

Abstract
The synthesis and characterization of a new water-soluble iminophosphorane ligand TPA=N-C(O)-2BrC6H4 (C,N-IM; TPA = 1,3,5-triaza-7-phosphaadamantane) 1 is reported. Oxidative addition of 1 to Pd2(dba)3 affords the orthopalladated dimer [Pd(μ-Br){C6H4(C(O)N=TPA-kC,N)-2}]2 (2) as a mixture of cis and trans isomers (1:1 molar ratio) where the iminophosphorane moeity behaves as a C,N-pincer ligand. By addition of different neutral or monoanionic ligands to 2, the bridging bromide can be cleaved and a variety of hydrophilic or water-soluble mononuclear organometallic palladium(II) complexes of the type [Pd{C6H4(C(O)N=TPA-kC,N)-2}(L-L)] (L-L = acac (3); S2CNMe2 (4); 4,7-Diphenyl-1,10-phenanthrolinedisulfonic acid disodium salt C12H6N2(C6H4SO3Na)2 (5)); [Pd{C6H4(C(O)N=TPA-kC,N)-2}(L)Br] (L = P(mC6H4SO3Na)3 (6); P(3-Pyridyl)3 (7)) and, [Pd(C6H4(C(O)N=TPA)-2}(TPA)2Br] (8) are obtained as single isomers. All new complexes were tested as potential anticancer agents and their cytotoxicity properties were evaluated in vitro against human Jurkat-T acute lymphoblastic leukemia cells, normal T-lymphocytes (PBMC) and DU-145 human prostate cancer cells. Compounds [Pd(μ-Br){C6H4(C(O)N=TPA-kC,N)-2}]2 (2) and [Pd{C6H4(C(O)N=TPA-kC,N)-2}(acac)] 3 (which has been crystallographically characterized) display the higher cytotoxicity against the above mentioned cancer cell lines while being less toxic to normal T-lymphocytes (peripheral blood mononuclear cells: PBMC). In addition, 3 is very toxic to cisplatin resistant Jurkat shBak indicating a cell death pathway that may be different to that of cisplatin. The interaction of 2 and 3 with plasmid (pBR322) DNA is much weaker than that of cisplatin pointing to an alternative biomolecular target for these cytotoxic compounds. All the compounds show an interaction with human serum albumin (HSA) faster than that of cisplatin.
Keywords: Palladium, water-soluble, iminophosphorane ligands, cytotoxic, leukemia, prostate cancer
INTRODUCTION
A significant fraction of currently used anticancer drugs are platinum compounds such as cisplatin, carboplatin (paraplatin™) and oxaliplatin (eloxatin™).1 However their effectiveness is still hindered by clinical problems, including acquired or intrinsic resistance, a limited spectrum of activity, and high toxicity leading to side effects.1,2 The search for anticancer agents with improved properties has focused on the synthesis of a new generation of platinum compounds3,4 which include Pt(IV) pro-drugs,5 multimetallic derivatives6 and the use of nanoparticles and oligonucleotides as delivery platforms.7 The synthesis of other metallo drugs (including organometallic complexes)8 of ruthenium,9 gold,10 titanium,11 osmium,12 copper,13 rhodium,14 iridium15 and iron16 has also attracted great interest. More recently, a number of heterometallic cytotoxic complexes17 have been reported, which show a synergic effect of two different metals with known antitumor properties. Palladium derivatives have been explored as an alternative18 to platinum-based compounds due to the obvious structural and thermodynamic analogy between platinum(II) and palladium(II) complexes. However, the ligand-exchange kinetics of platinum(II) and palladium(II) derivatives are quite different; hydrolysis of palladium (II) compounds is much more rapid leading to reactive species unable to reach their pharmacological targets. In addition, some Pd(II) compounds transform into inactive trans-derivatives. The stabilization of Pd(II) compounds by strongly coordinated nitrogen ligands and an appropriate leaving group has been exploited as a synthetic strategy to obtain palladium(II) antitumor complexes.18 Thus a variety of trans-compounds containing bulky monodentate ligands,19 complexes with bidentate ligands (N-N; P-P, or mixed N-O, N-S)20 and organometallic derivatives21 have been prepared. Since the seminal work by Navarro-Ranninger et al. on cytotoxic orthometallated palladium complexes22 (some containing thiosemicarbazone ligands) a few cyclopalladated compounds with potential anticancer activities have been described.23-25 The higher stability of cyclopalladated compounds in physiological media together with a lower toxicity to normal cells are promising features for their biological applications.18b Biphosphinic palladacycles of the type [Pd(C2,N-(S(-)dmap)(dppf)]Cl induced apoptotic cell death in human leukemia cells by rupture of lysosomal membrane and release of cathepsin B in the cytoplasm.26-28 Importantly, the related compound [Pd2(S(-)C2,N- dmpa)2(μ-dppe)Cl2] has been selected for its promising antitumor properties against murine and cisplatin-resistant human tumor cells both in vitro and in vivo29-31 for further preclinical studies,30 including a gene therapy protocol in conjunction with plasmids.31 Its mode of action seems to indicate DNA degradation and mitochondrial damage by interaction of the compound with thiol groups on the mitochondrial membrane proteins.29,30
We have reported that non-toxic iminophosphorane or iminophosphine compounds serve as stabilizing (C,N- or N,N) chelating ligands in the preparation of anticancer organometallic and coordination d8 metal complexes.32-34 Organogold(III) complexes containing iminophosphorane (C,N-IM) ligands of the type PPh3=NPh displayed a high cytotoxicity in vitro against human ovarian cancer and leukemia cell lines32,33 while being less toxic to normal T-lymphocytes by a non-cisplatin mode of action (involving mitochondrial production of reactive oxygen species).33 More recently, we have reported on the cytotoxicity of coordination Pd(II) and Pt(II) complexes with the first water-soluble iminophosphorane ligand TPA=N-C(O)-2-NC5H4 (N,N-IM) described to date.34 Interestingly, we found that the Pt(II) compound was more cytotoxic than cisplatin to leukemia cell lines (both T-Jurkat and cisplatin-resistant Jurkat sh-Bak) by mode of action different to that of cisplatin.34 The coordination Pd(II) derivative (slighty less cytotoxic than cisplatin to leukemia cell lines) displayed a mode of action similar to cisplatin.34 We report here on the synthesis of a related water-soluble iminophosphorane ligand TPA=N-C(O)-2BrC6H4 (C,N-IM; TPA = 1,3,5-triaza-7-phosphaadamantane) 1 which serves as an excellent precursor to organometallic Pd(II) derivatives with varying hydrophilic-lipophilic ratios. All new complexes have been tested as potential anticancer agents and their cytotoxicity properties were evaluated in vitro against human Jurkat-T acute lymphoblastic leukemia cells, normal T-lymphocytes (PBMC) and DU-145 human prostate cancer cells. The interactions of the most cytotoxic derivatives with plasmid (pBR322) DNA and human serum albumin (HSA) are reported.
RESULTS AND DISCUSSION
1. Synthesis and Characterization
Ligand 1 TPA=N-C(O)-2BrC6H4 (C,N-IM) can be conveniently prepared from 2-bromobenzamide and TPA through the Pomerantz method (Scheme 1).35 To the best of our knowledge, ligand 1 is one of the two examples of water-soluble iminophosphoranes prepared to date.34 It is well known that most IMs in contact with water or polar solvents break down to the corresponding amine and phosphine oxide.36 We have incorporated a CO group to stabilize the resulting water soluble IM ligands.34 Ligand 1 (solubility in H2O 10g/L or 28mM) is more stable in solution than ligand TPA=N-C(O)-2-NC5H4 (N,N-IM) prepared by our group to synthesize coordination compounds of d8 metals with potential anticancer properties.34 The half-life for 1 in D2O is 2 days while ligand TPA=N-C(O)-2-NC5H4 (N,N-IM) decomposed to 50% after 12 h.34 1 is stable in DMSO solution for over two weeks while in mixtures of d6-DMSO:D2O (1:1) the half-life is 14 days as opposed to 2 days for the N,N-IM ligand.34
Scheme 1.
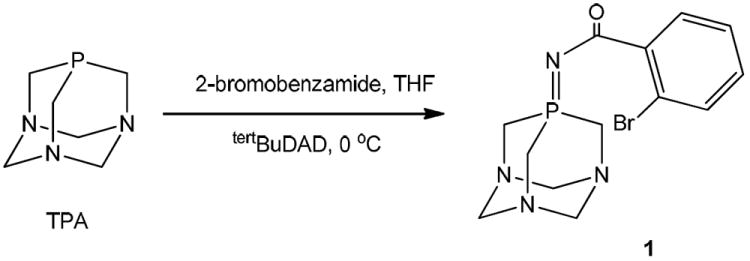
Preparation of water soluble ligand 1 by the Pomerantz22 method.
Oxidative addition of 1 to Pd2(dba)3 affords the orthopalladated dimer [Pd(μ-Br){C6H4(C(O)N=TPA-kC,N)-2}]2 (2) as a mixture of cis and trans isomers (1:1 molar ratio) where the iminophosphorane fragment behaves as a C,N-pincer ligand (Scheme 2 and experimental).
Scheme 2.
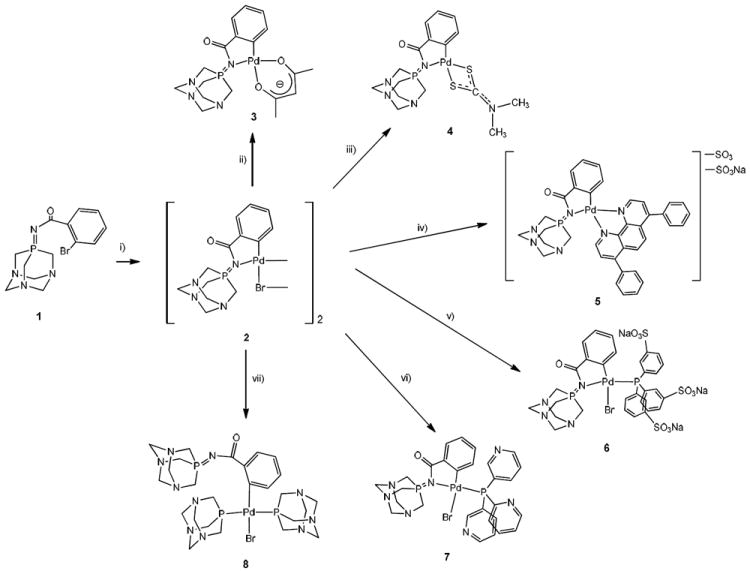
Preparation of di (2) and mononuclear (3-8) hydrophilic cyclopalladated iminophosphorane derivatives. i) Pd2(dba)3; ii) Tl(acac); iii) Na[S2CN(CH3)2]; iv) C12H6N2(C6H4SO3Na)2; v) P(mC6H4SO3Na)3; vi) P(3-Pyridyl)3; vii) TPA.
