

Abstract
 Human cytochrome P450 aromatase catalyzes with high specificity the synthesis of estrogens from androgens. Aromatase inhibitors (AIs) such as exemestane, 6-methylideneandrosta-1,4-diene-3,17-dione, are preeminent drugs for the treatment of estrogen-dependent breast cancer. The crystal structure of human placental aromatase has shown an androgen-specific active site. By utilization of the structural data, novel C6-substituted androsta-1,4-diene-3,17-dione inhibitors have been designed. Several of the C6-substituted 2-alkynyloxy compounds inhibit purified placental aromatase with IC50 values in the nanomolar range. Antiproliferation studies in a MCF-7 breast cancer cell line demonstrate that some of these compounds have EC50 values better than 1 nM, exceeding that for exemestane. X-ray structures of aromatase complexes of two potent compounds reveal that, per their design, the novel side groups protrude into the opening to the access channel unoccupied in the enzyme–substrate/exemestane complexes. The observed structure–activity relationship is borne out by the X-ray data. Structure-guided design permits utilization of the aromatase-specific interactions for the development of next generation AIs.
Human cytochrome P450 aromatase catalyzes with high specificity the synthesis of estrogens from androgens. Aromatase inhibitors (AIs) such as exemestane, 6-methylideneandrosta-1,4-diene-3,17-dione, are preeminent drugs for the treatment of estrogen-dependent breast cancer. The crystal structure of human placental aromatase has shown an androgen-specific active site. By utilization of the structural data, novel C6-substituted androsta-1,4-diene-3,17-dione inhibitors have been designed. Several of the C6-substituted 2-alkynyloxy compounds inhibit purified placental aromatase with IC50 values in the nanomolar range. Antiproliferation studies in a MCF-7 breast cancer cell line demonstrate that some of these compounds have EC50 values better than 1 nM, exceeding that for exemestane. X-ray structures of aromatase complexes of two potent compounds reveal that, per their design, the novel side groups protrude into the opening to the access channel unoccupied in the enzyme–substrate/exemestane complexes. The observed structure–activity relationship is borne out by the X-ray data. Structure-guided design permits utilization of the aromatase-specific interactions for the development of next generation AIs.
INTRODUCTION
Cytochrome P450 aromatase (CYP19A1) is the only enzyme in vertebrates known to catalyze the biosynthesis of estrogens from their androgenic precursors. The human enzyme uses with high specificity androstenedione (ASD), testosterone (TST), and 16α-hydroxytestosterone (HTST), all with the same androgen backbone, and converts them to estrone (E1), 17β-estradiol (E2), and 17β,16α-estriol (E3), respectively, by aromatization of the A-ring.1–3 The reaction is a three-step process, each requiring 1 mol of O2, 1 mol of NADPH, and coupling with its redox partner cytochrome P450 reductase (CPR) for the transfer of electrons. The human enzyme has a heme group, a single polypeptide chain of 503 amino acid residues, and is an integral membrane protein of the endoplasmic reticulum.4–7 Many of the human microsomal P450s catalyze the metabolism of a wide variety of endogenous and xenobiotic compounds and drugs with low substrate specificities. However, it is the narrow substrate selectivity and the third step (the aromatization step) that make aromatase unique.
Inhibition of estrogen biosynthesis by the third generation aromatase inhibitors (AIs) exemestane (EXM), letrozole (LTZ), and anastrozole (ANZ) (Figure 1) is the frontline therapy for postmenopausal estrogen-dependent breast cancer.8,9 AIs have also been effectively used in the treatments of endometriosis,10 ovarian,11 and lung12 cancers. The enzyme has been a subject of intense biochemical and biophysical investigations for the past 40 years.1–5,8,13–19 The recent crystal structure of aromatase in complex with the natural substrate ASD (androst-4-ene-3,17-dione, Figure 1) has provided unprecedented glimpses into the molecular basis for its substrate specificity, reaction mechanism, steroidal passage, vibrational modes, transmembrane integration, and oligomeric state.6,7,20,21 This structural insight has prompted the urgency for the incorporation of aromatase specificity into the inhibitors and revitalized new AI discovery research.22–24 The design that exploits the androgen-specific architecture of the active site and interactions exclusive to the substrate-binding pocket is likely to minimize cross-reactivities of the current AIs. ANZ and LTZ are known to cross-react with P450s such as CYP1A2, CYP2C9, CYP3A4,25 and CYP2A6,26,27 while EXM is metabolized by CYP3A4.8,9,28–30 Furthermore, EXM is weakly estrogenic via its agonistic interaction with estrogen receptor (ER).31
Figure 1.

Chemical structures of aromatase substrate and inhibitors. Androst-4-ene-3,17-dione (androstenedione, ASD) is the substrate, androsta-1,4-diene-3,17-dione (ASDD) a substrate analogue, and formestane a steroidal aromatase inhibitor. Exemestane (EXM) is the steroidal AI and letrozole (LTZ) and anastrozole (ANZ) are the nonsteroidal AIs used in the treatment of postmenopausal estrogen-dependent breast cancer. The color code is the following: carbon, black; nitrogen, blue; oxygen, red.
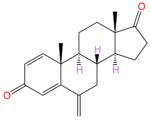
Here we report the synthesis and evaluation of the C6-substituted androsta-1,4-diene-3,17-dionealkynyloxy series of compounds whose designs have been guided by the active site architecture. The steroidal backbone of these new compounds is identical to that of EXM (also known as Aromasin), which is a C6-methylidene substituted androsta-1,4-diene-3,17-dione (ASDD, Figure 1).
RESULTS
Design and Synthesis of Steroidal Inhibitors
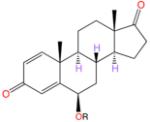
The design of the new inhibitors is based on analysis of the steroid-binding environments derived from the crystal structures of the ASD and EXM complexes of aromatase and is shown schematically in Figure 2. The steroid skeletons are completely surrounded by an envelope of hydrophobic residues and by proton donors to the 3- and 17-keto oxygens. The only exposed positions are C4 and C6, near the opening to the active site access channel (Figure 2) that begins at the protein–lipid bilayer interface. This lone open access to the active site from the ER membrane has been proposed to be the substrate entry and product exit pathways.6,7 Modeling analysis suggests that C6α-/C6β- and, to some extent, C4-substitutions could be accommodated in the available space and that a linear carbon chain would be most effecacious in penetrating the “hydrophobic clamp” (a tight entry point between Cβ of Ser478 and Cγ2 of Thr310)6 into the access channel opening. We test the molecular modeling predictions by evaluating a series of 6-substituted androsta-1,4-diene-3,17-dione containing variable alkoxy chains, with a particular emphasis on linear alkynyl functionalities. In order to secure a general approach to these compounds, we recently developed a silver catalyzed method for the selective etherification at the C6 position of the steroid.32 This method relies on an unusual carbene O–H insertion protocol in which the ether bond is selectively formed at the vinylogous position of the carbenoid rather than directly at the carbenoid site. We found that such reactions proceed smoothly using a catalytic amount of silver triflate in trifluorotoluene at room temperature (2–16 h). This new process is a powerful enabling technology that can be applied to the synthesis of a series of alkyne derivatives as illustrated in Scheme 1. The first two compounds are C6β-methoxy and -ethoxy derivates of ASDD (compounds 2 and 3, respectively; Scheme 1). The next five analogues, 4–8, all have a 2-alkynyloxy substitution at the C6β position with progressively longer alkyl chains (Scheme 1). The final compound 9 is a 2-butynyloxy derivative, similar to 4, except that it has a terminal hydroxy group on the side chain (Scheme 1).
Figure 2.

Design considerations for the new inhibitors derived from the binding interactions and exposure of the ligands to the enzyme interaction spaces: (a) ASD; (b) EXM. In (a) and (b) derived from the X-ray structures, the residues lining the binding pocket making hydrophobic and hydrogen-bonding contacts are shown (hydrophobic, green; acidic, red; basic, blue; polar, purple; sulfur-containing, yellow). Exposure at the C4 and C6 positions of the steroid to the access channel opening is indicated. Also shown schematically in (a) is a water molecule trapped between Asp309 and Arg192 side chains, postulated to have a role in the proton relay network and enolization of 3-keto.6
Scheme 1.

Synthesis of C6β-2-Alkynyloxy Derivatives of ASDDa
aShown are chemical structures of newly synthesized C6β-alkoxy-substituted androsta-1,4-diene-3,17-dione compounds 2–9.
