


Abstract
How does a cell respond to numerous external stresses with a limited number of internal molecular components? It has been observed that there are some common responses of yeast to various stresses, but most observations were based on gene-expression profiles and only some part of the common responses were intensively investigated. So far there has been no system-level analysis to identify commonly responsive or regulated genes against various stresses. In this study, we identified a core regulation module (CRM), a commonly involved regulation structure in the regulatory networks of yeast, which cells reuse in response to an array of environmental stresses. We found that regulators in the CRM constitute a hierarchical backbone of the yeast regulatory network and that the CRM is evolutionarily well conserved, stable against genetic variations and crucial for cell growth. All these findings were consistently held up to considerable noise levels that we introduced to address experimental noise and the resulting false positives of regulatory interactions. We conclude that the CRM of yeast might be an evolutionarily conserved information processing unit that endows a cell with enhanced robustness and efficiency in dealing with numerous environmental stresses with a limited number of internal elements.
INTRODUCTION
Cells have developed stress-responsive strategies which are dynamically implemented through molecular regulatory networks in order to survive the various stresses they may experience (1–3). A fundamental question arises as to how cells direct specific strategies against many possible stresses given a limited number of molecular components. Cells may realize stress-specific responses by combinatorial usage of regulatory molecules and their interactions, which suggests that common regulatory molecules might be involved in various specific responses.
Microarray experiments in yeast (4–5) have revealed that hundreds of genes, referred to as environmental stress response (ESR) or common environmental response (CER) genes, are commonly induced or repressed in response to a variety of stresses. These observations indicate that yeast has a common protective mechanism against various types of stress. However, very few regulatory molecules have been identified among ESR or CER genes, perhaps due to their tendency of inducing stable gene expressions. We should also note that many regulatory molecules play their roles in post-translational modifications. Thus, cellular responses may not be fully captured by gene-expression profiles (6), which suggests a fundamental limitation in identifying common regulators based on microarray experiments alone. On the other hand, other relevant studies revealed that signal transduction pathways such as the mitogen-activated protein kinase (MAPK) and cAMP/PKA pathways respond to a variety of stresses (7–10). However, these findings alone are also insufficient for the identification of common regulators because they lack genome-wide perspective.
To overcome the aforementioned limitations, we have employed in the present study the integrated information about genome, signaling networks and transcriptional regulatory networks of yeast and taken a network-based approach to identify common regulators. It is likely that common regulators would not work alone, but act in harmony with their interacting molecules. So, the common regulators would not be equally distributed in the whole yeast regulatory network but would probably exist as a sub-network by forming a core regulation module (CRM) (Figure 1). With this motivation and background, we have investigated such a CRM and identified its presence by solving a maximum-weight connected subgraph (MWCS) problem. From a network perspective, we have further investigated its topological structure and genetic properties. As a result, we found that the regulators in the CRM form a hierarchical backbone of the yeast-signaling regulatory network and that the CRM is evolutionarily well-conserved, robust against genetic variations and crucial for cell growth. From these, we infer that cells might have developed the CRM as a core information processing unit to respond to numerous stresses with a limited number of internal molecular components, which is an important organizing principle achieved evolutionarily.
Figure 1.

Schematic diagram of a CRM. Dotted red and blue squares indicate the SRN for stresses (A) and (B), respectively. Dotted red and blue ellipses denote the set of stress-responsive genes against stresses (A) and (B), respectively. ESR (or CER) genes are denoted at the intersection between dotted red and blue ellipses.
MATERIALS AND METHODS
Construction of the global regulatory network of yeast
We collected data describing a broad range of regulatory (signaling and transcriptional) networks of yeast (11–17). All the regulations were collected with stringent cutoff (P = 0.001) (17) or validated through the information from the literature (11–16). Using the regulatory information, we constructed a global regulatory network containing 523 nodes and 2093 links where all nodes are regulatory molecules and links are directed regulatory interactions. In the global regulatory network, regulatory molecules are defined as genes which have at least one out-degree. All the regulations are protein modifications except transcriptional regulations and therefore the regulatory molecules include transcription factors, kinases, phosphatases, guanine nucleotide exchange factor, GTPases, histone modifying enzymes, chromatin modifiers and ubiquitin modifiers.
Gold standards
We identified several stress-regulated pathways or stress-related functions for each of the seven stresses from literature (Supplementary Table S1). Based on this information, we further identified stress-specific gene ontology (GO) terms for each stress and then defined the gold standards of positives from these stress-specific GO terms (Supplementary Table S1). The rest of genes are considered as gold standards of negatives that are not responding to any specific stress (see Supplementary Material S1 for details).
Node scoring based on YeastNet
In order to identify stress-specific regulators, we first calculated the stress-specific node scores of all the yeast open reading frames (ORFs). To do this, we transformed functional linkage scores computed by integrating heterogeneous data (YeastNet) (18) into node scores based on a priori knowledge (Supplementary Table S1). The YeastNet score is more useful to infer a new biological insight than a single dataset due to its integrated nature. The functional linkage data in YeastNet provides us with the information about a network composed of genes (nodes) and the functional similarity (link) of each pair of genes. The link strength indicates the degree of functional similarity between genes. We have assigned the sum of link strengths to the ORF where only those links having gold standards of positives with the ORF are considered. Thus, an ORF having more strong functional linkages with gold standards of positives will have a larger stress-specific score.
The YeastNet is a functional similarity network which is an undirected graph. So, it does not include the information on molecular regulatory interactions. As our major goal is to understand how a cell processes information through complex molecular interactions, we have constructed a directed global regulatory network of yeast separately as described in the previous subsection. Note that we did not use YeastNet to construct our regulatory network of yeast, but only utilized it to calculate the node scores for each stress.
