

Abstract
Commitment to divide is one of the most crucial steps in the mammalian cell division cycle. It is critical for tissue and organismal homeostasis, and consequently is highly regulated. The vast majority of cancers evade proliferative control, further emphasizing the importance of the commitment step in cell cycle regulation. The Retinoblastoma (RB) tumor suppressor pathway regulates this decision-making step. Since being the subject of Knudson’s ‘two hit hypothesis’, there has been considerable interest in understanding pRB’s role in cancer. It is best known for repressing E2F dependent transcription of cell cycle genes. However, pRB’s role in controlling chromatin structure is expanding and bringing it into new regulatory paradigms. In this review we discuss pRB function through protein-protein interactions, at the level of transcriptional regulation of individual promoters and in organizing higher order chromatin domains.
Keywords: retinoblastoma, cell cycle, chromatin, transcription, senescence, E2F, differentiation, condensin
Introduction
The retinoblastoma protein (pRB) was the first tumor suppressor to be discovered, and it regulates the G1 to S phase transition at the beginning of the cell cycle.1 Deregulation of cell cycle control in cancer requires inactivation of this growth regulatory function.2 Not surprisingly, the RB1 gene and components of the pRB regulatory pathway are mutated or silenced in most human cancers.2,3 The RB protein is a member of the pocket protein family, which also includes similar proteins with overlapping functions: p107 and p130.4 However, the vast majority of tumor derived mutations identified in this family are found in the RB1 gene, suggesting a unique role for pRB among its siblings.4,5 For this reason, understanding the function of pRB is of considerable interest to cancer researchers.
pRB’s tumor suppressor property is generally attributed to its ability to repress transcription of cell cycle genes by binding to and inhibiting the E2F family of transcription factors.6-8 Upon growth factor stimulation, pRB is inactivated through phosphorylation by cyclin-dependent kinases (CDKs) releasing the E2F proteins to activate transcription of cell cycle genes.1 Viral oncoproteins, like E1A, bind to pRB, preventing it from interacting with E2Fs and thereby inducing cell proliferation.9,10 Similarly, in cancer cells, the pRB pathway is inactivated either by direct mutation of the RB1 gene, deregulation of CDKs or inactivation of cyclin-dependent kinase inhibitors such as p16INK4A. These mutational events serve to stably deregulate E2F transcription.2 In this model of pRB function, it is a local transcriptional repressor that regulates the expression of genes through direct interaction with the activation domain of E2F transcription factors. While this model provides a relatively simplistic and straight forward mechanistic basis for pRB function, pRB appears to be capable of exerting broader effects on transcriptional control and chromatin structure.
In addition to inhibiting E2Fs through direct interaction, pRB is also able to actively repress gene transcription mediated by neighboring transcription factors when recruited to promoters by E2Fs.11-13 These observations suggested that pRB can inhibit transcriptional activation throughout a gene’s promoter. In fact, pRB has been found to associate with a number of proteins that can regulate chromatin structure and transcription at E2F-responsive promoters. These findings have suggested that pRB is recruited to promoters by sequence-specific transcription factors such as E2Fs. In turn, pRB recruits co-repressors to these promoters, which can remodel chromatin in neighboring regions to silence transcription. Examples of co-repressors bound by pRB include histone deacetylases (HDAC1, HDAC2),14-16 histone demethylases (RBP2),17 DNA methyl transferases (DNMT1),18 helicases (Brg1, Brm),19,20 histone methyl transferases (Suv39h1, RIZ and Suv4–20h1/h2)21-23 and histone binding proteins like HP1.21,24 The ability of pRB to bring these chromatin-regulating activities to E2F-responsive promoters creates the opportunity to influence a broader genomic region than just the DNA footprint of the E2F transcription factor.
Beyond these two levels of regulatory control exerted by pRB at promoters, recent evidence suggests a genome-wide role for pRB in the regulation of large heterochromatin domains such as pericentric heterochromatin, telomeres and senescence-associated heterochromatic foci. The RB protein has been shown to interact with Suv4–20h1/h2 histone methyltransferases that regulate the trimethylation of histone H4 lysine 20 (H4K20) at pericentric heterochromatin.22 Fibroblasts that lack all three pRB family proteins or a knock in mutation in just pRB show a decrease in tri-methylation of H4K20 at this heterochromatin domain.22,25 Conditional knockout of pRB in fibroblasts also results in a similar reduction in H4K20 trimethylation at pericentric heterochromatin.26 Tri-methylation of H4K20 at telomeres is also found to be markedly reduced in cells lacking all three pRB family proteins.22 This could be one of the reasons for the elongated telomeres seen in these cells.27 In a similar manner, pRB has been shown to be important for heterochromatin assembly during cellular senescence, where it is required for the formation of senescence-associated heterochromatic foci (SAHF).28,29 In addition, pRB has also been implicated in regulating chromosome condensation during mitosis.30 The condensin II complex and pRB physically interact, and this is required for proper chromosome condensation during mitosis.31 Lack of this interaction causes defects in condensation that lead to lagging anaphase chromosomes and segregation defects. Condensation of mitotic chromosomes and SAHF formation represent chromatin organization at the broadest level, as they involve the compaction of entire chromosomes. Importantly, pRB’s role in chromosome condensation in mitosis is part of what makes it a tumor suppressor.31
Similar to this introduction, the focus of this review will be to highlight how our understanding of pRB function has evolved over time from being a local transcriptional repressor that relied on direct protein interactions at discreet genetic loci to a genome-wide regulator of chromatin structure. In addition to highlighting recent findings that have contributed to this evolution in thinking, we will also review the most recent findings on pRB function in senescence. We will focus on this paradigm of growth arrest, because it is a tumor-suppressive mechanism, and because it best exemplifies how pRB coordinates these three levels of regulation in a single cell cycle arrest event.
Here: pRB Mediates Positive and Negative Regulation of Transcription Through Direct Interactions
A number of pRB binding proteins include sequence-specific transcription factors that regulate the expression of genes involved in diverse physiological processes, including cell proliferation, differentiation and cell death. Depending on the transcription factor that is bound, and the physiological context, pRB has been reported to act both as a transcriptional activator and a repressor. This section will focus on current data that relates direct interactions between pRB and transcription factors with these two regulatory outputs.
Inhibition of E2F transcription by pRB binding
Analysis of pRB’s distribution across the genome in proliferating and senescent fibroblasts using chromatin immunoprecipitation and sequence analysis (ChIP-Seq) determined that the most abundant transcription factor binding sequence present in regions bound by pRB was E2F.29 This, along with considerable previous data, has placed pRB regulation of E2F-dependent transcription at the center of pRB’s function in cell cycle control and tumor suppression. The E2F family proteins that bind to pRB include E2Fs 1–4, and these proteins must heterodimerize with a DP family partner to bind DNA.6 The pRB binding domain of the E2Fs is their transcriptional activation domain.32 Thus a simple model that explains pRB’s E2F inhibition and transcriptional repressor role is that pRB blocks the transactivation function of E2Fs, thereby preventing the expression of the genes involved in DNA replication and cell cycle progression (see Fig. 1A for an illustration of this mechanism).
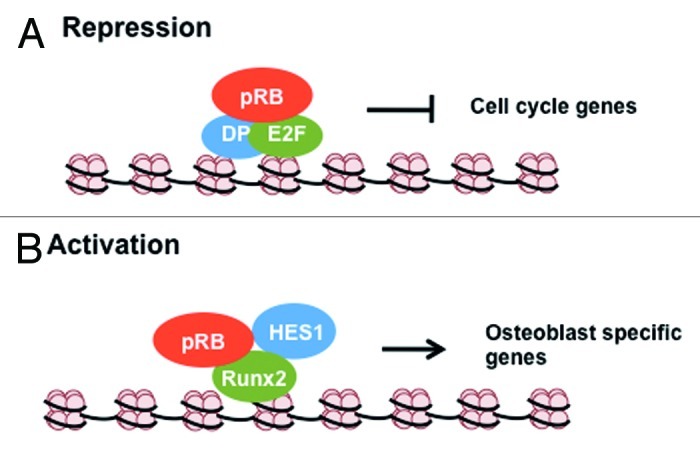
Figure 1. Transcriptional regulation by pRB through direct interactions. (A) During G0/G1, pRB inhibits cell cycle gene expression by directly blocking the trans-activating domain of E2F transcription factors. (B) During bone differentiation, pRB binds to osteoblast specific transcription factor Runx2 and co-operates with it and Hes1 in the activation of osteoblast specific genes like osteopontin and osteocalcin.