Inhibition of Aromatase Activity by New Compounds
The inhibitory properties of 2–9, along with those of LTZ and two steroidal inhibitors EXM and formestane, have been evaluated in human placental aromatase purified to homogeneity (Figure 3a). A composite of dose response curves demonstrating that these compounds inhibit aromatase in a concentration-dependent manner is given in Figure 3b. Table 1 summarizes the results from IC50 determination for all eight compounds and three controls, as well as the 95% confidence intervals of IC50 values. More detailed inhibition data and individual dose response curves for 2–9 and the controls are provided in Table S1 (Supporting Information).
Figure 3.

Measurement of inhibitory and antiproliferative activities of the novel C6-substituted androgens in purified aromatase and MCF-7a breast cancer cells. (a) SDS–PAGE analysis of a typical sample of purified human placental aromatase used for enzyme inhibition and complex crystallization experiments. (b) Enzyme inhibition activity of the novel inhibitors plotted as a function of concentration. Averaged data points and their standard deviations (SDs), resulting from quadruplicate measurements of three independent experiments, are shown for eight C6-substituted androgens (2–9) and three known inhibitors as controls. (c) Bar graphs representing E2-, ASD-, and TST-stimulated proliferation of MCF7a cells. The growth of MCF-7a cells is stimulated by 10 pM to 100 nM E2 and TST or by 1 nM to 100 nM ASD. The mean of three independent experiments and the SD are shown for each data point. (d) Inhibition of TST-induced proliferation of MCF-7a cells by the six most potent C6-substituted androgen inhibitors (4–9), as a function of concentration. The same symbol and color as (b) are used to designate each inhibitor. The data are normalized against the control cells treated with 1 nM TST alone. Each data point and the SD are the result of three independent experiments measured in triplicate.
Table 1.
Summary of IC50 and EC50 Measurements of Newly Designed C6β-Alkoxy/2-Alkynyloxy Series of Steroidal Aromatase Inhibitors and Three Known Inhibitors as Controlsa
| Compounds |
|
IC50 (nM) |
95% Confidence Interval for IC50 (nM) |
EC50 (nM) |
95% Confidence Interval for EC50 (nM) |
|---|---|---|---|---|---|
| R Group | |||||
| 2 | Me | 5200 | 4700 to 5500 | - | |
| 3 | Et | 18100 | 13100 to 25200 | - | |
| 4 |
|
112.3 | 78.2 to 161.3 | 1.7 | 1.2 to 2.2 |
| 5 |
|
11.8 | 9.3 to 14.9 | 0.03 | 0.02 to 0.06 |
| 6 |
|
83.0 | 74.5 to 93.9 | 3.4 | 2.4 to 4.7 |
| 7 |
|
181.1 | 164.0 to 200.0 | 5.4 | 3.2 to 7.3 |
| 8 |
|
2180 | 1750 to 2710 | 15.7 | 10.1 to 22.9 |
| 9 |

|
20.0 | 18.1 to 22.0 | 0.3 | 0.2 to 0.4 |
| Exemestane (EXM) |
|
50.1 | 40.9 to 61.4 | 5.6 | 2.7 to 6.5 |
| Formestane |
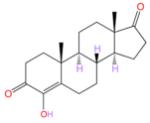
|
48.6 | 33.6 to 71.1 | - | - |
| Letrozole (LTZ) |

|
9.9 | 9.3 to 10.5 | 0.004 | 0.003 to 0.007 |
The aromatase specific activity is measured as number of moles of ASD converted to E1 per unit mass of the enzyme per unit time and typically ranges between 25 and 50 nmol min−1 mg−1.
The methoxy and ethoxy derivatives 2 and 3 have IC50 values of several micromolar, about 1000-fold less potent than the AIs. However, the IC50 drops to 112 nM for the next compound in the series, the butynyloxy derivative 4. This value is comparable to the 50 nM IC50 for the steroidal AI EXM and 49 nM for the known steroidal inhibitor formestane, measured as controls under the same experimental conditions (Table 1). The IC50 values for the two AIs and formestane from the current determination are very similar to the values reported by many groups over the past 20 years. For the 2-alkynyloxy derivatives 5 and 9, compounds with an additional methyl or a hydroxyl compared to 4, the IC50 values are 12 and 20 nM, respectively, quite similar to the in vitro potency of 10 nM for the most potent AI nonsteroidal LTZ. However, the compounds with progressively longer side groups, 6–8, have increasingly higher IC50 values of 83, 181, and 2180 nM, respectively, suggesting perhaps that a six-atom-long linear chain substitution at the C6β position as in derivatives 5 and 9 has the optimal size for the active site cleft. Direct validation of this structure–activity interrelation comes from the X-ray data described below.
Antiproliferative Activity of New Compounds
The six potent 2-alkynyloxy derivatives 4–9, as well as EXM and LTZ as controls, were assayed for their antiproliferative properties in the breast cancer cell MCF-7-Tet-off-3βHSD1-Arom (MCF-7a; see Experimental Section). The results are summarized in Figure 3c,d, and the EC50 values are given in Table 1. More detailed antiproliferation data and individual dose response curves for the new inhibitors and the controls are provided in Table S2 (Supporting Information). The proliferative activity of the MCF-7a cells is first evaluated by treating the cells with E2, TST, and ASD. All three compounds effectively stimulate the growth of the MCF-7a cells in a concentration-dependent manner (Figure 3c), confirming that these cells express both a functional estrogen receptor and an active aromatase. E2 (EC50 = 55 pM) is more potent than either TST (EC50 = 99 pM) or ASD (EC50 = 3.6 nM) in growth stimulation, indicating that there is a time lag for the synthesis of E2 from TST (via the aromatase pathway) and ASD (via the aromatase-17β-hydroxysteroid dehydrogenase type 1 pathway) in these breast cancer cells. The maximum concentration of E2 and TST required to stimulate MCF-7a cell proliferation in our cell-based assay system is determined to be 1 nM (Figure 3c); this concentration is used for subsequent proliferation inhibition assays.
Figure 3d summarizes the results from the antiproliferative activity assay of the 2-alkynyloxy aromatase inhibitors 4–9 in the MCF-7a cells in comparison with the activities of LTZ and EXM. The data show that the newly designed inhibitors abolish the TST-stimulated proliferation of MCF-7a cells in a dose dependent manner. The number of MCF-7a cells nearly doubles in response to 1 nM TST treatment. However, the addition of aromatase inhibitors 4–9 progressively inhibits the stimulatory activity of TST as a function of concentration (Figure 3d). The EC50 values of 4–9 are 1.7, 0.03, 3.4, 5.4, 15.7, and 0.3 nM, respectively (Table 1). Of these inhibitors, 4, 5, and 9 show 2.6-fold (p < 0.040), 119.6-fold (p < 0.010), and 14.7-fold (p < 0.012) antiproliferative activities, respectively, against TST-stimulated cell growth when compared to the steroidal AI EXM (EC50 = 5.6 nM). Thus, the structure–activity relationship of these compounds in the cell-based antiproliferation assay parallels their enzyme inhibitory properties in the cell-free system.
Aromatase–Androstenedione Complex Structure at 2.75 Å
The newly refined structure has yielded a better model than the 2.90 Å structure (PDB code 3EQM)6 in terms of overall quality and the refinement parameter statistics (Table S3, Supporting Information). Inclusion of the higher resolution data enabled rebuilding of some of the weakly defined loop regions and inclusion of additional solvent atoms into the model. The residues Ser267 to Cys275 in the G–H loop have clearer electron densities than the previous map and are rebuilt to better conformational geometries. The His459 side chain is modeled in two alternative conformations. The ΦΨ plot of the refined model has 95.6% of the residues in the favored regions and no outlier, as opposed to 94.4% and 0.4%, respectively, for the 2.90 Å model. This 2.75 Å structure of the androstenedione–complex is used as the reference for all structural data described in this work. The ASD binding site is schematically depicted in Figure 2a.
