal structure. The restraints were set so that stronger restraining forces were assigned to positions with lower B-factors:
F = - (2 [ 8 − int(B/30) ] ) • x,
where x is the deviation of the atom from its ideal (crystallographic) position, measured in Ångstroms, and B is the B-factor from the crystal structure, measured in Å2; the quantity in square brackets is the force constant, measured in kcal/(mol•Å2). The structure was then checked using the ADIT server (RCSB Protein Data Bank, http://deposit.rcsb.org/adit/) to identify any chirality and ring penetration problems. These were fixed manually, and the structure was re-minimized. After several cycles of manipulation and minimization, the structure was free of any stereochemical violations. This structure was minimized to convergence (~13,000 steps.)
The coordinates of the protein subunit that comprises the asymmetric unit were obtained from the crystal structure (PDB ID 1A34). This subunit contains 147 residues, and it is missing the first 12 residues of the N-terminus. To add the missing residues, we used Sybyl-X (Tripos, St. Louis, Missouri) to build a tail of 12 residues following the actual peptide sequence, attaching the C-terminus of the tail to the N-terminus of that part of the protein seen in the crystal structure. The whole capsid was generated from this 159-residue subunit using Oligomer Generator in VIPERdb (http://viperdb.scripps.edu/oligomer_multi.php). The protein capsid and RNA were then combined.
Since the protein and RNA models were developed separately, joining them together produced some inevitable steric overlaps, particularly between the RNA and the protein tails. We used the ADIT server as described above to identify those. We manually adjusted the positions of the appropriate tail residues to eliminate ring penetrations and serious RNA-protein overlaps. We then minimized the complete structure with some atoms restrained in a similar way as described earlier for the RNA alone (CHARMM force field, conjugate gradient, convergence in ~5000 steps). The restrained atoms included the P and C1’ atoms of the RNA, and the C, N and CA atoms of the protein residues obtained from the crystal structure (not those of amino acids in the protein tails). The tails were allowed free movement, since their densities were missing in the crystal structure. ADIT analysis of the final model indicated that it is free of any serious steric conflicts or stereochemical violations, and that the chirality is correct for all chiral centers.
The final model is shown in Figure 4. It contains every atom in all the protein subunits and the entire RNA genome. The model is free of any chirality problems and steric conflicts.
Figure 4.

Final model of STMV (gray: protein; magenta: RNA) A. Entire RNA with the protein capsid cut in half, to reveal the internal structure. B. A 50 Å section through the center of the final model.
2.2. Alternative Models
The secondary structure in Figure 2 has much less base pairing than would be predicted from thermodynamic considerations. We therefore generated a number of secondary structures based solely on thermodynamic considerations, using UNAFold (Markham and Zuker, 2008), to see if a structure with substantially lower free energy could be accommodated into the virus. Following the original proposal of Larson and McPherson (2001) and the lead of Schroeder et al. (2011), we did not permit long-range base pairs. We examined a series of folds in which the maximum base pairing distance was limited to 20, 30, 40, 50, or 60 nucleotides. From this ensemble of structures, we selected thirty stem-loops, each of which appeared in several of these folding exercises. A typical structure is shown in Figure S2.
This model was instantiated in three dimensions following the procedures described above.
2.3. Evaluation of the Models
We compared the two all-atom models described above with the crystal structure by correlation of electron density maps. We did the same evaluation of a model previously published by Larson and McPherson (Larson and McPherson, 2001) which was the basis of molecular dynamics (MD) simulations by Freddolino et al. (Freddolino et al., 2006). In the remainder of this section we describe the procedure in more detail.