Further evidence for the importance of the pRB-E2F interaction comes from the study of viral oncogenes, such as adenovirus E1A.9,10 E1A binds and dissociates E2Fs from pRB in a two-step mechanism. First, E1A interacts with the pRB pocket domain using a peptide motif called LXCXE.33,34 E1A then uses its conserved region 1 (CR1) domain to compete for the contact point on pRB occupied by the E2F transcriptional activation domain.35 The disruption of pRB-E2F interactions by this mechanism is required for activation of transcription by E1A.36 The LXCXE and CR1 domains are also essential for the cellular transforming activity of E1A.37 Additional evidence for the importance of this direct interaction comes from Rb1−/− cells derived from knockout mouse embryos. They display increased expression of E2F responsive genes like cyclin E and p107 and increased DNA synthesis under serum-free conditions.38,39 Taken together, these examples of E2F control by pRB demonstrate negative regulation of transcription that is mediated by pRB when it stably binds the transactivation domain of E2Fs.
Transcription factors activated in the presence of pRB
A number of transcription factors involved in cellular differentiation have been proposed to be activated upon association with pRB. These include MyoD during myogenesis,40 CBFA1/Runx2 during osteogenesis,41 and C/EBP and NF-IL6 during adipogeneis.42,43 It is important to emphasize that our mechanistic knowledge of how pRB regulates these is minimal in comparison to the negative regulation of E2F above. As outlined above there is clear structural, functional and genetic data that reveals how direct interaction between pRB and E2F is utilized in transcriptional control. In contrast, the interactions between these transcription factors and pRB have often been controversial, and the nature of their interactions is not understood beyond simple domain mapping experiments. Given that the emphasis of this part of our review is on transcriptional regulation mediated by direct protein-protein interactions, it is important to note that some of pRB’s influence on transcriptional activation is likely to be indirect. For example, pRB can bind and block the activity of differentiation inhibitors like EID-1,44,45 ID246 and RBP2.17 As a result the negative regulation of a negative regulator of differentiation results in the augmentation of transcription by factors like MyoD. Thus, even seemingly direct interactions with pRB may have somewhat indirect mechanisms that assist them. An example that gives some of the best insight into direct protein-protein interactions between pRB and a transcriptional activator is Runx2/CBFA1/OSF-2 and this will be our focus for this section of the review.
During osteogenesis pRB interacts with the osteoblast specific transcription factor Runx2 to induce differentiation into an osteogenic fate41 (see Fig. 1B for a diagram of this regulatory paradigm). Using a series of in vitro experiments, pRB has been shown to interact with the C terminus of Runx2 using its pocket domain. In addition, ChIP experiments have demonstrated that pRB associates with Runx2 at the osteoblast-specific promoters osteocalcin and osteopontin.41 Beyond recruiting pRB to this promoter, Runx2 also recruits HES1 as part of this transcriptional activation complex.47 Thus, it is thought that differentiation signals lead Runx2 to increase expression of p27KIP1 to inhibit cyclin dependent kinase activity and arrest the cell cycle. This, in turn, leads to dephosphorylation of pRB and it and HES1 interact with Runx2 to activate transcription of late markers of bone differentiation. Recent analysis of a bone-specific knockout of pRB in mice indicates that when complexes of Runx2 and pRB can’t form, cells dedifferentiate from an osteoblast fate to a more primitive mesenchymal cell type that is capable of differentiating into both adipocytes or osteogenic lineages.48,49
As stated above pRB-Runx2 interactions are the most thoroughly studied example of direct interaction between pRB and a transcription factor leading to transcriptional activation. The precise mechanism of how pRB acts as an activator of transcription is still not known. It could involve alterations in its structure that allow it to switch between activating and repressing functions. Alternatively, it could also be recruiting other co-factors that can activate transcription and in this way functions as an adaptor when activating transcription. Regardless of our current state of knowledge, this is an important issue for resolution in the future, because it will determine exactly how direct pRB’s involvement is in transcriptional activation.
Activation of transcription by pRB-E2F1 complexes
Recent work on pRB regulation of E2F1 and apoptosis has suggested that they can form a complex together in response to DNA damage signaling that can activate transcription.50 This is quite surprising given the depth of understanding of how pRB acts as a repressor of E2F transcription described above. Understanding how DNA damage can alter pRB-E2F1 complexes from one of negative transcriptional regulation to one of activation is likely to yield a great deal of insight into other transcriptional activation paradigms involving pRB, such as with Runx2.
Ianari et al. have recently reported that pRB-E2F1 complexes can be found at pro-apoptotic promoters such as TAp73 in response to DNA double-strand breaks by ChIP analysis.50 Furthermore, ChIP re-ChIP experiments reveal that histone tail modifications that are indicative of transcriptional activation are found at the same promoters occupied by pRB-E2F1 complexes and that TAp73 expression is increased at the same time. How can this be reconciled with prior experiments that demonstrate pRB binding and inhibition of E2F1? One clue comes from the identification of a second binding site on pRB that is used exclusively by E2F1.51 In other words, E2F1 can bind to pRB through two different configurations: one that is described above, where E2F1’s transactivation domain is masked by the pRB pocket when these proteins contact one another, and a second configuration that is mediated outside the activation domain on E2F1. This exclusive contact between pRB and E2F1 is maintained even when pRB is hyperphosphorylated and unable to bind to other E2Fs.52 This is important, because Ianari et al. described their pRB-E2F1 activation complex as containing phosphorylated pRB.50 Thus, it is tempting to speculate that DNA damage signaling somehow supports or augments pRB’s unique interaction with E2F1, and that this complex is simply organized differently than other pRB-E2F complexes and this allows E2F1 to activate transcription in the presence of pRB.
An additional clue as to how a pRB-E2F1 complex can have a net positive effect on transcription comes from studies on posttranslational modifications of E2F1 following DNA damage. Double-stranded breaks induce the phosphorylation of E2F1 at Ser364.53 E2F1 modified at this site is found to be predominantly in complex with hyperphosphorylated pRB following DNA damage, implying that this is a pRB-E2F1 “specific” complex.54 In addition, this modified form of E2F1 is found at the TA-p73 promoter during transcriptional activation and induction of apoptosis in response to DNA damage.54 It is possible that phosphorylation of E2F1 at Ser364 changes the binding confirmation of pRB-E2F1 complexes, switching them from repressive to activating configurations. Alternatively, Ser364 phosphorylation may be part of a signal that alleviates pRB-dependent transcriptional repression, thus creating an overall positive effect on transcriptional activation of TAp73.
Again, our knowledge of this mechanism is limited, as even the models proposed above have involved piecing together data from different studies utilizing different cell types and methodologies. It is also possible that the activation function of this complex relies on the recruitment of co-factors that activate transcription through mechanisms that exert a broader influence on the promoter. In this way seemingly direct effects of pRB on gene expression may involve a broader promoter-wide influence.
There: pRB Facilitates Transcriptional Regulation Across the Promoter by Altering Chromatin Structure
Apart from direct inhibition of transactivation by E2Fs described above, pRB has also been shown to actively repress transcription that is stimulated by factors elsewhere in the same promoter.11-13 This observation suggested that pRB might recruit co-repressors to these promoters to exert a broader effect on transcription of these genes. More recently it has been shown that pRB can influence chromatin structure at these promoters, and this is a likely explanation for its promoter-wide effects. Many co-repressor proteins that can bind to pRB have enzymatic activity and can alter post-translational modifications on histone tails to influence transcription of the associated genes. This part of the review will focus on the pRB protein interactions that affect local chromatin structure and how these modifications regulate gene expression (see Fig. 2 for an illustration of this type of regulation).
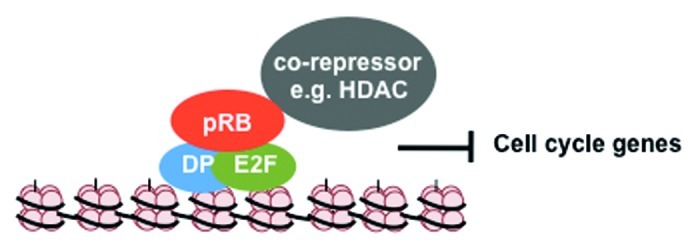
Figure 2. Transcriptional regulation by pRB through local chromatin changes. pRB can recruit co-repressors like HDAC to E2F responsive cell cycle gene promoters to deacetylate histones and compact chromatin, thereby stably repressing gene expression. In the case of cyclin E1 this impinges on a single histone octomer, but in other instances can target a broader region of the promoter.
As described in the introduction, there are a number of cellular proteins that pRB can recruit to promoters, where they act as co-repressors. While the list of histone deacetylases, histone demethylases, DNA methyl transferases, helicases and histone methyl transferases described in the introduction is quite impressive, the data that describes their respective roles in regulating chromatin structure and inhibiting transcription in conjunction with pRB is often limited to only a few papers. The examples of chromatin regulating mechanisms described below are selected because they offer the most complete picture of how pRB influences chromatin structure to provide promoter-wide regulation of transcription.