By addition of different ligands to 2 and subsequent cleavage of the bromide bridging system, a variety of hydrophilic or water-soluble mononuclear organometallic palladium(II) complexes of the type [Pd{C6H4(C(O)N=TPA-kC,N)-2}(L-L)] (L-L = acac (3); S2CNMe2 (4); 4,7-Diphenyl-1,10-phenanthrolinedisulfonic acid disodium salt C12H6N2(C6H4SO3Na)2 (5)); [Pd{C6H4(C(O)N=TPA-kC,N)-2}(L)Br] (L = P(mC6H4SO3Na)3 (6); P(3-Pyridyl)3 (7)) and, [Pd{C6H4(C(O)N=TPA)-2}(TPA)2Br] (8) are obtained (scheme 2) in moderate to high yields (see experimental). All complexes were characterized by means of NMR and IR spectroscopy, elemental analyses, mass spectrometry and conductivity measurements (see experimental). These techniques confirmed that the structures proposed for compounds 2-8 (scheme 2) are the most plausible ones. In the case of the reactions of 2 with one neutral phosphine (v) and vi) in Scheme 2) the single isomer in which the incoming ligand is coordinated trans to the nitrogen atom (6 and 7) is obtained (typical reactivity of C,N-cyclopalladated halide-dimers).37 The reaction of 2 with TPA gives exclusively derivative [Pd{C6H4(C(O)N=TPA)-2}(TPA)2Br] (8) with two TPA phosphines and with the iminophosphorane ligand coordinated solely through the C-atom. We have observed the same effect in the reaction of [Au{κ2-C,N-C6H4(PPh2=N(C6H5)-2}Cl(CH3CN)]PF6 with TPA which affords compound [Au{C6H4(PPh2=N(C6H5)-2}(TPA)2Cl]PF6 with two TPA phosphines.32
Compounds 2, 3 and 4 are not soluble in water but they are soluble in mixtures of DMSO:H2O (1:99) in concentrations ranging from 0.4 mM to 0.8 mM (which allows for their solubilization for further biological testing). Compounds 5-8 result more hydrophilic and can be dissolved in water (concentrations ranging from 0.5 to 1.0 mM). The stability of the compounds in solution can be easily ascertained by 31P{1H} NMR spectroscopy in d6-DMSO or mixtures d6-DMSO:D2O (50:50) (see supplementary information). Most compounds are quite stable in d6-DMSO solution with half-lives of several months (2-4, 6, 7) while others are stable over 2 days (5, 8). In mixtures d6-DMSO:D2O (50:50) the compounds are also very stable with half-lives of several months (3, 4, 6), 1-2 weeks (2, 7, 8) or above 4 days (5). The most unstable complex is that with the bathophenantroline ligand (5) but this compound is stable under biological testing conditions (24 h).
The crystal structure of 3 (determined by single crystal x-ray diffraction study) is depicted in Fig. 1. Selected bond lengths and angles are collected in Table 1. The geometry about the Pd(II) centers is pseudo-square planar with the C(18)-Pd(1)-N(1) angle of 81.35(16)° suggesting a rigid ‘bite’ angle of the chelating ligand. The Pd center is on an almost ideal plane with negligible deviations from the least-squares plane. The coordination plane for Pd and the metallocyclic plane are slightly twisted to give an angle of 5.5° between them.
Figure 1.
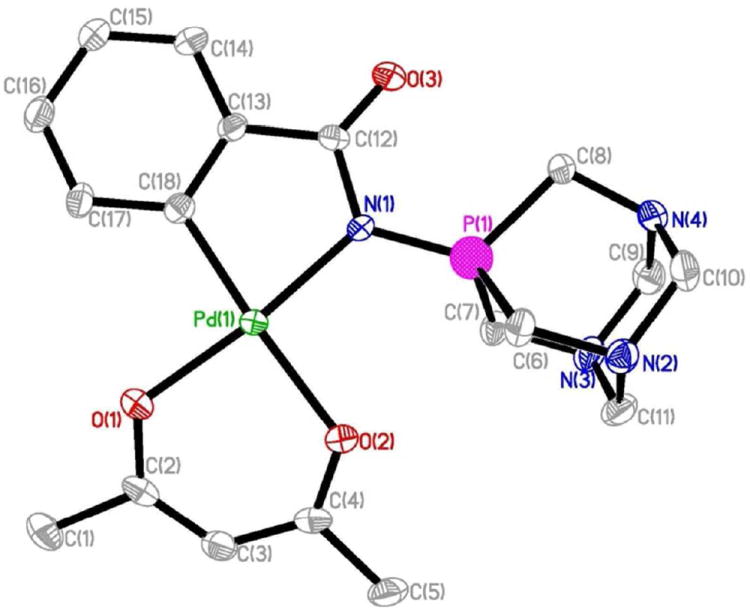
Molecular structure of the compound [Pd{C6H4(C(O)N=TPA-kC,N)-2}(acac-O,O’)] 3 with the atomic numbering scheme.
Table 1.
Selected Structural Parameters of complex 3 obtained from X-ray single crystal diffraction studies. Bond lengths in Å and angles in °.
| Pd(1)-C(18) | 1.966(4) | C(18)-Pd(1)-O(1) | 92.08(15) |
| Pd(1)-O(1) | 2.023(3) | C(18)-Pd(1)-N(1) | 81.35(16) |
| Pd(1)-N(1) | 2.037(4) | O(1)-Pd(1)-N(1) | 173.02(13) |
| Pd(1)-O(2) | 2.094(3) | C(18)-Pd(1)-O(2) | 175.51(14) |
| N(1)-P(1) | 1.643(4) | O(1)-Pd(1)-O(2) | 91.49(12) |
| O(1)-C(2) | 1.289(6) | N(1)-Pd(1)-O(2) | 94.96(13) |
| N(1)-C(12) | 1.394(6) | C(2)-O(1)-Pd(1) | 123.3(3) |
| C(12)-N(1)-P(1) | 120.5(3) | ||
| C(18)-Pd(1)-O(1) | 92.08(15) | C(12)-N(1)-Pd(1) | 115.8(3) |
| C(18)-Pd(1)-N(1) | 81.35(16) | P(1)-N(1)-Pd(1) | 123.4(2) |
The main distances Pd-N(1) and Pd-C(18) of 2.037(4) and 1.966(4) Å found for 3 are a little shorter or similar than those found in related carbonyl-stabilized iminophosphorane complexes where the cyclometalation occurs in the benzyl ring like [Pd{C6H4(C(O)N=P(p-tol)3-kC,N)-2}{acac-O,O’)] (2.0695(16) and 1.961(2) Å),38 [Pd(μ-Cl){C6H4(C(O)N=P(m-tol)3-kC,N)-2}]2 (2.085(2) and 1.979(2) Å)38 or in {Pd(μ-Cl){C6H3(Me-3)(C(O)N=PPh3-2}]2 (2.043(2) and 1.962(3) Å).39 The Pd1-O1 (2.023(3) Å) and Pd1-O2 (2.094(3) Å) bond distances fall in the usual range of distances found for chelating acac ligands.38 The P(1)-N(1) bond length in 3 of 1.643(4) Å is very similar to that in [Pd{C6H4(C(O)N=P(p-tol)3-kC,N)-2}{acac-O,O’)] (1.6457(18) Å),38 [Pd(μ-Cl){C6H4(C(O)N=P(m-tol)3-kC,N)-2}]2 (range 1.644-1.610 Å)28l (1.644(2) Å),38 [Pd(C6H4-2-PPh2=N-C(O)-2NC5H4-k-C,N,N)Cl] (1.649(2) Å)37 or in [PdCl2(TPA=N-C(O)-2-NC5H4)] (1.658(2) Å)34 but considerably longer than in other semi-stabilized cyclopalladated complexes.37 This elongation of the P-N bond is due not only to the coordination of the N atom to the palladium center but to the delocalization of charge density through the P-N-C-O system due to the presence of a CO group in the IM ligand.37
2. Biological Activity
2.1. Cytotoxicity Studies
Cytotoxicity results for ligand 1 and the new organometallic compounds (2-8) are collected in Table 2. The IC50 values for the decomposition product of ligand 1 (TPA=O) are also collected for comparison purposes. The cytotoxicity (by a modification of the MTT-reduction method, see Experimental) was evaluated against two selected cell lines: human Jurkat-T acute lymphoblastic leukemia cells and DU-145 human prostate cancer cells. Cells were incubated in the presence of the compounds for 24 h. The sensitivity of T-cell leukemia Jurkat cells to compounds cisplatin, 1-8, and TPA=O was compared to that of normal T-lymphocytes (peripheral blood mononuclear cells: PBMC). Ligand 1 and the phosphine oxide TPA had IC50 values above 500 micromolar in all cell lines studied and were considered as no toxic. The more cytotoxic derivatives are the more lipophilic palladium complexes 2 and 3. This behaviour has been also found for other cyclometallated compounds.25a We expected that 4 with the chemoprotectant ditiocarbamate ligand would display a higher cytotoxicity (as found for cycloaurated iminophosphorane complexes)32 but it was not the case. In general the IC50 values in DU145 are quite high and for 3 comparable to that of cisplatin (Table 2). 2 results more cytotoxic than cisplatin for this cell line. 2 and 3 have values of IC50 in T-cell leukemia Jurkat cells similar to those of cisplatin and very close to those obtained by previously reported tetranuclear cyclopalladated compounds with a p-isopropylbenzaldehyde thiosemicarbazone [Pd(p-is.TSCN)],22d dinuclear derivatives of the type [Pd2L2(μ-dppe)Cl2] (L = 1-methyl-5-phenyl-1H-1,4-benzodiazepin-2(3H)-one)25b and mononuclear [Pd(C2,N-S(-)dmpa)(dppf)]Cl.27 2 and 3 resulted three and two times more cytotoxic to T-cell leukemia Jurkat cells than the IM coordination complex [PdCl2(TPA=N-C(O)-2-NC5H4)].34 2 and 3 were 26 (2) or 11 times (3) less toxic to the normal lymphocytes (PMBC) than to the leukemia cell line. We have found, however, that cisplatin is 70 times less toxic to PMBC than to Jurkat cell lines.34
Table 2.