All these models were constructed with the crystal structure as the foundation, so they can be placed in a unit cell of P1 symmetry with cell parameters a=174.27, b=191.77, c=202.50 Å and α=β=γ=90° to match the cell of the crystal structure. For the constructed models, occupancies and B-factors for all atoms were assigned a value of 1.0 and 30 Å2, respectively. In the crystal structure, the RNA lies on both icosahedral and crystallographic two-fold axes. This makes it necessary to set the occupancies of the RNA atoms to 0.5 in the crystallographic model. The two-fold symmetry then creates a fully occupied site for every residue in each of the 30 RNA double helices. It is clear in the original crystal structure that the RNA segments at each of the two-fold axes are fully occupied, because the three central base pairs have B-factors that are only slightly elevated over the average of the protein B-factors, despite the inherent RNA disordering due to the variations in sequence between the different RNA duplexes.
Structure factors, Fc, were calculated for each model. The structure factors were then used to calculate Fc electron density maps that were subsequently 60-fold averaged using the icosahedral symmetry operators to simulate the disorder that occurs by virtue of the fact that the virion can incorporate into the lattice in 60 different orientations. We compared the map for each model with two 2Fo-Fc maps based on a new refinement of the model against the original data deposited in the Protein Data Bank (PDB ID 1A34) (Larson et al., 1998). These maps represent our best representation of the “true” electron distribution for the RNA. A 2Fo-Fc map was calculated from the structure factors obtained at the end of the refinement. A mask that covered the whole interior of the capsid was overlaid on the map, and all points in the map that were outside the mask were set to zero. The resulting map represents only the density inside the capsid, which is mainly RNA density. This map was averaged in the same manner as the model maps. Correlation coefficients were calculated between these two maps as a measure of agreement between the “true” RNA electron distribution and the model derived maps (both non-averaged and averaged) (see Table 1).
Table.1.
Correlation coefficients between the crystallographic electron density and the predicted electron densities from the RNA models (2Fo-Fc maps), with areas where the correlation coefficient exceeds 0.5 highlighted in grey.
| X-ray RNA model | Previous RNA model* | Current RNA model | ||||
|---|---|---|---|---|---|---|
| unaveraged | averaged | unaveraged | averaged | unaveraged | averaged | |
| Full model |
0.231 | 0.391 | 0.299 | 0.524 | ||
| Helices | 0.521 | 0.611 | 0.288 | 0.443 | 0.388 | 0.557 |
The previous RNA model is that from Larson and McPherson (2001), which was examined in molecular dynamics simulations byFreddolino et al.(2006).
The computer program CNS (Brunger et al., 1998) was used to calculate structure factors from the models, to calculate the electron density maps, and to perform map averaging and mask operations. The crystallographic refinement was performed with the program Refmac5 (Murshudov et al., 2011). The programs MAPMAN (Thimm et al., 2004) and MAMA (Kleywegt and Jones, 1999) were used to calculate correlation coefficients and create masks, respectively.
3. Results
The final model (Figure 4) contains every single residue for both the RNA and protein components of the virus, and it compares very favorably with the structural model obtained by X-ray crystallographic methods. The model is available in PDB format from http://harvey.gatech.edu/, or from the authors.
There is excellent agreement between our model and the crystal structure. To begin with, the RMSD between the phosphorus atoms of the model and those of the crystal structure is only 1.21 Å (Figure 5). This is excellent agreement for such a large structure. Second, the correlation coefficient of our model with the 2Fo-Fc map from the crystal structure (0.557 with icosahedral averaging) is 91% of the correlation coefficient (0.611) for the crystallographic model itself (PDB ID 1A34). The excellent agreement is clearly seen when the model is superposed on the 2Fo-Fc electron density map (Figure 6). Moreover, our model has a significantly higher correlation coefficient than does the model previously published by Larson and McPherson (Larson and McPherson, 2001) which had been examined in MD simulations (Freddolino et al., 2006). This is true both for the full RNA model and for the helical regions alone (Table 1).
Figure 5.

Hairpin loops in the model shown together with the helices in the crystal structure (magenta: model; blue: crystal structure). A. All 30 hairpins (loops are not shown for visual clarity). B. View along one of the 5-fold axes. C. view along one of the 3-fold axes.
Figure 6.

Two views of one helix from the current model, superimposed on the crystallographic 2Fo-Fc map. The model has thirty unique double helices, and the figure shows a typical level of agreement between the model and the experimental data.