Histone deacetylases
The RB protein was shown to bind to histone deacetylases through immunoprecipitation and western blot experiments, as well as co-precipitation followed by catalytic assays.14-16 Since these initial reports, a number of research groups have characterized histone acetylation levels and HDAC occupancy at E2F target genes. A key transcriptional target at the G1 to S-phase transition for pRB and E2Fs is cyclin E1.55,56 A repressor module containing pRB and E2F transcription factors binds an E2F site at the transcriptional start site of this gene.57 This binding site coincides with a single nucleosome whose acetylation levels are highly dynamic.58 Multiple histones in this nucleosome are acetylated when cyclin E1 is transcribed and deacetylated when the promoter is silenced.58 The upstream regions of this promoter have relatively stable histone acetylation, suggesting that a single nucleosome is the target of deacetylase activity recruited by pRB.58 As confirmation that regulation of acetylation is the key to controlling transcription at cyclin E1, the histone deacetylase inhibitor trichostatin A can inhibit pRB-dependent repression of this promoter.59 Similar observations have been made that pRB family proteins utilize HDACs to repress transcription of a broad group of E2F-responsive genes.60-62
The organization and structure of these repressive complexes that contain HDACs, pRB and E2Fs remains unclear. Initially, investigators pointed out that HDAC1 and 2 contain a peptide sequence that resembles the LXCXE motif found in viral oncoproteins like E1A, TAg and E7.14 This suggested a direct interaction between pRB and HDAC in a manner that is similar to the viral oncoproteins. However, there are important differences between the viral CR2 region that contacts pRB and the LXCXE-like sequence in HDAC1 and 2,63 and peptide sequences from HDACs fail to bind pRB with similar affinity as the viral-derived equivalent.64 It was also not immediately appreciated that HDAC proteins are found in large multimeric protein complexes and, thus, their interaction with pRB may not be direct. Specifically, Sin3 and CtBP complexes can contain HDACs and are known to bind to pRB, suggesting that there are a number of possible intermediaries.65-67 Regardless of the mechanism of recruitment, the LXCXE binding site on pRB appears to be the site of interaction for HDAC containing complexes. Peptide competitors containing the viral LXCXE sequence can compete for pRB’s ability to recruit HDAC activity in precipitation assays.15,68 This suggests that regardless of how HDACs are brought to pRB, their associated complex contacts pRB in a manner that at least overlaps with the viral LXCXE contact point on pRB.
Taken together, these experiments demonstrate pRB regulation of transcription by recruiting HDAC activity to E2F promoters (Fig. 2). Surprisingly, many promoters have only had their histone acetylation levels investigated in the immediate vicinity of the transcriptional start site. Studies of cyclin E1 repression of transcription by pRB and HDAC suggest it may not involve modifications to histones beyond the immediate transcriptional start site. However, by regulating histone deacetylation, pRB can create changes in local chromatin structure that have relatively broad effects on transcriptional activation across the entire promoter.
DNA-dependent helicases
The retinoblastoma protein has also been found to associate with the core components (Brm and Brg1) of the evolutionarily conserved SWI/SNF chromatin remodeling apparatus.19,20 Brm and Brg1 each use ATPase activity to mobilize nucleosomes along the DNA strand, remove histones from DNA and/or promote the exchange of histone variants. This activity alters nucleosomal structure such that accessibility to binding sites by transcription factors and other transcriptional machinery is increased.69-71 This generally leads to gene activation;72 however, there are examples of how Brm and Brg1 can promote transcriptional repression.73 In addition, there is considerable evidence to support roles for Brm and Brg1 in the negative regulation of proliferation in cell culture in a manner that is cooperative with pRB.19,74-76 Furthermore, ectopic proliferation is detectable in adult Brm−/− mice,77 and cancer susceptibility of Brg1+/− mice further suggests that they contribute to growth regulation in vivo.78 Brm and Brg1 were originally reported to interact with pRB in an LXCXE dependent manner;19,20,74 however, this is controversial, as more detailed analysis of this interaction fails to confirm this conclusion.64 Detection of endogenous interactions between pRB and Brm or Brg1 is also limited to only a two reports.19,20 Regardless of the precise mechanism of interaction between pRB and Brm or Brg1, there is strong genetic interdependence in proliferative control that links them.
The mechanism of how these ATPases cooperate with pRB in repressing E2F-dependent transcription also remains open ended. Thus far, using transcriptional reporter assays, it has been determined that the ATPase activity of these proteins is required for repression in cooperation with pRB, whereas the bromodomain is dispensable.76 C33A cells that are resistant to the expression of a constitutively active pRB mutant (PSM-RB) undergo arrest upon ectopic co-expression of Brg1, suggesting chromatin remodeling plays an important role in pRB-mediated arrest.75 Investigation of chromatin structure at the cyclin A promoter (an E2F target) demonstrates that it can’t be converted into a nuclease insensitive state without Brm.79 In this way, silencing of this key cell cycle target gene requires SWI/SNF to remodel chromatin and convert it into a compacted, restriction endonuclease resistant form. How this works is unclear, as it is unlikely to involve the recruitment of another DNA binding transcriptional repressor, since negative regulation of this promoter is largely E2F-dependent.80 One proposal is that SWI-SNF activity is necessary for loading a histone deacetylase containing complex.81
Taken together, this example of cooperation between pRB and Brm, or Brg1, further exemplifies how pRB recruits co-repressors to E2F-responsive promoters. The consequence is again similar to HDAC recruitment, in that the whole cyclin A promoter can be silenced by chromatin structure changes that occur in a pRB-dependent manner.
Histone methyl transferases
pRB has also been shown to direct the addition of histone methylation marks to repress transcription. In particular H3K9me3 and H4K20me3 can be pRB-dependent modifications. In this section, we will focus on H3K9me3 and the enzyme that adds it, because this modification is known to exist on pRB responsive gene promoters. The data for H4K20me3 suggests a broader role in heterochromatin organization beyond individual promoters and so it will be discussed later in this review.
In addition to regulating histone acetylation on E2F-responsive promoters such as cyclin E1, pRB is also able to recruit the histone methyl transferase Suv39h1 to trimethylate H3K9.21,24 Methylation of this residue creates a binding site for the chromo domain of the methyl lysine binding protein heterochromatin protein 1 (HP1).82 Importantly, endogenous pRB co-immunoprecipitates with Suv39h1 and HP1 in an LXCXE-dependent manner.21,24 In a striking parallel to HDAC function in repression of cyclin E1, H3K9me3 is found on the same nucleosome at the transcriptional start site that is deacetylated in an HDAC- and pRB-dependent manner. H3K9me3 is also pRB-dependent, as Rb1−/− mouse embryonic fibroblasts (MEFs) show drastically decreased H3K9 methylation and HP1 protein enrichment at the nucleosome positioned closest to the cyclin E1 transcriptional start site. Thus, a pathway of deacetylation of H3 preceding its methylation has emerged21 (Fig. 3A and B). Curiously, these early experiments did not offer insight into the physiological need for this gene silencing pathway. Histone methylation is a stable repressive mark, whereas cyclin E1 expression needs to be low through much of the G1 phase of the cell cycle and high at the G1-S boundary, indicating a need for reversibility with each cell division cycle. This pathway will be discussed again in a later section of this review on senescence, a permanent form of cell cycle exit where histone methylation is prevalent.
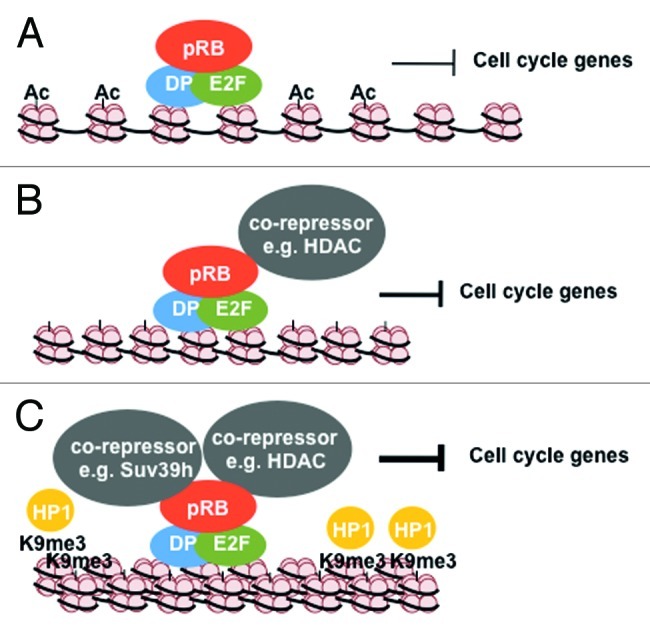
Figure 3. Multi-level regulation by pRB during cellular senescence. (A) pRB inhibits cell cycle gene transcription by directly blocking trans-activation by E2Fs. (B) pRB recruits co-repressor proteins like HDAC’s that deacetylate histones at the promoter regions thereby further repressing these genes. (C) pRB can also recruit chromatin regulating proteins like Suv39h1, and HP1 that can propogate heterochromatin and lead to large scale chromatin compaction resulting in permanent silencing of proliferative genes.