IC50 (μM) of cisplatin, ligand 1, phosphine oxide TPA=O and metal complexes 2-8 complexes in human cell lines.a All compounds were dissolved in 1% of DMSO and diluted with water before addition to cell culture medium for a 24 h incubation period. Cisplatin was dissolved in water.
| Jurkat | PBMC | DU-145 | |
|---|---|---|---|
| Cisplatinb | 7.4±2.1 | 500±18 | 79.2±15.8 |
| TPA=O | >500 | >500 | >500 |
| 1 | >500 | >500 | >500 |
| 2 | 8.1±1.1 | 209±11 | 49.3±3.1 |
| 3 | 10.5±1.8 | 118±11 | 96.2±12 |
| 4 | 142±9 | 186±9 | 135±27 |
| 5 | 89±18 | >500 | 187±16 |
| 6 | 322±29 | >500 | 318±20 |
| 7 | 101±20 | 298±14 | 299±25 |
| 8 | 374±40 | >500 | 448±38 |
Data are expressed as mean ± SD (n =3).
Values obtained by the same MTT assay conditions (ref 34).
We also studied the activity of compounds 2, 3 and cisplatin against apoptosis-resistant cells (Fig. 2). Multidomain proapoptotic proteins Bax and Bak are essential for the onset of mitochondrial permeabilization and apoptosis through the intrinsic pathway. Importantly, we observed that compounds 2 and, especially 3, exhibited toxicity to cisplatin resistant Jurkat shBak cells (Bax/Bak-deficient Jurkat cells). We have demonstrated previously that cisplatin at 25 μM caused death of around 60% of Jurkat control (CN) cells while Jurkat-shBak cells were very resistant to cisplatin (less than 8% death).33 Figure 2 depicts that 2 and 3 (at 50 μM) caused death of around 25% (2) and 75% (3) of Jurkat-shBak cells. This could be an advantage over cisplatin, whose activity is totally dependent on the presence of functional Bax and Bak.
Figure 2.
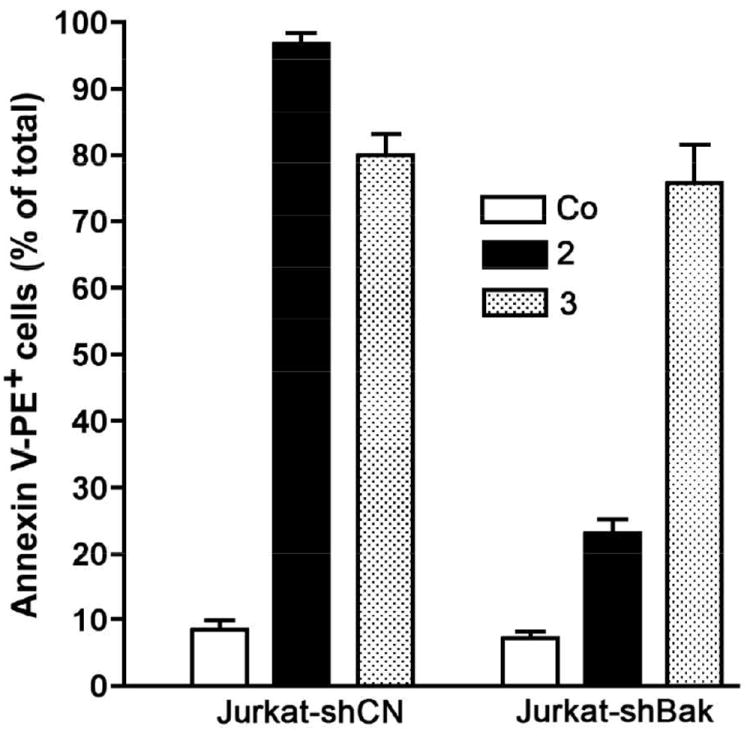
Jurkat-CN or Jurkat-shBak cells were treated with 2 or 3 (50 μM) for 24h and PS exposure was evaluated by flow cytometry after labeling with annexinV-PE. Co (control): cells cultured in the presence of 1% DMSO.
Bax-independent apoptosis has been proposed as the death mechanism induced by a biphosphinic palladacycle complex in K562 leukemic cells.27 Toxicity to Bax/Bak-deficient Jurkat cells has also been found for iminophosphorane organogold(III)33 and coordination Pt(II) complexes.34 In contrast, the coordination palladium compound [PdCl2(TPA=N-C(O)-2-NC5H4)] did not cause death of Jurkat-shBak cells.34 Cyclopalladation seems to have a positive effect on the cytotoxicity of IM-Pd complexes on cisplatin-sensitive and cisplatin-resistant leukemia cell lines.
2.2. Interactions of 2 and 3 with DNA and with human serum albumin (HSA)
Since DNA replication is a key event for cell division, it is among critically important targets in cancer chemotherapy. Most cytotoxic platinum drugs form strong covalent bonds with the DNA bases.40 However, a variety of platinum compounds act as DNA intercalators upon coordination to the appropriate ancillary ligands.41 There are also reports on palladium derivatives interacting with DNA in covalent42,43 and noncovalent ways.22d,44 We performed agarose gel electrophoresis studies on the effects of compounds 2 and 3 on plasmid (pBR322) DNA (Fig. 3). This plasmid has two main forms: OC (open circular or relaxed form, Form II) and CCC (covalently closed or supercoiled form, Form I).
Fig. 3.
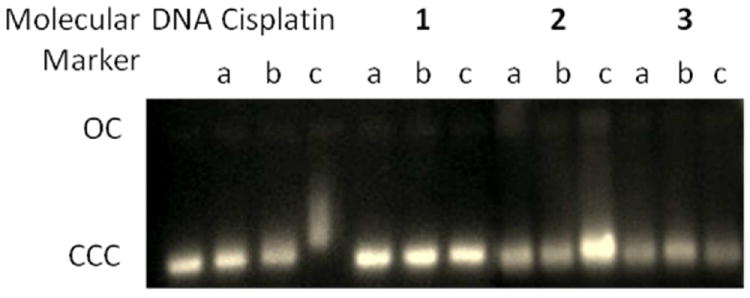
Electrophoresis mobility shift assays for cisplatin, ligand 1 and compounds 2 and 3 (see Experimental for details). DNA refers to untreated plasmid pBR322. a, b, and c correspond to metal/DNA ratios of 0.25, 0.5, and 1.0 respectively.
Changes in electrophoretic mobility of both forms are usually taken as evidence of metal-DNA binding. Generally, the larger the retardation of supercoiled DNA (CCC, Form I), the greater the DNA unwinding produced by the drug.45 Binding of cisplatin to plasmid DNA, for instance results in a decrease in mobility of the CCC form and an increase in mobility of the OC form (see lanes a, b and c for cisplatin in Fig. 3).45 Treatment with increasing amounts of ligand 1 does not cause any shift for either form, consistent with no unwinding or other change in topology under the chosen conditions. Treatment with increasing amounts of compound 3 has a similar effect. Treatment with increasing amounts of compound 2 barely retards the mobility of the faster-running supercoiled form (Form I) at high molar ratios (c) but to a much lesser extent than the retardation produced by cisplatin. Organogold(III) compounds containing iminophosphorane ligands did not interact with DNA.32 Coordination iminophosphorane compounds of platinum(II) and palladium(II) of the type [MCl2(TPA=N-C(O)-2-NC5H4)] showed a retardation of the faster-running supercoiled form (Form I) especially at high molar ratios in a way different from that produced by cisplatin.34
In conclusion, the experiments probing DNA-drug interactions showed that the new cyclopalladated iminophosphorane biologically active complexes have no (3) or very little (2) interaction with plasmid (pBR322) DNA pointing to an alternative biomolecular target for these compounds. Mitochodrial damage by interaction of some cyclopalladated complexes containing phosphines with membrane proteins26-28 and inhibition of cathepsin B25,26 have been reported as the plausible causes of their toxicity to cancer cell lines. In contrast, orthometallated complexes with thiosemicarbazones were found to have an enhanced capacity to form DNA interstrand cross-links in comparison with cisplatin.22e-f
Human serum albumin (HSA) is the most abundant carrier protein in plasma and is able to bind a variety of substrates including metal cations, hormones and most therapeutic drugs. It has been demonstrated that the distribution, the free concentration and the metabolism of various drugs can be significantly altered as a result of their binding to the protein.46 HSA possesses three fluorophores, these being tryptophan (Trp), tyrosine (Tyr) and phenylalanine (Phe) residues, with Trp214 being the major contributor to the intrinsic fluorescence of HSA. This Trp fluorescence is sensitive to the environment and binding of substrates or changes in conformation that can result in quenching (either dynamic or static). The fluorescence spectra of HSA in the presence of increasing amounts of ligand 1, compounds 2-8 and cisplatin were recorded in the range of 300-450 nm upon excitation of the tryptophan residue at 295 nm (Fig. 4). The compounds caused a concentration dependent quenching of fluorescence without changing the emission maximum or shape of the peaks (2,3, 6-8), as seen in Fig. 4 for compound 3. For compound 5 a shift of the emission maximum was observed (from 340 nm to 390 nm). All this data indicates an interaction of the palladium compounds with HSA. The fluorescence data was analyzed by the Stern-Volmer equation. While a linear Stern-Volmer plot is indicative of a single quenching mechanism, either dynamic or static, the positive deviation observed in the plots of F0/F versus [Q] of our compounds (Fig. 4) is indicative of the presence of different binding sites in the protein.47 In this graph higher quenching by the iminophosphorane complexes was observed compared to that of cisplatin under the chosen conditions. This is probably an indication of the faster hydrolysis in aqueous solution of our compounds compared to that of cisplatin.
Figure 4.