As pointed out above, the Schroeder secondary structure used in our model (Figure 2) has much less base pairing than a similar structure based on thermodynamic predictions (Figure S2.) This is a consequence of Schroeder's requirement that the two sides of all stems must be of equal length. Although it has a much lower free energy than the Schroeder structure (-302 kcal/mol vs. - 160 kcal/mol), the thermodynamically favored model presents serious problems for three-dimensional modeling. The stem-loops in Figure S2 are much more varied in length than those in Figure 2, and there are many more asymmetric bulges and bent stems than in the Schroeder structure. An all-atom model using this alternative secondary structure gives a correlation coefficient with the crystallographic electron density of only 0.12, vs. 0.56 for the model using the Schroeder secondary structure.
Twelve residues of the positively charged tails at the amino terminus of the capsid proteins are not visible in the crystal structure, presumably because the 60 tails have a variety of conformations; they are statically disordered. There is not sufficient experimental information to allow us to predict those conformations with any accuracy, but the tails in our model do penetrate through the array of RNA double helices just under the capsid, reaching toward the center of the virus (Figure 7). In building our earlier model of PaV (Devkota et al., 2009), we forcibly stretched the tails toward the center of the virus. We made no such effort in the STMV model reported here, nor did we attempt to position the tails so their positive charges would maximize neutralization of the RNA charge. As a consequence, the results shown in Figure 7 represent a first-order approximation to the tails' positions, not a set of specific predictions.
Figure 7.
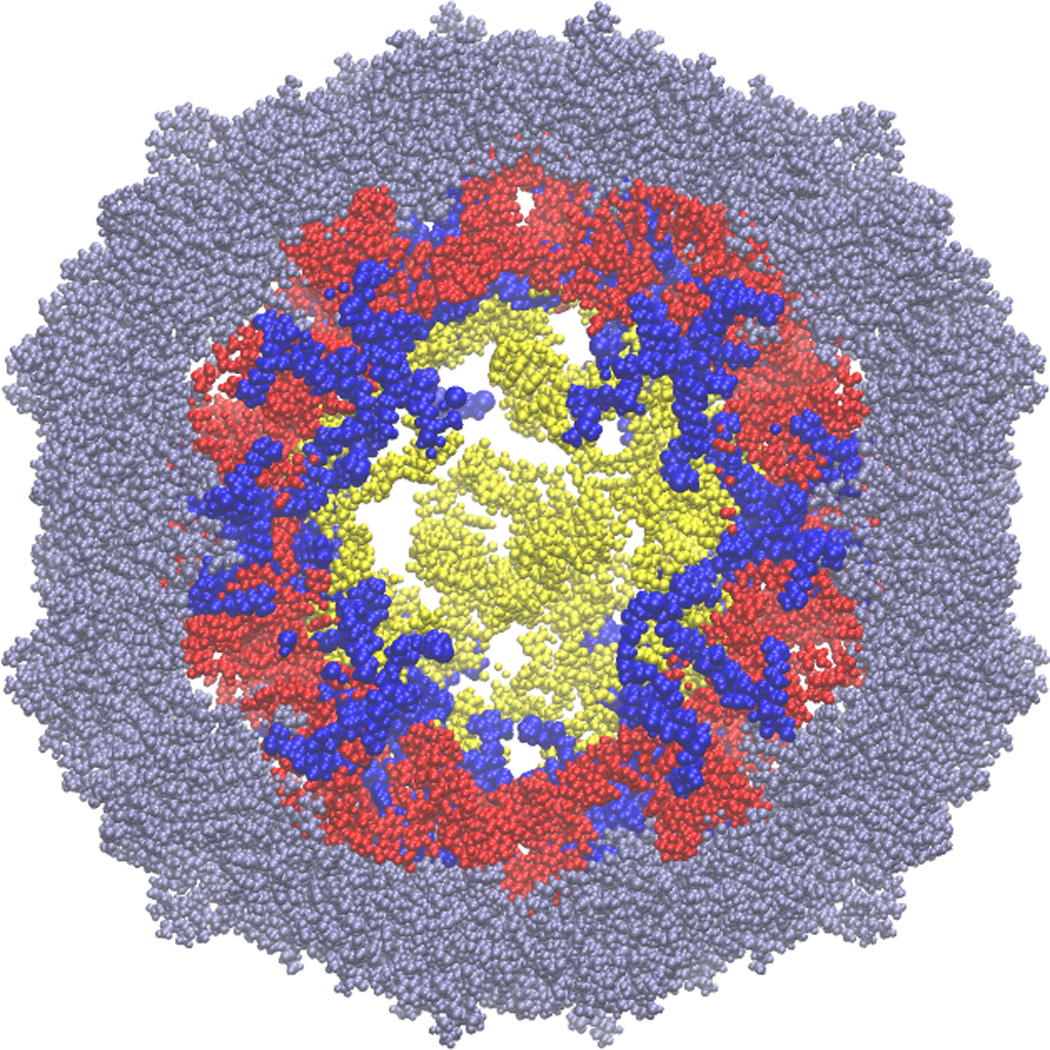
The 12 residues of the N-terminal protein tails (blue) pass through those double-helical regions of the model RNA (red) that correspond to the double helices seen in the crystal structure. The figure shows a slice (section) through the center of the virus; the RNA section is 50Å thick, while the protein section is 60Å thick, to facilitate visualization of the tails. The crystallographically observed regions of the capsid proteins are grey, and those parts of the RNA model that do not correspond to regions in the crystal structure are yellow. For scale, the virus is about 165Å in diameter. We emphasize that there is no experimental information on how deeply the protein tails reach into the center of the virus, and that we have not attempted to stretch the tails. Fully extended, 12 amino acids could just reach the center of the virus.