In addition to regulating histone methylation at the transcriptional start site of cyclin E1, pRB could also play a role in propagating H3K9me3 to neighboring histone octamers. For example methylation of histone H3 lysine 9 by Suv39h1 creates a binding site for HP1. HP1 can, in turn, recruit more of these methylases to modify neighboring histones. This, in turn, would allow new HP1 molecules to bind and for the process to be repeated. Importantly, HDACs have also been shown to recruit HP1 proteins and Suv39h1 to promoters, suggesting that H3K9 could be deacetylated to facilitate trimethylation and the spread of heterochromatin83 (Fig. 3C). Hence, pRB localization to a specific gene locus, and recruitment of HDAC, HP1 and Suv39h1, can potentially lead to spreading of this repressive histone mark. This suggests that pRB may exert broader regulation of gene expression across promoters through histone methylation; however, analysis of the size of H3K9me3 islands at these promoters has yet to be reported.
Everywhere: pRB Influences Chromatin Structure of Large Heterochromatin Domains and Whole Chromosomes
In addition to its regulation of transcription at specific loci, recent studies suggest a role for pRB in influencing larger genomic regions and even entire chromosomes. This function of pRB has a direct effect on cell division and the maintenance of genome stability. This section will highlight recent literature linking pRB compaction of large-scale heterochromatin regions to their consequences on the genome.
pRB regulation of heterochromatin at repetitive sequences throughout the genome
The RB protein and its family members have been implicated in the organization of repetitive elements at telomeres, long interspersed nuclear elements (LINE) and pericentromeric heterochromatin.22,27,84 The consequences of misregulation of these domains are lengthened telomere repeats, deregulated expression of LINE transcripts and altered centromere structure leading to mitotic abnormalities, respectively. Specifically, RB family-deficient mouse fibroblasts show hypocondensed chromatin and display butterfly chromosomes, whereby centromeres become joined, thus preventing separation in anaphase.22 A similar phenotype is also described in cells that express a mutant form of pRB that is uniquely deficient for LXCXE-type interactions called Rb1∆L 25. This implies that pRB is the pocket protein that is most responsible for organizing chromatin structure in this region.
Cells lacking all three RB-family proteins display decreased levels of H4K20me3 in each of the three heterochromatin domains described above.22,84 Perhaps not surprisingly, pRB and its related family members can each physically interact with Suv4–20h1/h2, the histone methyltransferases that trimethylate histone H4K20.22 Acute loss of pRB expression in fibroblasts from Rb1lox/lox mice also results in a reduction in H4K20me3 at pericentric heterochromatin.26 Similarly, Rb1∆L/∆L cells have reduced H4K20me3 at pericentromeric DNA.25 The exact role of pRB in H4K20 histone methylation is not known. Despite the reports of Suv4–20h1/h2 physically interacting with RB family proteins, these enzymes are still targeted correctly to pericentric heterochromatin in their absence.22 Previous work studying H4K20 methylation suggests that H3K9me3 recruitment of HP1 is essential for localization of Suv4–20h1/h2 and addition of H4K20me3.85 This leaves in the question of how pRB facilitates H4K20me3. One potential clue comes from the analysis of chromatin from Suv4–20h1/h2 double deficient mouse cells. Cells devoid of these enzymes lack H4K20me3 at telomeres and pericentromeric chromatin domains, and this leads to longer telomeres but not the centromere fusions that best characterize Rb1 mutant cells.86 The differences in mitotic phenotypes between Suv4–20h and Rb1 mutants suggest that diminished H4K20me3 in Rb1 mutants could be a secondary consequence of altered chromatin structure, rather than a direct role for pRB in the catalytic addition of this modification. For example, conditional deletion of Brg1 in mouse fibroblasts leads to dispersion of H3K9me3 and disruption of H4K20me3 from pericentromeric heterochromatin and lagging anaphase chromosomes.87 It is possible that pRB partially mediates Brg1 function in the assembly of pericentromeric heterochromatin, and in its absence, the effects on histone tail modifications are a downstream effect.
Chromatin condensation mediated by pRB
In addition to influencing H4K20me3 levels at centromeres, there are other potential explanations for defective pericentromeric heterochromatin assembly. Recently, the fruit fly ortholog of pRB (Rbf) has been shown to be able to regulate chromosome condensation through interactions with the Condensin II complex.30 Condensin complexes promote chromosome compaction during prophase.88,89 Rbf mutant chromosomes appear diffuse in comparison with control chromosomes during prophase and prometaphase in neuroblast chromosome spreads. Deficiency for the Drosophila ortholog of Cap-D3, a condensin II subunit, shows a similar phenotype in this and other assays of chromosome condensation. Longworth et al. also demonstrate that pRB interacts specifically with condensin II, and that this interaction is mediated through its LXCXE binding cleft.30 Furthermore, Drosophila Cap-D3 requires Rbf for efficient localization to chromatin.30 Mammalian cells expressing a pRB mutant defective in LXCXE interactions also display decreased loading of condensin II on chromatin, whereas cohesin and condensin I chromatin levels remain normal.31 Importantly, condensin II can co-immunoprecipitate with pRB but not the Rb1∆L mutant protein.31 Condensin II staining patterns reveal that it is most highly concentrated at the centromere,90 a region that is abnormal in Rb1∆L/∆L mouse chromosomes. Similar to the mitotic phenotypes of Rb1∆L/∆L cells, diminished expression of condensin II components such as Cap-D3 results in delayed progression to metaphase and lagging anaphase chromosomes.31,88,89
Taken together, pRB has a clear contribution to heterochromatin organization in relatively gene poor, repetitive regions of the genome. Loss of chromatin structure in the pericentromeric region is particularly noteworthy because of the mitotic defects that it causes. Comparison of spontaneous tumors in Rb1∆L/∆L; Trp53−/− and Trp53−/− controls suggests that maintenance of genome stability through chromosome segregation by pRB is part of what makes it a tumor suppressor gene.31 This demonstrates that pRB makes critical contributions to chromatin organization both at large heterochromatin domains and individual target genes. Cancer susceptibility studies cited above suggest that pRB’s regulation of higher order chromatin structure may be no less important in its role as a tumor suppressor than its other functions that are more closely focused on transcriptional control of individual genes.
All Three Levels of Control Rolled into One, pRB Regulation of Cellular Senescence
Cellular senescence is a stable form of cell cycle arrest wherein cells exit the cell cycle and can remain post-mitotic for an indefinite period of time.91 Senescence can be triggered by telomere attrition, activated oncogenes, or other genotoxic stresses.91,92 It is now believed that senescence acts as a barrier to cellular transformation by blocking proliferation of pre-cancerous cells before they can evolve to acquire malignant traits.93,94
Phenotypically, senescent cells show a number of morphological changes as well as characteristic changes in gene expression and chromatin structure.95 In general, they show downregulation of proliferative genes,95 upregulation of anti proliferative genes29 and an increase in expression of inflammatory genes known as the senescence associated secretory phenotype.96 At the chromatin level, senescence is associated with global changes in heterochromatin organization. Senescent cells often display facultative heterochromatin structures in the nucleus that are called senescence associated heterochromatic foci (SAHF).28 These structures are a result of the compaction of individual chromosomes.97,98 SAHFs are linked to transcriptional repression of proliferative genes as they become enclosed in these foci leading to stable silencing. A causal role for SAHFs during senescence has not been established, but it has been proposed that SAHFs contribute to the long-term stability of senescent arrest by stably repressing the expression of proliferative genes.99
Early experiments in cancer cells lacking pRB suggested a critical role for it in senescence. Reintroduction of pRB into cancer cells can induce a senescent arrest.100 Conversely, acute loss of pRB in senescent mouse embryonic fibroblasts (MEFs) results in increased DNA synthesis, cell cycle re-entry and subsequent reversal of cellular senescence.101 Rb1−/− MEF’s do arrest in culture with features of senescence, but they escape from this arrest and immortalize sooner than control cells expressing the wild type protein.102,103 These studies suggested a key role for pRB in establishing the stability of senescent cell cycle arrest.
The retinoblastoma protein is capable of influencing senescence cell cycle arrest at various levels. First, pRB represses the transcription of genes involved in DNA replication by directly binding to and inhibiting E2F transcription factors and through histone deacetylation of their respective promoters.28 The mechanism occurs essentially as described in earlier sections of this review (see Fig. 3A and B). Indeed, pRB is found to be enriched on E2F target gene promoters during senescence.28,29,104 Acute knock down of pRB in primary human fibroblasts that are induced to senesce with oncogenic ras (HrasV12) show deregulated DNA synthesis as reflected in continued incorporation of BrdU and deregulated E2F transcription.29 Use of Rb1∆L/∆L fibroblasts in senescence induction demonstrates that early events in proliferative arrest and downregulation of E2F transcriptional targets take place.104 However, these mutant cells ultimately re-enter the cell cycle and can resume proliferation, indicating that later pRB-dependent steps in establishing a senescent arrest are critical.