Log likelihood ratio
We have employed a log likelihood ratio to normalize the various stress-specific scores [mRNA expression data, growth fitness defect score data and YeastNet (18) based score data] and to evaluate the sensitivity and specificity. Log likelihood ratio, L is defined as follows:
where f, positive and negative indicate the target dataset, gold standards of positives and gold standards of negatives, respectively. If the log likelihood ratio of a gene is larger (smaller) than zero, the gene can be considered active (inactive, respectively) given the specific stress condition. L is computed from contingency tables by binning the dataset values into N intervals (19). So, scores of the likelihood ratio can strongly depend on how the score interval is divided. To avoid this problem, we calculated the average likelihood ratio over 100 times of randomly divided score intervals (see Supplementary Material S1 for details). Here, we set N to 5 as it showed a smooth likelihood ratio distribution while maintaining the data specificity (data not shown).
For overall assessment of the log likelihood ratio, we have divided the gold standards into a training set and a test set. We conducted the 7-fold cross-validation (19). Then, we evaluated the sensitivity and specificity of the log likelihood ratio. We have also computed the sensitivity and specificity of mRNA expression data and growth fitness defect score data (Supplementary Figure S1). We found that the log likelihood ratio based on YeastNet is much more reliable than other log likelihood ratio based on mRNA or growth fitness defect score data (Supplementary Figure S1). This indicates that our stress-specific score faithfully reflects the stress-specific responses. In total, we obtained 6243 ORF specific log likelihood ratios as node scores for each stress condition (adenine dropout, DNA damage, glycerol, H2O2, heat shock, NaCl and sorbitol treatment).
MWCS problem formulation and optimization
Given a connected and undirected node-weighted graph G = (V, E, w) with nodes V, edges E and weights w, the MWCS problem is defined as finding a connected subgraph T = (VT, ET) of G, VT ∈ V, ET ∈ E that maximizes the score w(T):=∑V∈VT w(v) (20). In case both positive and negative node weights are present, solving the MWCS problem is nontrivial. It is known as an NP-complete problem, but a mathematical programming-based algorithm to search for an optimal solution has been proposed (20,21). Therefore, we can obtain an optimal sub-network by solving the MWCS problem using this algorithm. We obtained seven stress-specific regulatory networks (SRNs) based on seven stress-specific regulator scores and a global regulatory network (N = 523, E = 2093) where all nodes are regulatory molecules and links are directed regulatory interactions. We employed the algorithm provided by (21), and IBM ILOG CPLEX callable library version 12.1 was used for programming.
This approach provides us with a global optimal solution and therefore it is advantageous compared to other heuristic approaches that can only identify non-unique local optimal solutions (22,23). For instance, if we employ heuristic approaches, we cannot assess whether the obtained solutions are globally optimal. Moreover, if a poor solution is obtained, we cannot determine whether it is caused by optimality gap, parameter setting, or score function (20). On the other hand, by solving the MWCS problem, we can easily evaluate whether the scoring function is adequate for obtaining biologically relevant solution.
Identification of a CRM against various noise levels
It is hard to determine the most biologically relevant SRN since every experimental measurement contains a certain level of inherent noise. To identify SRNs that are irrelevant of different noise levels, we added a random noise following the normal distribution ∼ N(0, σ2) to the score of each node where σ denotes a noise level (σ = 0.30 in Figure 2). Next, we computed SRNs by formulating the MWCS problem. Finally, we found out the CRM from the identified seven SRNs (Figure 2). The CRM is the intersection of both nodes and edges of seven SRNs. The whole procedure is described in Supplementary Figure S2 in detail. We examined whether the gold standards of positives for each stress are overlapped and found that none of them overlaps with each other. This means that the genes in the CRM we defined are not simply originated from the overlapping of gold standards of positives.
Figure 2.

The CRM identified from the global regulatory network of yeast. S1–7 indicate the seven SRNs. The CRM is determined by the intersection of seven SRNs. Nodes are named according to Saccharomyces Genome Database nomenclature (73). The CRM includes 31 ‘Cell cycle (GO:0007049)’ genes and 39 ‘Response to stress (GO:0033554)’ genes. ‘Both’ indicates those genes having both GO terms. The network is created by Cytoscape (74).
We defined the regulators in the CRM as CRs (core regulators) and the regulators not in the CRM as NCRs (non-core regulators). This classification has been done by conservative selection of only consistent results over 100 times repeated simulations. Then we further examined whether all the topological and genetic properties of the CRM are still consistent against different CRs/NCRs classifications. For this purpose, we carried out the same statistical tests for different CRs/NCRs classifications under both node and link perturbations. It turns out that most of the properties are consistently significant (Supplementary Tables S2–S7). Thus, we confirm that the properties of the CRM do not depend on particular CRs/NCRs classifications.
Computation of the hierarchical order and hierarchy destruction score
Hierarchical order of each node is computed based on the global regulatory network using the vertex sort algorithm (24). If a node does not have a specific hierarchical order, we assigned a median hierarchical order to it. Hierarchy destruction score of node X (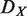 ) is defined as follows:
) is defined as follows:
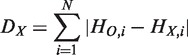 |
where 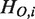 ,
, 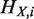 and N denote the hierarchical order of node i in the original network, that in the network after removing node X, and the number of nodes in the network, respectively. Removed and isolated nodes are not considered for computation of
and N denote the hierarchical order of node i in the original network, that in the network after removing node X, and the number of nodes in the network, respectively. Removed and isolated nodes are not considered for computation of 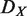 (see Supplementary Figure S3 for details). Nodes which are included in the hierarchical backbone are expected to have larger
(see Supplementary Figure S3 for details). Nodes which are included in the hierarchical backbone are expected to have larger 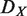 .
.