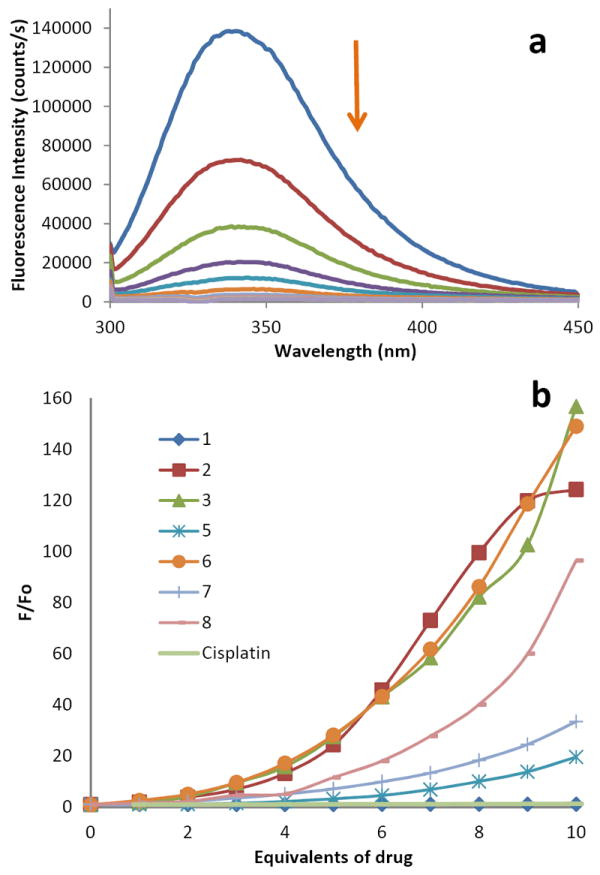
(a) Fluorescence titration curve of HSA with compound 2. Arrow indicates the increase of quencher concentration. (b) Stern-Volmer plot for quenching with ligand 1 and compounds 2, 3, 5-8 and cisplatin. In the measurements of the fluorescence titration curve of HAS with compound 4 the appearance of a precipitate prevented us to obtain a valid reading and therefore the Stern-Volmer plot was not included in graph b).
We have found a similar behaviour for coordination iminophosphorane complexes of d8 metals for which we also observed a dependent concentration quenching.34 In the case of [MCl2(TPA=N-C(O)-2-NC5H4)] (M = Pd, Pt) isothermal titration calorimetry (ITC)34 showed two different binding interactions which explained the lack of linearity observed in the fluorescence quenching studies, as the Stern-Volmer method assumes all binding sites to be equivalent. We believe that a similar behaviour is observed for the cyclopalladated compounds described here. For [PtCl2(TPA=N-C(O)-2-NC5H4)] we were able to demonstrate by circular dichroism studies that its binding to HSA was reversed by addition of a chelating agent (citric acid) and by a decrease in pH. In the case of the palladium compound34 and compounds 2,3 described here, the CD reversibility experiments were not conclusive. Further analysis by a quantitative technique (atomic absorption spectrometry) is underway since studies of the reversibility of the binding of metallodrugs to HSA are very relevant to asses their potential efficacy in vivo.
Conclusion
We have prepared a new water-soluble iminophosphorane which serve as an excellent precursor to iminophosphorane cyclopalladated complexes with varying hydrophilic:lipophilic ratios stable in DMSO and DMSO:H2O solutions under biological testing conditions. We found that the most lipophilic compounds from the series, [Pd(μ-Br){C6H4(C(O)N=TPA-kC,N)-2}]2 (2) and [Pd{C6H4(C(O)N=TPA-kC,N)-2}(acac)] 3 display the higher cytotoxicity against T-Jurkat leukemia cell while being less toxic to normal T-lymphocytes (PBMC). The IC50 values were similar to those reported for other cyclopalladated compounds with promising antitumor properties and higher than an iminophosphorane coordination palladium complex [PdCl2(TPA=N-C(O)-2-NC5H4)],34 previously described by us. Importantly, 2, and especially 3 showed toxicity against cisplatin resistant Jurkat sh-Bak cells (Bax/Bak-deficient Jurkat cells). This could be an advantage over cisplatin, whose activity is totally dependent on the presence of functional Bax and Bak. It was demonstrated that these compounds have no (3) or very little (2) interaction with plasmid (pBR322) DNA pointing to an alternative biomolecular target for their cytotoxic effect. All the compounds show an interaction with human serum albumin (HSA) faster than that of cisplatin. We envision that an appropriate modification of IM ligands and subsequent orthopalladation can afford new cyclopalladated complexes with improved anticancer properties. In addition, some of the water-soluble non-toxic organometallic palladium complexes described here (such as [Pd{C6H4(C(O)N=TPA-kC,N)-2}(L)Br] (L = P(mC6H4SO3Na)3 (6); P(3-Pyridyl)3 (7)) could serve as stable homogeneous catalysts in water.
EXPERIMENTAL SECTION
All manipulations involving air-free syntheses were performed using standard Schlenk-line techniques under a nitrogen atmosphere or in a glove-box MBraun MOD System. Solvents were purified by use of a PureSolv purification unit from Innovative Technology, Inc. The substrates TPA and 4,7-Diphenyl-1,10-phenanthrolinedisulfonic acid disodium salt were purchased from Sigma-Aldrich, Pd2(dba)3 was purchased from Strem chemicals and used without further purification. Phosphine P(3-pyridyl)3 was prepared by a reported method.48 Phosphine TPPTS P(mC6H4SO3Na)3 was generously donated by Prof. László T. Mika (Eötvös University, Budapest, Hungary). NMR spectra were recorded in a Bruker AV400 (1H NMR at 400MHz, 13C NMR at 100.6 MHz, 31P NMR at 161.9 MHz). Chemical shifts (δ) are given in ppm using CDCl3, d6-DMSO or D2O as solvent, unless otherwise stated. 1H and 13C chemical shifts were measured relative to solvent peaks considering TMS = 0 ppm; 31P{1H} was externally referenced to H3PO4 (85%). Infrared spectra (4000-250 cm-1) were recorded on a Nicolet 6700 FT-IR spectrophotometer on KBr pellets. Elemental analyses were performed on a Perkin Elmer 2400 CHNS/O Analyzer, Series II. Mass spectra (ESI) were performed on an Agilent Analyzer or a Bruker Analyzer. Conductivity was measured in an OAKTON pH/conductivity meter in CH3CN, acetone or H2O solutions (10-3M). Circular Dichroism spectra were recorded using a Chirascan CD Spectrometer equipped with a thermostated cuvette holder. Electrophoresis experiments were carried out in a Bio-Rad Mini sub-cell GT horizontal electrophoresis system connected to a Bio-Rad Power Pac 300 power supply. Photographs of the gels were taken with an Alpha Innotech FluorChem 8900 camera. Fluorescence intensity measurements were carried out on a PTI QM-4/206 SE Spectrofluorometer (PTI, Birmingham, NJ) with right angle detection of fluorescence using a 1 cm path length quartz cuvette.
Synthesis
TPA=N-C(O)-2BrC6H4 (C,N-IM) (1)
TPA (0.157 g, 1.0 mmol) and 2-bromobenzamide (0.200 g, 1.0 mmol) were placed in a Schenck flask under nitrogen. Dry and degassed THF (15 mL) was added and to this solution, tBuDAD (0.230 g, 1.0 mmol) in dry and degassed THF (4 mL) was added dropwise at 0 °C. The reaction was left stirring to warm up for 15 hours. After this period the yellow reaction mixture had become colorless. The solvent was reduced in vacuo to a minimum (< 2 mL) and Et2O (10 mL) was added, giving a white solid that was filtered off and dried in vacuo. Yield: 0.30 g (86%). Anal. Calcd for C13H16BrN4OP (354.02): C, 43.96; H, 4.54; N, 15.77. Found: C, 43.81; H, 4.31; N, 15.93. MS (ESI+) [m/z]: 355.03 [M+H]+. 31P{1H} NMR: δ -31.2 (s, CDCl3), -29.8 (s, DMSO-d6), -25.1 (s, D2O). 1H NMR (CDCl3): δ 7.66 (1H, d, 3JHH 7 Hz, H6, C6H4), 7.56 (1H, d, 3JHH 7 Hz, H3, C6H4), 7.28 (1H, dd, H5, 3JHH 8 Hz, 3JHH 3 Hz, C6H4), 7.18 (1H, t, 3JHH 7 Hz, H4, C6H4), 4.34 (6H, d, JPH 4 Hz, PCH2N), 4.40 (AB system, 6H, NCH2N). 13C{1H} NMR (CDCl3): δ 173.1 (s, C=O), 133.5 (s, C6H4), 130.2 (s, C6H4), 129.8 (s, C6H4), 127.0 (s, C6H4), 72.8 (d, JPC 9 Hz, NCH2N), 52.5 (d, JPC 45 Hz, PCH2N). IR (cm-1): ν 1574 (C=O), 1340 (P=N). Conductivity (MeCN): Λ = 1.3 μS/cm. Solubility: 28 mM or 10 mg/mL (H2O).
[Pd(μ-Br){C6H4(C(O)N=TPA-kC,N)-2}]2 (2)
To a suspension of Pd2(dba)3 (0.457 g, 0.50 mmol) in dry and degassed CH2Cl2 (15 mL), iminophosphorane 1 (0.354 g, 1.0 mmol) was added and the resulting mixture was stirred for 15 h at room temperature. The initial purple suspension evolved to a yellowish solution with some Pd0 in suspension, which was filtered over Celite. The clear yellow solution was evaporated to a small volume (< 2 mL) and Et2O (20 mL) was added. By continuous stirring, 2 was obtained as a yellow solid. Complex 2 was characterized by NMR as a mixture (1/1 molar ratio) of the geometric cis and trans isomers. Yield: 0.170 g (82%). Anal. Calcd for C26H32Br2N8O2P2Pd2 (922.86): C, 33.81; H, 3.49; N, 12.14. Found: C, 34.33; H, 3.39; N, 12.13. Although these results are outside the range viewed as establishing analytical purity, they are provided to illustrate the best values obtained to date. The 31P, 13C and 1H NMR spectra of 2 appear in the Supporting Information. MS (ESI+) [m/z]: 945.09 [M+Na]+. 31P{1H} NMR: δ -17.2 and -17.3 (cis and trans isomers, CDCl3), -16.2 and -13.0 (cis and trans isomers, DMSO-d6). 1H NMR (DMSO-d6): δ 7.68 (2H, br, H5), 7.11 (2H, br, H2), 7.01 (4H, br, H3+H4), 4.58 (12H, d, JPH 9 Hz, PCH2N), 4.30 (AB system, 12H, NCH2N). 13C{1H} NMR (DMSO-d6): δ 188.9 (s, C=O), 143.3 (s, C1, C6H4), 134.8 (s, C2, C6H4), 130.5 (s, C6H4), 129.1 (s, C6H4), 128.2 (s, C6H4), 125.4 (s, C6H4), 72.5 (d, 3JPC 9 Hz, NCH2N), 54.4 (d, 1JPC 41 Hz, PCH2N). IR (cm-1): ν 1634 (C=O), 1294 (P=N). Conductivity (MeCN): Λ = 12.6 μS/cm. Solubility: 0.4 mM or 1.1 mg/mL (1:99 DMSO:H2O).