Second, pRB is required for the enrichment of repressive histone methylation (H3K9me3) and the removal of activating methylation (H3K4me3) on E2F target gene promoters during senescence.28,104,105 Addition of H3K9me3 is defective in Rb1∆L/∆L fibroblasts, implicating it in long-term stability of senescence.104 Presumably, histone methylation is through recruitment of the histone methyl transferase Suv39h1 by pRB to E2F regulated promoters; however, differences in senescence between Rb1∆L/∆L and Suv39h1−/− mouse cells suggest that other methyltransferases, such as RIZ1, may also cooperate with pRB.94,104 Regardless of the precise mechanism of H3K9me3 deposition, this further illustrates how pRB-dependent chromatin regulation can exert its influence on transcriptional repression across E2F-responsive promoters (Fig. 3C).
Lastly, pRB plays a role in the formation of SAHFs themselves and thus also influences higher order chromatin structure in senescence as well. Knock down of pRB results in decreased formation of SAHFs.28,29 Since SAHFs represent the compaction of entire individual chromosomes,97,98 these structures represent considerable reorganization of higher order chromatin structure that is pRB-dependent. The exact signals that trigger this compaction and the mechanism of chromosome condensation that facilitates their formation is only beginning to be elucidated. Promyelocytic leukemia (PML) bodies appear to be one component in the pathway to assembling SAHFs, and they have recently been shown to co-localize to genes that are silenced in a pRB-dependent manner.106 Furthermore, PML participates in the incorporation of the repressive histone variant macroH2A1; however, whether pRB is involved at this particular step or an earlier one is unclear. Other broad changes to chromatin during the formation of SAHFs are the ejection of histone H197 and the incorporation of HMGA proteins.107 A logical expectation is that these changes facilitate DNA bending during SAHF formation. Again, it is unclear if pRB’s actual function in the induction of chromosome condensation to form SAHFs mediates these events, or if they are merely downstream of earlier pRB-dependent steps. It is difficult to envision SAHF formation being driven purely by pRB-dependent H3K9me3; however, as discussed above there are many steps in SAHF formation and our knowledge of how they take place remains limited.
In the context of pRB’s ability to regulate chromatin and gene expression on many levels, it may not be surprising that the multi-level regulation of gene expression and chromatin structure needed in senescence can be controlled by pRB. Senescence, unlike other cell cycle exit paradigms, has distinguished itself as having a bona fide tumor-suppressive role, and pRB may need to use its complete arsenal of functions in order to maintain the fidelity of this arrest. Since loss of pRB results in deregulated gene expression, DNA synthesis and eventual escape from senescence, it is imperative that we investigate further the steps in gene silencing and higher order chromatin assembly that are controlled by pRB. In this way, we will come to a thorough understanding of pRB function as a tumor suppressor protein.
Conclusions and Future Perspectives
Since being cloned in 1986, the retinoblastoma susceptibility gene (Rb1) and its product, the retinoblastoma protein (pRB), has been a subject of intense scientific research. Thanks to a considerable body of work that encompasses diverse model organisms, our understanding of pRB function has grown and evolved during this time. A number of studies, both biochemical and genetic, have shown pRB to be a multidimensional protein with multiple binding partners that are involved in diverse physiological functions. Regulation of gene transcription has emerged as pRB’s most popular function. This regulation is exerted both by directly interacting with transcription factors and also by recruiting co-repressors/activators to sequence-specific transcription factors, further influencing gene expression. Furthermore, pRB interacts and co-operates with proteins that regulate chromatin structure of large genomic regions, including entire chromosomes. Together, pRB is capable of influencing gene transcription on a very broad scale.
Despite the vast research that has shaped our understanding of pRB, many questions remain. Is every aspect of pRB function in transcriptional control and chromatin regulation essential for cell cycle control in development and differentiation? Are all of these functions of pRB necessary for it to be a tumor suppressor, or are they just select aspects of its overall function? Answers to these open questions will ultimately help us understand what makes pRB a tumor suppressor. One possible interpretation of the complexity of pRB function is that it ultimately only has a few functions, and when these are lost, many secondary consequences can be detected experimentally. From this perspective, the real question that looms in understanding how the original tumor suppressor works is whether it is truly multifunctional, or if we are still searching for an elusive unifying mechanism of action.
Acknowledgments
The authors wish to thank lab members and colleagues for stimulating discussions on R.B.’s role in cell cycle and chromatin regulation. S.T. wishes to thank the strategic training program in cancer research and technology transfer for stipend support. F.A.D. is the Wolfe Senior Research Fellow in Tumor Suppressor Genes and is funded by operating grants from the Canadian Institutes of Health Research and the Canadian Cancer Society. The authors have no conflict of interest with the publication of this review.
Footnotes
Previously published online: www.landesbioscience.com/journals/cc/article/21263
References
- 1.Classon M, Harlow E. The retinoblastoma tumour suppressor in development and cancer. Nat Rev Cancer. 2002;2:910–7. doi: 10.1038/nrc950. [DOI] [PubMed] [Google Scholar]
- 2.Sherr CJ, McCormick F. The RB and p53 pathways in cancer. Cancer Cell. 2002;2:103–12. doi: 10.1016/S1535-6108(02)00102-2. [DOI] [PubMed] [Google Scholar]
- 3.Sherr CJ. Cancer cell cycles. Science. 1996;274:1672–7. doi: 10.1126/science.274.5293.1672. [DOI] [PubMed] [Google Scholar]
- 4.Mulligan G, Jacks T. The retinoblastoma gene family: cousins with overlapping interests. Trends Genet. 1998;14:223–9. doi: 10.1016/S0168-9525(98)01470-X. [DOI] [PubMed] [Google Scholar]
- 5.Dick FA. Structure-function analysis of the retinoblastoma tumor suppressor protein - is the whole a sum of its parts? Cell Div. 2007;2:26. doi: 10.1186/1747-1028-2-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dyson N. The regulation of E2F by pRB-family proteins. Genes Dev. 1998;12:2245–62. doi: 10.1101/gad.12.15.2245. [DOI] [PubMed] [Google Scholar]
- 7.Trimarchi JM, Lees JA. Sibling rivalry in the E2F family. Nat Rev Mol Cell Biol. 2002;3:11–20. doi: 10.1038/nrm714. [DOI] [PubMed] [Google Scholar]
- 8.Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000;100:57–70. doi: 10.1016/S0092-8674(00)81683-9. [DOI] [PubMed] [Google Scholar]
- 9.Whyte P, Buchkovich KJ, Horowitz JM, Friend SH, Raybuck M, Weinberg RA, et al. Association between an oncogene and an anti-oncogene: the adenovirus E1A proteins bind to the retinoblastoma gene product. Nature. 1988;334:124–9. doi: 10.1038/334124a0. [DOI] [PubMed] [Google Scholar]
- 10.Whyte P, Williamson NM, Harlow E. Cellular targets for transformation by the adenovirus E1A proteins. Cell. 1989;56:67–75. doi: 10.1016/0092-8674(89)90984-7. [DOI] [PubMed] [Google Scholar]
- 11.Hamel PA, Gill RM, Phillips RA, Gallie BL. Transcriptional repression of the E2-containing promoters EIIaE, c-myc, and RB1 by the product of the RB1 gene. Mol Cell Biol. 1992;12:3431–8. doi: 10.1128/mcb.12.8.3431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Weintraub SJ, Prater CA, Dean DC. Retinoblastoma protein switches the E2F site from positive to negative element. Nature. 1992;358:259–61. doi: 10.1038/358259a0. [DOI] [PubMed] [Google Scholar]