Binding Modes of EXM and the 2-Alkynyloxy Derivatives 4 and 5 from the Crystal Structures of Their Aromatase Complexes
The crystal structures of inhibited complexes of aromatase with EXM, 4, and 5 have been determined at 3.21, 3.48, and 3.90 Å, respectively. The initial atomic model of each inhibitor is generated first by fitting within its unbiased difference electron density map ((∣Fobs∣ – ∣Fcal∣) map before addition of the inhibitor to the model for phase calculation). The atomic model of the complex is then refined against the diffraction data. The data collection and refinement results are summarized in Table S3. The unbiased difference electron density maps calculated before the inclusion of the inhibitors or solvent molecules in the models and their respective refined structures are shown in parts a, b, and c of Figure 4, respectively.
Figure 4.

Unbiased difference (∣Fobs∣ – ∣Fcal∣) electron density maps, calculated before inclusion of the inhibitors in the models. Shown are the refined atomic models of the aromatase complexes with (a) EXM, 3.21 Å resolution contoured at 4.5σ (PDB code 3S7S), (b) 2-butynyloxy derivative 4, 3.48 Å at 4.0σ (PDB code 4GL5), and (c) 2-pentynyloxy derivative 5, 3.90 Å at 2.7σ (PDB code 4GL7). The opening toward the active site access channel is indicated with an arrow in (a). The C6-substituted alkyne side groups of the 2-alkynoxy derivatives 4 and 5 protrude into the channel space. Some residues referenced in the text are marked. Carbon atoms are in green, nitrogen blue, and oxygen red. The viewing direction is roughly the same in all three parts.
All three inhibitors bind at the active site with their steroid backbones similar to the binding mode of the substrate ASD.6 The C6-methylidene group of the EXM molecule is accommodated within the bulge of the density at the C6 position (Figures 4a and 5a). A difference electron density map calculated without the methylidene carbon shows the missing density (Figure S1, Supporting Information). The root-meansquare deviation (rmsd) from the superposition of the ASD and EXM structures is 0.3 Å for the backbone atoms and 0.2 Å for the Cα atoms, within the limits of the random positional error. The Cα rmsd values of 0.5 Å and higher are associated with various loops and at the N and C termini and correlate well with high X-ray isotropic B factors. In the active site cavity, residue Asp309 has the highest rmsd of 0.6 Å, followed by Phe221 from the active site access channel cavity and two residues from the β8–β9 loop (in standard P450 nomenclature, β4 loop), Leu477 and Ser478 (Figure 5). The segments that undergo some conformational movements are located at the entrance from the access channel to the active site (Figures 4a and 5a,b). These changes are likely to be responsible for the accommodation of different ligands. Several concerted adjustments between the active site residues and EXM in the catalytic cleft that serve to accommodate the C6-methylidene group of exemestane are the following: (1) The backbone A-ring of EXM shifts 0.5 Å away from the access channel, while the D-ring positions of both steroids are similar when compared with the ASD complex (Figure 5b). The two steroid backbones are superimposable with an overall backbone rmsd of ~0.3 Å. (2) Cγ of Thr310 moves slightly away from the C6-methylidene group to maintain a van der Waals contact distance of 3.4 Å. (3) The protonated Asp309 side chain adjusts its orientation toward the 3-keto oxygen of exemestane (Figure 5b). These movements result in marginal increase in the length of the hydrogen bond between 3-keto oxygen and HOδAsp309 from 2.7 to 2.9 Å, leaving the ones between the 17-keto oxygen and NH1 of Arg 115 (~3.4 Å) and 17-keto oxygen and backbone amide of Met374 (~2.8 Å) virtually unchanged.
Figure 5.

Closeup views of the catalytic clefts of aromatase complexes. (a) The steroid-binding pocket of the bound EXM molecule and its unbiased (before inclusion of any small molecule or solvent in the model) difference (∣Fobs∣ – ∣Fcal∣) electron density at 4.5σ (PDB code 3S7S). The surrounding protein environment is shown and labeled. The protein backbone is rendered in rainbow color (N terminus, blue; C terminus, red): carbon, gray; nitrogen, blue; oxygen, red. (b) The superimposed catalytic clefts of the ASD and EXM complexes illustrating the deviation between two steroid-binding modes and the active site residues. The ASD complex is shown in blue backbone and blue carbon (PDB code 3S79). The complexes of the C6-substituted androgens 4/5 also have similar deviations. The van der Waals contact distance (3.4 Å) between the C6-methylidene carbon and Cγ of Thr310 is indicated by a black solid line. The hydrogen bonds between the exemestane and active site residues are drawn as dashed lines. (c) View of the catalytic cleft from the access channel perspective. A hydrophobic crevice formed by Thr310-Cγ, Val370-Cγ2, and Ser478-Cβ firmly grips the C6-methylidene group of exemestane. The van der Waals surface of the catalytic cleft for the aromatase–EXM complex is rendered in light gray.
The overall structures of the complexes with 4 and 5 have the EXM complex-like deviations from the ASD complex but with higher rmsd values for the backbone atoms, perhaps owing to the lower resolution of the data. The active sites also undergo similar small adjustments as seen in the EXM complex to accommodate the inhibitors. The steroid skeletons in all four complexes bind similarly. The hydrogen bond distances between 3-keto and HOδAsp309, as well as between 17-keto and the main chain amide of Met374/Arg115 side chain (Figure 4), are virtually the same given the positional error limits at the resolutions of the crystal structures. Furthermore, the binding modes of the side groups in 4 and 5 are very similar except that compound 5 has the extra C24 methyl group (Scheme 1, Figure 6a). The active site pocket and the binding environment of 5 are schematically depicted in Figure 6a. The protein van der Waals interaction surface of the active site pouch that encompasses the steroid-binding pocket and the heme group in the aromatase-compound 5 structure is shown in Figure 6b. The alkyne side chains of the novel inhibitors protrude into the access channel cavity; the C20 atom is wedged between CβSer478 (4.4 Å) and Cγ2Thr310 (3.3 Å) (Figure 6a,b). C23 is at or near the van der Waals distances from Cγ2 Val313 (4.2 Å), OγSer478 (3.4 Å), and CγHis480 (4.7 Å). The C24 methyl end of the alkyne chain of the 2-pentynyloxy derivative 5 packs against Cγ2 Val313 (4.2 Å), the main chain carbonyl of Asp309 (3.4 Å), and the arene ring of Phe221 and His480 side chain are roughly at distances 3.9 and 4.6 Å, respectively, to the ring centers. The tip is thus near the end of the channel, roughly 5 Å away from the salt-bridged Arg192–Glu483 pair and only about 3.2 Å from the water molecule trapped between Asp309 and Arg192 side chains in the ASD complex.6 It is therefore likely that this water molecule, which was proposed to have catalytic roles in the enolization and proton relay via Arg192–Asp309 network,6 is displaced by the binding of 5. Any further elongation of the side group by one or more carbon atoms as in the higher homologues 6-8 could conceivably result in steric clashes at this end of the access channel. However, having a hydroxyl at position 24, as in the hydroxy derivative 9, or at position 25, in lieu of a methyl group, could elicit more favorable polar contacts.
Figure 6.

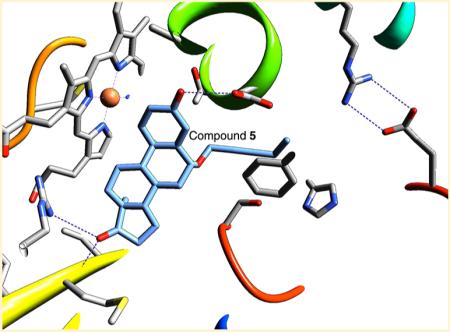
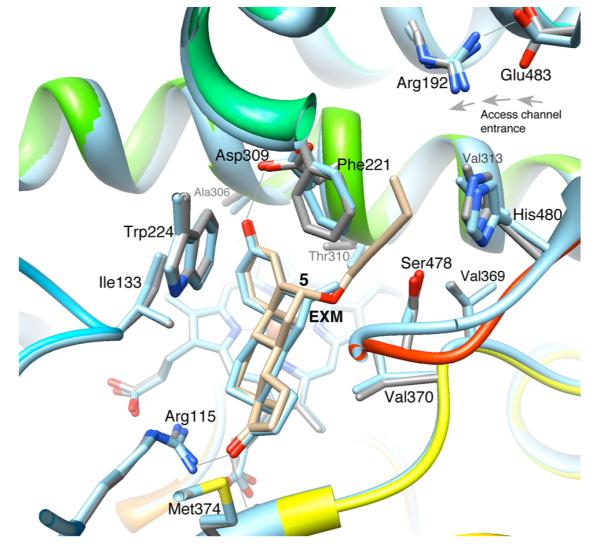
Binding mode of the novel aromatase inhibitor C6β-2-alkynyloxy derivative 5. (a) Schematic diagram depicting the tight hydrophobic binding pocket for the steroid skeleton, the proton donors at the 3- and 17-keto positions, and the 6β-alkoxy-substituted alkyne side group that nearly fills the access channel. The color codes are the same as in previous figures. (b) View of the catalytic pocket and the access channel residues from the crystal structure of the aromatase-C6β-2-alkynyloxy derivative 5 complex (PDB code 4GL7). The bound molecule 5 (carbon atoms in light blue) is shown within the active site “pouch”, which results from the rendering of the van der Waals protein surface in semitransparent gray color. The access channel entrance at the lipid interface is the only opening of the “pouch” as indicated. See text for the details of interaction of the alkyne side group with access channel residues. (c) View along access channel entrance roughly normal to the viewing direction in (b).