Genetic measurements
Evolutionary rate indicates the rank of the protein evolutionary rates (25) where a low (high) rank means a low (high, respectively) evolutionary rate. Interstrain variance was calculated as the variance of gene expressions across different yeast Saccharomyces cerevisiae strains with and without stress (26–28). From these data, we excluded clinical and laboratory strains because they have very different phenotypic outcomes among yeast strains and thus can be considered as phenotypic extremes (28,29). From this regard, we used the gene-expression differences with respect to YPS163 (neither clinical nor laboratory strain) in (27). Synthetic lethal pairs were obtained from (30) and only the dataset with stringent cutoff was considered. We computed the growth fitness defect as the average ratio of the mean control intensity to the chemical treatment intensity of yeast strains with homozygous gene deletions (31). Each gene is assigned with a growth fitness defect score which indicates how slow the cell growth will be if the gene is deleted. If the score of a gene is large, then the gene is essential against various stresses. In the computation, we excluded essential genes since their growth rates cannot be measured.
RESULTS
Identifying stress-specific regulatory networks in yeast
Since not all regulators respond to each stress, sub-network identification methods (20,22–23) were used to identify connected sets of stress-specific regulatory molecules. We identified seven SRNs by formulating an MWCS problem based on the global regulatory network and stress-specific regulator scores (see ‘Materials and Methods’ section for details). The SRNs we identified are mathematically optimal, but they might not be biologically optimal since all experimental measurements employed here to determine the stress-specific regulator scores could contain inherent noise and this might have induced a certain level of bias. To exclude such a bias, we extracted only those SRNs that are not affected by different noise levels (see ‘Materials and Methods’ section for details). The seven stresses considered in this study are adenine dropout, DNA damage, glycerol, H2O2, heat shock, NaCl and sorbitol treatment.
Validation of the CRM based on biological evidences
From the intersection of the SRNs, we identified a CRM (Figure 2) composed of 79 regulators and 210 regulatory interactions (isolated regulators are excluded). In order to validate our finding, we have investigated the elements of the CRM and examined whether they coincide with the established knowledge. We found that the CRM includes the most well-known general stress responsive regulators, MSN2 and MSN4. We have identified a set of genes related to the MAPK-signaling pathway (e.g. FUS3, HOG1 and SLT2) which is known to be central in response to oxidative, osmotic and chemical stresses and in diverse functions such as osmoadaptation, filamentous growth and cell-cycle control (32–36). We have also identified those genes related to the cAMP/PKA pathways (e.g. BCY1, TPK1 and TPK2) that are primarily associated with the response to environmental nutrient or growth factor signaling and with the regulation of cell-cycle, growth and stress responses (10,37,38). We have further identified those genes related to the AMPK-signaling pathway (e.g. SNF1 and ELM1) which controls energy homeostasis and various ESRs (39). Other identifications include 31 ‘regulation of cell cycle (GO:0051726)’ genes (P = 0.001) and 39 ‘response to stress (GO:0033554)’ genes (P = 0.001) which were significantly enriched in the CRM (Figure 2 and Supplementary Table S2). In particular, the cell-cycle machinery was found closely linked to the aforementioned general stress-regulating pathways. We note that yeast is known to control the cell cycle for energy balance when it encounters environmental stresses (40).
If a cell encounters an environmental stress, it could gain tolerance against subsequent stresses of not only the same type but also different ones (41). This phenomenon called ‘cross-protection’ leads us to infer that a cell might have a common strategy against various types of environmental stresses. To examine this, we analyzed whether the target genes of the CRM have specific GO terms. We found that most of the target genes have ‘cell cycle (GO:0007049)’ (P = 6E–11) and ‘negative regulation of biological process (GO:0048519)’ (P = 5E–10) (Supplementary Table S3). This implicates that, when a cell perceives a stress signal, it would first shut down all the processes including cell-cycle progression which is the most crucial event of a living organism. This result has in common with the fact that one of the main responses against various stresses is the cell-cycle arrest (41). So, we confirm that our findings are well supported from previous evidenced knowledge.
Regulators in the CRM form a hierarchical backbone of the global regulatory network
What is the characteristic feature of the structural organization of CRM in the global regulatory network of yeast? We investigated the topological basis of CRM as it is closely related to transmitting and processing of information through multiple molecular interactions. Hierarchy is one of the crucial topological characteristics of directed networks since the hierarchical order is known to be strongly related to many biological properties (24,42). We have constructed the hierarchical structure of the global regulatory network by using the ‘vertex sort algorithm’ (24). We identified first the strongly connected components (SCC) defined as a set of nodes connected through feedback regulation. Next, we collapsed each SCC into a single node to transform the complex global regulatory network into a directed acyclic graph (Figure 3A). Then we assigned the hierarchical order to each node of the network. We classified the nodes into five groups: Middle, Top–middle, Top, Bottom–middle and Bottom defined, respectively, as a giant SCC, nodes that only regulate the SCC, the remaining nodes which control other nodes, nodes which are only regulated by the SCC, and the rest of the nodes which are regulated by other nodes (Figure 3A). From this transformed network, we identified that the global regulatory network has a bow-tie structure with the Middle group as a knot (43). In this structure, the knot can integrate various inputs and regulate multiple outputs, so the Middle group is a central regulator of yeast. Intriguingly, we found that most of genes in the CRM are located in the Middle (Figure 3B) and this is very significant compared to random samples (Figure 3C). Therefore, we conclude that the CRM locates in the center of the global regulatory network, which leads us to infer that the CRM might tune the relationship between input (=environmental stress) and output (=cellular response) as a unified information processing unit of the yeast regulatory system.
Figure 3.

Topological properties of the CRM. (A) A schematic view of the global regulatory network of yeast. All the SCC are reduced to single nodes. Node size is proportional to the number of genes included in each group. Red and grey links denote the regulation of the genes in the Middle group and those in the others, respectively. (B) The ratio of nodes in the CRM for each group (the inlet shows those of the global regulatory network of yeast). (C) The number of Middle nodes in the CRM (Observed) and randomly selected sub-networks (histogram). (D) Hierarchy destruction score of CRs and NCRs. (E), (F) the numbers of two-node feedback loops and feedforward loops within the CRM and those in randomly selected sub-networks, respectively. In (C), (E) and (F), empirical P-values were computed using 1000 random samples. In (D), P-value was computed using Wilcoxon’s rank-sum test.