4. Discussion
As far as we are aware, this is the first all-atom model for any virus that is based on the actual sequence of the genome. The model demonstrates that the secondary structure proposed by Schroeder et al. (Schroeder et al., 2011) can be realized in three dimensions, providing support both for that structure and for the original suggestion that the genomic RNA is organized in a series of 30 stem-loops (Larson and McPherson, 2001). The very high correlation between the model and crystallographic electron density maps argues that we have captured the essential features of the actual structure.
We also examined an alternative secondary structure based on thermodynamic predictions. It has a lower folding free energy, but it also has much less regular stem-loop structures than does the Schroeder model, so it has a much lower correlation with the experimental electron density (0.12 vs. 0.56). This result emphasizes that RNA folds with asymmetric stems are incompatible with the crystallographic data. It also suggests that the secondary structure of the packaged RNA may be different from that of the free RNA, and that the capsid could play a substantial role in directing RNA secondary structure.
The secondary structure in our model does not include a possible tRNA-like structure at the 3’-end of the genome. Such structures have been found at the 3’-end of the genomes in several RNA viruses, including tobacco mosaic virus and turnip yellow mosaic virus (Dreher, 2009). Aminoacylation experiments and sequence analysis suggest that the 3’-terminal 188 nucleotides of the STMV RNA also fold into a tRNA-like structure (Felden et al., 1994). Those authors argued that this part of the structure involves multiple pseudoknots and is terminated with an acceptor stem. However, the most energetically stable secondary structure with pseudoknots, predicted by pknotsRG (Reeder et al., 2007), is completely different from this structure. Furthermore, this proposed set of pseudoknots is not consistent with the experimental data (Schroeder et al., 2011): chemical probing showed 25 nucleotides to be single-stranded within this 188 nt region, but in the tRNA-like model, 7 of those 25 nucleotides are basepaired. This suggests that, if a tRNA-like structure exists in the 3’-region of the STMV RNA, it must undergo re-folding during packaging. There is evidence that the RNA secondary structure inside a mature virus can be different from the unpackaged RNA (Rodriguez-Alvarado and Roossinck, 1997).
This model raises three challenges for future research.