The inhibition and antiproliferation data presented in Table 1 are reflective of the structural results. Starting with the 2-methoxy derivative 2, which has a small methyl side group at the 6β-position, progressively longer alkyne groups yield more potent inhibitors, reaching the optimal length for the 2-alkynoxy derivatives 5 and 9. The crystal structure of this inhibitor complex provides the rationale and the molecular basis for the observed inhibition and antiproliferation results. The structural data also demonstrate that any longer alkyne groups would be increasingly detrimental to favorable interactions within the access channel and destabilizing to inhibitor binding, in agreement with the activity data for 6–8 in Table 1.
Docking of the 2-Pentynyloxy Derivative 5 into Other P450s and ERα
In order to assess the likelihood of cross-reactivity of the new inhibitors with other P450s and ERα, we have attempted to dock 5 at the ligand-binding sites of the crystal structures of CYP3A4, CYP2A6, CYP2C9, CYP2D6, CYP17A1, and ERα. In the self-docking experiments, crystallo-graphically observed ligands are docked to their respective cocrystal apo structures (Figure S2, Supporting Information). The best docking poses in all cases are nearly identical to the respective crystal structures with low rmsd values, validating the docking process (Figure S2, and Table S4, Supporting Information). The docking of EXM, which has been suggested to be weakly estrogenic, to the E2 site of ERα suggests favorable interactions based on the free energy of EXM binding (Table S4). However, no favorable docking pose for 5 binding at the E2 site of ERα can be found, as the alkyne chain at C6 causes severe steric clashes (Figure S3, Supporting Information). Similarly, for the CYP receptors, the best docking poses of 5 suffer from severe steric clashes and/or lack of favorable polar/hydrophobic interactions as evident from higher empirical free energy than for the corresponding ligand in the X-ray structure (Table S3 and Figure S3, Supporting Information). Furthermore, we have utilized receptor–ligand complex structure-based pharmacophore queries to screen for potential receptor–5 complex poses (see Experimental Section for description). The pharmacophore filtering method for docking of 5 into ERα, CYP3A4, CYP2A6, CYP2C9, CYP2D6, and CYP17A1 also fails to yield meaningful poses for 5 (Table S4), implying that the docked poses of the query ligand (compound 5) fail to utilize the binding interactions of the ligand in the X-ray structure. These docking calculations, however, use the static X-ray structure as obtained by the PDB coordinates and do not take into account possible structural flexibility of the ligand binding sites, which many of the xenobiotic-metabolizing P450s are known to have.
DISCUSSION AND CONCLUSION
We have shown that the newly developed steroidal compounds by structure-guided approach are potent inhibitors of human aromatase. The potencies of some of these inhibitors, based on both purified aromatase inhibition and prevention of breast cancer cell proliferation assays, are at least comparable to those of the most effective AIs being used today in postmenopausal hormonal breast cancer therapy. The results presented also establish that the most potent of these new inhibitors optimally utilize the androgen-specific active site cleft and bind in competition with the substrate. Furthermore, the observed structure–activity relationship of the current series of steroidal inhibitors can be rationalized by the data at the molecular level.
Our crystallographic and computational results have already established that the aromatase structure is stable and has a rigid core and that the active site is small, compact, and not easily flexed by ligand binding,6,7 unlike many other P450s.33–35 This rigid core property of aromatase appears to be intrinsic as the normal-mode analysis of the aromatase molecule suggests.7 We have also shown that membrane integration would not affect the structural rigidity of the aromatase active site.7 Nevertheless, this limited flexibility of the active site is utilized in accommodating ligands by modest adjustments of the catalytic cavity, as we see in the complexes of all three inhibitors. For EXM, the additional hydrophobic interactions via the C6-methylidene group with the hydrophobic crevice surrounded by Thr310-Cγ, Val370-Cγ2, and Ser478-Cβ (Figures 4 and 5) could add to its binding affinity. The crevice, acting like a “hydrophobic clamp”, holds firmly the C6-methylidene group of exemestane, which has a better shape complementarity to aromatase than the substrate ASD (Jiang and Ghosh, unpublished data). The C6 methylidene group rests at the mouth of the access channel at the van der Waals distance from the nearest atom. No covalent bond formation between the exemestane molecule and catalytic active site residues is found, despite reports of exemestane being a mechanism-based inhibitor that binds irreversibly.9,29,36 However, any such covalent bond formation would require coupling with CPR and steps through the catalytic cycle, which the crystallized aromatase–EXM complex had not undergone. Our experimental data show that in the absence of CPR, exemestane is held at the C6-methylidene by the “hydrophobic clamp”, reducing the mobility of Thr310 critical for the hydroxylation reaction.6 Thus, a “tighter” binding and “immobilization” of the catalytic machinery could also be the means of inactivation by EXM.
The chemical nature and orientation of the unsaturated C6-methylidene group in EXM are different from those of the C6β-alkoxy group, which is the basis for all new inhibitors with alkyne side chains. This is illustrated by the least-squares superposition of the EXM and 5 complex structures in Figure 7. It is thus possible that the mechanism of inhibition of this series of compounds is different from that of the EXM. Interestingly, 2 and 3 with small methyl or ethyl side chains are less potent than EXM. Nevertheless, the C6β-alkoxy group is suitably oriented to pierce through the “hydrophobic clamp” to bury the longer side chains within the access channel (Figure 7), thereby immobilizing the catalytic residues as well. Any significant conformational change between the superimposed active sites occurs in the β8-to-β9 loop region containing Ser478 at the mouth of the access channel (Figure 7) and could result from the accommodation of the C6β-alkynyloxy side chain. The structural data also reveal that the compounds with a 2-alkynyloxy side chains fit tightly within the hydrophobic environment of the channel. Both the structural and functional results are consistent in that 5 and 9 possess the right dimensions to traverse the access channel. The side group of 5 nearly extends to the polar residues Arg192 and Glu483 at the channel entrance, which is presumed to be at the membrane–protein interface (Figure 6b). It is likely that the terminal C24 methyl of the 2-pentynyloxy derivative 5, which also has a favorable hydrophobic surrounding, interferes with the water molecules trapped in the channel by polar residues Ser478, Arg192, and Asp309.6 These water molecules were proposed to be in the proton relay network that presumably plays a critical role in the A-ring aromatization chemistry.6 The hydroxy derivative 9 has a hydroxyl group at this position, which may promote polar interactions with the polar moieties, and thus appears to be just as potent as 5. These observations are indicative of an optimal size/nature of the side group lying between the 2-alkynoxy derivatives 5/9 and 6, leaving room for design improvement.
Figure 7.
Superposition of the structures of EXM and compound 5 complexes, illustrating that the orientation of the unsaturated C6-methylidene group in EXM is different from that of the C6β-alkoxy group. EXM complex is shown in light blue carbon and backbone (PDB code 3S7S). Compound 5 (PDB code 4GL7) is in beige color. Side chain carbons are in gray, and the backbone is in rainbow.