To unravel the topological importance of each node, we computed the hierarchy destruction score defined as how much each node perturbs the original hierarchical order when it is removed (see ‘Materials and Methods’ section). We found that nodes in the CRM (CRs) are more structurally crucial than nodes outside the CRM (NCRs). This suggests that the CRM constitutes a hierarchical backbone of the yeast regulatory system (Figure 3D). In addition, we investigated the enrichment of feedback and feedforward network motifs in the CRM as they are important basic building blocks in constructing SCC and multi-layered hierarchical order, respectively, and therefore are crucial in determining the hierarchy of a network. We found that the CRM contains a significant number of these network motifs compared to random samples (Figure 3E, F, Supplementary Tables S4 and S5), which also supports that the CRM is a structural core. Taken together, we infer that the regulatory system of yeast might have been designed to let all the environmental signals flow into the CRM, a core decision processor, such that common stress responses can be induced and utilized.
CRM is well conserved, robust against genetic variations and crucial for cell growth
We compared various genetic properties (see ‘Materials and Methods’ section) between CRs and NCRs to find out functional characteristics of the CRM. We assessed whether CRs or NCRs are evolutionarily more conserved by comparing the evolutionary rate of each group, and found that CRs tend to be more conserved than NCRs (Figure 4A). In addition, we found that CRs have less variation in their gene expressions than NCRs across different wine yeast strains (Figure 4B). We further compared the gene-expression variance of different yeast strains and found the same result (Supplementary Figure S4) (27). However, we noted that all these expression profiles were measured without stress. So, we have further analyzed the gene-expression profiles of various yeast strains under stress (28). We found again that the expressions of CRs are significantly less variable than those of NCRs after adaptation to new environments (Supplementary Figure S5). Therefore, we conclude that a cell can generate robust steady-state responses through CRs under stress. These results indicate that CRs might have undergone stronger selective pressure to maintain their gene-expression levels during evolution. Overall, we conclude that CRs are evolutionarily well conserved in terms of molecular function.
Figure 4.

Genetic properties of the CRM. In (A), (B) and (D), box plots show a five-number summary of data: the 25th, 50th, 75th percentiles of the samples, and the two most extreme values within 1.5 times of the interquartile range (distance between 25th and 75th percentiles). P-values were computed using Wilcoxon’s rank-sum test. In (C), P-value was computed using Fisher’s exact test.
We further investigated whether the synthetic lethal pairs are enriched in the CRM and found that the ratio of synthetic lethal pairs within the CRM is >2-fold of that within the non-CRM (Figure 4C). Finally, we found that deletion of CRs leads to slower cell growth than the deletion of NCRs upon exposure to various stresses (Figure 4D). In order to further examine whether CRs are crucial against multiple stress, we have investigated whether MDR (multiple drug resistance) genes are enriched in CRs or NCRs. MDR genes are defined as essential genes to grow in the presence of multiple drugs (31). We found that MDR genes are significantly enriched in CRs (P = 0.034, Hypergeometric test), but are not overlapped with NCRs (Supplementary Figure S6). All the above results are consistent with various CRs/NCRs classifications (Supplementary Tables S6 and S7). Therefore, we can conclude that deletion of CRs leads to slower cell growth than the deletion of NCRs upon exposure to various stresses. These are the direct evidences showing that knockout mutants of CRs result in greatly increased sensitivity to a wide range of stress conditions. It also leads us to infer that CRs are more critical to cell growth than NCRs. Taken together, the genes in the CRM are not only evolutionarily conserved, but also they are central regulators of cellular function.
DISCUSSION
In this article, we have identified the CRM of S. cerevisiae and uncovered its topological and genetic properties. Biological data used for determination of gene function and construction of a molecular interaction network usually contain various inherent noises. So, in this study, we intentionally added random perturbations to the node scoring as well as to the construction of a network and identified only those properties consistently held irrespective of different noise levels (see Supplementary Tables S2–S7 for details). Therefore, we confirm that our results do not depend on such random biases that might be introduced during the integration of uncertain biological data.
We wondered whether the genes in the CRM might be overlapped with the ESR genes, but such an overlapping was very little. This is because most ESR genes are not regulatory molecules. In addition, we noted that the functional role of ESR genes and that of the genes in the CRM are considerably different. In contrast with the genes in the CRM, cell-cycle genes were not identified in the ESR genes which are mostly involved with the transcriptional (e.g. RNA processing), translational (e.g. ribosome biogenesis), or other cellular processes. ESR genes perform cellular protection by preserving energy, balancing internal osmolarity and stabilizing biomolecular structure (2). However, these functions were not represented in the CRM. How can we interpret such differential functions? For instance, we can think that cell-cycle control is the most crucial event regulated by the CRM and the ESR gene expressions are the resulting responses. In other words, the genes in the CRM are involved in the stress-induced cell-cycle arrest and the ESR gene expressions are the subsequent processes followed by the growth arrest. Brauer etal. also suggested that many ESR genes may not be reacting to stress but responding to a growth reduction (44). In summary, our network-based approach provides a complementary insight into the common stress responses of yeast compared to previous studies on genome-wide gene expressions.
What might be the reason why yeast cells have developed the CRM? There is no doubt that the most prominent activity of yeast as a unicellular organism is to determine whether it should run a cell-cycle or not in consideration of all environmental stresses. For a wild yeast, to survive under full of environmental stresses, there might be a necessity to develop some central information processing unit like the CRM that is to be frequently and robustly utilized. We note that the expression variation of those genes in the CRs is lower than that in the NCRs. From this, we speculate that yeast cells can have constant transmission of various kinds of environmental information as the molecular level of CRs is kept invariant across various yeast strains. This also suggests that the CRM might be designed to maintain the robustness of cellular information processing. Furthermore, we infer that the CRM is the most important information processing unit of a cell achieved evolutionarily since the CRM is both topologically and genetically essential parts of the yeast regulatory network. Taken together, we summarize that a cell can efficiently interpret numerous external inputs using the CRM, a unified information processor, and thereby the cell can choose an appropriate decision from the most important and problematic choices of yeast: to run a cell cycle, or not.