First is the issue of structural heterogeneity. It is not known if all STMV genomes in the mature virus have essentially identical secondary structures. Schroeder et al. (2011) emphasized that the secondary structure model in Figure 2 is only one of many possibilities. They suggested that a multiplicity of possible secondary structures might be advantageous to the virus, from an evolutionary standpoint, and that a single RNA conformation "would be a brittle solution to the RNA folding problem". All of the secondary structures in the Schroeder ensemble are similar in the sizes and symmetries of the stem-loops, so any of these should satisfy the constraints from the crystal structure equally well. Even if we assume that all viral RNAs have secondary structures identical to that shown in Figure 2, the organization shown in Figure 3 is not unique, so the structure might vary from virion to virion.
The second challenge is to understand the structural, thermodynamic and kinetic aspects of viral assembly. We have previously proposed an assembly mechanism for small, icosahedral RNA viruses like STMV (Devkota et al., 2009; Harvey et al., 2009). We suggested that the positively charge tails of the capsid proteins bind nonspecifically to the RNA genome. When a sufficient fraction of the negative charge on the RNA has been neutralized, the complex of RNA and protein tails collapses into a dense state in a process reminiscent of DNA condensation by polyvalent cations. This squeezes the proteins' globular domains into a shell surrounding the dense collapsed structure, where their concentration becomes sufficient to promote the specific protein-protein interactions that are required for formation of the mature capsid.
In addition, the structural heterogeneity mentioned above could be advantageous: many different assembly pathways would be available for the formation of the mature virus, if they do not all have to reach the same end point. If this is true, then viral assembly could be fundamentally different from many other biological processes, where unique structures, interactions and processes are required to generate specificity.
The third challenge is to determine how the STMV capsid protein recognizes the viral RNA. In some viruses, recognition of the viral genome requires specific sequences in the RNA (Pollack and Ganem, 1993), or the capsid protein (Lago et al., 1998; Schneemann and Marshall, 1998), or both (Aldovini and Young, 1990). In other viruses, the capsid proteins are promiscuous and will package a variety of non-viral RNAs, giving virus-like particles that resemble the mature virus (Tihova et al., 2004). There are no known packaging signals in the STMV RNA, and it is not known if the capsid proteins are capable of packaging RNAs other than the STMV genome.
These issues merit experimental attack, and we are developing computational models for simulating the assembly process and examining these questions.
5. Conclusions
The final model of STMV contains every single atom in the virus, including every residue of all the protein subunits, and the entire genome of 1058 nt. It is available as indicated above. The RMSD between the phosphorus atoms of the model and those of the crystal structure is only 1.21 Å. Our model is based on the crystal structure, and it extrapolates beyond the crystallographic data, including, as it does, those parts of the RNA genome that are not seen in the crystal structure. It satisfies all standards of RNA stereochemistry and has a better correlation to the X-ray based maps than the earlier model of Larson and McPherson, built from an artificial sequence. The model shows how the genomic sequence, when folded into Schroeder’s secondary structure, is able to form the mature icosahedral structure. This supports earlier arguments that the protein capsid plays an important part in RNA refolding, and that the resulting RNA structure facilitates viral assembly (Larson and McPherson, 2001).
We emphasize that this is only one of many possible three-dimensional arrangements of the RNA that satisfy all of the available data. Following the argument of Schroeder et al. (2011) that heterogeneity in secondary structures might be evolutionarily advantageous to the virus, we suggest that heterogeneity in both secondary structure and three-dimensional structure might facilitate virus assembly. If a variety of RNA conformations can be packaged, this would reduce the entropic barrier to the assembly process. The model presented here is representative of the possible structures.