Although the mechanism of inactivation cannot be ascertained from the present data, structural results are suggestive of competitive inactivation by complementing hydrophobic and polar interactions, a telltale sign of reversibility. The residues with the largest shift among active site regions are Asp309, Phe221, Leu477, and Ser478. These are the residues that either constitute the access channel opening or reside next to them. The inherent flexibility of the access channel makes it possible for the modest ligand-induced conformational changes. Channel breathing is a likely means for substrate and product transport between the active site and the membrane interior via the access channel.6,7
In conclusion, we demonstrate that the new steroidal compounds are highly potent in both enzyme inhibition and breast cancer cell viability assays. The X-ray structures of the aromatase complexes of two new inhibitors, in comparison with those of ASD and EXM, reveal that the novel side groups occupy the access channel-pocket as their design intended. Results from the docking of the 2-pentynyloxy derivative 5, the most potent of the current series, into ligand-binding sites of ERα and other P450s indicate that these inhibitors of aromatase are less likely to have binding affinity for these targets. However, given the known flexibility of some of the xenobiotic-metabolizing P450 enzymes, it is difficult to predict cross-reactivity from docking analysis alone. Nonetheless, structure-guided design permits optimal utilization of the enzyme-specific interactions exclusive to human aromatase.
EXPERIMENTAL SECTION
Chemistry
All reactions were conducted in oven-dried glassware (100 °C, >12 h) under an atmosphere of argon. Commercially available reagents were used as received unless otherwise stated. Solvents were purified by passage through a bed of activated alumina (Grubbs-type solvent purifier). NMR spectra were recorded on Varian INOVA 400 and 600 MHz NMR spectrometers. 1H NMR data are presented as follows: chemical shift (in ppm on the δ scale relative to the residual solvent peaks), multiplicity (s = singlet, d = doublet, t = triplet, q = quartet, m = multiplet, br = broad, app = apparent), coupling constant (J in Hz), integration, assignment. Coupling constants were taken directly from the spectra. 13C spectra were recorded at 100 MHz, and all chemical shift values are reported in ppm on the δ scale relative to the residual solvent peaks. Flash chromatography was performed on silica gel 60 Å (230–400 mesh). Melting points were obtained with an Electrothermal melting point apparatus and are uncorrected. Infrared spectral data are reported in units of cm–1. Mass spectrometry was performed on a JEOL JMS-SX102/SX102A/E mass spectrometer using ACPI as an ionization method. Infrared spectra were recorded on a Thermo Scientific Nicolet iS10 and reported in units of cm–1. The optical rotation was determined using a Jasco P-2000 polarimeter.
All compounds possess a purity superior or equal to 95%. The purity was checked by HPLC using a Dynamax 60A column and isopropanol/hexanes as eluting solvent. The purity of individual compounds was determined from the peak areas in the chromatogram of the sample solution.
(8 R, 9 S, 10 R, 13 S, 14 S)-4-Diazo-10, 13-dimethyl-7,8,9,10,11,12,13,14,15,16-decahydro-3H-cyclopenta[a]-phenanthrene-3,17(4H)-dione (1)
An oven-dried 250 mL round-bottomed flask was charged with deconjugated androgen32 (1.0 equiv, 3.52 mmol, 1.00 g) and 3-(azidosulfonyl)benzoic acid37 (1.2 equiv, 4.19 mmol, 951 mg). Anhydrous acetonitrile (100 mL) was added, and the mixture was stirred rapidly under an atmosphere of Ar for 20 min at ambient temperature. Diazabicycloundecene (1.5 equiv, 5.28 mmol, 800 μL) was added dropwise over 10 min. The mixture changed from clear pale yellow to orange and cloudy. The mixture was stirred at ambient temperature for 2 h. The mixture was diluted with Et2O (50 mL), and the reaction was quenched with a saturated solution of aqueous ammonium chloride (50 mL). The mixture was separated, extracted with Et2O (3 × 50 mL) and the combined organic phase washed with brine (2 × 50 mL), dried over magnesium sulfate, and concentrated under reduced pressure to afford an orange solid. The steroidal diazo was isolated using the Isolera purification system (SNAP 25 g cartridge, gradient of 90:10 hexane/EtOAc to 5:95 hexane/EtOAc over 10 column volumes), as an orange solid (720 mg, 67%). 1H NMR (400 MHz, CDCl3) δ 6.81 (d, 1H, J = 10.4 Hz, 1-CH), 5.95 (d, J = 10.4 Hz, 1H, 2-CH), 5.36 (dd, J = 4.8 and 2.5 Hz, 1H, 6-CH), 2.48 (dd, J = 19.2 and 8.9 Hz, 1H, 16-CHα), 2.41–2.31 (m, 1H), 2.11 (dt, J = 19.2 and 9.4 Hz, 1H, 16-CHβ), 2.01–1.85 (m, 5H), 1.64–1.50 (m, 2H), 1.46–1.30 (m, 3H), 1.26 (s, 3H, 19-CH3), 0.92 (s, 3H, 18-CH3); 13C NMR (100 MHz, CDCl3) δ 220.3, 182.4, 150.1, 126.8, 126.7, 118.0, 51.7, 47.8, 44.6, 39.6, 35.9, 31.7, 31.4, 29.9, 23.7, 21.9, 20.6, 13.9; IR (film) 2944, 2078 (N2), 1735 (C═O), 1659 (C═O), 1282, 729; m/z (APCI) 311.2 (25%, M + H), 299.2 (100%), 284.2 (39%); HRMS-APCI m/z 311.1757 (C19H23N2O2 requires 311.1754).
General Procedure for the O–H Insertion of Alcohols with Steroid Diazo Compound (1)
An oven-dried round-bottomed flask was charged with a solution of ROH (10 equiv) in degassed trifluorotoluene (5 mL), to which was added AgOTf (5–10 mol %), and the mixture was stirred at room temperature for 10 min. A solution of steroid diazo 1 (1 equiv) in degassed trifluorotoluene (3 mL) was added dropwise over 1 h. The progress of the reaction was monitored by TLC, and upon consumption of the steroidal diazo starting material (between 2 and 16 h), the mixture was concentrated under reduced pressure. The product was purified by flash chromatography (eluting with hexanes/EtOAc, 80:20).
(6R,8R,9S,10R,13S,14S)-6-Methoxy-10,13-dimethyl-7,8,9,10,11,12,13,14,15,16-decahydro-3H-cyclopenta[a]-phenanthrene-3,17(6H)-dione (2)
2 was derived from steroid diazo 1 (103 mg, 0.33 mmol, 1.0 equiv), methanol (133 μL, 3.30 mmol, 10 equiv), and AgOTf (9 mg, 10 mol %) and isolated as an off-white solid (48 mg, 46%). Mp 150–152 °C; 1H NMR (600 MHz, CDCl3) δ 7.03 (1H, d, J = 10.1 Hz, H-1), 6.2 (1H, dd, J = 10.1 and 1.9 Hz, H-2), 6.18 (1H, d, J = 1.9 Hz, H-4), 3.89 (1H, app t, J = 2.9 Hz, H-6), 3.23 (3H, s, OCH3), 2.45 (1H, dd, J = 19.4 and 8.9 Hz, Hβ-16), 2.22–2.16 (1H, m, Hβ-7), 2.15–2.09 (1H, m, Hα-7), 2.10–2.00 (1H, m, Hα-16), 1.97–1.89 (1H, m, Hα-15), 1.88–1.77 (2H, m, Hα-11 and Hβ-12), 1.74 (1H, ddd, J = 22.0, 13.1, and 4.1 Hz, Hβ-8), 1.66–1.59 (1H, m, Hβ-15), 1.38 (3H, s, CH3-19), 1.34–1.20 (3H, m, Hβ-11, Hα-12 and Hα-14), 1.10 (1H, dt, J = 12.2 and 4.1 Hz, Hα-9), 0.95 (3H, s, CH3-18); 13C NMR (100 MHz, CDCl3) δ 220.2, 186.1, 162.9, 156.9, 128.3, 127.1, 82.5, 56.7, 52.0, 50.7, 47.9, 43.9, 38.1, 35.9, 31.3, 30.6, 22.0, 21.9, 19.1, 14.1; IR (film) 902, 1216, 1374, 1661, 1742, 2848, 2916; m/z (ES) 315.2 (45%, M + H), 283.2 (100%), 265.2 (15%); HRMS-ES m/z 315.1955 (C20H27O3 requires 315.1955); [α]20D +74.4 (c 0.96, CHCl3); 96% pure by HPLC (Dynamax-60A, 3% i-PrOH/hexanes, tR = 48.6 min).
