

Abstract
Changes in the peptide and MHC molecules have been extensively examined for how they alter T cell activation, but many fewer studies have examined the TCR. Structural studies of how TCR differences alter T cell specificity have focused on broad variation in the CDR3 loops. However, changes in the CDR1 and 2 loops can also alter TCR recognition of pMHC. In this study we focus on two mutations in the CDR1α loop of the TCR that increased the affinity of a TCR for agonist Hb(64-76)/I-Ek by increasing the on-rate of the reaction. These same mutations also conferred broader recognition of altered peptide ligands. TCR transgenic mice expressing the CDR1α mutations had altered thymic selection, as most of the T cells were negatively selected compared to T cells expressing the wildtype TCR. The few T cells that escaped negative selection and were found in the periphery were rendered anergic, thereby avoiding autoimmunity. T cells with the CDR1α mutations were completely deleted in the presence of Hb(64-76) as an endogenous peptide. Interestingly, the wildtype T cells were not eliminated, identifying a threshold affinity for negative selection where a 3-fold increase in affinity is the difference between incomplete and complete deletion. Overall, these studies highlight how small changes in the TCR can increase the affinity of TCR:pMHC but with the consequences of skewing selection and producing an unresponsive T cell.
Keywords: Anergy, T cell, CDR1, CD4, Affinity, Selection
1. Introduction
TCR:pMHC complex formation needs to exceed a specific binding energy to achieve a T cell response (Davis-Harrison et al., 2007; Gakamsky et al., 2004; Garcia et al., 2001; Holler and Kranz, 2003; Krogsgaard et al., 2003). Structural changes in the TCR are often required to form a stable TCR:pMHC complex, but it is not clear if such conformational changes are necessary for productive signaling through the TCR (Borg et al., 2005; Burrows et al., 2010; Davis et al., 1998; Holler and Kranz, 2004; Qi et al., 2006). It has long been known that the variable CDR loops form the binding footprint for TCR to contact the pMHC complex (Garcia et al., 1999; Huseby et al., 2006). More recently it has been proposed that the TCR maintains germline-encoded affinity for MHC by interactions between key residues on the MHC helices and CDRs 1 and 2 (Adams et al., 2011; Marrack et al., 2008). CDRs 1 and 2 differ in the V gene segments, and it has been shown that introduction of mutations in these CDR loops can result in generation of high affinity TCRs (Chlewicki et al., 2005; Manning et al., 1999), potentially as a consequence of an optimal binding conformation or enhanced TCR:pMHC stability (Adams et al., 2011; Burrows et al., 2010; Dai et al., 2008; Holler and Kranz, 2004; Willcox et al., 1999).
The TCR has inherent specificity for an agonist peptide but can retain binding to some variants of the agonist (Kersh and Allen, 1996). The strength of recognition of an altered peptide ligand (APL) regulates the level of T cell response (Evavold et al., 1992). Some TCR flexibility is critical during T cell development in the thymus as it ensures recognition of foreign antigens during an infection in addition to self peptides during selection. While flexible recognition of pMHC is advantageous, highly promiscuous T cells can inappropriately recognize self-peptides and cause autoimmune disease (Basu et al., 2001; Garcia et al., 2001). Therefore, it remains important to understand the process by which a T cell discriminates between peptides to generate a productive and appropriate immune response.
There is an affinity continuum of a TCR for endogenous pMHC that regulates selection of T cells in the thymus (Daniels et al., 2006). For T cells to be selected, they must have sufficient affinity for endogenous pMHC above the threshold for positive selection but below the threshold for negative selection (Hogquist and Bevan, 1996; Kosmrlj et al., 2008). A complex set of distinct signals regulates positive and negative selection (Alberola-Ila et al., 1996; Gascoigne and Palmer, 2011), tuning T cell responsiveness in the periphery. Negative selection results in apoptosis of T cells that are highly responsive to self pMHC in the process of central tolerance (Klein et al., 2009; Williams et al., 1999). While the majority of self-reactive T cells are deleted to generate central tolerance, it is thought that regulatory T cells can develop from these self-reactive, high affinity TCRs (Yu et al., 2008). Treg development is not merely a consequence of a high affinity TCR, as specific environmental cues and TCR sequences may also be required (Bautista et al., 2009; Lathrop et al., 2008).
Negative selection is an incomplete process and some high affinity T cells escape to the periphery where they can cause autoimmune disease (Zehn and Bevan, 2006). While failure to eliminate these cells breaks mechanisms of central tolerance, a back up system is in place to prevent autoimmunity. Potentially autoreactive T cells can be rendered unresponsive to antigenic stimulation (De Boer et al., 2003) or deleted in the periphery to establish peripheral tolerance (Williams et al., 1999). Anergy, a state of hyporesponsiveness characterized by low IL-2 production and inhibited proliferation (De Boer et al., 2003), has been induced in naive T cells through lack of costimulation (Schwartz, 1996), and exposure to APLs (Evavold et al., 1993; Klein et al., 2009; Sloan-Lancaster et al., 1993). Anergized T cells downregulate TCR and costimulatory receptors to maintain the hyporeactive state. Because of the dual role of the TCR:pMHC interaction in selection and activation, the propensity for T cells to undergo tolerance may be set in the thymus by the affinity of the expressed TCR for self pMHC.
To understand how slight structural changes affect T cell development and sensitivity, we used a system that compares two TCRs recognizing the same cognate antigen, Hb(64-76)/I-Ek. In the mouse, the Hb protein exists in two naturally occurring allelic variants, Hbd and Hbs, such that T cells can develop normally in Hbs strains and be reactive to cells from Hbd mice. The n3.L2 TCR was generated against the Hbd allele of the (64-76) peptide (Evavold et al., 1992). We previously generated a series of mutants of the n3.L2 TCR using a yeast display system (Weber et al., 2005). These mutants were selected for increased surface stability on yeast or increased binding to Hb(64-76)/I-Ek. Mutations selected for increased affinity to Hb(64-76)/I-Ek were generated in the CDR3 of the n3.L2 TCR. Surprisingly, one mutant, called M2, which had two mutations in the CDR1α loop (K25E and T28S), was selected only for increased surface levels on yeast and not for increased affinity, yet it exhibited an increase in affinity. Here we verify that the two CDR1α mutations in M2 resulted in a 3.7 stronger affinity due to a faster on-rate, as measured by surface plasmon resonance. We generated and used hybridomas and transgenic mice expressing the n3.L2 and M2 TCRs to determine how such a moderate change in the CDR1α loop could alter TCR specificity. The M2 TCR has a broader and more sensitive response to altered peptide ligands of the Hb(64-76) peptide. In addition, the M2 TCR has a stronger association with the I-Ek molecule, relying solely on contacts with the MHCIIβ chain for pMHC recognition. As a consequence of these changes, T cell selection in the thymus was also altered. M2 T cells were negatively selected at a higher rate and completely eliminated when exposed to Hbd(64-76) as self antigen. Interestingly, the few peripheral M2 T cells that escaped negative selection were rendered anergic. These studies show how subtle changes in the TCR structure that modestly increase the TCR:pMHC affinity can have a profound effect on T cell development and activation.
2. Materials and Methods
2.1 Mice and cells
In addition to the new mouse line described below, n3.L2/B6.K TCR transgenic mice, B6.K, Rag1−/−, and Hbd congenic mice were used in these studies. The n3.L2/B6.K mouse was previously generated in the laboratory (Kersh et al., 1998) and was crossed to the Rag1−/− and separately to the Hbd mice for the studies presented in this manuscript. All mice were bred and maintained in a pathogen-free barrier facility within Washington University in St Louis following a protocol approved by and in accordance with guidelines from the Washington University Division of Comparative Medicine. In addition to the new hybridoma cell lines described below, CH27 B cells were used to as antigen presentation cells in some of the hybridoma response experiments. CH27 cells were maintained in RPMI + 10% FCS + 1% L-glutamine + 5×10−5M β-2-mercaptoethanol + 0.5% gentamycin at 37°C and 5% CO2.
2.2 Generation of the M2 TCR transgenic mouse line
The M2 mouse was generated using the method described for the n3.L2 mouse (Kersh et al., 1998). The n3.L2 V-Jα plasmid was mutated by PCR to express the two amino acid changes in the M2 CDR1α chain. The M2α chain was cloned into the TCR shuttle vector. TCRα and β minigene constructs were coinjected into C57Bl/6 pronuclei in the Washington University Department of Pathology and Immunology’s Transgenic Core Facility. Transgenic mice were identified by PCR amplification of the M2α and β transgenes from tail DNA. Expression of the M2α chain was confirmed by sequencing the founders’ genomic DNA. One founder expressed both the M2α and β transgenes and was bred to the B6.K strain to provide the selecting MHC. Peripheral CD4 T cells in the M2/B6.K mouse stained with the clonotypic antibody, CAb. M2 and n3L.2 mice were further crossed to a Rag-1−/− background producing n3.L2/B.6K/Rag1−/− and M2/B6.K/Rag1−/− mice. M2/B6.K mice were also crossed to Hbd congenic mice. Mice were used at 4-8 weeks of age in these studies, unless otherwise noted. All mice were bred and maintained in a pathogen-free barrier facility within Washington University in St Louis following a protocol approved by and in accordance with guidelines from the Washington University Division of Comparative Medicine.