[Pd{C6H4(C(O)N=TPA-kC,N)-2}(acac-O,O’)] (3)
To a solution of 2 (0.060 g, 0.064 mmol) in CH2Cl2 (15 mL), Tl(acac) (0.039 g, 0.128 mmol) was added. The resulting suspension was stirred for 1h at rt, and then filtered over Celite. The clear solution was evaporated to dryness, and the residue was treated with cold n–hexane (15 mL), giving 3 as an off-white solid. It was filtered off and dried in vacuo. Yield: 0.034 g (55%). Anal. Calcd for C18H23N4O3PPd (480.57): C, 44.95; H, 4.82; N, 11.66. Found: C, 44.38; H, 4.56; N, 11.20. Although these results are outside the range viewed as establishing analytical purity, they are provided to illustrate the best values obtained to date. The 31P, 13C and 1H NMR spectra of 3 appear in the Supporting Information. MS (ESI+) [m/z]: 481.0619 [M+H]+. 31P{1H} NMR (CDCl3): δ -18.6 (s). 1H NMR (CDCl3): δ 7.56 (1H, d, 3JHH 8 Hz, H6, C6H4), 7.29 (1H, d, 3JHH 8 Hz, H3, C6H4), 7.20 (1H, t, 3JHH 7 Hz, H5, C6H4), 7.06 (1H, t, 3JHH 8 Hz, H4, C6H4), 5.39 (s, CH, acac), 4.68 (6H, d, JPH 9 Hz, PCH2N), 4.44 (AB system, 6H, NCH2N), 2.06 (s, 3H, CH3, acac), 1.60 (s, 3H, CH3, acac). 13C{1H} NMR (CDCl3): δ 187.2 (s, C=O, acac), 187.1 (s, C=O, acac), 146.4 (s, C1, C6H4), 139.2 (d, JPC 12 Hz, C2, C6H4), 130.6 (s, C6H4), 129.5 (s, C6H4), 127.2 (d, 4JPC 3 Hz, C6H4), 124.4 (s, C6H4), 100.3 (s, CH, acac), 71.4 (d, 3JPC 10 Hz, NCH2N), 52.2 (d, 1JPC 39 Hz, PCH2N), 29.2 (s, Me, acac), 28.0 (s, Me, acac). IR (cm-1): ν 1617 (C=O), 1570 (acac), 1518 (acac), 1302 (P=N). Conductivity (MeCN): Λ = 2.1 μS/cm. Solubility: 0.8 mM or 1.2 mg/mL (1:99 DMSO:H2O).
[Pd{C6H4(C(O)N=TPA-kC,N)-2}(S2CNMe2)] (4)
To a suspension of 2 (0.050 g, 0.054 mmol) in MeOH (35 mL) was added sodium dimethyldithiocarbamate as a solid. The suspension changed immediately from yellow to colorless. After stirring for 1 hour at room temperature, the solvent was removed in vacuo to dryness. Acetone (35 mL) was added and the resulting suspension was filtered through celite to remove the NaBr formed. The solvent was reduced in vacuo to a minimum (~2 mL) and Et2O (15 mL) was added affording an off-white solid (4) that was filtered off and dried in vacuo. Yield: 0.047 g (79%). Anal. Calcd for C16H22N5OPPdS2·2H2O: C, 35.72; H, 4.87; N, 13.02. Found: C, 36.19; H, 4.33; N, 12.88. MS (ESI+) [m/z]: 502.02 [M+H]+ (Calcd for C16H22N5OPPdS2 (501.00)). 31P{1H} NMR: δ -12.9 (s, DMSO-d6), -15.4 (s, Acetone-d6). 1H NMR (Acetone-d6): δ 7.27 (1H, d, 3JHH 7 Hz, H6, C6H4), 7.09 (1H, t, 3JHH 7 Hz, C6H4), 7.02 (1H, t, 3JHH 7 Hz, C6H4), 7.37 (1H, d, 3JHH 7 Hz, C6H4), 4.48 (6H, d, JPH 9 Hz, PCH2N), 4.37 (AB system, 6H, NCH2N), 3.29 (3H, s, Me), 3.25 (3H, s, Me). 13C{1H} NMR (Acetone-d6): δ 207.9 (s, S2CN), 184.0 (s, C=O), 151.0 (s, C1, C6H4), 138.5 (d, JPC 14 Hz, C2, C6H4), 132.2 (s, C6H4), 131.1 (s, C6H4), 128.1 (s, C6H4), 123.7 (s, C6H4), 70.9 (d, 3JPC 10 Hz, NCH2N), 51.6 (d, 1JPC 41 Hz, PCH2N), 39.4 (s, Me), 39.1 (s, Me). IR (cm-1): ν 1610 (C=O), 1294 (P=N). Conductivity (acetone): Λ = 1.7 μS/cm. Solubility: 0.7 mM or 1.0 mg/mL (1:99 DMSO:H2O).
[Pd{C6H4(C(O)N=TPA-kC,N)-2}{C12H6N2(C6H4SO3Na)2}] (5)
Sulphonated bathophenantroline (0.059 g, 0.10 mmol) was dissolved in MeOH (3 mL) and the minimum amount of water to obtain a clear solution that was then added slowly to a solution of 2 (0.046 g, 0.050 mmol) in CH2Cl2 (7 mL). The resulting mixture was stirred for 1 h at rt, after which all solvents were evaporated to dryness. The resulting residue was redissolved in MeOH and filtered through celite to remove the byproduct NaBr. The solvent was removed in vacuo and the resulting pale yellow residue 5 was washed with Et2O, filtered off and dried in vacuo. Yield: 0.072 g (78%). Anal. Calcd for C39H38N6NaO7PPdS2.7H2O: C, 44.47; H, 4.13; N, 7.98; S, 6.09. Found: C, 44.63; H, 3.72; N, 7.80; S, 6.80. MS (ESI+) [m/z]: 737.20 [M+Na]+ (Calcd for M=C37H32N6OPPd (714.08)). 31P{1H} NMR (DMSO-d6): δ -26.0 (s). 1H NMR (DMSO-d6): δ 9.29 (2H, s, Ar bathoPhen), 8.21 (2H, s, Ar bathoPhen), 8.07 (2H, s, Ar bathoPhen), 7.99 (2H, s, Ar bathoPhen), 7.91 (2H, s, Ar bathoPhen), 7.70 (2H, s, Ar bathoPhen), 7.41 (1H, d, 3JHH 7 Hz, H6, C6H4), 7.14 (1H, d, 3JHH 7 Hz, H3, C6H4), 7.03 (1H, t, 3JHH 5 Hz, H5, C6H4), 6.93 (1H, t, 3JHH 5 Hz, H4, C6H4), 4.47 (6H, d, JPH 9 Hz, PCH2N), 4.42 (6H, s, NCH2N). 13C{1H} NMR (DMSO-d6): δ 181.2 (s, C=O), 150.4 (d, J 11 Hz, C1, C6H4), 148.6 (d, JPC 10 Hz, Ar bathoPhen), 136.7 (d, JPC 18 Hz, Ar bathoPhen), 136. 1 (d, J 14 Hz,), 133.0 (s, C2, C6H4), 132.1 (s, C6H4), 132.0 (s, Ar bathoPhen), 132.0 (s, C6H4), 131.6 (s, Ar bathoPhen), 131.3 (s, Ar bathoPhen), 130.7 (s, Ar bathoPhen), 129.8 (s, C6H4), 129.0 (s, Ar bathoPhen), 128.9 (s, C6H4), 128.9 (s, Ar bathoPhen), 128.2 (d, JPC 11 Hz, Ar bathoPhen), 124.2 (s, Ar bathoPhen), 71.5 (d, 3JPC 9 Hz, NCH2N), 54.2 (d, 1JPC 41 Hz, PCH2N). IR (cm-1): ν 1620 (C=O), 1187 (SO3 anti), 1031 (SO3 sym). Conductivity (H2O): Λ = 145.3 μS/cm. Solubility: 1.0 mM or 0.90 mg/mL (H2O).
[Pd{C6H4(C(O)N=TPA-kC,N)-2}{P(mC6H4SO3Na)3}Br] (6)
TPPTS (0.072 g, 0.128 mmol) was dissolved in MeOH (3 mL) and the minimum amount of water to obtain a clear solution that was then added slowly to a solution of 2 (0.060 g, 0.064 mmol) in CH2Cl2 (7 mL). The resulting mixture was stirred for 3 h at rt, after which all solvents were evaporated to dryness, and the residue was treated with Et2O (15 mL), affording 6 as an off-white solid that was filtered off and dried in vacuo. Yield: 0.092 g (70%). Anal. Calcd for C31H28BrN4Na3O10P2PdS3.6H2O: C, 32.72; H, 3.54; N, 4.92; S, 8.45. Found: C, 32.43; H, 3.45; N, 5.16; S, 8.07. MS (ESI+) [m/z]: 882.9577 [(M-H)-HBr] (M= C31H31BrN4O10P2PdS3). 31P{1H} NMR (DMSO-d6): δ 43.4 (s, TPPTS) and -14.0 (s, 1). 1H NMR (DMSO-d6): δ 7.93 (3H, d, 3JHH 10 Hz, Ar TPPTS), 7.73 (3H, d, 3JHH 8 Hz, Ar TPPTS), 7.62 (3H, t, 3JHH 8 Hz, Ar TPPTS), 7.42 (3H, t, 3JHH 6 Hz, Ar TPPTS), 7.27 (1H, d, 3JHH 7 Hz, H6, C6H4), 6.84 (1H, t, 3JHH 7 Hz, C6H4), 6.51 (1H, t, 3JHH 7 Hz, C6H4), 6.32 (1H, t, 3JHH 7 Hz, C6H4), 4.92 (6H, d, JPH 10 Hz, PCH2N), 4.36 (6H, s, NCH2N). 13C{1H} NMR (DMSO-d6): δ 181.2 (s, C=O), 150.5 (s, C1, C6H4), 148.2 (d, JPC 10 Hz, Ar TPPTS), 140.9 (d, JPC 18 Hz, C2, C6H4), 136.7 (d, JPC 18 Hz, Ar TPPTS), 136.0 (d, JPC 13 Hz, Ar TPPTS), 131.6 (s, Ar TPPTS), 131.3 (s, C6H4), 131.2 (s, C6H4), 128.8 (s, Ar TPPTS), 128.6 (s, C6H4), 128.1 (d, JPC 16 Hz, Ar TPPTS), 124.2 (s, C6H4), 71.4 (d, JPC 15 Hz, NCH2N), 54.2 (d, JPC 41 Hz, PCH2N). IR (cm-1): ν 1613 (C=O), 1309 (P=N), 1198 (SO3 anti), 1038 (SO3 sym). Conductivity (H2O): Λ = 241.3 μS/cm. Solubility: 0.9 mM or 0.9 mg/mL (H2O).