(6R,8R,9S,10R,13S,14S)-6-Ethoxy-10,13-dimethyl-7,8,9,10,11,12,13,14,15,16-decahydro-3H-cyclopenta[a]-phenanthrene-3,17(6H)-dione (3)
3 was derived from steroid diazo 1 (78 mg, 0.25 mmol, 1.0 equiv), ethanol (146 μL, 2.5 mmol, 10 equiv), and AgOTf (7 mg, 10 mol %) and was isolated as a white solid (34 mg, 41%). Mp 156–158 °C; 1H NMR (600 MHz, CDCl3) δ 6.99 (1H, d, J = 10.2 Hz, H-1), 6.18 (1H, dd, J = 10.2 and 1.4 Hz, H-2), 6.13 (1H, d, J = 1.4 Hz, H-4), 3.99 (1H, app t, J = 2.9 Hz, Hα-6), 3.39 (1H, dq, J = 9.2 and 7.0 Hz, Hα-20), 3.31 (1H, dq, J = 9.2 and 7.0 Hz, Hβ-20), 2.43 (1H, dd, J = 19.3 and 9.0 Hz, Hβ-16), 2.19–2.11 (2H, m, Hβ-7 and Hβ-11), 2.05 (1H, ddd, J = 19.3, 9.6, and 9.0 Hz, Hα-16), 1.95–1.90 (1H, m, Hβ-15), 1.86–1.79 (2H, m, Hα-11 and Hβ-12), 1.70 (1H, dd, J = 13.0 and 4.0 Hz, Hβ-8), 1.62 (1H, ddd, J = 12.4, 9.2, and 3.2 Hz, Hα-15), 1.36 (3H, s, CH3-19), 1.30–1.19 (3H, m, Hα-14, Hα-12, and Hα-7), 1.14 (3H, t, J = 7 Hz, CH3-21), 1.06 (1H, ddd, J = 12.0, 10.9, and 4.0 Hz, Hα-9), 0.93 (3H, s, CH3-18); 13C NMR (100 MHz, CDCl3) δ 220.3, 186.2, 163.7, 157.0, 127.9, 127.1, 80.4, 64.3, 52.3, 50.9, 47.9, 43.8, 38.3, 35.9, 31.4, 30.6, 22.1, 22.0, 19.1, 15.2, 14.2; IR (neat) 2941, 1738, 1664, 1403, 1186, 1092, 1010; m/z (FTMS) 329.2 (100%, M + H), 283.2 (20%); HRMS–FTMS m/z 329.2107 (C21H29O3 requires 329.2111); [α]20D +52.7 (c 0.31, CHCl3); 95% pure by HPLC (Dynamax-60A, 3% i-PrOH/hexanes, tR = 33.5 min).
(6R,8R,9S,10R,13S,14S)-6-(But-2-ynyloxy)-10,13-dimethyl-7,8,9,10,11,12,13,14,15,16-decahydro-3H-cyclopenta[a]-phenanthrene-3,17(6H)-dione (4)
4 was derived from steroid diazo 1 (62 mg, 0.20 mmol, 1.0 equiv), but-2-yn-1-ol (150 μL, 2.00 mmol, 10 equiv), and AgOTf (5 mg, 10 mol %) and was isolated as an off-white solid (44 mg, 61%). 1H NMR (600 MHz, CDCl3) δ 7.02 (1H, d, J = 9.9 Hz, H-1), 6.22 (1H, d, J = 1.9 Hz, H-4), 6.21 (1H, dd, J = 9.9 and 1.9 Hz, H-2), 4.29 (1H, t, J = 3 Hz, Hα-6), 4.09 (1H, dq, J = 15.2 and 2.4 Hz, Hα-20), 3.88 (1H, dq, J = 15.2 and 2.4 Hz, Hβ-20), 2.45 (1H, dd, J = 19.4 and 8.6 Hz, Hβ-16), 2.22 (1H, dt, J = 14.0 and 3.0 Hz, Hβ-7), 2.14 (1H, ddd, J = 12.0, 10.3, and 3.4 Hz, Hβ-8), 2.06 (1H, ddd, J = 19.4, 18.0, and 9.0 Hz, Hα-16), 1.97–1.85 (3H, m, Hα-15, Hα-11, Hβ-12), 1.83 (3H, t, J = 1.9 Hz, H-23), 1.71 (1H, dd, J = 13.5 and 4.8 Hz, Hβ-11), 1.61 (1H, ddd, J = 18.0, 12.4, and 9.0 Hz, Hβ-15), 1.35 (3H, s, CH3-19), 1.33–1.19 (3H, m, Hα-7, Hα-14, and Hα-12), 1.09 (1H, ddd, J = 11.8, 10.2, and 4.0 Hz, Hα-9), 0.93 (3H, s, CH3-18); 13C NMR (100 MHz, CDCl3) (C-17 carbonyl signal out of the spectra range) δ 186.2, 162.6, 157.1, 128.7, 127.2, 83.5, 78.7, 74.6, 56.5, 52.1, 50.8, 47.9, 43.8, 38.9, 38, 35.9, 31.4, 30.6, 29.9, 22.1, 22, 19.3, 14.1; IR (neat) 2943, 1735, 1661, 1266, 1183, 1054, 731; m/z (FTMS) 353.2 (100%, M + H), 283.2 (10%); HRMS–FTMS m/z 353.2110 (C23H29O3 requires 353.2111); 98% pure by HPLC (Dynamax-60A, 3% i-PrOH/hexanes, tR = 14.3 min).
(6R,8S,9S,10R,13S,14S)-10,13-Dimethyl-6-(pent-2-yn-1-yloxy)-7,8,9,10,11,12,13,14,15,16-decahydro-3H-cyclopenta-[a]phenanthrene-3,17(6H)-dione (5)
5 was derived from steroid diazo 1 (96 mg, 0.30 mmol, 1.0 equiv), pent-2-yn-1-ol (270 μL, 3.00 mmol, 10 equiv), and AgOTf (8 mg, 10 mol %) and was isolated as a white solid (81 mg, 71%). 1H NMR (400 MHz, CDCl3) δ 7.02 (1H, d, J = 9.8 Hz, H-1), 6.22 (1H, s, H-4), 6.20 (1H, dd, J = 9.8 and 2.0 Hz, 1H, H-2), 4.30 (1H, t, J = 3 Hz, H-6), 4.11 (1H, dt, J = 15.3 and 3.0 Hz, Hα-20), 3.90 (1H, dt, J = 15.3 and 3.0 Hz, Hβ-20), 2.44 (1H, dd, J = 19.3 and 8.5 Hz, Hβ-16), 2.25–2.09 (4H, m), 2.05 (1H, dt, J = 19.3 and 9.3 Hz, Hα-16), 1.96–1.78 (3H, m), 1.70 (1H, ddd, J = 13.5, 12.6, and 4.6 Hz, H-8), 1.60 (1H, dt, J = 9.2 and 3.3 Hz), 1.35 (3H, s, CH3-18), 1.35–1.18 (3H, m), 1.11 (3H, t, J = 7.4 Hz, CH3-24), 1.08 (1H, dt, J = 12.1 and 4.5 Hz, H-9), 0.92 (3H, s, CH3-19); 13C NMR (100 MHz, CDCl3) (C-17 carbonyl signal out of the spectra range) δ 186.1, 162.4, 156.9, 128.7, 127.2, 89.3, 78.6, 78.5, 74.7, 56.5, 52.1, 50.8, 47.9, 43.7, 38.0, 35.9, 31.4, 30.6, 22.1, 22.0, 19.3, 14.1, 14.0, 12.7; IR (film) 2939, 2245, 1736, 1662, 1453, 1052; m/z (FTMS) 367.2 (100%, M + H), 283.2 (17%); HRMS–FTMS m/z 367.2265 (C24H31O3 requires 367.2268); IR (film) 2939, 2245, 1736, 1662, 1453, 1052; m/z (APCI) 367.2 (100%, M + H), 283.2 (17%); HRMS-APCI m/z 367.2265 (C24H31O3 requires 367.2268); [α]20D +31.0 (c 0.25, CHCl3); 99% pure by HPLC (Dynamax-60A, 3% i-PrOH/hexanes, tR = 11.2 min).