In order to identify the CRM for a specific species, we need three kinds of information: a genome-wide functional association network, gene lists that are known to respond to specific stresses, and a molecular interaction network. Since the first (e.g. SPRING (45)) and third (e.g. Bio-GRID (46)) are well-established and widely available in general, our method can be applied to many other species with different physiological conditions.
A similar concept of the CRM has also been proposed in developmental regulatory networks (47), in which the developmental role and regulatory interactions of a CRM have been argued to be most impervious to change during evolutionary processes and suggested to affect the planning of major body morphology. Why does a cell have such a regulatory system? Reuse of the CRM in response to various stimuli allows cells to minimize resource utilization while the robustness of the CRM maximizes the efficiency and accuracy of information processing. Thus, the CRM endows a cell with an enhanced efficiency in dealing with numerous external stimuli with a limited number of internal molecular components. We speculate that the CRM might be a ubiquitous organizing principle of various regulatory systems by shaping central responses in an efficient and accurate manner.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online: Supplementary Tables 1–7, Supplementary Figures 1–6, Supplementary Data and Supplementary References [48–72].
FUNDING
Funding for open access charge: National Research Foundation of Korea (NRF) grants funded by the Korea Government, the Ministry of Education, Science and Technology (MEST) [2009-0086964 and 2010-0017662]; NRF grants funded by the MEST through the WCU (World Class University) program [R32-2008-000-10218-0].
Conflict of interest statement. None declared.
Supplementary Material
ACKNOWLEDGEMENTS
The authors thank Jeong-Rae Kim for his valuable comments on this article. The authors also thank Ivana Ljubić for the provision of software and Namsu An for his helpful technical support.
REFERENCES
- 1.Estruch F. Stress-controlled transcription factors, stress-induced genes and stress tolerance in budding yeast. FEMS Microbiol. Rev. 2000;24:469–486. doi: 10.1111/j.1574-6976.2000.tb00551.x. [DOI] [PubMed] [Google Scholar]
- 2.Gasch AP. The environmental stress response: a common yeast response to diverse environmental stresses. Top. Curr. Genet. 2003;1:11–70. [Google Scholar]
- 3.Gasch AP. Comparative genomics of the environmental stress response in ascomycete fungi. Yeast. 2007;24:961–976. doi: 10.1002/yea.1512. [DOI] [PubMed] [Google Scholar]
- 4.Gasch AP, Spellman PT, Kao CM, Carmel-Harel O, Eisen MB, Storz G, Botstein D, Brown PO. Genomic expression programs in the response of yeast cells to environmental changes. Mol. Biol. Cell. 2000;11:4241–4257. doi: 10.1091/mbc.11.12.4241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Causton HC, Ren B, Koh SS, Harbison CT, Kanin E, Jennings EG, Lee TI, True HL, Lander ES, Young RA. Remodeling of yeast genome expression in response to environmental changes. Mol. Biol. Cell. 2001;12:323–337. doi: 10.1091/mbc.12.2.323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Birrell GW, Brown JA, Wu HI, Giaever G, Chu AM, Davis RW, Brown JM. Transcriptional response of Saccharomyces cerevisiae to DNA-damaging agents does not identify the genes that protect against these agents. Proc. Natl Acad. Sci. USA. 2002;99:8778–8783. doi: 10.1073/pnas.132275199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Schaeffer HJ, Weber MJ. Mitogen-activated protein kinases: specific messages from ubiquitous messengers. Mol. Cell. Biol. 1999;19:2435–2444. doi: 10.1128/mcb.19.4.2435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Thevelein JM, de Winde JH. Novel sensing mechanisms and targets for the cAMP-protein kinase A pathway in the yeast Saccharomyces cerevisiae. Mol. Microbiol. 1999;33:904–918. doi: 10.1046/j.1365-2958.1999.01538.x. [DOI] [PubMed] [Google Scholar]
- 9.Ikner A, Shiozaki K. Yeast signaling pathways in the oxidative stress response. Mutat. Res. 2005;569:13–27. doi: 10.1016/j.mrfmmm.2004.09.006. [DOI] [PubMed] [Google Scholar]
- 10.Zurita-Martinez SA, Cardenas ME. Tor and cyclic AMP-protein kinase A: two parallel pathways regulating expression of genes required for cell growth. Eukaryot. Cell. 2005;4:63–71. doi: 10.1128/EC.4.1.63-71.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Breitkreutz BJ, Stark C, Reguly T, Boucher L, Breitkreutz A, Livstone M, Oughtred R, Lackner DH, Bahler J, Wood V, et al. The BioGRID Interaction Database: 2008 update. Nucleic Acids Res. 2008;36:D637–D640. doi: 10.1093/nar/gkm1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fiedler D, Braberg H, Mehta M, Chechik G, Cagney G, Mukherjee P, Silva AC, Shales M, Collins SR, van Wageningen S, et al. Functional organization of the S. cerevisiae phosphorylation network. Cell. 2009;136:952–963. doi: 10.1016/j.cell.2008.12.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Henrik Dohlman JES. Pheromone signaling pathways in yeast. Sci. Signal. 2009 doi: 10.1126/stke.3642006cm6. (Connections Map in the Database of Cell Signaling), http://stke.sciencemag.org/cgi/cm/stkecm;CMP_13999 (21 June 2012, date last accessed) [DOI] [PubMed] [Google Scholar]