2.3 Flow cytometry
Antibodies used in flow cytometry are commercially available except for the CAb antibody, which was previously generated in the laboratory. CAb is a clonotypic antibody for the n3.L2 TCR. CAb was conjugated with AlexaFluor-647 using an antibody conjugation kit (Invitrogen). FITC-, PE-, PE-Cy7-, PerCP-, APC-, APC-Cy7-, Pacific Blue-, and Pacific Orange-labeled antibodies were used in various combinations. Intracellular labeling of FoxP3 was performed using a kit from eBioscience. Cells were permeabilized and fixed for 30 minutes followed by washing with permeabilization buffer and antibody labeling with PE-anti-FoxP3. Data collection was performed using a BD FACSCalibur, a FACSCanto, and a customized FACSLSR II. Data analysis was performed using FlowJo software.
2.4 Generation of n3.L2 and M2 hybridomas
n3.L2 and M2 TCRα and β chains were cloned into a p2A retroviral vector with an IRES-GFP tag (pMIIG) developed by the Vignali lab (Holst et al., 2006), which places the α and β chains as a single polypeptide linked by the p2A peptide. No stability mutations were added into the sequence. The 2A peptide (p2A) is cleaved posttranslationally, ensuring equal expression of the α and β chains and resulting in efficient expression of transduced αβ TCRs. PlatE cells were transformed with lipofectamine + 30μg plasmid DNA. Supernatants from PlatE cells containing packaged retrovirus were used to spinfect the 58 α−β− CD4+ hybridoma cell line. M2 and n3.L2 expressing hybridomas were generated simultaneously, sorted for comparable high GFP expression, and equal expression of the n3.L2 and M2 TCRs. Cells were sorted a second time to generate a population with stable TCR expression. Equivalent expression levels of CD3, CD4, and TCR between n3.L2 and M2 hybridomas were confirmed by flow cytometry. Surface TCR levels were assessed with CAb, Vβ8.3, and H57. Hybridomas were cultured in RPMI + 10% FCS + 1% L-glutamine + 5×10−5M β-2-mercaptoethanol + 0.5% gentamycin at 37°C and 5% CO2.
2.5 T cell IL-2 production and proliferation assays
To assess T cell hybridoma responses, 5×105 hybridomas were cultured overnight at 37°C 5% CO2 in wells of a 96-well plate with 2×104 CH27 B cell APCs and varying concentrations of the Hb(64-76) peptide or APLs from 0.0001-100μM. Peptides were synthesized in the lab and purified by HPLC prior to use. 18-20 hrs after culture, supernatants were assayed using an IL-2 ELISA. Briefly, IL-2 was captured using an anti-IL-2 antibody (BioXCell) and detected with a second, biotinylated anti-IL-2 antibody (BioLegend). Biotin-labeled IL-2 was detected by a streptavidin conjugated horseradish peroxidase antibody (Southern Biotech) followed by TMB substrate for accurate detection of 5-500 pg/mL of IL-2 (1-step Ultra TMB ELISA, Thermo Scientific). After stopping the reaction with 2M sulfuric acid, absorbance was read at 450nm. Absorbance levels were converted into amount of IL-2 based on a regression analysis of IL-2 standards using GraphPad Prism (GraphPad Software). For the APL studies, the amount of peptide required for 50% maximal IL-2 production (EC50) was calculated to determine relative levels of activation. For other experiments, the maximal IL-2 production and minimum stimulatory dose were compared.
For isolated T cell stimulation, single cell suspensions were made from pooled lymph nodes and spleen from transgenic mice. CD4+ cells were isolated by negative selection using Miltenyi CD4 II isolation kit (Miltenyi Biotec). Non-CD4+ cells were depleted by binding to a Miltenyi AutoMACS column. Purity was tested by flow cytometry analysis for percent CD4+ CAb+. To account for minor variations in the purity between n3.L2 and M2 isolation, cell numbers were normalized to the percent CD4+ CAb+ cells to ensure the same number of specific CD4 cells were plated in each well of the IL-2 or proliferation assays. 1×105 specific CD4 cells were plated in IMDM + 10% FCS with 5×105 irradiated B6.K splenocytes and varying concentrations of peptides. IL-2 production was measured as described. To measure proliferation, CD4 T cells were isolated and cultured as in the IL-2 production assay. After 72 hrs in culture at 37°C 5% CO2, each well was pulsed with 0.4μCi 3H thymidine and maintained in culture for an additional 24 hrs. Plates were then harvested and measured for 3H thymidine incorporation.
2.6 I-Ek Ig dimers
I-Ek dimers were produced in Drosophila S2 cells, as previously described (Masteller et al., 2003). The I-Ek dimers have some weakly bound peptide that can easily be exchanged in vitro making them ideal for stimulation of T cells with specific pMHC complexes. Amino acid residues chosen for mutagenesis were located on the top of the MHCII α and β helices as potential TCR contact residues. To generate mutant I-Ek dimers, mutations were introduced into I-Ek constructs at one of 4 MHCIIα and 3 MHCIIβ residues chosen from a subset of mutants expressed in CHO cells (Felix et al., 2007). Wildtype and mutant I-Ek-Ig dimer constructs were transfected into Drosophila S2 cells. Dimers were isolated from culture supernatants by binding to Protein A. Dimers were exposed to acidic pH to remove the endogenous, weakly binding peptides and maintained in excess amounts of soluble peptide to substitute the desired peptide into the peptide binding groove. To assay the TCR:pMHC binding footprint, 96 well plates were coated overnight with Hb(64-76)-loaded I-Ek Ig dimers. After 20 hrs, plates were washed to remove unbound dimer, hybridomas were added, and activation was assayed using IL-2 production as described above.
2.7 Surface Plasmon Resonance
We used established lab protocols to measure binding affinities for n3.L2 and M2 single chain TCR (scTCR) to Hb(64-76)/I-Ek (Weber et al., 2005). 2500-3000 response units of Hb(64-76)-loaded I-Ek Ig dimers were directly coupled to a CM5 sensor chip by amine coupling. Previously, refolded Hb(64-76)/I-Ek was generated from E. coli inclusion bodies for use in surface plasmon resonance studies. Both ligands were tested in this system and the affinity measurements were the same using either the refolded monomer or Ig dimer and maintained a 1:1 TCR:MHC binding ratio ((Persaud et al., 2010), data not shown). Data presented are based on measurements obtained using only peptide loaded I-Ek dimers. Refolded, soluble single chain n3.L2 or M2 TCR (Vα-linker-Vβ) (Holst et al., 2006; Shusta et al., 1999) was purified by fast protein liquid chromatography (FPLC), concentrated in PBS, and injected over flow cells with coupled I-Ek at a rate of 30μL/min. scTCR was injected in duplicate at increasing concentrations from 0-100μM at 25°C. Moth cytochrome C peptide loaded I-Ek was used as a negative control for binding. Moth cytochrome C sensograms were subtracted from experimental Hb/I-Ek sensograms to eliminate nonspecific binding artifacts. Measurements were performed using a Biacore 2000. BiaEval version 4.1 software (Biacore AB) was used to generate 1:1 Langmuir models of sensograms to determine KD, koff and kon. The Langmuir model was adjusted until a Chi2 value below 50 was obtained, indicating the best approximation of data. KD values were confirmed by Scatchard analysis using GraphPad Prism (GraphPad Software). Statistical significance was measured by Student’s t-test for differences between n3.L2 and M2 parameters.