(6R,8R,9S,10R,13S,14S)-6-(Hex-2-yn-1-yloxy)-10,13-dimeth-yl-7,8,9,10,11,12,13,14,15,16-decahydro-3H-cyclopenta[a]-phenanthrene-3,17(6H)-dione (6)
6 was derived from steroid diazo 1 (270 mg, 0.86 mmol, 1.0 equiv), hex-2-yn-1-ol (950 μL, 8.65 mmol, 10 equiv), and AgOTf (20 mg, 10 mol %) and was isolated as a white solid (81 mg, 25%). 1H NMR (400 MHz, CDCl3) δ 7.03 (1H, d, J = 9.8 Hz, H-1), 6.24 (1H, s, H-4), 6.21 (1H, dd, J = 9.8 and 2.0 Hz, H-2), 4.33 (1H, t, J = 3 Hz, H-6), 4.14 (1H, dt, J = 15.3 and 3.0 Hz, Hα-20), 3.92 (1H, dt, J = 15.3 and 3.0 Hz, Hβ-20), 2.47 (1H, dd, J = 19.1 and 9.7 Hz, Hβ-16), 2.26–2.13 (3H, m), 2.08 (1H, dt, J = 19.1 and 10.0 Hz, Hα-16), 1.98–1.79 (3H, m), 1.73 (1H, ddd, J = 13.4, 13.0, and 4.7 Hz, H-8), 1.62 (1H, dt, J = 9.3 and 3.2 Hz), 1.58–1.46 (3H, m), 1.37 (3H, s, CH3–18), 1.35–1.20 (3H, m), 1.10 (1H, ddd, J = 12.3, 6.3, 5.8 Hz, H-9), 0.97 (3H, t, J = 7.3 Hz, CH3-25), 0.94 (3H, s, CH3-19); 13C NMR (100 MHz,CDCl3) (C-17 carbonyl signal out of the spectra range) δ 186.1, 162.4, 156.9, 128.7, 127.2, 87.9, 78.5, 75.5, 56.4, 52.1, 50.9, 47.9, 43.7, 38.0, 35.9, 31.4, 30.6, 22.2, 22.1, 22.0, 21.0, 19.4, 14.1, 13.7; IR (film) 2937, 1737, 1664, 1074, 1055, 897; m/z (APCI) 381.24 (100%, M + H), 283.17 (6%); HRMS-APCI m/z 381.2418 (C25H33O3 requires 381.2424); 95% pure by HPLC (Dynamax-60A, 3% i-PrOH/hexanes, tR = 20.6 min).
(6R,8R,9S,10R,13S,14S)-6-(Hept-2-yn-1-yloxy)-10,13-dimeth-yl-7,8,9,10,11,12,13,14,15,16-decahydro-3H-cyclopenta[a]-phenanthrene-3,17(6H)-dione (7)
7 was derived from steroid diazo 1 (270 mg, 0.86 mmol, 1.0 equiv), hept-2-yn-1-ol (1.10 mL, 8.65 mmol, 10 equiv), and AgOTf (20 mg, 10 mol %) and was isolated as a white solid (74 mg, 22%). 1H NMR (400 MHz, CDCl3) δ 7.02 (1H, d, J = 10.2 Hz, H-1), 6.22 (1H, s, H-4), 6.21 (1H, d, J = 10.2 Hz, H-2), 4.31 (1H, app t, J = 1.5 Hz, H-6), 4.12 (1Hd, J = 15.3 Hz, Hα-20), 3.91 (1H, d, J = 15.3 Hz, Hβ-20), 2.45 (1H, dd, J = 18.9 and 9.0 Hz, Hβ-16), 2.24–2.12 (4H, m), 2.06 (1H, dt, J = 18.9 and 9.2 Hz, Hα-16), 1.96–1.79 (3H, m), 1.71 (1H, ddd, J = 14.0, 12.5, and 5.0 Hz, H-8), 1.60 (1H, ddt, J = 9.3, 8.1, and 3.5 Hz), 1.51–1.41 (2H, m), 1.40–1.35 (2H, m), 1.35 (3H, s, CH3-18), 1.30–1.16 (3H, m), 1.10 (1H, dt, J = 12.5 and 5.4 Hz, H-9), 0.93 (3H, s, CH3-19), 0.88 (3H, t, J = 7.3 Hz, CH3-26); 13C NMR (100 MHz, CDCl3) (C-17 carbonyl signal out of the spectra range) δ 186.1, 162.4, 156.9, 128.7, 127.2, 88.0, 78.5, 78.4, 75.3, 56.4, 52.1, 50.9, 47.9, 43.7, 38.0, 35.9, 31.4, 30.8, 30.6, 22.2, 22.1, 22.0, 19.4, 18.7, 14.1, 13.8; IR (film) 2934, 2859, 1737, 1663, 1625, 1055, 897; m/z (APCI) 395.26 (100%, M + H), 283.17 (30%); HRMS APCI m/z 395.2575 (C26H35O3 requires 395.2581); 96% pure by HPLC (Dynamax-60A, 3% i-PrOH/hexanes, tR = 19.3 min).
(6R,8R,9S,10R,13S,14S)-6-(Dec-2-yn-1-yloxy)-10,13-dimethyl-7,8,9,10,11,12,13,14,15,16-decahydro-3H-cyclopenta[a]-phenanthrene-3,17(6H)-dione (8)
8 was derived from steroid diazo 1 (270 mg, 0.86 mmol, 1.0 equiv), dec-2-yn-1-ol (1.56 mL, 8.65 mmol, 10 equiv), and AgOTf (20 mg, 10 mol %) and was isolated as an off-white solid (112 mg, 30%). 1H NMR (400 MHz, CDCl3) δ 7.00 (1H, d, J = 10.5 Hz, H-1), 6.21 (1H, s, H-4), 6.19 (1H, dd, J = 10.5 and 2.0 Hz, H-2), 4.30 (1H, app t, J = 2.5 Hz, H-6), 4.10 (1H, dd, J = 15.3 and 2.5 Hz, Hα-20), 3.89 (1H, dd, J = 15.3 and 2.5 Hz, Hβ-20), 2.43 (1H, app dd, J = 18.5 and 9.0 Hz, Hβ-16), 2.25–2.11 (4H, m), 2.09–1.98 (1H, m), 1.94–1.77 (2H, m), 1.69 (1H, dd, J = 13.7, 12.4, and 3.6 Hz, H-8), 1.63–1.52 (1H, m), 1.52–1.42 (1H, m), 1.34 (3H, s, CH3-18), 1.32–1.18 (12H, m), 1.08 (1H, dt, J = 10.6 and 4.1 Hz, H-9), 0.91 (3H, s, CH3-19), 0.86–0.79 (4H, m); 13C NMR (100 MHz, CDCl3) (C-17 carbonyl signal out of the spectra range) δ 186.1, 162.4, 156.9, 128.7, 127.2, 88.1, 78.5, 78.4, 75.3, 56.1, 52.1, 50.8, 47.9, 43.7, 37.9, 35.9, 31.9, 31.4, 30.5, 29.0, 28.9, 28.7, 22.8, 22.1, 21.9, 19.3, 19.0, 14.3, 14.1; IR (film) 2928, 1737, 1663, 1088, 1054, 897; m/z (APCI) 437.3 (100%, M + H), 283.17 (30%); HRMS-APCI m/z 437.3042 (C29H41O3 requires 437.3050); 95% pure by HPLC (Dynamax-60A, 3% i-PrOH/hexanes, tR = 16.6 min).