- 14.Jeremy Thorner DMT, Garrenton LS. Filamentous growth pathway in yeast. Sci. Signal. 2009 (Connections Map in the Database of Cell Signaling), http://stke.sciencemag.org/cgi/cm/stkecm;CMP_14554 (21 June 2012, date last accessed) [Google Scholar]
- 15.Jeremy Thorner PJW, Ballon DR. High osmolarity glycerol (HOG) pathway in yeast. Sci. Signal. 2009 (Connections Map in the Database of Cell Signaling), http://stke.sciencemag.org/cgi/cm/stkecm;CMP_14620 (21 June 2012, date last accessed) [Google Scholar]
- 16.Kaizu K, Ghosh S, Matsuoka Y, Moriya H, Shimizu-Yoshida Y, Kitano H. A comprehensive molecular interaction map of the budding yeast cell cycle. Mol. Syst. Biol. 2010;6:415. doi: 10.1038/msb.2010.73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.MacIsaac KD, Wang T, Gordon DB, Gifford DK, Stormo GD, Fraenkel E. An improved map of conserved regulatory sites for Saccharomyces cerevisiae. BMC Bioinformatics. 2006;7:113. doi: 10.1186/1471-2105-7-113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lee I, Li Z, Marcotte EM. An improved, bias-reduced probabilistic functional gene network of baker's yeast, Saccharomyces cerevisiae. PLoS One. 2007;2:e988. doi: 10.1371/journal.pone.0000988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jansen R, Yu H, Greenbaum D, Kluger Y, Krogan NJ, Chung S, Emili A, Snyder M, Greenblatt JF, Gerstein M. A Bayesian networks approach for predicting protein-protein interactions from genomic data. Science. 2003;302:449–453. doi: 10.1126/science.1087361. [DOI] [PubMed] [Google Scholar]
- 20.Dittrich MT, Klau GW, Rosenwald A, Dandekar T, Muller T. Identifying functional modules in protein-protein interaction networks: an integrated exact approach. Bioinformatics. 2008;24:i223–i231. doi: 10.1093/bioinformatics/btn161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ljubić I, Weiskircher R, Pferschy U, Klau GW, Mutzel P, Fischetti M. An algorithmic framework for the exact solution of the prize-collecting Steiner tree problem. Math. Program. 2006;105:427–449. [Google Scholar]
- 22.Ideker T, Ozier O, Schwikowski B, Siegel AF. Discovering regulatory and signalling circuits in molecular interaction networks. Bioinformatics. 2002;18(Suppl. 1):S233–S240. doi: 10.1093/bioinformatics/18.suppl_1.s233. [DOI] [PubMed] [Google Scholar]
- 23.Guo Z, Wang L, Li Y, Gong X, Yao C, Ma W, Wang D, Zhu J, Zhang M, Yang D, et al. Edge-based scoring and searching method for identifying condition-responsive protein-protein interaction sub-network. Bioinformatics. 2007;23:2121–2128. doi: 10.1093/bioinformatics/btm294. [DOI] [PubMed] [Google Scholar]
- 24.Jothi R, Balaji S, Wuster A, Grochow JA, Gsponer J, Przytycka TM, Aravind L, Babu MM. Genomic analysis reveals a tight link between transcription factor dynamics and regulatory network architecture. Mol. Syst. Biol. 2009;5:294. doi: 10.1038/msb.2009.52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Xia Y, Franzosa EA, Gerstein MB. Integrated assessment of genomic correlates of protein evolutionary rate. PLoS Comput. Biol. 2009;5:e1000413. doi: 10.1371/journal.pcbi.1000413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Townsend JP, Cavalieri D, Hartl DL. Population genetic variation in genome-wide gene expression. Mol. Biol. Evol. 2003;20:955–963. doi: 10.1093/molbev/msg106. [DOI] [PubMed] [Google Scholar]
- 27.Kvitek DJ, Will JL, Gasch AP. Variations in stress sensitivity and genomic expression in diverse S. cerevisiae isolates. PLoS Genet. 2008;4:e1000223. doi: 10.1371/journal.pgen.1000223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Carreto L, Eiriz MF, Domingues I, Schuller D, Moura GR, Santos MA. Expression variability of co-regulated genes differentiates Saccharomyces cerevisiae strains. BMC Genomics. 2011;12:201. doi: 10.1186/1471-2164-12-201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Warringer J, Zorgo E, Cubillos FA, Zia A, Gjuvsland A, Simpson JT, Forsmark A, Durbin R, Omholt SW, Louis EJ, et al. Trait variation in yeast is defined by population history. PLoS Genet. 2011;7:e1002111. doi: 10.1371/journal.pgen.1002111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Costanzo M, Baryshnikova A, Bellay J, Kim Y, Spear ED, Sevier CS, Ding H, Koh JL, Toufighi K, Mostafavi S, et al. The genetic landscape of a cell. Science. 2010;327:425–431. doi: 10.1126/science.1180823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hillenmeyer ME, Fung E, Wildenhain J, Pierce SE, Hoon S, Lee W, Proctor M, St Onge RP, Tyers M, Koller D, et al. The chemical genomic portrait of yeast: uncovering a phenotype for all genes. Science. 2008;320:362–365. doi: 10.1126/science.1150021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bermejo C, Rodriguez E, Garcia R, Rodriguez-Pena JM, Rodriguez de la Concepcion ML, Rivas C, Arias P, Nombela C, Posas F, Arroyo J. The sequential activation of the yeast HOG and SLT2 pathways is required for cell survival to cell wall stress. Mol. Biol. Cell. 2008;19:1113–1124. doi: 10.1091/mbc.E07-08-0742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Escote X, Zapater M, Clotet J, Posas F. Hog1 mediates cell-cycle arrest in G1 phase by the dual targeting of Sic1. Nat. Cell Biol. 2004;6:997–1002. doi: 10.1038/ncb1174. [DOI] [PubMed] [Google Scholar]