3. Results
3.1 CDR1α mutations increase the affinity of TCR:pMHC through a faster kon
The n3.L2 TCR is specific for the Hbd(64-76) peptide presented on the I-Ek MHC class II molecule (Evavold et al., 1992). Previously, the n3.L2 receptor was mutagenized using a yeast display system to generate a series of higher affinity mutants (Weber et al., 2005). Mutants were isolated for enhanced stability measured by increased surface levels on yeast. One TCR mutant, M2, contained two point mutations in the CDR1α chain (K25E and T28S). Although it had not been selected for higher affinity binding to Hb(64-76)/I-Ek, M2 was shown to have several fold improved affinity over the n3.L2 TCR. Soluble single chain TCR molecules (Vα-linker-Vβ; scTCR) were generated for the n3.L2 and M2 T cell receptors (Holst et al., 2006), containing several additional stabilizing mutations in framework regions (Shusta et al., 1999). These scTCR stabilizing mutations were needed to produce the soluble scTCR, but not for expression of the mutants in T cells (Persaud et al., 2010). The scTCRs were used previously to measure binding affinity of the series of mutants to Hb(64-76)/I-Ek by surface plasmon resonance. To confirm and extend these studies, we performed similar surface plasmon resonance studies. n3.L2 had an affinity of 16.6μM for Hb(64-76)/I-Ek (Figure 1A, B). M2 had a 3.7 fold higher affinity for Hb(64-76)/I-Ek (4.3μM) due to an equivalent change in kon, without a significant change in koff (Figure 1A, B). Sensograms of increasing concentrations of scTCR up to 100μM were modeled for a 1:1 binding ratio to determine kinetic measurements of n3.L2 and M2 scTCR binding to Hb(64-76)/I-Ek dimers (Figure 1C). The KD values were confirmed by Scatchard analysis (Figure 1D). These kinetic values were consistent with what was originally reported for n3.L2 and M2 (Weber et al., 2005). Therefore, the M2 TCR has an increased affinity for Hb(64-76)/I-Ek as the result of two CDR1α mutations.
Figure 1. M2 TCR has higher affinity for Hb(64-76) due to a change in kon.

A and B. Kinetics of n3.L2, M2, K25E, and T28S were measured by surface plasmon resonance using scTCRs binding to Hb(64-76)/I-Ek dimers. Values in arrows indicate fold change above n3.L2 for kon measurements. Measurements of n3.L2 and M2 kinetics are averages of 5 independent experiments on separate CM5 chips. Measurements of K25E and T28S kinetics are averages of 3 independent experiments on separate CM5 chips. Statistical differences were determined using Student’s t test. C. Representative sensograms are shown for scTCR injections over bound I-Ek Ig dimer at the indicated concentrations. Kinetic constants were calculated by fitting sensograms to a 1:1 Langmuir binding model. D. Equilibrium responses at each scTCR concentration were used to generate Scatchard plots of n3.L2 and M2 to confirm KD values predicted from 1:1 modeling. The response was highly linear, with r2 values of 0.8.
We next wanted to identify the individual contribution of the two amino acid differences between n3.L2 and M2. We were interested to determine if one residue had a dominant effect on increasing n3.L2’s affinity for Hb(64-76)/I-Ek or if the two mutations were synergistic. Somewhat unexpectedly, the conservative substitution of a threonine to a serine at position 28 (T28S) increased the affinity of the TCR by 1.9 fold over n3.L2, specifically by changing the kon (Figure 1A, B). The non-conservative single substitution of lysine to glutamic acid at position 25 (K25E) maintained the wildtype n3.L2 affinity of the TCR for Hb(64-76)/I-Ek (Figure 1A, B). Although the single substitution at position 25 did not have an effect on affinity, it must act cooperatively with the substitution at position 28 to achieve the 3.7 fold increase in affinity. The n3.L2 and M2 residues at position 28 are highly conserved in many TCRs with published crystal structures, suggesting the importance of this residue in TCR stability and/or MHC interactions (Manning et al., 1999; Nalefski et al., 1990).
3.2 M2 and n3.L2 hybridomas have the same response to Hb(64-76)/I-Ek
The M2 TCR had not been previously tested functionally. Because the M2 mutations altered the affinity of the TCR for pMHCII, we predicted M2 T cells would differ in their response (sensitivity or specificity) to the Hb(64-76) peptide in comparison with n3.L2 T cells. To measure the sensitivity of n3.L2 and M2 TCRs to Hb(64-76)/I-Ek, we generated T cell hybridomas expressing either the n3.L2 or M2 TCR. Sorted n3.L2 and M2 hybridomas expressed equivalent levels of the n3.L2 or M2 TCR and CD4 (Supplemental Figure 1). Hybridomas were stimulated by CH27 cells pulsed with varying doses of purified Hb(64-76) peptide. The n3.L2 and M2 response to agonist stimulation was measured by IL-2 production. The response curves for the n3.L2 and M2 hybridomas were the same, with the same maximal response and sensitivity to the Hb(64-76) peptide (Figure 2). A similar result was obtained for a related, higher affinity TCR, M15 (Weber et al., 2005). Therefore, the M2 TCR is fully functional and equivalent to the n3.L2 TCR in its ability to recognize and be stimulated by Hb(64-76)/I-Ek independent of effects from development.
Figure 2. Response of n3.L2 and M2 hybridomas to Hb(64-76) peptide.
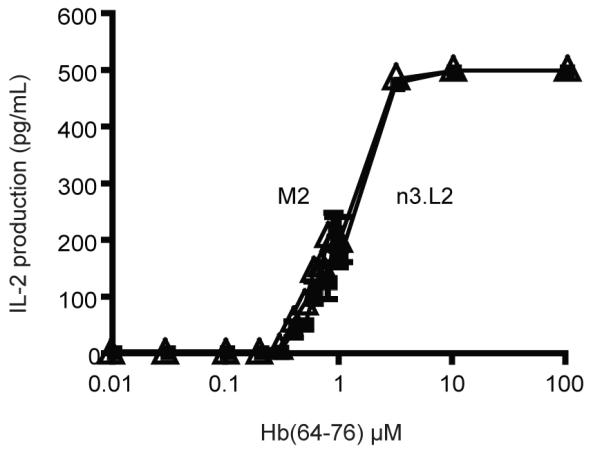
IL-2 production by n3.L2 (closed square) and M2 (open triangle) hybridomas in response to wild-type Hb(64-76). Hybridomas were cultured in triplicate for 18-20 hrs with peptide loaded CH27 APCs. Mean + SEM is presented for triplicates at each concentration in the dose curve, which is representative of 6 independent experiments.
3.3 CDR1α of M2 stabilizes the TCR to maintain activation despite mutations in the MHCIIα chain
We wanted to begin to understand the structural basis for how subtle changes in the CDR1α loop could alter TCR recognition of pMHC. We used a set of mutant I-Ek dimers, each containing a single amino acid substitution at a potential TCR contact site, to measure changes in the TCR:pMHCII energetic footprint. Mutant I-Ek dimers were loaded with Hb(64-76) peptide and used to stimulate IL-2 production by the n3.L2 and M2 hybridomas. Mutations that completely inhibited IL-2 production (> 90% reduction) were considered critical in contributing to the energy required for TCR stimulation by pMHCII. All 3 mutations in the MHCIIβ chain completely inhibited IL-2 secretion for both n3.L2 and M2 hybridomas (Figure 3 A, B) suggesting interactions with the MHCIIβ are highly specific and critical for stimulation through the TCR:pMHCII complex. The n3.L2 hybridomas failed to recognize α53, α55, and α61 mutations, indicating these MHCIIα residues are critical for the n3.L2 interaction (Figure 3 A, B). Interestingly, M2 hybridomas maintained stimulation by all MHCIIα chain mutants except α61, which was critical for both TCRs (Figure 3A). The CDR1α loop may stabilize the M2 TCR into an optimized conformation for MHCII recognition. It is unclear whether the enhanced recognition of MHC dictated by CDR1α is directly through generation of new contacts or through enhanced interactions with key amino acid residues.
Figure 3. Binding of n3.L2 and M2 TCRs to Hb(64-76)/I-Ek dimers.

A. Plate bound Hb-loaded mutant I-Ek dimers were used to stimulate IL-2 production by n3.L2 and M2 hybridomas. We tested the response to 4 mutations in the I-Ek α chain and 3 mutations in the I-Ek β chain. MHC residues were considered critical if mutation at that position inhibited >90% of IL-2 production (highlighted by the dashed line). Representative plot of mean + S.D. IL-2 production normalized to wildtype levels. B. Critical contact residues are highlighted by red circles on a surface model of the crystal structure of Hb bound to I-Ek (Protein DataBank accession code, 1FNG) to identify the TCR binding footprint. The MHCIIα mutation at position 57 was not critical for IL-2 production by n3.L2 or M2 and is not indicated on the model. Green ovals represent the general contact area for TCR CDR1α loops, where the two amino acid differences between n3.L2 and M2 are located. Summarized results are based on 3 independent experiments.