Supplementary Material
Acknowledgments
Supported by grants from the US National Institutes of Health (GM70785 to SCH, GM83621 to CEH and GM80412 to AM). We are indebted to the referees for their thorough and thoughtful reviews, which led to several significant improvements in the final version of this paper.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Aldovini A, Young RA. Mutations of RNA and protein sequences involved in human immunodeficiency virus type 1 packaging result in production of noninfectious virus. J Virol. 1990;64:1920–1926. doi: 10.1128/jvi.64.5.1920-1926.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunger AT, Adams PD, Clore GM, DeLano WL, Gros P, Grosse-Kunstleve RW, Jiang JS, Kuszewski J, Nilges M, Pannu NS, Read RJ, Rice LM, Simonson T, Warren GL. Crystallography & NMR system: A new software suite for macromolecular structure determination. Acta Crystallographica Section D-Biological Crystallography. 1998;54:905–921. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- Devkota B, Petrov AS, Lemieux S, Boz MB, Tang L, Schneemann A, Johnson JE, Harvey SC. Structural and electrostatic characterization of Pariacoto virus: Implications for viral assembly. Biopolymers. 2009;91:530–538. doi: 10.1002/bip.21168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dreher TW. Role of tRNA-like structures in controlling plant virus replication. Virus Res. 2009;139:217–229. doi: 10.1016/j.virusres.2008.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Felden B, Florentz C, McPherson A, Giege R. A histidine accepting tRNA- like fold at the 3'-end of satellite tobacco mosaic virus RNA. Nucleic Acids Res. 1994;22:2882–2886. doi: 10.1093/nar/22.15.2882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freddolino PL, Arkhipov AS, Larson SB, McPherson A, Schulten K. Molecular dynamics simulations of the complete satellite tobacco mosaic virus. Structure. 2006;14:437–449. doi: 10.1016/j.str.2005.11.014. [DOI] [PubMed] [Google Scholar]
- Harvey SC, Petrov AS, Devkota B, Boz MB. Viral assembly: a molecular modeling perspective. Phys Chem Chem Phys. 2009;11:10553–10564. doi: 10.1039/b912884k. [DOI] [PubMed] [Google Scholar]
- Humphrey W, Dalke A, Schulten K. VMD: Visual Molecular Dynamics. J. Mol. Graphics. 1996;14:33–38. doi: 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
- Jonikas MA, Radmer RJ, Altman RB. Knowledge-based instantiation of full atomic detail into coarse-grain RNA 3D structural models. Bioinformatics. 2009a;25:3259–3266. doi: 10.1093/bioinformatics/btp576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jonikas MA, Radmer RJ, Laederach A, Das R, Pearlman S, Herschlag D, Altman RB. Coarse-grained modeling of large RNA molecules with knowledge-based potentials and structural filters. RNA. 2009b;15:189–199. doi: 10.1261/rna.1270809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleywegt GJ, Jones TA. Software for handling macromolecular envelopes. Acta Crystallographica Section D-Biological Crystallography. 1999;55:941–944. doi: 10.1107/s0907444999001031. [DOI] [PubMed] [Google Scholar]
- Kuznetsov YG, Dowell JJ, Gavira JA, Ng JD, McPherson A. Biophysical and atomic force microscopy characterization of the RNA from satellite tobacco mosaic virus. Nucleic Acids Res. 2010;38:8284–8294. doi: 10.1093/nar/gkq662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lago H, Fonseca SA, Murray JB, Stonehouse NJ, Stockley PG. Dissecting the key recognition features of the MS2 bacteriophage translational repression complex. Nucleic Acids Res. 1998;26:1337–1344. doi: 10.1093/nar/26.5.1337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larson SB, McPherson A. Satellite tobacco mosaic virus RNA: structure and implications for assembly. Current Opinion in Structural Biology. 2001;11:59–65. doi: 10.1016/s0959-440x(00)00166-4. [DOI] [PubMed] [Google Scholar]