- 13.Weintraub SJ, Chow KNB, Luo RX, Zhang SH, He S, Dean DC. Mechanism of active transcriptional repression by the retinoblastoma protein. Nature. 1995;375:812–5. doi: 10.1038/375812a0. [DOI] [PubMed] [Google Scholar]
- 14.Magnaghi-Jaulin L, Groisman R, Naguibneva I, Robin P, Lorain S, Le Villain JP, et al. Retinoblastoma protein represses transcription by recruiting a histone deacetylase. Nature. 1998;391:601–5. doi: 10.1038/35410. [DOI] [PubMed] [Google Scholar]
- 15.Brehm A, Miska EA, McCance DJ, Reid JL, Bannister AJ, Kouzarides T. Retinoblastoma protein recruits histone deacetylase to repress transcription. Nature. 1998;391:597–601. doi: 10.1038/35404. [DOI] [PubMed] [Google Scholar]
- 16.Luo RX, Postigo AA, Dean DC. Rb interacts with histone deacetylase to repress transcription. Cell. 1998;92:463–73. doi: 10.1016/S0092-8674(00)80940-X. [DOI] [PubMed] [Google Scholar]
- 17.Benevolenskaya EV, Murray HL, Branton P, Young RA, Kaelin WGJ., Jr. Binding of pRB to the PHD protein RBP2 promotes cellular differentiation. Mol Cell. 2005;18:623–35. doi: 10.1016/j.molcel.2005.05.012. [DOI] [PubMed] [Google Scholar]
- 18.Robertson KD, Ait-Si-Ali S, Yokochi T, Wade PA, Jones PL, Wolffe AP. DNMT1 forms a complex with Rb, E2F1 and HDAC1 and represses transcription from E2F-responsive promoters. Nat Genet. 2000;25:338–42. doi: 10.1038/77124. [DOI] [PubMed] [Google Scholar]
- 19.Dunaief JL. RB and BRG1 form a complex and cooperate to induce mitotic arrest. Diss Abstr Int. 1994;55:2087. [Google Scholar]
- 20.Singh P, Coe J, Hong W. A role for retinoblastoma protein in potentiating transcriptional activation by the glucocorticoid receptor. Nature. 1995;374:562–5. doi: 10.1038/374562a0. [DOI] [PubMed] [Google Scholar]
- 21.Nielsen SJ, Schneider R, Bauer UM, Bannister AJ, Morrison A, O’Carroll D, et al. Rb targets histone H3 methylation and HP1 to promoters. Nature. 2001;412:561–5. doi: 10.1038/35087620. [DOI] [PubMed] [Google Scholar]
- 22.Gonzalo S, García-Cao M, Fraga MF, Schotta G, Peters AH, Cotter SE, et al. Role of the RB1 family in stabilizing histone methylation at constitutive heterochromatin. Nat Cell Biol. 2005;7:420–8. doi: 10.1038/ncb1235. [DOI] [PubMed] [Google Scholar]
- 23.Steele-Perkins G, Fang W, Yang XH, Van Gele M, Carling T, Gu J, et al. Tumor formation and inactivation of RIZ1, an Rb-binding member of a nuclear protein-methyltransferase superfamily. Genes Dev. 2001;15:2250–62. doi: 10.1101/gad.870101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Vandel L, Nicolas E, Vaute O, Ferreira R, Ait-Si-Ali S, Trouche D. Transcriptional repression by the retinoblastoma protein through the recruitment of a histone methyltransferase. Mol Cell Biol. 2001;21:6484–94. doi: 10.1128/MCB.21.19.6484-6494.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Isaac CE, Francis SM, Martens AL, Julian LM, Seifried LA, Erdmann N, et al. The retinoblastoma protein regulates pericentric heterochromatin. Mol Cell Biol. 2006;26:3659–71. doi: 10.1128/MCB.26.9.3659-3671.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Siddiqui H, Fox SR, Gunawardena RW, Knudsen ES. Loss of RB compromises specific heterochromatin modifications and modulates HP1alpha dynamics. J Cell Physiol. 2007;211:131–7. doi: 10.1002/jcp.20913. [DOI] [PubMed] [Google Scholar]
- 27.García-Cao M, Gonzalo S, Dean D, Blasco MA. A role for the Rb family of proteins in controlling telomere length. Nat Genet. 2002;32:415–9. doi: 10.1038/ng1011. [DOI] [PubMed] [Google Scholar]
- 28.Narita M, Nũnez S, Heard E, Narita M, Lin AW, Hearn SA, et al. Rb-mediated heterochromatin formation and silencing of E2F target genes during cellular senescence. Cell. 2003;113:703–16. doi: 10.1016/S0092-8674(03)00401-X. [DOI] [PubMed] [Google Scholar]
- 29.Chicas A, Wang X, Zhang C, McCurrach M, Zhao Z, Mert O, et al. Dissecting the unique role of the retinoblastoma tumor suppressor during cellular senescence. Cancer Cell. 2010;17:376–87. doi: 10.1016/j.ccr.2010.01.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Longworth MS, Herr A, Ji JY, Dyson NJ. RBF1 promotes chromatin condensation through a conserved interaction with the Condensin II protein dCAP-D3. Genes Dev. 2008;22:1011–24. doi: 10.1101/gad.1631508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Coschi CH, Martens AL, Ritchie K, Francis SM, Chakrabarti S, Berube NG, et al. Mitotic chromosome condensation mediated by the retinoblastoma protein is tumor-suppressive. Genes Dev. 2010;24:1351–63. doi: 10.1101/gad.1917610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Helin K, Harlow E, Fattaey A. Inhibition of E2F-1 transactivation by direct binding of the retinoblastoma protein. Mol Cell Biol. 1993;13:6501–8. doi: 10.1128/mcb.13.10.6501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ikeda MA, Nevins JR. Identification of distinct roles for separate E1A domains in disruption of E2F complexes. Mol Cell Biol. 1993;13:7029–35. doi: 10.1128/mcb.13.11.7029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Fattaey AR, Harlow E, Helin K. Independent regions of adenovirus E1A are required for binding to and dissociation of E2F-protein complexes. Mol Cell Biol. 1993;13:7267–77. doi: 10.1128/mcb.13.12.7267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Liu X, Marmorstein R. Structure of the retinoblastoma protein bound to adenovirus E1A reveals the molecular basis for viral oncoprotein inactivation of a tumor suppressor. Genes Dev. 2007;21:2711–6. doi: 10.1101/gad.1590607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bagchi S, Raychaudhuri P, Nevins JR. Adenovirus E1A proteins can dissociate heteromeric complexes involving the E2F transcription factor: a novel mechanism for E1A trans-activation. Cell. 1990;62:659–69. doi: 10.1016/0092-8674(90)90112-R. [DOI] [PubMed] [Google Scholar]
- 37.Whyte P, Ruley HE, Harlow E. Two regions of the adenovirus early region 1A proteins are required for transformation. J Virol. 1988;62:257–65. doi: 10.1128/jvi.62.1.257-265.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Herrera RE, Sah VP, Williams BO, Mäkelä TP, Weinberg RA, Jacks T. Altered cell cycle kinetics, gene expression, and G1 restriction point regulation in Rb-deficient fibroblasts. Mol Cell Biol. 1996;16:2402–7. doi: 10.1128/mcb.16.5.2402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hurford RK, Jr., Cobrinik D, Lee MH, Dyson N. pRB and p107/p130 are required for the regulated expression of different sets of E2F responsive genes. Genes Dev. 1997;11:1447–63. doi: 10.1101/gad.11.11.1447. [DOI] [PubMed] [Google Scholar]
- 40.Gu W, Schneider JW, Condorelli G, Kaushal S, Mahdavi V, Nadal-Ginard B. Interaction of myogenic factors and the retinoblastoma protein mediates muscle cell commitment and differentiation. Cell. 1993;72:309–24. doi: 10.1016/0092-8674(93)90110-C. [DOI] [PubMed] [Google Scholar]
- 41.Thomas DM, Carty SA, Piscopo DM, Lee JS, Wang WF, Forrester WC, et al. The retinoblastoma protein acts as a transcriptional coactivator required for osteogenic differentiation. Mol Cell. 2001;8:303–16. doi: 10.1016/S1097-2765(01)00327-6. [DOI] [PubMed] [Google Scholar]
- 42.Chen PL, Riley DJ, Chen-Kiang S, Lee WH. Retinoblastoma protein directly interacts with and activates the transcription factor NF-IL6. Proc Natl Acad Sci USA. 1996;93:465–9. doi: 10.1073/pnas.93.1.465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chen P-L, Riley DJ, Chen Y, Lee W-H. Retinoblastoma protein positively regulates terminal adipocyte differentiation through direct interaction with C/EBPs. Genes Dev. 1996;10:2794–804. doi: 10.1101/gad.10.21.2794. [DOI] [PubMed] [Google Scholar]