3.4 n3.L2 and M2 hybridomas differ in response to Hb(64-76) APLs
Subtle changes in peptide and MHC sequence can alter a T cell response. We wanted to understand how moderate changes in the TCR could also affect specificity. We examined TCR specificity for peptide by testing the ability of Hb APLs to stimulate the n3.L2 and M2 hybridomas. A panel of 76 peptides was generated by singly substituting each amino acid at the 4 TCR contact residues and each APL was tested for the ability to stimulate IL-2 production. Of the 76 APLs, 13 APLs stimulated IL-2 production by the n3.L2 hybridomas (Figure 4A), whereas, 25 of the 76 peptides were agonists for M2 hybridomas. M2 recognized changes at each of the 4 TCR contact residues. Additionally, M2 hybridomas were more sensitive, with a lower EC50 value for IL-2 production, to APLs that were agonistic for both n3.L2 and M2 hybridomas (Figure 4B). For example, the M69 APL stimulated both n3.L2 and M2 hybridomas. The EC50 for M2 was 5μM with a maximal IL-2 production at 350 pg/mL (Figure 4B, left). M69 was able to stimulate n3.L2 hybridomas at concentrations as low as 3.1μM but did not produce maximal IL-2 at 100μM, the highest concentration testable. A similar difference was seen with the D70 APL. M2 hybridomas produced more IL-2 in response to D70 but the sensitivity of the two hybridomas was similar (Figure 4B, right). Thus, as a consequence of stabilizing the TCR, the M2 CDR1α structural changes led to enhanced recognition of a broader number of APLs.
Figure 4. Response of n3.L2 and M2 hybridomas to Hb(64-76) APLs.

A. All 20 amino acids were substituted individually into each of the 4 TCR contact positions in the Hb(64-76) peptide (P2, P3, P5 and P8). Wildtype Hb(64-76) residues are noted under the label for each position. APLs that induced IL-2 production are shaded. Each APL was tested in triplicate up to 100μM in at least 3 independent experiments. B. Stimulation of IL-2 production by n3.L2 (closed square) and M2 (open triangle) with M69 (left) and D70 (right) as examples of differences in APL response. Mean + SEM is presented for the full dose curve which is representative of 4 independent experiments.
3.5 Generation of M2/B6.K/Rag1−/− TCR transgenic mice
T cell hybridomas are immortalized cell lines and only require TCR signals for activation. We wanted to determine how changes in the TCR also affected T cell development and normal function. Therefore, we generated an M2 TCR transgenic mouse. The n3.L2 TCR transgenic mouse has previously been described (Kersh et al., 1998). The M2 mouse was generated by coinjection of TCR α and β chains onto the B6 background. One founder was generated that co-expressed the M2α and β chains. The M2αβ founder was crossed to the B6.K/Rag1−/− background to provide the proper selecting environment for M2 T cells and eliminate any potential complications from secondary TCRα rearrangements. The n3.L2 mouse was simultaneously crossed to a B6.K/Rag1−/− background and the thymocytes and T cells of both mice were analyzed by flow cytometry (Figure 5).
Figure 5. Comparison of the n3.L2 and M2 Rag1−/− TCR transgenic mice.

A. Flow cytometry analysis of the transgenic CD4 population in the spleens of age matched n3.L2 (left) and M2 (right) transgenic mice. Plots were gated on live lymphocytes. The percentage of specific transgenic CD4 (CAb+ CD4+) cells in the lymphocyte population is noted on each dot plot. Representative plots from 10 mice. Flow cytometry analysis was used to calculate the total number of CD4+ cells from the spleens of n3.L2 and M2 mice. n = 10 mice for each group. P values determined by Student’s t-test. B. Histogram overlays showing TCR, CD3 and CD4 levels on the gated CAb+ CD4+ population in panel A from n3.L2 (black line) and M2 (filled histogram) TCR transgenic mice. C. Labeling of representative thymus from age matched n3.L2 (left) and M2 (right) transgenic mice for CD4 and CD8 expression. Percentage of DN, DP, CD4 and CD8 SP populations are listed. Bar graph indicates total thymus size for mice aged 4-6 weeks. p < 0.0001 measured by Student’s t test. n = 10 mice for each group. D. Analysis of the DN population from the thymus plot and gating shown in panel C. E. Intracellular staining for the transgenic Vβ8.3 TCR β chain in n3.L2 (black) and M2 (filled) DN cells. Representative plots from 5 mice in 3 independent experiments. F. Overlays of TCR (CAb), CD3, and CD4 levels on gated n3.L2 (black line) and M2 (filled histogram) CD4 SP populations from panel C.
Mature M2 Rag1−/− CD4+ T cells developed and were present in the periphery of the M2 transgenic mouse (Figure 5A). These cells were completely mature and expressed high levels of TCR, CD3, and CD4 (Figure 5B). However, the CD4+ cell number was greatly reduced in the M2 mouse in comparison to n3.L2 (Figure 5A). The decreased CD4 population suggested a difference in T cell selection. M2 mice had dramatically reduced thymus cellularity in comparison to n3.L2 (Figure 5C), also indicative of a difference in T cell selection. The M2 thymus had an increased proportion of DN cells (Figure 5C), arrested at the DN3 stage of development (Figure 5D). While this block indicated inefficient development of the M2 TCR, the transgenic Vβ8.3 was highly expressed on a small population at the same level as in the n3.L2 mouse (Figure 5E). The same TCRβ chain was expressed on a larger population in the n3.L2 Rag1−/− mice (Figure 5E), suggesting the TCRβ transgene was not efficiently expressed in the M2 Rag1−/− mouse, potentially as a consequence of a transgene affect.
Despite the inefficient development of the M2 T cells, the DN population that did express Vβ8.3 developed into DP, SP, and peripheral T cells, permitting further analysis of those populations. In the thymus, both the M2 and n3.L2 mice had a mature CD4 SP population (Figure 5C). The CD4 SP populations expressed the same levels of TCR, CD4, CD3 and maturation markers in the thymus and periphery (Figure 5B, F). Very few Treg cells developed from the transgenic T cells either in the thymus or by peripheral conversion (Supplemental Figure 2A, B). Therefore, M2 CD4+ T cells developed and persisted in the periphery in M2/B6.K/Rag1−/− mice. While these cells appeared to be phenotypically normal, we predicted the consequences of changes in selection in the thymus would alter CD4 function in response to cognate antigen.
3.6 M2 T cells are deleted when exposed to Hb(64-76) as an endogenous peptide
The Hb model, involving two alleleic forms of the antigen, afforded us the possibility to determine thymic selection changes in response to Hb(64-76) as an endogenous ligand. A strong signal during T cell selection can result in deletion of T cells by negative selection (Mirshahidi et al., 2004; Williams et al., 1999). We proposed that the slight increased affinity of the M2 TCR for Hb(64-76) would result in enhanced negative selection, despite the identical in vitro responses of n3.L2 and M2 to Hb(64-76). As a control, neither T cell was reactive to the Hbs (64-76) peptide (data not shown). A B6.K/Hbd congenic line was bred to the Rag sufficient n3.L2 and M2 TCR transgenic mice. Due to the high level of agonist antigen present in these mice, we predicted that n3.L2 and M2 T cells would be deleted by negative selection. Surprisingly, a small number of n3.L2 CD4+ T cells were present in the periphery of the n3.L2 × Hbd mice (Figure 6A). While some specific T cells were deleted due to recognition of Hbd(64-76) as self peptide, the n3.L2 affinity for Hbd(64-76) was apparently not above the threshold for complete negative selection. By 4 weeks of age, these mice developed autoimmune hemolytic anemia as measured by the presence of anti RBC antibodies (Figure 6B). In the n3.L2 × Hbd mice, we suggest that the small population of Hb specific T cells induce production of anti-Hbd antibodies by B cells in the spleen. The Rag1+/+ transgenic mice were used for Hbd crosses to preserve the B cell population required to induce the autoimmune response against endogenous Hbd(64-76). Anti-RBC antibodies are then deposited on red blood cells resulting in their lysis and the mice develop autoimmune hemolytic anemia. We are then able to use antibody labeling of RBCs as a readout for the onset of anemia.
Figure 6. Selection of n3.L2 and M2 T cells by endogenous Hbd(64-76).

A. n3.L2 and M2 Rag+/+ transgenic mice were crossed to congenic Hbd mice, which express the agonist Hbd (64-76) as a self peptide. Thymi and spleens from the crosses were assessed for presence of CAb+ CD4+ cells. Percent CAb+ CD4+ is listed for representative n3.L2, M2, n3.L2 × Hbd, M2 × Hbd, and transgenic negative Hbd littermate mice, aged 6 weeks. 6 n3.L2 × Hbd and 6 M2 × Hbd mice were characterized for development of transgenic T cells. B. n3.L2 × Hbd and M2 × Hbd mice were monitored for development of anemia by deposition of antibodies on RBC. Peripheral blood from 5 week old n3.L2 × Hbd (black line) and M2 × Hbd (filled histogram) mice was labeled with a PE-pan Ig antibody. Overlays show levels of anti-Ig on gated RBCs for 3 n3.L2 × Hbd and 3 M2 × Hbd mice.