- Larson SB, Day J, Greenwood A, McPherson A. Refined structure of satellite tobacco mosaic virus at 1.8Å resolution. J Mol Biol. 1998;277:37–59. doi: 10.1006/jmbi.1997.1570. [DOI] [PubMed] [Google Scholar]
- Larson SB, Koszelak S, Day J, Greenwood A, Dodds JA, McPherson A. Double-helical RNA in satellite tobacco mosaic virus. Nature. 1993a;361:179–182. doi: 10.1038/361179a0. [DOI] [PubMed] [Google Scholar]
- Larson SB, Koszelak S, Day J, Greenwood A, Dodds JA, McPherson A. Three-dimensional structure of satellite tobacco mosaic virus at 2.9Å resolution. J Mol Biol. 1993b;231:375–391. doi: 10.1006/jmbi.1993.1289. [DOI] [PubMed] [Google Scholar]
- Markham NR, Zuker M. UNAFold: software for nucleic acid folding and hybridization. Methods Mol Biol. 2008;453:3–31. doi: 10.1007/978-1-60327-429-6_1. [DOI] [PubMed] [Google Scholar]
- Mirkov TE, Mathews DM, Du Plessis DH, Dodds JA. Nucleotide sequence and translation of satellite tobacco mosaic virus RNA. Virology. 1989;170:139–146. doi: 10.1016/0042-6822(89)90361-9. [DOI] [PubMed] [Google Scholar]
- Murshudov GN, Skubak P, Lebedev AA, Pannu NS, Steiner RA, Nicholls RA, Winn MD, Long F, Vagin AA. REFMAC5 for the refinement of macromolecular crystal structures. Acta Crystallographica Section D- Biological Crystallography. 2011;67:355–367. doi: 10.1107/S0907444911001314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parisien M, Major F. The MC-Fold and MC-Sym pipeline infers RNA structure from sequence data. Nature. 2008;452:51–55. doi: 10.1038/nature06684. [DOI] [PubMed] [Google Scholar]
- Phillips JC, Braun R, Wang W, Gumbart J, Tajkhorshid E, Villa E, Chipot C, Skeel RD, Kale L, Schulten K. Scalable molecular dynamics with NAMD. J Comput Chem. 2005;26:1781–1802. doi: 10.1002/jcc.20289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pollack JR, Ganem D. An RNA stem-loop structure directs hepatitis B virus genomic RNA encapsidation. J Virol. 1993;67:3254–3263. doi: 10.1128/jvi.67.6.3254-3263.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reeder J, Steffen P, Giegerich R. pknotsRG: RNA pseudoknot folding including near-optimal structures and sliding windows. Nucleic Acids Res. 2007;35:W320–W324. doi: 10.1093/nar/gkm258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez-Alvarado G, Roossinck MJ. Structural analysis of a necrogenic strain of cucumber mosaic cucumovirus satellite RNA in planta. Virology. 1997;236:155–166. doi: 10.1006/viro.1997.8731. [DOI] [PubMed] [Google Scholar]
- Schneemann A, Marshall D. Specific encapsidation of nodavirus RNAs is mediated through the C terminus of capsid precursor protein alpha. J Virol. 1998;72:8738–8746. doi: 10.1128/jvi.72.11.8738-8746.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schroeder SJ, Stone JW, Bleckley S, Gibbons T, Mathews DM. Ensemble of secondary structures for encapsidated satellite tobacco mosaic virus RNA consistent with chemical probing and crystallography constraints. Biophys J. 2011;101:167–175. doi: 10.1016/j.bpj.2011.05.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang L, Johnson KN, Ball LA, Lin T, Yeager M, Johnson JE. The structure of pariacoto virus reveals a dodecahedral cage of duplex RNA. Nature Struct Biol. 2001;8:77–83. doi: 10.1038/83089. [DOI] [PubMed] [Google Scholar]
- Thimm O, Blasing O, Gibon Y, Nagel A, Meyer S, Kruger P, Selbig J, Muller LA, Rhee SY, Stitt M. MAPMAN: a user-driven tool to display genomics data sets onto diagrams of metabolic pathways and other biological processes. Plant Journal. 2004;37:914–939. doi: 10.1111/j.1365-313x.2004.02016.x. [DOI] [PubMed] [Google Scholar]
- Tihova M, Dryden KA, Le TV, Harvey SC, Johnson JE, Yeager M, Schneemann A. Nodavirus coat protein imposes dodecahedral RNA structure independent of nucleotide sequence and length. J Virol. 2004;78:2897–2905. doi: 10.1128/JVI.78.6.2897-2905.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.