- 34.Haghnazari E, Heyer WD. The Hog1 MAP kinase pathway and the Mec1 DNA damage checkpoint pathway independently control the cellular responses to hydrogen peroxide. DNA Repair. 2004;3:769–776. doi: 10.1016/j.dnarep.2004.03.043. [DOI] [PubMed] [Google Scholar]
- 35.Proft M, Mas G, de Nadal E, Vendrell A, Noriega N, Struhl K, Posas F. The stress-activated Hog1 kinase is a selective transcriptional elongation factor for genes responding to osmotic stress. Mol. Cell. 2006;23:241–250. doi: 10.1016/j.molcel.2006.05.031. [DOI] [PubMed] [Google Scholar]
- 36.Schwartz MA, Madhani HD. Principles of MAP kinase signaling specificity in Saccharomyces cerevisiae. Annu. Rev. Genet. 2004;38:725–748. doi: 10.1146/annurev.genet.39.073003.112634. [DOI] [PubMed] [Google Scholar]
- 37.Gimeno CJ, Ljungdahl PO, Styles CA, Fink GR. Unipolar cell divisions in the yeast S. cerevisiae lead to filamentous growth: regulation by starvation and RAS. Cell. 1992;68:1077–1090. doi: 10.1016/0092-8674(92)90079-r. [DOI] [PubMed] [Google Scholar]
- 38.Rohde JR, Bastidas R, Puria R, Cardenas ME. Nutritional control via Tor signaling in Saccharomyces cerevisiae. Curr. Opin. Microbiol. 2008;11:153–160. doi: 10.1016/j.mib.2008.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hedbacker K, Carlson M. SNF1/AMPK pathways in yeast. Front. Biosci. 2008;13:2408–2420. doi: 10.2741/2854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lai LC, Kissinger MT, Burke PV, Kwast KE. Comparison of the transcriptomic “stress response” evoked by antimycin A and oxygen deprivation in Saccharomyces cerevisiae. BMC Genomics. 2008;9:627. doi: 10.1186/1471-2164-9-627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lu C, Brauer MJ, Botstein D. Slow growth induces heat-shock resistance in normal and respiratory-deficient yeast. Mol. Biol. Cell. 2009;20:891–903. doi: 10.1091/mbc.E08-08-0852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bhardwaj N, Kim PM, Gerstein MB. Rewiring of transcriptional regulatory networks: hierarchy, rather than connectivity, better reflects the importance of regulators. Sci. Signal. 2010;3:ra79. doi: 10.1126/scisignal.2001014. [DOI] [PubMed] [Google Scholar]
- 43.Kitano H. Biological robustness. Nat. Rev. Genet. 2004;5:826–837. doi: 10.1038/nrg1471. [DOI] [PubMed] [Google Scholar]
- 44.Brauer MJ, Huttenhower C, Airoldi EM, Rosenstein R, Matese JC, Gresham D, Boer VM, Troyanskaya OG, Botstein D. Coordination of growth rate, cell cycle, stress response, and metabolic activity in yeast. Mol. Biol. Cell. 2008;19:352–367. doi: 10.1091/mbc.E07-08-0779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Szklarczyk D, Franceschini A, Kuhn M, Simonovic M, Roth A, Minguez P, Doerks T, Stark M, Muller J, Bork P, et al. The STRING database in 2011: functional interaction networks of proteins, globally integrated and scored. Nucleic. Acids Res. 2011;39:D561–D568. doi: 10.1093/nar/gkq973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Stark C, Breitkreutz BJ, Chatr-Aryamontri A, Boucher L, Oughtred R, Livstone MS, Nixon J, Van Auken K, Wang X, Shi X, et al. The BioGRID Interaction Database: 2011 update. Nucleic Acids Res. 2011;39:D698–D704. doi: 10.1093/nar/gkq1116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Davidson EH, Erwin DH. Gene regulatory networks and the evolution of animal body plans. Science. 2006;311:796–800. doi: 10.1126/science.1113832. [DOI] [PubMed] [Google Scholar]
- 48.Barrett T, Troup DB, Wilhite SE, Ledoux P, Rudnev D, Evangelista C, Kim IF, Soboleva A, Tomashevsky M, Edgar R. NCBI GEO: mining tens of millions of expression profiles—database and tools update. Nucleic Acids Res. 2007;35:D760–D765. doi: 10.1093/nar/gkl887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Berdichevsky A, Guarente L. A stress response pathway involving sirtuins, forkheads and 14-3-3 proteins. Cell Cycle. 2006;5:2588–2591. doi: 10.4161/cc.5.22.3513. [DOI] [PubMed] [Google Scholar]
- 50.Delaunay A, Pflieger D, Barrault MB, Vinh J, Toledano MB. A thiol peroxidase is an H2O2 receptor and redox-transducer in gene activation. Cell. 2002;111:471–481. doi: 10.1016/s0092-8674(02)01048-6. [DOI] [PubMed] [Google Scholar]
- 51.Demeter J, Beauheim C, Gollub J, Hernandez-Boussard T, Jin H, Maier D, Matese JC, Nitzberg M, Wymore F, Zachariah ZK, et al. The Stanford Microarray Database: implementation of new analysis tools and open source release of software. Nucleic Acids Res. 2007;35:D766–D770. doi: 10.1093/nar/gkl1019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Erlich RL, Fry RC, Begley TJ, Daee DL, Lahue RS, Samson LD. Anc1, a protein associated with multiple transcription complexes, is involved in postreplication repair pathway in S. cerevisiae. PLoS One. 2008;3:e3717. doi: 10.1371/journal.pone.0003717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Fry RC, DeMott MS, Cosgrove JP, Begley TJ, Samson LD, Dedon PC. The DNA-damage signature in Saccharomyces cerevisiae is associated with single-strand breaks in DNA. BMC Genomics. 2006;7:313. doi: 10.1186/1471-2164-7-313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Grauslund M, Lopes JM, Ronnow B. Expression of GUT1, which encodes glycerol kinase in Saccharomyces cerevisiae, is controlled by the positive regulators Adr1p, Ino2p and Ino4p and the negative regulator Opi1p in a carbon source-dependent fashion. Nucleic Acids Res. 1999;27:4391–4398. doi: 10.1093/nar/27.22.4391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hao N, Behar M, Parnell SC, Torres MP, Borchers CH, Elston TC, Dohlman HG. A systems-biology analysis of feedback inhibition in the Sho1 osmotic-stress-response pathway. Curr. Biol. 2007;17:659–667. doi: 10.1016/j.cub.2007.02.044. [DOI] [PubMed] [Google Scholar]