In contrast to the n3.L2 × Hbd mice, the M2 × Hbd mice completely lacked M2 specific cells in the periphery, though CAb+ cells were found in the thymus (Figure 6A). M2 × Hbd mice failed to develop anemia due to complete elimination of the M2 population (Figure 6B), since the percentage of CD4 cells found in the periphery of M2 × Hbd mice was not different from non-transgenic controls (Figure 6A). The M2 × Hbd mice did not develop hemolytic anemia up to 10 weeks of age. The increased affinity of the M2 TCR for Hbd leads to elimination of M2 cells by negative selection as a consequence of stronger selection signals. These findings demonstrate the sensitivity of negative selection to minor changes in the TCR:pMHC interaction, and establish a threshold for complete negative selection.
3.7 M2 Rag1−/− CD4 T cells are hyporesponsive to Hb(64-76) in the periphery
The immune system has developed multiple mechanisms to ensure self tolerance in the periphery in order to prevent autoimmunity. High affinity T cells that escape negative selection can become unresponsive in the periphery (Kirberg et al., 1993; Macian et al., 2004) with decreased cytokine production and inhibited proliferation. Isolated M2 and n3.L2 Rag1−/− CD4+ T cells had the same sensitivity in producing IL-2 in response to Hb(64-76)-I-Ek presented on irradiated B6.K splenocytes (Figure 7A). However, the M2 T cells produced significantly lower levels of IL-2 in comparison to n3.L2 T cells (Figure 7A). Interestingly, isolated M2 T cells failed to proliferate in response to Hb(64-76) stimulation in contrast to the strong proliferation of n3.L2 T cells (Figure 7B). The reduced ability to produce IL-2 and inhibition of proliferation is a classic pattern of T cell anergy (Sloan-Lancaster et al., 1993).
Figure 7. M2 CD4 T cells are hyporesponsive to Hb(64-76).

A. Rag1−/− CAb+ CD4+ cells isolated from n3.L2 (closed squares) and M2 (open triangle) mice were cultured in triplicate overnight with irradiated B6.K splenocytes pulsed with indicated concentrations of purified Hb(64-76) peptide. After 18-20 hrs, IL-2 production was assayed by ELISA. Dose curves are mean + SEM of triplicate samples from 3 independent experiments. B. The same cells assayed for IL-2 production in panel A were maintained in culture for an additional 72 hrs to measure proliferation of isolated n3.L2 (closed squares) and M2 (open triangle) cells by 3H incorporation. Dose curves are mean + SEM of triplicate samples from 3 independent experiments. C. Levels of costimulatory and inhibitory molecules on the surface of naive CAb+ CD4+ cells from the spleen of n3.L2 (black line) and M2 (filled histogram) TCR transgenic mice. Representative plots from 5 mice in 3 independent experiments.
An anergic state is often accompanied by downregulation of the TCR or costimulatory molecules on the T cell surface. M2 and n3.L2 T cells retained high TCR levels in the periphery of the Rag1−/− transgenic mice (Figure 5B). CD3 and CD4 levels were the same on n3.L2 and M2 cells (Figure 5B), suggesting the unresponsive nature of M2 T cells is not due to an inability to receive activating signals. Levels of CD25, CD28, PD-1, and CTLA-4 were also similar on n3.L2 and M2 CD4 cells (Figure 7C). As the induction of anergy did not appear to be due to decreased costimulation or increased inhibitory signals, M2’s hyporesponsive phenotype may be set in the thymus as a result of different T cell selection in comparison to n3.L2. Alternatively, the anergic state could be set in the periphery as a consequence of altered activation. In either case, the TCR structure is fine tuned for T cell activation. Slight changes in the TCR that improve TCR:pMHC affinity, such as the CDR1α mutations resulting in the M2 TCR, do not necessarily result in enhanced T cell function. Selection in the thymus determines productive pMHC engagement and what structural changes improve peptide recognition by a T cell.
4. Discussion
The affinity of a TCR for specific pMHC is determined by the strength of contacts between the TCR and pMHC. We have generated a TCR, M2, that has a stronger affinity for Hb(64-76)/I-Ek than the wildtype TCR, n.3L2. The same mutations in the CDR1α loop that result in M2’s increased affinity alter the TCR:pMHC binding footprint. The M2 TCR has increased sensitivity to and broader recognition of APLs. The increased breadth and strength of response to APLs may contribute to the high level of negative selection observed in the M2 TCR transgenic mice. M2 T cells undergo complete negative selection when exposed to agonist Hbd(64-76) endogenously as a consequence of the stronger affinity. Instead of improving the T cell response, M2 T cells that escape negative selection are anergic. Therefore, subtle changes in the TCR that enhance binding to pMHC can lead to generation of unresponsive T cells.
What is unique about our system is that the M2 TCR has a higher affinity due to two amino acid changes in the CDR1α loop. In the traditional view of TCR:pMHC binding, the CDR3 loops are the primary regions that interact with the peptide, conferring the cross-reactive property of T cells, and therefore have been the main focus in determining what makes a T cell potentially autoreactive (Reiser et al., 2003; Yin et al., 2011). A few additional studies have directly mutagenized CDR 1, 2, and 3 loops and seen large changes in antigen specificity (Jones et al., 2008; Zhao et al., 2007) and selection (Sim et al., 1996). With M2, mutations in the CDR1α loop not only generate a higher affinity TCR (Chlewicki et al., 2005) but also regulate activation through the TCR. The stability of the TCR or the TCR:pMHC complex conferred by the CDR1α mutations may maintain an affinity closer to the threshold for T cell activation. As a result, M2 T cells are stimulated to produce IL-2 with a broad range of APLs and can potentially recognize a larger number of selecting peptides in the thymus. Previously the two amino acids at positions 26 and 28 were shown to affect binding pMHC for arsenic specific TCRs (Nalefski et al., 1990). These same residues were individually important in contributing to stronger affinity and enhanced TCR:pMHC energetics (Pierce et al., 2010), though these residues may not be important for all TCR:pMHC complexes. While CDR1α loops in general have a flexible conformation (Armstrong et al., 2008), we cannot determine if changes in M2’s peptide fine specificity are due to greater flexibility or merely the result of stronger affinity interactions through alterations in peptide or MHC contact by CDR1α or changes in the structure of other CDRs.
The increased level of negative selection of M2 cells may be a consequence of broader recognition of selecting peptides or stronger interactions with self peptides that select n3.L2. By increasing the affinity of the TCR for self pMHC complexes, peptides which were low affinity, inducing positive selection, would have high enough affinity to overcome the threshold for negative selection. Therefore, the M2 CDR1α mutations may in fact be generating a TCR with stronger recognition of MHCII in the thymus. While we were unable to identify a positively selecting peptide, this does not disprove M2’s stronger interaction with endogenous pMHCII, as positively selecting peptides are highly specific and do not reflect the structural features of agonists (Dao et al., 2003; Hogquist et al., 1997; Lo et al., 2009).
The threshold between thymocyte survival and death by negative selection is very narrow. Previous work using n3.L2 T cells selected in the presence of Hb(64-76) APLs identified that interactions longer than two seconds (t1/2) led to complete negative selection (Williams et al., 1999). Based on our new findings using n3.L2 × Hbd and M2 × Hbd T cell development, this selection threshold for CD4 T cells needs to be adjusted. Both n3.L2 and M2 exceeded the two second half life for binding Hb/I-Ek but differed in the level of negative selection by Hbd. N3.L2 T cells bound Hb(64-76)/I-Ek with an affinity of 16.6μM. While developing n3.L2 T cells were subjected to low levels of negative selection in the presence of endogenous Hbd(64-76), a large proportion of the n3.L2 CD4 T cells did persist in the periphery of the n3.L2 × Hbd mice. By increasing the TCR:pMHC affinity 3.7-fold to 4μM, M2 thymocytes were completely eliminated by negative selection in the M2 × Hbd mice. This value is consistent with the threshold measured for CD8 T cells (Naeher et al., 2007). The difference between n3.L2 and M2 clearly defines the negative selection threshold for CD4 T cells and shows that the overall affinity, not the t1/2, dictates this threshold.