[Pd{C6H4(C(O)N=TPA-kC,N)-2}{P(3py)3}Br] (7)
To a solution of 2 (0.050 g, 0.054 mmol) in CH2Cl2 (7 mL) in a Schlenck flask under nitrogen was added tris-3-pyridylphosphine (0.029 g, 0.109 mmol) in CH2Cl2 (1 mL). The reaction mixture was stirred at rt for 2 h, after which the solvent was reduced to a minimum and Et2O was added affording 7 as a pale brown solid that was filtered off and dried in vacuo. Yield: 0.070 g (87%). Anal. Calcd for C28H28BrN7OP2Pd.0.5Et2O: C, 47.17; H, 4.35; N, 12.84. Found: C, 47.51; H, 4.14; N, 12.90. MS (ESI+) [m/z]: 646.0867 [(M+H)-HBr](Calcd for C28H28BrN7OP2Pd (725.0049)). 31P{1H} NMR: δ -15.9 (s, 1) and 30.9 (br, PPy3) (CDCl3), -14.1 (s, 1) and 29.0 (br, PPy3) (DMSO-d6). 1H NMR (CDCl3): δ 8.88 (3H, d, 3JHH 4 Hz, Py), 8.73, (3H, s), 8.07 (3H, t, 3JHH 9 Hz, Py), 7.51 (1H, d, 3JHH 8 Hz, H6, C6H4), 7.38 (3H, t, 3JHH 4 Hz, Py), 6.96 (1H, t, 3JHH 7 Hz, C6H4), 6.60 (1H, t, 3JHH 8 Hz, C6H4), 6.27 (1H, d, 3JHH 8 Hz, C6H4), 4.95 (6H, d, JPH 9 Hz, PCH2N), 4.43 (6H, s, NCH2N). 13C{1H} NMR (CDCl3): δ 158.2 (s, PPy3), 154.4 (d, 1JPC 11 Hz, PPy3), 151.9 (s, PPy3), 150.5 (s, C1, C6H4), 142.5 (s, C2, C6H4), 136.4 (s, PPy3), 131.0 (s, C6H4), 129.7 (s, C6H4), 124.7 (s, C6H4), 123.4 (s, C6H4), 123.2 (s, PPy3), 72.5 (d, 3JPC 10 Hz, NCH2N), 55.1 (d, 1JPC 42 Hz, PCH2N). IR (cm-1): ν 1640 (C=O), 1297 (P=N). Conductivity (MeCN): Λ = 11.7 μS/cm. Solubility: 0.5 mM or 1.0 mg/mL (H2O).
[Pd{C6H4(C(O)N=TPA)-2}(TPA)2Br] (8)
To a solution of 2 (0.060 g, 0.065 mmol) in CH2Cl2 (7 mL) was added TPA (0.041 g, 0.261mmol) as a solid. The reaction was left stirring at rt for 30 min after which the solvent was reduced to a minimum and Et2O was added affording an off-white solid (8) that was filtered off and dried in vacuo. Yield: 0.082 g (82%) Anal. Calcd for C25H40BrN10OP3Pd.CH2Cl2 (774.0818): C, 36.28; H, 4.92; N, 16.27. Found: C, 36.74; H, 5.18; N, 16.04. MS (ESI+) [m/z]: 776.08 [M+H]+ (Calcd for C25H40BrN10OP3Pd (775.8918)). 31P{1H} NMR: δ -30.5 (s, 1) and -68.7 (br, TPA) (CDCl3). 1H NMR (CDCl3): δ 7.87 (1H, dd, 3JHH 8, JPH 2 Hz, H6, C6H4), 8.22 (1H, d, 3JHH 7 Hz, H3, C6H4), 7.14 (1H, td, 3JHH 7 Hz, JPH 2 Hz, H5, C6H4), 7.03 (1H, d, 3JHH 8 Hz, H4, C6H4), 4.45 (AB system, 6H, NCH2N 1), 4.39 (6H, d, JPH 8 Hz, PCH2N 1), 4.35 (12 H, virtual t, PCH2N TPA), 3.89 (12H, s, NCH2N TPA). 13C{1H} NMR (CDCl3): δ 180.7 (d, JPC 9 Hz, C=O), 148.8 (s, C1, C6H4), 141.3 (s, JPC 16 Hz, C2, C6H4), 137.3 (s, C6H4), 130.7 (s, C6H4), 130.5 (s, C6H4), 123.7 (s, C6H4), 72.1 (s, NCH2N, TPA), 72.7 (d, 3JPC 9 Hz, NCH2N), 53.2 (d, 1JPC 44 Hz, PCH2N), 51.1 (s, PCH2N, TPA). IR (cm-1): ν 1575 (C=O), 1321 (P=N). Conductivity (MeCN): Λ = 22.0 μS/cm. Solubility: 0.8 mM or 1.2 mg/mL (H2O).
X-Ray crystallography
Single crystals of 3 (see details in Table 2) were mounted on a glass fiber in a random orientation. Data collection was performed at room temperature on a Kappa CCD diffractometer using graphite monochromated Mo-Kα radiation (λ=0.71073 Å). Space group assignments were based on systematic absences, E statistics and successful refinement of the structures. The structures were solved by direct methods with the aid of successive difference Fourier maps and were refined using the SHELXTL 6.1 software package. All non-hydrogen atoms were refined anisotropically. Hydrogen atoms were assigned to ideal positions and refined using a riding model. Details of the crystallographic data are given in Table 2. These data can be obtained free of charge from The Cambridge Crystallographic Data Center via www.ccdc.cam.ac.uk/data_request/cif. (CCDC 887963) or in the supporting information. 3: Crystals of 3 (colorless prisms with approximate dimensions 0.14 × 0.16 × 0.22 mm) were obtained from a solution of 3 in CHCl3/n-hexane by slow diffusion of Et2O at RT.
Determination of Compound Cytotoxicity
The human T-cell leukemia Jurkat (clone E6.1) from the ATCC collection and the prostate carcinoma DU-145 were routinely cultured in RPMI 1640 medium supplemented with 5% FCS, L-glutamine and penicillin/streptomycin (hereafter, complete medium). Blood samples from healthy donors were obtained from the Banco de Sangre y Tejidos de Aragón. All subjects gave written informed consent and the Ethical Committee of Aragón approved the study. Normal Tlymphocytes (peripheral blood mononuclear cells: PBMC) were freshly isolated by Ficoll-Paque density centrifugation. After isolation, normal PBMC were kept in RPMI 1640 medium supplemented with 10% decomplemented FCS, L-glutamine and penicillin/streptomycin. Jurkat cells lacking Bak were generated using RNA interference techniques, as described previously.49 For toxicity assays, Jurkat cells (5 × 105 cells/ml), DU145 cells (1 × 105 cells/ml) or normal T-lymphocytes PBMC (3 × 106 cells/ml) were seeded in flat-bottom 96-well plates (100 μl/well) in complete medium. DU145 cells were allowed to attach prior to addition of cisplatin or tested compounds. Compounds were added at different concentrations in triplicate. Cells were incubated with cisplatin or compounds for 24 h and then cell proliferation was determined by a modification of the MTT-reduction method.50 Total cell number and cell viability were determined by the Trypan-blue exclusion test.
Interaction of 1 and metal complexes 2,3 with plasmid (pBR322) DNA by Electrophoresis (Mobility Shift Assay)
10 μL aliquots of pBR322 plasmid DNA (20 μg/mL) in buffer (5 mM Tris/HCl, 50 mM NaClO4, pH = 7.39) were incubated with molar ratios between 0.25 and 4.0 of the compounds (1-3) at 37 °C for 20 h in the dark. Samples of free DNA and cisplatin-DNA adduct were prepared as controls. After the incubation period, 2 μL of loading dye were added to the samples and of these mixtures, 7 μL were finally loaded onto the 1 % agarose gel. The samples were separated by electrophoresis for 1.5 h at 80 V in Tris-acetate/EDTA buffer (TAE). Afterwards, the gel was stained for 30 min with a solution of GelRed Nucleic Acid stain.
Interaction of metal complexes with HSA by Fluorescence Spectroscopy
The excitation wavelength was set to 295 nm, and the emission spectra were recorded at room temperature in the range of 300 to 450 nm. The fluorescence intensities of the metal compounds 2-8, the buffer and the DMSO are negligible under these conditions, and so is the effect of additions of pure DMSO on the fluorescence of HSA. An 8 mM solution of each compound in DMSO was prepared and ten aliquots of 2.5 μL were added successively to a solution of HSA (10 μM) in phosphate buffer (pH = 7.39), the fluorescence being measured 240 s after each addition. The data was analyzed using the classical Stern-Volmer equation F0/F = 1 + KSV[Q].