- 44.MacLellan WR, Xiao G, Abdellatif M, Schneider MD. A novel Rb- and p300-binding protein inhibits transactivation by MyoD. Mol Cell Biol. 2000;20:8903–15. doi: 10.1128/MCB.20.23.8903-8915.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Miyake S, Sellers WR, Safran M, Li X, Zhao W, Grossman SR, et al. Cells degrade a novel inhibitor of differentiation with E1A-like properties upon exiting the cell cycle. Mol Cell Biol. 2000;20:8889–902. doi: 10.1128/MCB.20.23.8889-8902.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Iavarone A, Garg P, Lasorella A, Hsu J, Israel MA. The helix-loop-helix protein Id-2 enhances cell proliferation and binds to the retinoblastoma protein. Genes Dev. 1994;8:1270–84. doi: 10.1101/gad.8.11.1270. [DOI] [PubMed] [Google Scholar]
- 47.Lee JS, Thomas DM, Gutierrez G, Carty SA, Yanagawa S, Hinds PW. HES1 cooperates with pRb to activate RUNX2-dependent transcription. J Bone Miner Res. 2006;21:921–33. doi: 10.1359/jbmr.060303. [DOI] [PubMed] [Google Scholar]
- 48.Calo E, Quintero-Estades JA, Danielian PS, Nedelcu S, Berman SD, Lees JA. Rb regulates fate choice and lineage commitment in vivo. Nature. 2010;466:1110–4. doi: 10.1038/nature09264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Gutierrez GM, Kong E, Sabbagh Y, Brown NE, Lee JS, Demay MB, et al. Impaired bone development and increased mesenchymal progenitor cells in calvaria of RB1-/- mice. Proc Natl Acad Sci USA. 2008;105:18402–7. doi: 10.1073/pnas.0805925105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ianari A, Natale T, Calo E, Ferretti E, Alesse E, Screpanti I, et al. Proapoptotic function of the retinoblastoma tumor suppressor protein. Cancer Cell. 2009;15:184–94. doi: 10.1016/j.ccr.2009.01.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Dick FA, Dyson N. pRB contains an E2F1-specific binding domain that allows E2F1-induced apoptosis to be regulated separately from other E2F activities. Mol Cell. 2003;12:639–49. doi: 10.1016/S1097-2765(03)00344-7. [DOI] [PubMed] [Google Scholar]
- 52.Cecchini MJ, Dick FA. The biochemical basis of CDK phosphorylation-independent regulation of E2F1 by the retinoblastoma protein. Biochem J. 2011;434:297–308. doi: 10.1042/BJ20101210. [DOI] [PubMed] [Google Scholar]
- 53.Stevens C, Smith L, La Thangue NB. Chk2 activates E2F-1 in response to DNA damage. Nat Cell Biol. 2003;5:401–9. doi: 10.1038/ncb974. [DOI] [PubMed] [Google Scholar]
- 54.Carnevale J, Palander O, Seifried LA, Dick FA. DNA damage signals through differentially modified E2F1 molecules to induce apoptosis. Mol Cell Biol. 2012;32:900–12. doi: 10.1128/MCB.06286-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Duronio RJ, O’Farrell PH. Developmental control of the G1 to S transition in Drosophila: cyclin Eis a limiting downstream target of E2F. Genes Dev. 1995;9:1456–68. doi: 10.1101/gad.9.12.1456. [DOI] [PubMed] [Google Scholar]
- 56.Duronio RJ, Brook A, Dyson N, O’Farrell PH. E2F-induced S phase requires cyclin E. Genes Dev. 1996;10:2505–13. doi: 10.1101/gad.10.19.2505. [DOI] [PubMed] [Google Scholar]
- 57.Le Cam L, Polanowska J, Fabbrizio E, Olivier M, Philips A, Ng Eaton E, et al. Timing of cyclin E gene expression depends on the regulated association of a bipartite repressor element with a novel E2F complex. EMBO J. 1999;18:1878–90. doi: 10.1093/emboj/18.7.1878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Morrison AJ, Sardet C, Herrera RE. Retinoblastoma protein transcriptional repression through histone deacetylation of a single nucleosome. Mol Cell Biol. 2002;22:856–65. doi: 10.1128/MCB.22.3.856-865.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Polanowska J, Fabbrizio E, Le Cam L, Trouche D, Emiliani S, Herrera R, et al. The periodic down regulation of Cyclin E gene expression from exit of mitosis to end of G(1) is controlled by a deacetylase- and E2F-associated bipartite repressor element. Oncogene. 2001;20:4115–27. doi: 10.1038/sj.onc.1204514. [DOI] [PubMed] [Google Scholar]
- 60.Rayman JB, Takahashi Y, Indjeian VB, Dannenberg JH, Catchpole S, Watson RJ, et al. E2F mediates cell cycle-dependent transcriptional repression in vivo by recruitment of an HDAC1/mSin3B corepressor complex. Genes Dev. 2002;16:933–47. doi: 10.1101/gad.969202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ferreira R, Naguibneva I, Mathieu M, Ait-Si-Ali S, Robin P, Pritchard LL, et al. Cell cycle-dependent recruitment of HDAC-1 correlates with deacetylation of histone H4 on an Rb-E2F target promoter. EMBO Rep. 2001;2:794–9. doi: 10.1093/embo-reports/kve173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Nicolas E, Roumillac C, Trouche D. Balance between acetylation and methylation of histone H3 lysine 9 on the E2F-responsive dihydrofolate reductase promoter. Mol Cell Biol. 2003;23:1614–22. doi: 10.1128/MCB.23.5.1614-1622.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Dick FA, Sailhamer E, Dyson NJ. Mutagenesis of the pRB pocket reveals that cell cycle arrest functions are separable from binding to viral oncoproteins. Mol Cell Biol. 2000;20:3715–27. doi: 10.1128/MCB.20.10.3715-3727.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Singh M, Krajewski M, Mikolajka A, Holak TA. Molecular determinants for the complex formation between the retinoblastoma protein and LXCXE sequences. J Biol Chem. 2005;280:37868–76. doi: 10.1074/jbc.M504877200. [DOI] [PubMed] [Google Scholar]
- 65.Meloni AR, Smith EJ, Nevins JR. A mechanism for Rb/p130-mediated transcription repression involving recruitment of the CtBP corepressor. Proc Natl Acad Sci USA. 1999;96:9574–9. doi: 10.1073/pnas.96.17.9574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Lai A, Kennedy BK, Barbie DA, Bertos NR, Yang XJ, Theberge MC, et al. RBP1 recruits the mSIN3-histone deacetylase complex to the pocket of retinoblastoma tumor suppressor family proteins found in limited discrete regions of the nucleus at growth arrest. Mol Cell Biol. 2001;21:2918–32. doi: 10.1128/MCB.21.8.2918-2932.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Chinnadurai G. Transcriptional regulation by C-terminal binding proteins. Int J Biochem Cell Biol. 2007;39:1593–607. doi: 10.1016/j.biocel.2007.01.025. [DOI] [PubMed] [Google Scholar]
- 68.Ferreira R, Magnaghi-Jaulin L, Robin P, Harel-Bellan A, Trouche D. The three members of the pocket proteins family share the ability to repress E2F activity through recruitment of a histone deacetylase. Proc Natl Acad Sci USA. 1998;95:10493–8. doi: 10.1073/pnas.95.18.10493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Imbalzano AN, Kwon H, Green MR, Kingston RE. Facilitated binding of TATA-binding protein to nucleosomal DNA. Nature. 1994;370:481–5. doi: 10.1038/370481a0. [DOI] [PubMed] [Google Scholar]
- 70.Wang W, Côté J, Xue Y, Zhou S, Khavari PA, Biggar SR, et al. Purification and biochemical heterogeneity of the mammalian SWI-SNF complex. EMBO J. 1996;15:5370–82. [PMC free article] [PubMed] [Google Scholar]
- 71.Schnitzler G, Sif S, Kingston RE. Human SWI/SNF interconverts a nucleosome between its base state and a stable remodeled state. Cell. 1998;94:17–27. doi: 10.1016/S0092-8674(00)81217-9. [DOI] [PubMed] [Google Scholar]
- 72.Martens JA, Winston F. Recent advances in understanding chromatin remodeling by Swi/Snf complexes. Curr Opin Genet Dev. 2003;13:136–42. doi: 10.1016/S0959-437X(03)00022-4. [DOI] [PubMed] [Google Scholar]
- 73.Ooi L, Belyaev ND, Miyake K, Wood IC, Buckley NJ. BRG1 chromatin remodeling activity is required for efficient chromatin binding by repressor element 1-silencing transcription factor (REST) and facilitates REST-mediated repression. J Biol Chem. 2006;281:38974–80. doi: 10.1074/jbc.M605370200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Strober BE, Dunaief JL, Guha S, Goff SP. Functional interactions between the hBRM/hBRG1 transcriptional activators and the pRB family of proteins. Mol Cell Biol. 1996;16:1576–83. doi: 10.1128/mcb.16.4.1576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Strobeck MW, Knudsen KE, Fribourg AF, DeCristofaro MF, Weissman BE, Imbalzano AN, et al. BRG-1 is required for RB-mediated cell cycle arrest. Proc Natl Acad Sci USA. 2000;97:7748–53. doi: 10.1073/pnas.97.14.7748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Trouche D, Le Chalony C, Muchardt C, Yaniv M, Kouzarides T. RB and hbrm cooperate to repress the activation functions of E2F1. Proc Natl Acad Sci USA. 1997;94:11268–73. doi: 10.1073/pnas.94.21.11268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Reyes JC, Barra J, Muchardt C, Camus A, Babinet C, Yaniv M. Altered control of cellular proliferation in the absence of mammalian brahma (SNF2alpha) EMBO J. 1998;17:6979–91. doi: 10.1093/emboj/17.23.6979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Bultman S, Gebuhr T, Yee D, La Mantia C, Nicholson J, Gilliam A, et al. A Brg1 null mutation in the mouse reveals functional differences among mammalian SWI/SNF complexes. Mol Cell. 2000;6:1287–95. doi: 10.1016/S1097-2765(00)00127-1. [DOI] [PubMed] [Google Scholar]