Despite evidence supporting an affinity model for selection, studies have implicated signaling differences regulate positive versus negative selection. Even so, the quality and quantity of pMHC-specific signaling received by a thymocyte during development must still be regulated by the affinity of the TCR:pMHC complex. Low affinity interactions have maintained both selection and activation of specific 2C CD8 T cells to induce target lysis (Sykulev et al., 1994; Udaka et al., 1992). This same functionality was based on the number of pMHC complexes available to TCRs on the cell surface (Delaney et al., 1998). The ability to form multiple TCR:pMHC interactions, either with neighboring molecules or through rebinding, can determine the level of phosphorylation of downstream signaling molecules and the level of T cell function. Rebinding is dependent upon the on-rate (Huang et al., 2010; Huppa et al., 2010) and therefore could be a determining parameter for the difference between n3.L2 and M2 T cells. It would be interesting to identify signaling differences between n3.L2 and M2 thymocytes that could explain the skewed selection phenotype observed in vivo. The affect is not the same as was described by Sim et al. (Sim et al., 1996) where mutation of CDR1 skewed CD4 development to CD8, however the increased level of negative selection of M2 thymocytes could also be a consequence of altered MHC recognition. Levels of costimulatory and inhibitory molecules can modulate thymus and peripheral T cell reactivity to cytokines and induction of apoptosis resulting in negative selection and anergy (Xing and Hogquist, 2012). While surface expression of costimulatory and inhibitory molecules was identical between the two TCRs, the functionality of these receptors was not tested and could contribute to the skewed selection profile and induction of anergy for M2 T cells. Further dissection of signaling pathways downstream of the TCR may uncover a mechanism for how altering CDR1α and increasing TCR affinity for pMHC changes the T cell response to selecting peptides and peripheral antigens.
As a mechanism to prevent autoimmune disease, high affinity TCRs can be tolerized, leaving a population of autoreactive T cells unresponsive to agonist stimuli. M2 T cells develop in an environment lacking the Hbd agonist peptide and do not respond to Hbs stimulation, both indicators that M2 (and n3.L2) T cells are unlikely to become autoreactive in the transgenic mouse. Even so, M2 T cells are anergized in the periphery. Weak agonist TCR:pMHC interactions, such as is measured with some APLs, often result in incomplete T cell activation and induction of anergy (Evavold et al., 1993; Garbi et al., 2010; Lyons et al., 1996; Rabinowitz et al., 1996; Sloan-Lancaster et al., 1993; Zehn et al., 2009). Rechallenge with an agonist after anergy induction by APL stimulation leads to an inability to proliferate but restored IL-2 production both in vitro and in vivo (Edwards and Evavold, 2010). Because of broadened peptide recognition due to the CDR1α mutations, M2 T cells may recognize weakly stimulatory peptides that are null for n3.L2. M2 T cells that escape negative selection following selection by a weakly stimulatory peptide would be hyporesponsive when exposed to Hb(64-76) agonist. Early signaling events downstream of the hyporesponsive M2 TCR may be insufficient to trigger full IL-2 production and proliferation, as has been seen with cytolytic activity of CD8 T cells (Beal et al., 2009). The developmental programming imposed on these cells in the thymus leads to an attenuated response, despite enhanced contact with agonist pMHC for full activation.
The optimized M2 TCR structure has a faster on-rate for binding Hb(64-76) I-Ek. The CDR1α region stabilizes the TCR with pMHC by contributing energetically to the overall complex as a result of contact with MHC or by directly influencing the CDR3α contact with peptide (Armstrong et al., 2008; Borg et al., 2005; Burrows et al., 2010). Interestingly, in one study position 28 of the TCRα chain was important in increasing the association rate of TCRs with additional mutations through enhancing interaction between residues on CDR1α and CDR3α loops (Pierce et al., 2010). Since mutations on the I-Ek α chain maintained sufficient interaction with M2 for IL-2 production but not n3.L2, the M2 CDR1α mutations may result in new or stronger contacts with the MHCII, decreasing the requirement for others. An altered binding orientation is unlikely, as structures of high affinity variants in the 2C system do not show altered footprints (Varani et al., 2007). Instead the faster kon of M2 binding pMHC may reflect a more optimal conformation for association with MHC, maintaining affinity above the threshold for T cell activation with a broad range of peptides. The M2 CDR1α loop may be more flexible, which would contribute to an altered ability to be in a “ready” conformation for faster pMHC binding. Alternatively, the CDR1α mutations may change pMHC contact directly or indirectly by altering the orientation of the CDR1α or other CDR loops of the n3.L2 TCR, as has been seen for other systems (Krogsgaard et al., 2003; Manning et al., 1999; Pierce et al., 2010; Reiser et al., 2003). A crystal structure of the M2: Hb/I-Ek co-complex would identify the exact structural changes induced by the M2 CDR1α mutations. What is clear, however, is that a TCR with enhanced binding to pMHC does not always produce a better overall T cell.
Supplementary Material
Acknowledgments
We thank Darren Kreamalmeyer for maintenance and breeding of all mice used in this manuscript; Terri Sherlinski, and the Washington University Transgenic Mouse Core for generation of the M2 TCR transgenic mouse; Wan-Lin Lo for generating the mutant I-Ek Ig dimers; Stephen Horvath, Kazuo Omi, and Stephen Persaud for assistance with various aspects of the experimental studies in this manuscript. We thank Ted Hansen, Chyi Hseih, and Yina Huang for review of early versions of this manuscript and constructive advice. These studies were supported by the NIH grants AI21457 (P.M.A.), and CA097296 (D.M.K.) and The Rheumatic Diseases Core Center grant (AR48335).
References
- Adams JJ, Narayanan S, Liu B, Birnbaum ME, Kruse AC, Bowerman NA, Chen W, Levin AM, Connolly JM, Zhu C, Kranz DM, Garcia KC. T cell receptor signaling is limited by docking geometry to peptide-major histocompatibility complex. Immunity. 2011;35:681–93. doi: 10.1016/j.immuni.2011.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alberola-Ila J, Hogquist KA, Swan KA, Bevan MJ, Perlmutter RM. Positive and negative selection invoke distinct signaling pathways. J. Exp. Med. 1996;184:9–18. doi: 10.1084/jem.184.1.9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armstrong KM, Piepenbrink KH, Baker BM. Conformational changes and flexibility in T-cell receptor recognition of peptide-MHC complexes. Biochem. J. 2008;415:183–96. doi: 10.1042/BJ20080850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basu D, Horvath S, O’Mara L, Donermeyer D, Allen PM. Two MHC surface amino acid differences distinguish foreign peptide recognition from autoantigen specificity. J. Immunol. 2001;166:4005–11. doi: 10.4049/jimmunol.166.6.4005. [DOI] [PubMed] [Google Scholar]
- Bautista JL, Lio CW, Lathrop SK, Forbush K, Liang Y, Luo J, Rudensky AY, Hsieh CS. Intraclonal competition limits the fate determination of regulatory T cells in the thymus. Nat. Immunol. 2009;10:610–7. doi: 10.1038/ni.1739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beal AM, Anikeeva N, Varma R, Cameron TO, Vasiliver-Shamis G, Norris PJ, Dustin ML, Sykulev Y. Kinetics of early T cell receptor signaling regulate the pathway of lytic granule delivery to the secretory domain. Immunity. 2009;31:632–42. doi: 10.1016/j.immuni.2009.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borg NA, Ely LK, Beddoe T, Macdonald WA, Reid HH, Clements CS, Purcell AW, Kjer-Nielsen L, Miles JJ, Burrows SR, McCluskey J, Rossjohn J. The CDR3 regions of an immunodominant T cell receptor dictate the ‘energetic landscape’ of peptide-MHC recognition. Nat. Immunol. 2005;6:171–80. doi: 10.1038/ni1155. [DOI] [PubMed] [Google Scholar]