Supplementary Material
Acknowledgments
We thank the financial support of a grant from the National Institute of General Medical Sciences (NIGMS), SC2GM082307 (M.C.) and a grant from the Spanish Ministry of Science and Innovation SAF2010-14920 (I.M.). We thank Dr. Esteban P. Urriolabeitia for useful discussions on the synthesis of ligand 1 and undergraduate LSAMP student Farrah Benoit for her assistance with some of the fluorescence spectroscopy measurements. We thank Prof. László T. Mika for his generous donation of phosphine TPPTS P(mC6H4SO3Na)3. This paper is dedicated to Prof. Paul G. Pringle on the occasion of his 55th birthday.
Footnotes
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
Supporting Information. Supporting information available: 1H and 13C{1H} NMR spectra of compounds 2 and 3. Studies on the stability of ligand 1 and compounds 2-8 in d6-DMSO solution and d6-DMSO:D2O (50:50) mixtures overtime by 31P{1H} NMR spectroscopy and selected 31P{1H} NMR spectra of the decomposition of ligand 1 and compound 5 overtime. Table with the crystal data and structure refinement for complex 3. CIF file for the X-ray crystal structure of compound 3. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Thayer AM. Platinum drugs take their roll. Chem Eng News. 2010;88(26):24. [Google Scholar]
- 2.Kelland L. Nat Rev Cancer. 2007;7:573. doi: 10.1038/nrc2167. [DOI] [PubMed] [Google Scholar]
- 3.(a) Sava G, Bergamo A, Dyson PJ. Dalton Trans. 2011;40:9069. doi: 10.1039/c1dt10522a. [DOI] [PubMed] [Google Scholar]; (b) van Rijt SH, Sadler PJ. Drug Discov Today. 2009;14:1089. doi: 10.1016/j.drudis.2009.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dhar S, Lippard SJ. Current status and mechanism of action of platinum-based drugs. In: Alessio E, editor. Bioinorganic Medicinal Chemistry. Ch 3. Wiley-VCH; 2011. p. 79.. Wang X, Guo Z. New trends and future developments of platinum-based antitumor drugs. In: Alessio E, editor. Bioinorganic Medicinal Chemistry. Ch 4. Wiley-VCH; 2011. p. 97.. Selected review: Olszewski U, Hamilton G. Anti-Cancer Agents in Med Chem. 2010;10:320. doi: 10.2174/187152010791162306. and refs therein.
- 5.Selected examples: Alemán J, del Solar V, Alvarez-Valdés A, Ríos-Luci C, Padrón JM, Navarro-Ranninguer C. MedChemComm. 2011;2:789. and refs. therein. Cubo L, Groessl M, Dyson PJ, Quiroga AG, Navarro-Ranninguer C, Casini A. ChemMedChem. 2010;5:1335. doi: 10.1002/cmdc.201000104.. Montero EI, Zhang J, Moniodis JJ, Berners-Price SJ, Farrell N. Chem A Eur J. 2010;16:9175. doi: 10.1002/chem.200903578.
- 6.(a) Berners-Price SJ. Angew Chem Int Ed. 2011;50:804. doi: 10.1002/anie.201004552. [DOI] [PubMed] [Google Scholar]; (b) Farrer NJ, Woods JA, Salassa L, Zhao Y, Robinson KS, Clarkson G, Mackay FS, Sadler PJ. Angew Chem Int Ed. 2010;49:1. doi: 10.1002/anie.201003399. [DOI] [PubMed] [Google Scholar]
- 7.(a) Dhar S, Daniel WL, Giljohann DA, Mirkin CA, Lippard SJ. J Am Chem Soc. 2009;131:14652. doi: 10.1021/ja9071282. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Dhar S, Liu Z, Thomale J, Lippard SJ. J Am Chem Soc. 2008;130:11467. doi: 10.1021/ja803036e. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Dhar S, Gu FX, Langer R, Farokhzad OC, Lippard SJ. Proc Natl Acad Sci USA. 2008;105:17356. doi: 10.1073/pnas.0809154105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Noffke AL, Habtemariam A, Pizarro AM, Sadler PJ. Chem Commun. 2012;48:5219. doi: 10.1039/c2cc30678f. [DOI] [PubMed] [Google Scholar]
- 9.Selected recent review: Ang W-H, Casini A, Sava G, Dyson PJ. J Organomet Chem. 2011;696:989. and refs. therein.
- 10.Selected recent reviews: Berners-Price SJ. Gold-based therapeutic agents. A new perspective. In: Alessio E, editor. Bioinorganic Medicinal Chemistry. Ch 7. Wiley-VCH; 2011. p. 197.. Nobili S, Mini E, Landini I, Gabbiani C, Casini A, Messori L. Med Res Revs. 2010;30:550. doi: 10.1002/med.20168.. Pacheco E, Tiekink E, Whitehouse M. Gold compounds and their applications in medicine. In: Mohr F, editor. Gold Chemistry. Ch 6. Wiley-VCH; 2009. p. 283.. Sun RWY, Che CM. Coord Chem Revs. 2009;253:1682.
- 11.Selected recent examples and reviews: Olzewski U, Clafeey J, Hogan M, Tacke M, Zeillinger R, Bednarski P, Hamilton G. Invest New Drugs. 2011;29:607. doi: 10.1007/s10637-010-9395-5.. Cuffe S, Dowling CM, Claffey J, Pampillon C, Hogan M, Fitzpatrick JM, Carty MP, Tacke M, Watson RWG. The Prostate. 2011;71:11. doi: 10.1002/pros.21227.. Strohfeldt K, Tacke M. Chem Soc Rev. 2008;37:11. doi: 10.1039/b707310k.
- 12.Selected example: Shyder SD, Fu Y, Habtemariam A, van Rijt SH, Cooper PA, Loadman PM, Sadler PJ. Med Chem Commun. 2011;2:666. and refs. therein.
- 13.Some selected recent examples: Sathyadevi P, Krishnamoorthy P, Butorac RR, Cowley AH, Dharmaraj N. Metallomics. 2012;4:498. doi: 10.1039/c2mt00004k.. Loganathan R, Ramakrishnan S, Suresh E, Riyasdeen A, Akbarsha MA, Palaniandavar M. Inorg Chem. 2012;51:5512. doi: 10.1021/ic2017177.. Gandin V, Pellei M, Tisato F, Porchia M, Santini C, Marzano C. J Cell Mol Med. 2012;16:142. doi: 10.1111/j.1582-4934.2011.01292.x.
- 14.Selected recent examples and reviews: Hackenberg F, Oehninger L, Alborzinia H, Can S, Kitanovic I, Geldmacher Y, Kokoschka M, Woelfl S, Ott I, Sheldrick WS. J Inorg Biochem. 2011;105:991. doi: 10.1016/j.jinorgbio.2011.04.006.. Geldmacher Y, Kitanovic I, Alborzinia H, Bergerhoff K, Rubbiani R, Wefelmeier P, Prokop A, Gust R, Ott I, Woelfl S. ChemMedChem. 2011;6:429. doi: 10.1002/cmdc.201000517.. Top S, Efremenko I, Rager MN, Vessieres A, Yaswen P, Jaouen G, Fish RH. Inorg Chem. 2011;50:271. doi: 10.1021/ic1019372.. Kostova I. Int J Curr Chem. 2010;1:81.
- 15.Selected recent examples: Kastl A, Wilbuer A, Merkel AL, Feng L, Di Fazio P, Ocker M, Meggers E. Chem Commum. 2012;48:1863. doi: 10.1039/c1cc15378a.. Lee PK, Law WH-T, Hua-Wei L, Kenneth K-W. Inorg Chem. 2011;50:8570. doi: 10.1021/ic201153d.. Liu Z, Habtemariam A, Pizarro AM, Clarkson G, Sadler PJ. Organometallics. 2011;30:4702.. Liu Z, Habtemariam A, Pizarro AM, Fletcher SA, Kisova A, Vrana O, Salassa L, Bruijnincx PCA, Clarkson G, Brabec V, Sadler PJ. J Med Chem. 2011;54:3011. doi: 10.1021/jm2000932.
- 16.Selected review including a revision of ferrocene-derived anticancer agents: Gasser G, Ott I, Metzler-Nolte N. J Med Chem. 2011;54:3. doi: 10.1021/jm100020w. and refs therein.
- 17.For example: Nieto D, González-Vadillo AM, Bruna S, Pastor CJ, Ríos-Luci C, León LG, Padrón JM, Navarro-Ranninger C, Cuadrado I. Dalton Trans. 2012;41:432. doi: 10.1039/c1dt11358e.. González-Pantoja JF, Stern M, Jarzecki AA, Royo E, Robles-Escajeda E, Varela-Ramírez A, Aguilera RJ, Contel M. Inorg Chem. 2011;50:11099. doi: 10.1021/ic201647h.. Wenzel M, Bertrand B, Eymin M-J, Comte V, Harvey JA, Richard P, Groessl M, Zava O, Amrouche H, Harvey PD, Le Gendre P, Picquet M, Casini A. Inorg Chem. 2011;50:9472. doi: 10.1021/ic201155y.. Pelletier F, Comte V, Massard A, Wenzel M, Toulot S, Richard P, Picquet M, Le Gendre P, Zava O, Edafe F, Casini A, Dyson PJ. J Med Chem. 2010;53:6923. doi: 10.1021/jm1004804.
- 18.Selected reviews: Abu-Surrah AS, Al-Sa’doni HH, Abdalla MY. Cancer Ther. 2008;6:1.. Favero Caires AC. Anti-Cancer Agents in Med Chem. 2007;7:484. doi: 10.2174/187152007781668661.. Abu-Surrah AS, Kettunen M. Curr Med Chem. 2006;13:1337. doi: 10.2174/092986706776872970.
- 19.For example: Abu-Surrah AS, Al-Allaf R, Rashan L, Klinga M, Leskela M. Eur J Med Chem. 2002;37:919. doi: 10.1016/s0223-5234(02)01415-0. and refs. therein.
- 20.Selected recent examples: Kalaivani P, Prabhakaran R, Dallemer F, Poornima P, Vaishnavi E, Ramachandran E, Padma VV, Renganathan R, Natarajan K. Metallomics. 2012;4:101. doi: 10.1039/c1mt00144b.. Gao EJ, Lei L, Zhu MC, Huang Y, Feng G, Gao XN, Zhang M, Wang L, Zhang WZ, Sun YG. Inorg Chem. 2011;50:4732. doi: 10.1021/ic102142j.. Radim V, Pavel S, Zdenek D, Zdenek T. J Inorg Biochem. 2010;104:1130.. Valentini A, Conforti F, Crispini A, De Martino A, Condello R, Stellitano C, Rotilio G, Ghedini M, Federici G, Bernardini S, Pucci D. J Med Chem. 2009;52:484. doi: 10.1021/jm801276a.. Keter FK, Kanyanda S, Lyantagaye SSL, Darkwa J, Rees DJG, Meyer M. Cancer Chem Pharmacol. 2008;63:127. doi: 10.1007/s00280-008-0721-y.. Aldinucci D, Cattaruzza L, Lorenzon D, Giovagnini L, Fregona D, Colombatti A. Oncol Res. 2008;17:103. doi: 10.3727/096504008785055558.