- 79.Coisy M, Roure V, Ribot M, Philips A, Muchardt C, Blanchard JM, et al. Cyclin A repression in quiescent cells is associated with chromatin remodeling of its promoter and requires Brahma/SNF2alpha. Mol Cell. 2004;15:43–56. doi: 10.1016/j.molcel.2004.06.022. [DOI] [PubMed] [Google Scholar]
- 80.Schulze A, Zerfass K, Spitkovsky D, Middendorp S, Bergès J, Helin K, et al. Cell cycle regulation of the cyclin A gene promoter is mediated by a variant E2F site. Proc Natl Acad Sci USA. 1995;92:11264–8. doi: 10.1073/pnas.92.24.11264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Gunawardena RW, Fox SR, Siddiqui H, Knudsen ES. SWI/SNF activity is required for the repression of deoxyribonucleotide triphosphate metabolic enzymes via the recruitment of mSin3B. J Biol Chem. 2007;282:20116–23. doi: 10.1074/jbc.M701406200. [DOI] [PubMed] [Google Scholar]
- 82.Bannister AJ, Zegerman P, Partridge JF, Miska EA, Thomas JO, Allshire RC, et al. Selective recognition of methylated lysine 9 on histone H3 by the HP1 chromo domain. Nature. 2001;410:120–4. doi: 10.1038/35065138. [DOI] [PubMed] [Google Scholar]
- 83.Vaute O, Nicolas E, Vandel L, Trouche D. Functional and physical interaction between the histone methyl transferase Suv39H1 and histone deacetylases. Nucleic Acids Res. 2002;30:475–81. doi: 10.1093/nar/30.2.475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Montoya-Durango DE, Liu Y, Teneng I, Kalbfleisch T, Lacy ME, Steffen MC, et al. Epigenetic control of mammalian LINE-1 retrotransposon by retinoblastoma proteins. Mutat Res. 2009;665:20–8. doi: 10.1016/j.mrfmmm.2009.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Schotta G, Lachner M, Sarma K, Ebert A, Sengupta R, Reuter G, et al. A silencing pathway to induce H3-K9 and H4-K20 trimethylation at constitutive heterochromatin. Genes Dev. 2004;18:1251–62. doi: 10.1101/gad.300704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Benetti R, Gonzalo S, Jaco I, Schotta G, Klatt P, Jenuwein T, et al. Suv4-20h deficiency results in telomere elongation and derepression of telomere recombination. J Cell Biol. 2007;178:925–36. doi: 10.1083/jcb.200703081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Bourgo RJ, Siddiqui H, Fox S, Solomon D, Sansam CG, Yaniv M, et al. SWI/SNF deficiency results in aberrant chromatin organization, mitotic failure, and diminished proliferative capacity. Mol Biol Cell. 2009;20:3192–9. doi: 10.1091/mbc.E08-12-1224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Ono T, Losada A, Hirano M, Myers MP, Neuwald AF, Hirano T. Differential contributions of condensin I and condensin II to mitotic chromosome architecture in vertebrate cells. Cell. 2003;115:109–21. doi: 10.1016/S0092-8674(03)00724-4. [DOI] [PubMed] [Google Scholar]
- 89.Hirota T, Gerlich D, Koch B, Ellenberg J, Peters JM. Distinct functions of condensin I and II in mitotic chromosome assembly. J Cell Sci. 2004;117:6435–45. doi: 10.1242/jcs.01604. [DOI] [PubMed] [Google Scholar]
- 90.Ono T, Fang Y, Spector DL, Hirano T. Spatial and temporal regulation of Condensins I and II in mitotic chromosome assembly in human cells. Mol Biol Cell. 2004;15:3296–308. doi: 10.1091/mbc.E04-03-0242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Campisi J. Senescent cells, tumor suppression, and organismal aging: good citizens, bad neighbors. Cell. 2005;120:513–22. doi: 10.1016/j.cell.2005.02.003. [DOI] [PubMed] [Google Scholar]
- 92.Dimri GP. What has senescence got to do with cancer? Cancer Cell. 2005;7:505–12. doi: 10.1016/j.ccr.2005.05.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Collado M, Gil J, Efeyan A, Guerra C, Schuhmacher AJ, Barradas M, et al. Tumour biology: senescence in premalignant tumours. Nature. 2005;436:642. doi: 10.1038/436642a. [DOI] [PubMed] [Google Scholar]
- 94.Braig M, Lee S, Loddenkemper C, Rudolph C, Peters AH, Schlegelberger B, et al. Oncogene-induced senescence as an initial barrier in lymphoma development. Nature. 2005;436:660–5. doi: 10.1038/nature03841. [DOI] [PubMed] [Google Scholar]
- 95.Campisi J, d’Adda di Fagagna F. Cellular senescence: when bad things happen to good cells. Nat Rev Mol Cell Biol. 2007;8:729–40. doi: 10.1038/nrm2233. [DOI] [PubMed] [Google Scholar]
- 96.Kuilman T, Michaloglou C, Vredeveld LC, Douma S, van Doorn R, Desmet CJ, et al. Oncogene-induced senescence relayed by an interleukin-dependent inflammatory network. Cell. 2008;133:1019–31. doi: 10.1016/j.cell.2008.03.039. [DOI] [PubMed] [Google Scholar]
- 97.Funayama R, Saito M, Tanobe H, Ishikawa F. Loss of linker histone H1 in cellular senescence. J Cell Biol. 2006;175:869–80. doi: 10.1083/jcb.200604005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Zhang R, Chen W, Adams PD. Molecular dissection of formation of senescence-associated heterochromatin foci. Mol Cell Biol. 2007;27:2343–58. doi: 10.1128/MCB.02019-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Beauséjour CM, Krtolica A, Galimi F, Narita M, Lowe SW, Yaswen P, et al. Reversal of human cellular senescence: roles of the p53 and p16 pathways. EMBO J. 2003;22:4212–22. doi: 10.1093/emboj/cdg417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Xu HJ, Zhou Y, Ji W, Perng GS, Kruzelock R, Kong CT, et al. Reexpression of the retinoblastoma protein in tumor cells induces senescence and telomerase inhibition. Oncogene. 1997;15:2589–96. doi: 10.1038/sj.onc.1201446. [DOI] [PubMed] [Google Scholar]
- 101.Sage J, Miller AL, Pérez-Mancera PA, Wysocki JM, Jacks T. Acute mutation of retinoblastoma gene function is sufficient for cell cycle re-entry. Nature. 2003;424:223–8. doi: 10.1038/nature01764. [DOI] [PubMed] [Google Scholar]
- 102.Sage J, Mulligan GJ, Attardi LD, Miller A, Chen S, Williams B, et al. Targeted disruption of the three Rb-related genes leads to loss of G(1) control and immortalization. Genes Dev. 2000;14:3037–50. doi: 10.1101/gad.843200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Dannenberg J-H, van Rossum A, Schuijff L, te Riele H. Ablation of the retinoblastoma gene family deregulates G(1) control causing immortalization and increased cell turnover under growth-restricting conditions. Genes Dev. 2000;14:3051–64. doi: 10.1101/gad.847700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Talluri S, Isaac CE, Ahmad M, Henley SA, Francis SM, Martens AL, et al. A G1 checkpoint mediated by the retinoblastoma protein that is dispensable in terminal differentiation but essential for senescence. Mol Cell Biol. 2010;30:948–60. doi: 10.1128/MCB.01168-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Chicas A, Kapoor A, Wang X, Aksoy O, Evertts AG, Zhang MQ, et al. H3K4 demethylation by Jarid1a and Jarid1b contributes to retinoblastoma-mediated gene silencing during cellular senescence. Proceedings of the National Academy of Sciences of the United States of America 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Vernier M, Bourdeau V, Gaumont-Leclerc MF, Moiseeva O, Bégin V, Saad F, et al. Regulation of E2Fs and senescence by PML nuclear bodies. Genes Dev. 2011;25:41–50. doi: 10.1101/gad.1975111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Narita M, Narita M, Krizhanovsky V, Nuñez S, Chicas A, Hearn SA, et al. A novel role for high-mobility group a proteins in cellular senescence and heterochromatin formation. Cell. 2006;126:503–14. doi: 10.1016/j.cell.2006.05.052. [DOI] [PubMed] [Google Scholar]