- Burrows SR, Chen Z, Archbold JK, Tynan FE, Beddoe T, Kjer-Nielsen L, Miles JJ, Khanna R, Moss DJ, Liu YC, Gras S, Kostenko L, Brennan RM, Clements CS, Brooks AG, Purcell AW, McCluskey J, Rossjohn J. Hard wiring of T cell receptor specificity for the major histocompatibility complex is underpinned by TCR adaptability. Proc. Natl. Acad. Sci. U S A. 2010;107:10608–13. doi: 10.1073/pnas.1004926107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chlewicki LK, Holler PD, Monti BC, Clutter MR, Kranz DM. High-affinity, peptide-specific T cell receptors can be generated by mutations in CDR1, CDR2 or CDR3. J. Mol. Biol. 2005;346:223–39. doi: 10.1016/j.jmb.2004.11.057. [DOI] [PubMed] [Google Scholar]
- Dai S, Huseby ES, Rubtsova K, Scott-Browne J, Crawford F, Macdonald WA, Marrack P, Kappler JW. Crossreactive T Cells spotlight the germline rules for alphabeta T cell-receptor interactions with MHC molecules. Immunity. 2008;28:324–34. doi: 10.1016/j.immuni.2008.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daniels MA, Teixeiro E, Gill J, Hausmann B, Roubaty D, Holmberg K, Werlen G, Hollander GA, Gascoigne NR, Palmer E. Thymic selection threshold defined by compartmentalization of Ras/MAPK signalling. Nature. 2006;444:724–9. doi: 10.1038/nature05269. [DOI] [PubMed] [Google Scholar]
- Dao T, Blander JM, Sant’Angelo DB. Recognition of a specific self-peptide: self-MHC class II complex is critical for positive selection of thymocytes expressing the D10 TCR. J. Immunol. 2003;170:48–54. doi: 10.4049/jimmunol.170.1.48. [DOI] [PubMed] [Google Scholar]
- Davis MM, Boniface JJ, Reich Z, Lyons D, Hampl J, Arden B, Chien Y. Ligand recognition by alpha beta T cell receptors. Annu. Rev. Immunol. 1998;16:523–44. doi: 10.1146/annurev.immunol.16.1.523. [DOI] [PubMed] [Google Scholar]
- Davis-Harrison RL, Insaidoo FK, Baker BM. T cell receptor binding transition states and recognition of peptide/MHC. Biochemistry. 2007;46:1840–50. doi: 10.1021/bi061702p. [DOI] [PubMed] [Google Scholar]
- De Boer RJ, Homann D, Perelson AS. Different dynamics of CD4+ and CD8+ T cell responses during and after acute lymphocytic choriomeningitis virus infection. J. Immunol. 2003;171:3928–35. doi: 10.4049/jimmunol.171.8.3928. [DOI] [PubMed] [Google Scholar]
- Delaney JR, Sykulev Y, Eisen HN, Tonegawa S. Differences in the level of expression of class I major histocompatibility complex proteins on thymic epithelial and dendritic cells influence the decision of immature thymocytes between positive and negative selection. Proc. Natl. Acad. Sci. U S A. 1998;95:5235–40. doi: 10.1073/pnas.95.9.5235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards LJ, Evavold BD. A unique unresponsive CD4+ T cell phenotype post TCR antagonism. Cell. Immunol. 2010;261:64–8. doi: 10.1016/j.cellimm.2009.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evavold BD, Sloan-Lancaster J, Allen PM. Tickling the TCR: selective T-cell functions stimulated by altered peptide ligands. Immunol. Today. 1993;14:602–9. doi: 10.1016/0167-5699(93)90200-5. [DOI] [PubMed] [Google Scholar]
- Evavold BD, Williams SG, Hsu BL, Buus S, Allen PM. Complete dissection of the Hb(64-76) determinant using T helper 1, T helper 2 clones, and T cell hybridomas. J. Immunol. 1992;148:347–53. [PubMed] [Google Scholar]
- Felix NJ, Donermeyer DL, Horvath S, Walters JJ, Gross ML, Suri A, Allen PM. Alloreactive T cells respond specifically to multiple distinct peptide-MHC complexes. Nat. Immunol. 2007;8:388–97. doi: 10.1038/ni1446. [DOI] [PubMed] [Google Scholar]
- Gakamsky DM, Luescher IF, Pecht I. T cell receptor-ligand interactions: a conformational preequilibrium or an induced fit. Proc. Natl. Acad. Sci. U S A. 2004;101:9063–6. doi: 10.1073/pnas.0402840101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garbi N, Hammerling GJ, Probst HC, van den Broek M. Tonic T cell signalling and T cell tolerance as opposite effects of self-recognition on dendritic cells. Curr. Opin. Immunol. 2010;22:601–8. doi: 10.1016/j.coi.2010.08.007. [DOI] [PubMed] [Google Scholar]
- Garcia KC, Radu CG, Ho J, Ober RJ, Ward ES. Kinetics and thermodynamics of T cell receptor-autoantigen interactions in murine experimental autoimmune encephalomyelitis. Proc. Natl. Acad. Sci. U S A. 2001;98:6818–23. doi: 10.1073/pnas.111161198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia KC, Teyton L, Wilson IA. Structural basis of T cell recognition. Annu. Rev. Immunol. 1999;17:369–97. doi: 10.1146/annurev.immunol.17.1.369. [DOI] [PubMed] [Google Scholar]
- Gascoigne NR, Palmer E. Signaling in thymic selection. Curr. Opin. Immunol. 2011;23:207–12. doi: 10.1016/j.coi.2010.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hogquist KA, Bevan MJ. The nature of the peptide/MHC ligand involved in positive selection. Semin. Immunol. 1996;8:63–8. doi: 10.1006/smim.1996.0009. [DOI] [PubMed] [Google Scholar]
- Hogquist KA, Tomlinson AJ, Kieper WC, McGargill MA, Hart MC, Naylor S, Jameson SC. Identification of a naturally occurring ligand for thymic positive selection. Immunity. 1997;6:389–99. doi: 10.1016/s1074-7613(00)80282-4. [DOI] [PubMed] [Google Scholar]
- Holler PD, Kranz DM. Quantitative analysis of the contribution of TCR/pepMHC affinity and CD8 to T cell activation. Immunity. 2003;18:255–64. doi: 10.1016/s1074-7613(03)00019-0. [DOI] [PubMed] [Google Scholar]
- Holler PD, Kranz DM. T cell receptors: affinities, cross-reactivities, and a conformer model. Mol. Immunol. 2004;40:1027–31. doi: 10.1016/j.molimm.2003.11.013. [DOI] [PubMed] [Google Scholar]
- Holst J, Szymczak-Workman AL, Vignali KM, Burton AR, Workman CJ, Vignali DA. Generation of T-cell receptor retrogenic mice. Nat. Protoc. 2006;1:406–17. doi: 10.1038/nprot.2006.61. [DOI] [PubMed] [Google Scholar]
- Huang J, Zarnitsyna VI, Liu B, Edwards LJ, Jiang N, Evavold BD, Zhu C. The kinetics of two-dimensional TCR and pMHC interactions determine T-cell responsiveness. Nature. 2010;464:932–6. doi: 10.1038/nature08944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huppa JB, Axmann M, Mortelmaier MA, Lillemeier BF, Newell EW, Brameshuber M, Klein LO, Schutz GJ, Davis MM. TCR-peptide-MHC interactions in situ show accelerated kinetics and increased affinity. Nature. 2010;463:963–7. doi: 10.1038/nature08746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huseby ES, Crawford F, White J, Marrack P, Kappler JW. Interface-disrupting amino acids establish specificity between T cell receptors and complexes of major histocompatibility complex and peptide. Nat. Immunol. 2006;7:1191–9. doi: 10.1038/ni1401. [DOI] [PubMed] [Google Scholar]
- Jones LL, Colf LA, Bankovich AJ, Stone JD, Gao YG, Chan CM, Huang RH, Garcia KC, Kranz DM. Different thermodynamic binding mechanisms and peptide fine specificities associated with a panel of structurally similar high-affinity T cell receptors. Biochemistry. 2008;47:12398–408. doi: 10.1021/bi801349g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kersh GJ, Allen PM. Essential flexibility in the T-cell recognition of antigen. Nature. 1996;380:495–8. doi: 10.1038/380495a0. [DOI] [PubMed] [Google Scholar]
- Kersh GJ, Donermeyer DL, Frederick KE, White JM, Hsu BL, Allen PM. TCR transgenic mice in which usage of transgenic alpha- and beta-chains is highly dependent on the level of selecting ligand. J. Immunol. 1998;161:585–93. [PubMed] [Google Scholar]
- Kirberg J, Swat W, Rocha B, Kisielow P, von Boehmer H. Induction of tolerance in immature and mature T cells. Transplant. Proc. 1993;25:279–80. [PubMed] [Google Scholar]
- Klein L, Hinterberger M, Wirnsberger G, Kyewski B. Antigen presentation in the thymus for positive selection and central tolerance induction. Nat. Rev. Immunol. 2009;9:833–44. doi: 10.1038/nri2669. [DOI] [PubMed] [Google Scholar]
- Kosmrlj A, Jha AK, Huseby ES, Kardar M, Chakraborty AK. How the thymus designs antigen-specific and self-tolerant T cell receptor sequences. Proc. Natl. Acad. Sci. U S A. 2008;105:16671–6. doi: 10.1073/pnas.0808081105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krogsgaard M, Prado N, Adams EJ, He XL, Chow DC, Wilson DB, Garcia KC, Davis MM. Evidence that structural rearrangements and/or flexibility during TCR binding can contribute to T cell activation. Mol Cell. 2003;12:1367–78. doi: 10.1016/s1097-2765(03)00474-x. [DOI] [PubMed] [Google Scholar]