- 21.For example: Butsch K, Gust R, Klein A, Ott I, Romanski M. Dalton Trans. 2010;39:43331. doi: 10.1039/b926233d.. Klein A, Luning A, Ott I, Hamel L, Neugebauer M, Butsch K, Lingen V, Heinrich F, Elmas S. J Organomet Chem. 2010;695:1898.. Teyssot M-L, Jarrousse A-S, Manin M, Chevry A, Roche S, Norre F, Beaudoin C, Morel L, Boyer D, Mahiou R, Gautier A. Dalton Trans. 2009:6894. doi: 10.1039/b906308k. and refs. therein. Ruíz J, Villa MD, Cutillas N, López G, de Haro C, Bautista D, Moreno V, Valencia L. Inorg Chem. 2008;47:4490. doi: 10.1021/ic701873b.
- 22.(a) Navarro-Ranninger C, López-Solera I, Pérez JM, Masaguer JR, Alonso C. Appl Organomet Chem. 1993;7:57. [Google Scholar]; (b) Navarro-Ranninger C, López-Solera I, Pérez JM, Rodrigues J, García-Ruano JL, Raitby PR, Masaguer JR, Alonso C. J Med Chem. 1993;36:3795. doi: 10.1021/jm00076a006. [DOI] [PubMed] [Google Scholar]; (c) Navarro-Ranninguer C, López-Solera I, González VM, Pérez JM, Alvarez-Valdes A, Martín A, Raithby PR, Masaguer JR, Alonso C. Inorg Chem. 1996;35:5181. [Google Scholar]; (d) Quiroga A, Pérez JM, López-Solera I, Masaguer JR, Luque A, Román P, Edwards A, Alonso C, Navarro-Ranninguer C. J Med Chem. 1998;41:1399. doi: 10.1021/jm970520d. [DOI] [PubMed] [Google Scholar]; (e) Quiroga AD, Pérez JM, López-Solera I, Montero EI, Masaguer JR, Alonso C, Navarro-Ranninger C. J Inorg Biochem. 1998;69:275. doi: 10.1016/s0162-0134(98)00003-8. [DOI] [PubMed] [Google Scholar]; (f) Quiroga AD, Pérez JM, Montero EI, Masaguer JR, Alonso C, Navarro-Ranninger C. J Inorg Biochem. 1998;70:117. doi: 10.1016/s0162-0134(98)10007-7. [DOI] [PubMed] [Google Scholar]
- 23.Pucci D, Bloise R, Bellusci A, Bernardini S, Ghedini M, Pirillo S, Valentini A, Crispini A. J Inorg Biochem. 2007;101:1013. doi: 10.1016/j.jinorgbio.2007.03.006. [DOI] [PubMed] [Google Scholar]
- 24.Gao E-J, Wang K-H, Zhu M-C, Liu L. Eur J Med Chem. 2010;45:2784. doi: 10.1016/j.ejmech.2010.02.061. [DOI] [PubMed] [Google Scholar]
- 25.(a) Spencer J, Casini A, Zava O, Rathnam RR, Velhanda SK, Pfeffer M, Callear SK, Hursthouse MB, Dyson PJ. Dalton Trans. 2009:10371. doi: 10.1039/b912096c. [DOI] [PubMed] [Google Scholar]; (b) Spencer J, Rathnam RR, Motukuri M, Kotha AK, Richardason SCW, Hazrati A, Hartley JA, Male L, Hursthouse MB. Dalton Trans. 2009:4299. doi: 10.1039/b819061e. [DOI] [PubMed] [Google Scholar]
- 26.Bincoletto C, Tersariol ILS, Oliveira CR, Dreher S, Fausto DM, Soufen AS, Nascimento FD, Caires ACF. Bioorg Med Lett. 2005;13:3047. doi: 10.1016/j.bmc.2005.01.057. [DOI] [PubMed] [Google Scholar]
- 27.Barbosa CMV, Oliveira CR, Nascimiento FD, Smith MCM, Fausto DM, Soufen MA, Sena E, Araujo RC, Tersariol ILS, Bincoletto C, Caires ACF. Eur J Pharmacol. 2006;542:37. doi: 10.1016/j.ejphar.2006.06.004. [DOI] [PubMed] [Google Scholar]
- 28.Oliveira CR, Barbosa CMV, Nascimento FD, Lanetzki CS, Meneghin MB, Pereira FEV, Paredes-Gamero EJ, Ferreira AT, Rodrigues T, Queiroz MLS, Caires ACF, Tersariol ILS, Bincoletto C. Chem Biol Interact. 2009;177:181. doi: 10.1016/j.cbi.2008.10.034. [DOI] [PubMed] [Google Scholar]
- 29.Rodrigues EG, Silva LS, Fausto DM, Hayash MS, Dreher S, Santos EL, Pesquero JB, Travassos LR, Caires ACF. Int J Cancer. 2003;107:498. doi: 10.1002/ijc.11434. [DOI] [PubMed] [Google Scholar]
- 30.Hebeler-Barbosa F, Rodrigues EG, Puccia R, Caires ACF, Travassos LR. Tras Oncol. 2008;1:110. doi: 10.1593/tlo.08115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Serrano FA, Matsuo AL, Monteforte PT, Bechara A, Smaili SS, Santana DP, Rodrigues T, Pereira FV, Silva LS, Machado J, Jr, Santos EL, Pesquero JB, Martins RM, Travassos LR, Caires ACF, Rodrigues EG. BMC Cancer. 2011;11:296. doi: 10.1186/1471-2407-11-296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Shaik N, Martínez A, Augustin I, Giovinazzo H, Varela A, Aguilera R, Sanaú M, Contel M. Inorg Chem. 2009;48:1577. doi: 10.1021/ic801925k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Vela L, Contel M, Palomera L, Azaceta G, Marzo I. J Inorg Biochem. 2011;105:1306. doi: 10.1016/j.jinorgbio.2011.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Carreira M, Calvo-Sanjuán R, Sanaú M, Zhao X, Magliozzo RS, Marzo I, Contel M. J Inorg Biochem. 2012 doi: 10.1016/j.jinorgbio.2012.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bittner S, Assaf Y, Krief P, Pomerantz M, Ziemnicka BT, Smith CG. J Org Chem. 1985;50:1712. [Google Scholar]
- 36.van Berkel SS, van Eldijk MB, van Hest JCM. Angew Chem Int Ed. 2011;50:8806. doi: 10.1002/anie.201008102. and refs. therein.
- 37.Bielsa R, Larrea A, Navarro R, Soler T, Urriolabeitia EP. Eur J Inorg Chem. 2005:1724. and refs therein.
- 38.Aguilar D, Bielsa R, Contel M, Lledós A, Navarro R, Soler T, Urriolabeitia EP. Organometallics. 2008;27:2929. [Google Scholar]
- 39.Aguilar D, Aragüés MA, Bielsa R, Serrano E, Navarro R, Urriolabeitia EP. Organometallics. 2007;26:3541. [Google Scholar]
- 40.(a) Cardonna JP, Kippard SJ, Gait MJ, Singh M. J Am Chem Soc. 1982;104:5793. [Google Scholar]; (b) Dabrowiak JC. Metals in medicine. John Wiley and Sons; 2009. [Google Scholar]
- 41.Liu H-K, Sadler P. Acc Chem Res. 2011;44:349. doi: 10.1021/ar100140e. [DOI] [PubMed] [Google Scholar]
- 42.For example: Ruíz J, Cutillas N, Vicente C, Villa MD, López G, Lorenzo J, Avilés FX, Moreno V, Bautista D. Inorg Chem. 2005;44:7365. doi: 10.1021/ic0502372.
- 43.Gao E, Zhu M, Liu L, Huang Y, Wang L, Shi S, Chuyue Z, Sun WY. Inorg Chem. 2010;49:3261. doi: 10.1021/ic902176e. [DOI] [PubMed] [Google Scholar]
- 44.(a) Gao E-J, Wang K-H, Zhu M-C, Liu L. Eur J Med Chem. 2010;45:2784. doi: 10.1016/j.ejmech.2010.02.061. [DOI] [PubMed] [Google Scholar]; (b) Mansori-Torshizi H, I-Moghaddam M, Divsalar A, Saboury A-A. Biorg Med Chem. 2008;16:9616. doi: 10.1016/j.bmc.2008.08.021. [DOI] [PubMed] [Google Scholar]; (c) Paul AK, Mansuri-Torshizi H, Srivastava TS, Chavan SJ, Chitnis MP. J Inorg Biochem. 1993;50:9. doi: 10.1016/0162-0134(93)80010-7. [DOI] [PubMed] [Google Scholar]
- 45.Fox K. Drug-DNA Interact Protocols. 1997;90:95. [Google Scholar]
- 46.Timerbaev AR, Hartinger CG, Aleksenko SS, Keppler BK. Chem Rev. 2006;106:2224. doi: 10.1021/cr040704h. [DOI] [PubMed] [Google Scholar]
- 47.Lacowicz JR. Principles of Fluorescence Spectroscopy. Ch 8. Springer; 2006. p. 238. [Google Scholar]
- 48.Kluwer AM, Ahmad I, Reek JNH. Tetrahedron Lett. 2007;48:2999. [Google Scholar]
- 49.López-Rayuela N, Pérez-Galán P, Galán-Malo P, Yuste VJ, Anel A, Susín SA, Naval J, Marzo I. Biochem Pharmacol. 2010;79:1746. doi: 10.1016/j.bcp.2010.02.010. [DOI] [PubMed] [Google Scholar]
- 50.Alley MC, Scudiero DA, Monks A, Hursey ML, Czerwinski MJ, Fine DL, Abbott BJ, Mayo JG, Shoemaker RH, Boyd MR. Cancer Res. 1998;48:589. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.