- 56.Kuranda K, Leberre V, Sokol S, Palamarczyk G, Francois J. Investigating the caffeine effects in the yeast Saccharomyces cerevisiae brings new insights into the connection between TOR, PKC and Ras/cAMP signalling pathways. Mol. Microbiol. 2006;61:1147–1166. doi: 10.1111/j.1365-2958.2006.05300.x. [DOI] [PubMed] [Google Scholar]
- 57.Lee I, Date SV, Adai AT, Marcotte EM. A probabilistic functional network of yeast genes. Science. 2004;306:1555–1558. doi: 10.1126/science.1099511. [DOI] [PubMed] [Google Scholar]
- 58.Levin DE. Cell wall integrity signaling in Saccharomyces cerevisiae. Microbiol. Mol. Biol. Rev. 2005;69:262–291. doi: 10.1128/MMBR.69.2.262-291.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Lopez-Mirabal HR, Winther JR, Kielland-Brandt MC. Oxidant resistance in a yeast mutant deficient in the Sit4 phosphatase. Curr. Genet. 2008;53:275–286. doi: 10.1007/s00294-008-0184-z. [DOI] [PubMed] [Google Scholar]
- 60.Medicherla B, Goldberg AL. Heat shock and oxygen radicals stimulate ubiquitin-dependent degradation mainly of newly synthesized proteins. J. Cell. Biol. 2008;182:663–673. doi: 10.1083/jcb.200803022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Raychaudhuri S, Stuart JM, Altman RB. Principal components analysis to summarize microarray experiments: application to sporulation time series. Pac. Symp. Biocomput. 2000:455–466. doi: 10.1142/9789814447331_0043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Rebora K, Desmoucelles C, Borne F, Pinson B, Daignan-Fornier B. Yeast AMP pathway genes respond to adenine through regulated synthesis of a metabolic intermediate. Mol. Cell. Biol. 2001;21:7901–7912. doi: 10.1128/MCB.21.23.7901-7912.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Servant G, Pennetier C, Lesage P. Remodeling yeast gene transcription by activating the Ty1 long terminal repeat retrotransposon under severe adenine deficiency. Mol. Cell. Biol. 2008;28:5543–5554. doi: 10.1128/MCB.00416-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Shalem O, Dahan O, Levo M, Martinez MR, Furman I, Segal E, Pilpel Y. Transient transcriptional responses to stress are generated by opposing effects of mRNA production and degradation. Mol. Syst. Biol. 2008;4:223. doi: 10.1038/msb.2008.59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Shaner L, Gibney PA, Morano KA. The Hsp110 protein chaperone Sse1 is required for yeast cell wall integrity and morphogenesis. Curr. Genet. 2008;54:1–11. doi: 10.1007/s00294-008-0193-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Shrivastav M, De Haro LP, Nickoloff JA. Regulation of DNA double-strand break repair pathway choice. Cell Res. 2008;18:134–147. doi: 10.1038/cr.2007.111. [DOI] [PubMed] [Google Scholar]
- 67.Silva RD, Sotoca R, Johansson B, Ludovico P, Sansonetty F, Silva MT, Peinado JM, Corte-Real M. Hyperosmotic stress induces metacaspase- and mitochondria-dependent apoptosis in Saccharomy ces cerevisiae. Mol. Microbiol. 2005;58:824–834. doi: 10.1111/j.1365-2958.2005.04868.x. [DOI] [PubMed] [Google Scholar]
- 68.Tirosh I, Weinberger A, Carmi M, Barkai N. A genetic signature of interspecies variations in gene expression. Nat. Genet. 2006;38:830–834. doi: 10.1038/ng1819. [DOI] [PubMed] [Google Scholar]
- 69.Toivari MH, Salusjarvi L, Ruohonen L, Penttila M. Endogenous xylose pathway in Saccharomyces cerevisiae. Appl. Environ. Microbiol. 2004;70:3681–3686. doi: 10.1128/AEM.70.6.3681-3686.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Winkler A, Arkind C, Mattison CP, Burkholder A, Knoche K, Ota I. Heat stress activates the yeast high-osmolarity glycerol mitogen-activated protein kinase pathway, and protein tyrosine phosphatases are essential under heat stress. Eukaryot. Cell. 2002;1:163–173. doi: 10.1128/EC.1.2.163-173.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Ye T, Elbing K, Hohmann S. The pathway by which the yeast protein kinase Snf1p controls acquisition of sodium tolerance is different from that mediating glucose regulation. Microbiology. 2008;154:2814–2826. doi: 10.1099/mic.0.2008/020149-0. [DOI] [PubMed] [Google Scholar]
- 72.Yoshimoto H, Saltsman K, Gasch AP, Li HX, Ogawa N, Botstein D, Brown PO, Cyert MS. Genome-wide analysis of gene expression regulated by the calcineurin/Crz1p signaling pathway in Saccharomyces cerevisiae. J. Biol. Chem. 2002;277:31079–31088. doi: 10.1074/jbc.M202718200. [DOI] [PubMed] [Google Scholar]
- 73.Hong EL, Balakrishnan R, Dong Q, Christie KR, Park J, Binkley G, Costanzo MC, Dwight SS, Engel SR, Fisk DG, et al. Gene Ontology annotations at SGD: new data sources and annotation methods. Nucleic Acids Res. 2008;36:D577–D581. doi: 10.1093/nar/gkm909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Smoot ME, Ono K, Ruscheinski J, Wang PL, Ideker T. Cytoscape 2.8: new features for data integration and network visualization. Bioinformatics. 2011;27:431–432. doi: 10.1093/bioinformatics/btq675. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.