- Lathrop SK, Santacruz NA, Pham D, Luo J, Hsieh CS. Antigen-specific peripheral shaping of the natural regulatory T cell population. J. Exp. Med. 2008;205:3105–17. doi: 10.1084/jem.20081359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lo WL, Felix NJ, Walters JJ, Rohrs H, Gross ML, Allen PM. An endogenous peptide positively selects and augments the activation and survival of peripheral CD4+ T cells. Nat. Immunol. 2009;10:1155–61. doi: 10.1038/ni.1796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyons DS, Lieberman SA, Hampl J, Boniface JJ, Chien Y, Berg LJ, Davis MM. A TCR binds to antagonist ligands with lower affinities and faster dissociation rates than to agonists. Immunity. 1996;5:53–61. doi: 10.1016/s1074-7613(00)80309-x. [DOI] [PubMed] [Google Scholar]
- Macian F, Im SH, Garcia-Cozar FJ, Rao A. T-cell anergy. Curr. Opin. Immunol. 2004;16:209–16. doi: 10.1016/j.coi.2004.01.013. [DOI] [PubMed] [Google Scholar]
- Manning TC, Parke EA, Teyton L, Kranz DM. Effects of complementarity determining region mutations on the affinity of an alpha/beta T cell receptor: measuring the energy associated with CD4/CD8 repertoire skewing. J. Exp. Med. 1999;189:461–70. doi: 10.1084/jem.189.3.461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marrack P, Rubtsova K, Scott-Browne J, Kappler JW. T cell receptor specificity for major histocompatibility complex proteins. Curr. Opin. Immunol. 2008;20:203–7. doi: 10.1016/j.coi.2008.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masteller EL, Warner MR, Ferlin W, Judkowski V, Wilson D, Glaichenhaus N, Bluestone JA. Peptide-MHC class II dimers as therapeutics to modulate antigen-specific T cell responses in autoimmune diabetes. J. Immunol. 2003;171:5587–95. doi: 10.4049/jimmunol.171.10.5587. [DOI] [PubMed] [Google Scholar]
- Mirshahidi S, Ferris LC, Sadegh-Nasseri S. The magnitude of TCR engagement is a critical predictor of T cell anergy or activation. J. Immunol. 2004;172:5346–55. doi: 10.4049/jimmunol.172.9.5346. [DOI] [PubMed] [Google Scholar]
- Naeher D, Daniels MA, Hausmann B, Guillaume P, Luescher I, Palmer E. A constant affinity threshold for T cell tolerance. J. Exp. Med. 2007;204:2553–9. doi: 10.1084/jem.20070254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nalefski EA, Wong JG, Rao A. Amino acid substitutions in the first complementarity-determining region of a murine T-cell receptor alpha chain affect antigen-major histocompatibility complex recognition. J. Biol. Chem. 1990;265:8842–6. [PubMed] [Google Scholar]
- Persaud SP, Donermeyer DL, Weber KS, Kranz DM, Allen PM. High-affinity T cell receptor differentiates cognate peptide-MHC and altered peptide ligands with distinct kinetics and thermodynamics. Mol. Immunol. 2010;47:1793–801. doi: 10.1016/j.molimm.2010.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pierce BG, Haidar JN, Yu Y, Weng Z. Combinations of affinity-enhancing mutations in a T cell receptor reveal highly nonadditive effects within and between complementarity determining regions and chains. Biochemistry. 2010;49:7050–9. doi: 10.1021/bi901969a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qi S, Krogsgaard M, Davis MM, Chakraborty AK. Molecular flexibility can influence the stimulatory ability of receptor-ligand interactions at cell-cell junctions. Proc. Natl. Acad. Sci. U S A. 2006;103:4416–21. doi: 10.1073/pnas.0510991103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rabinowitz JD, Beeson C, Wulfing C, Tate K, Allen PM, Davis MM, McConnell HM. Altered T cell receptor ligands trigger a subset of early T cell signals. Immunity. 1996;5:125–35. doi: 10.1016/s1074-7613(00)80489-6. [DOI] [PubMed] [Google Scholar]
- Reiser JB, Darnault C, Gregoire C, Mosser T, Mazza G, Kearney A, van der Merwe PA, Fontecilla-Camps JC, Housset D, Malissen B. CDR3 loop flexibility contributes to the degeneracy of TCR recognition. Nat. Immunol. 2003;4:241–7. doi: 10.1038/ni891. [DOI] [PubMed] [Google Scholar]
- Schwartz RH. Models of T cell anergy: is there a common molecular mechanism? J. Exp. Med. 1996;184:1–8. doi: 10.1084/jem.184.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shusta EV, Kieke MC, Parke E, Kranz DM, Wittrup KD. Yeast polypeptide fusion surface display levels predict thermal stability and soluble secretion efficiency. J. Mol. Biol. 1999;292:949–56. doi: 10.1006/jmbi.1999.3130. [DOI] [PubMed] [Google Scholar]
- Sim BC, Zerva L, Greene MI, Gascoigne NR. Control of MHC restriction by TCR Valpha CDR1 and CDR2. Science. 1996;273:963–6. doi: 10.1126/science.273.5277.963. [DOI] [PubMed] [Google Scholar]
- Sloan-Lancaster J, Evavold BD, Allen PM. Induction of T-cell anergy by altered T-cell-receptor ligand on live antigen-presenting cells. Nature. 1993;363:156–9. doi: 10.1038/363156a0. [DOI] [PubMed] [Google Scholar]
- Sykulev Y, Brunmark A, Jackson M, Cohen RJ, Peterson PA, Eisen HN. Kinetics and affinity of reactions between an antigen-specific T cell receptor and peptide-MHC complexes. Immunity. 1994;1:15–22. doi: 10.1016/1074-7613(94)90005-1. [DOI] [PubMed] [Google Scholar]
- Udaka K, Tsomides TJ, Eisen HN. A naturally occurring peptide recognized by alloreactive CD8+ cytotoxic T lymphocytes in association with a class I MHC protein. Cell. 1992;69:989–98. doi: 10.1016/0092-8674(92)90617-l. [DOI] [PubMed] [Google Scholar]
- Varani L, Bankovich AJ, Liu CW, Colf LA, Jones LL, Kranz DM, Puglisi JD, Garcia KC. Solution mapping of T cell receptor docking footprints on peptide-MHC. Proc. Natl. Acad. Sci. U S A. 2007;104:13080–5. doi: 10.1073/pnas.0703702104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber KS, Donermeyer DL, Allen PM, Kranz DM. Class II-restricted T cell receptor engineered in vitro for higher affinity retains peptide specificity and function. Proc. Natl. Acad. Sci. U S A. 2005;102:19033–8. doi: 10.1073/pnas.0507554102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willcox BE, Gao GF, Wyer JR, Ladbury JE, Bell JI, Jakobsen BK, van der Merwe PA. TCR binding to peptide-MHC stabilizes a flexible recognition interface. Immunity. 1999;10:357–65. doi: 10.1016/s1074-7613(00)80035-7. [DOI] [PubMed] [Google Scholar]
- Williams CB, Engle DL, Kersh GJ, Michael White J, Allen PM. A kinetic threshold between negative and positive selection based on the longevity of the T cell receptor-ligand complex. J. Exp. Med. 1999;189:1531–44. doi: 10.1084/jem.189.10.1531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xing Y, Hogquist KA. T-cell tolerance: central and peripheral. Cold Spring Harb Perspect Biol. 2012;1:4. doi: 10.1101/cshperspect.a006957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin Y, Li Y, Kerzic MC, Martin R, Mariuzza RA. Structure of a TCR with high affinity for self-antigen reveals basis for escape from negative selection. Embo J. 2011;30:1137–48. doi: 10.1038/emboj.2011.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu P, Haymaker CL, Divekar RD, Ellis JS, Hardaway J, Jain R, Tartar DM, Hoeman CM, Cascio JA, Ostermeier A, Zaghouani H. Fetal exposure to high-avidity TCR ligand enhances expansion of peripheral T regulatory cells. J. Immunol. 2008;181:73–80. doi: 10.4049/jimmunol.181.1.73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zehn D, Bevan MJ. T cells with low avidity for a tissue-restricted antigen routinely evade central and peripheral tolerance and cause autoimmunity. Immunity. 2006;25:261–70. doi: 10.1016/j.immuni.2006.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zehn D, Lee SY, Bevan MJ. Complete but curtailed T-cell response to very low-affinity antigen. Nature. 2009;458:211–4. doi: 10.1038/nature07657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Y, Bennett AD, Zheng Z, Wang QJ, Robbins PF, Yu LY, Li Y, Molloy PE, Dunn SM, Jakobsen BK, Rosenberg SA, Morgan RA. High-affinity TCRs generated by phage display provide CD4+ T cells with the ability to recognize and kill tumor cell lines. J. Immunol. 2007;179:5845–54. doi: 10.4049/jimmunol.179.9.5845. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.