(6R,8R,9S,10R,13S,14S)-6-((4-Hydroxybut-2-yn-1-yl)oxy)-10,13-dimethyl-7,8,9,10,11,12,13,14,15,16-decahydro-3H-cyclopenta[a]phenanthrene-3,17(6H)-dione (9)
9 was derived from steroid diazo 1 (60 mg, 0.19 mmol, 1.0 equiv), 4-((tert-butyldimethylsilyl)oxy)but-2-yn-1-ol38 (384 mg, 1.92 mmol, 10 equiv), and AgOTf (5 mg, 10 mol %). The crude product was filtered through a short plug of silica (eluting with hexanes/EtOAc, 90:10 to 70:30), and the solvent was concentrated under reduced pressure. The resulting silyl ether was dissolved in dry DMF (1.0 mL) at room temperature, and tris(dimethylamino)sulfonium difluorotrimethylsilicate (TASF) (52 mg, 0.19 mmol, 1.0 equiv) was added. The mixture was stirred at room temperature for 30 min. The mixture was then diluted with EtOAc (5 mL) and poured into a separating funnel containing a pH 7 phosphate buffer solution (5 mL). The layers were separated, and the aqueous layer was extracted with ethyl acetate (2 × 5 mL). The organics were dried over MgSO4, concentrated under reduced pressure, and purified by flash chromatography on silica gel (eluting with hexanes/EtOAc, 60:40 to 50:50). The titled compound was isolated as a white solid (11 mg, 16% over two steps). Mp 46–48 °C; 1H NMR (400 MHz, CDCl3) δ 7.03 (1H, d, J = 10.0 Hz, H-1), 6.24 (1H, d, J = 2.0 Hz, H-4), 6.21 (1H, dd, J = 10.0 and 2.0 Hz, H-2), 4.31 (1H, app t, J = 2.8 Hz, Hα-6), 4.30 (2H, app t, J = 1.6 Hz, CH2-23), 4.18 (1H, dt, J = 15.6 and 2.0 Hz, Hα-20), 3.97 (1H, dt, J = 15.6 and 2.0 Hz, Hβ-20), 2.46 (1H, dd, J = 19.2 and 8.8 Hz, Hβ-16), 2.22 (1H, dt, J = 13.6 and 3.2 Hz, Hβ-7), 2.01–2.16 (2H, m, Hβ-8, Hα-16), 1.79–1.97 (3H, m, Hα-15, Hα-11, Hβ-12), 1.58–1.76 (2H, m, Hβ-11, Hβ-15), 1.35 (3H, s, CH3-19), 1.34–1.21 (3H, m, Hα-7, Hα-14 and Hα-12), 1.10 (1H, ddd, J = 12.5, 10.4, and 4.4 Hz, Hα-9), 0.93 (3H, s, CH3-18); 13C NMR (100 MHz, CDCl3) δ 220.3, 186.2, 162.1, 157.0, 128.8, 127.2, 85.7, 81.0, 78.9, 56.2, 52.0, 51.2, 50.8, 47.9, 43.7, 37.9, 35.9, 31.4, 30.5, 22.1, 22.0, 19.4, 14.1; IR (film) 3427, 2942, 1736, 1662, 1057; m/z (APCI) 369.2 (57%, M + H), 283.17 (100%); HRMS-APCI m/z 369.2058 (C23H29O4 requires 369.2060); [α]20D +159.2 (c 0.25, CHCl3); 95% pure by HPLC (Dynamax-60A, 8% i-PrOH/hexanes, tR = 37.0 min).
Measurement of Inhibitory Activity in Purified Aromatase
The aromatase activity was measured according to the established 3H-water method,19 using 3H1β-ASD as the substrate. Human placental aromatase was purified to homogeneity by immunoaffinity chromatography as previously described.19 The purified enzyme was highly active with a specific activity typically between 25 and 50 nmol min−1 mg−1. Placental aromatase at 40 nM was incubated with the 2-alkoxy derivative series of compounds for approximately 16 h, and the controls (EXM, LTZ, and formestane) were at 10 μM, 9.1 μM, 1 μM, 500 nM, 100 nM, or 10 nM, depending on the potency. All inhibitors were dissolved in 100% PEG400/PEG550 to prepare the stock solutions. The final concentration of PEG in the purified enzyme was always 10%. Inhibition was normalized against the specific activity of native placental aromatase in the presence of 10% PEG400/PEG550. Table S1 (Supporting Information) shows the percent inhibition for different concentrations of inhibitor, which represents the mean of at least three independent experiments. Each data point was measured in quadruplicate. For the purpose of IC50 determination, 0.01 nM inhibitor was set as the blank value (the inhibitor concentration at which there was no inhibition). The potency of an inhibitor was determined by fitting all data to a dose–response curve with a standard slope in the GraphPad Prism software.39 The IC50 values were interpolated using a nonlinear regression analysis to generate the best-fit value for IC50. The uncertainty or error in the IC50 calculation is expressed as the 95% confidence interval computed by the GraphPad Prism software.39 The 95% confidence interval is calculated by using the antilog of the standard error of log IC50 as a multipler of the computed IC50.39
Measurement of Antiproliferative Activity in a Breast Cancer Cell Line
The MCF-7 breast cancer cell line MCF-7-Tet-off-3βHSD1-Arom (MCF-7a) that stably expresses aromatase was obtained as a gift from Prof. James Thomas, Macon, GA.40 The MCF-7a cells were cultured in RPMI-1640 medium without phenol red (Mediatech, Manassas, VA, U.S.), containing 10% FBS (Atlanta Biological, Lawrenceville, GA, U.S.), 2 mM glutamine, 100 IU/mL penicillin, and 100 μg/mL streptomycin (Cellgro, Mediatech Inc., Manassas, VA), at 37 °C in a humidified atmosphere containing 5% CO2. Cells were selected by incubation with 10 ng/mL doxycycline (Fisher BioReagents, U.S.), 0.2 mg/mL Geneticin (G418, Invitrogen, San Diego, CA, U.S.), 0.1 mg/mL hygromycin (Fisher Scientific, Fair Lawn, NJ, U.S.), and 100 μg/mL zeocin (Invivogen, San Diego, CA, U.S.) as previously described.40
The MCF-7a cells were plated in 96-well plates at 2000 cells/well using RPMI-1640 medium without phenol red, containing 10% charcoal dextran-stripped FBS (Gemini Bio-Products, West Sacramento, CA, U.S.). Other components were the same as the culture medium but without doxycycline. Stimulation of the growth of the MCF-7a cells was measured following addition of different concentrations of TST (0.01–100 nM). E2 and ASD were also used to stimulate the cell growth in order to compare the proliferation rates. Inhibition of cell proliferation by aromatase inhibitors was measured by adding 1 nM TST and various concentrations of LTZ, EXM, compounds 4–9 (0.01–100 nM) to the cultures. LTZ and EXM were used as positive controls in the inhibition of this TST (1 nM) stimulated breast cancer cell proliferation assay. The medium and the treatments were refreshed every 3 days. On day 10 after cell plating, the cell viability was determined using a CellTiter-Blue assay kit (Promega, U.S.) according to the manufacturer’s instructions. Briefly, 20 L of CellTiter-Blue reagent was added to each well and cells were incubated for 2–4 h at 37 °C. Afterward, fluorescence (540 ± 35 nm excitation, 600 ± 40 nm emission) was recorded using a fluorescence plate reader (BioTek, Synergy 2). Assays were performed in triplicate for each experiment; mean cell viability was compared to vehicle-treatment (0.005% PEG550) controls. Each experimental data point was the average of at least three independent experiments. The antiproliferation potency of an inhibitor (EC50: concentration for 50% growth suppression) was determined by fitting all data to a dose–response curve using the GraphPad Prism software.39
Preparation and Crystallization of Aromatase–EXM Complex
Human placental aromatase was purified to homogeneity as previously described.19 The specific activity of the purified aromatase in β-d-dodecylmaltopyranoside (BDM) was 7328 pmol min−1 mg−1. To the purified protein solution in 100 mM KPO4, pH 7.4, containing 20% glycerol, 0.5 μM ASD, 0.1 mM EDTA, and 1.0 mM n-dodecyl-β-d-maltopyranoside (BDM) were adde 20 mM DTT and 400 μM of EXM, and the mixture was then incubated overnight at 4 °C. At the final crystallization setup concentration, the enzyme was 96% inhibited. Solutions of inhibited aromatase–EXM complex exhibited the Soret peak at 394 nm, similar to the Soret peak at 392 nm exhibited by the oxidized high spin ferric ASD complex.
The aromatase–EXM complex was then concentrated to about 34 mg/mL. Sitting droplets of protein solutions were vapor-diffused against reservoir solutions of 24–32% polyethylene glycol 4000 containing 0.5 M NaCl in 50 mM Tris-HCl, pH 8.5. Each droplet contained variable ratios of protein to reservoir solutions ranging from 1:1 to 4:1. Crystals appeared in about 1–2 weeks and continued to grow up to 4 weeks.
Preparation and Crystallization of Aromatase Complexes of 4/5
Human placental aromatase was freshly purified according to the previously described protocol.19 The enzyme–inhibitor complexes were prepared by the addition from 20 mM stock solutions of 4/5 in PEG550 to 18 μM (~1 mg/mL) of aromatase to give a final inhibitor concentration of 300 μM. The mixture was incubated overnight at 4 °C in 100 mM potassium phosphate buffer, pH 7.4, containing 20% glycerol, 20 mM dithiothreitol, 0.5 μM ASD, and 1 mM BDM. The complex was then concentrated to 25–30 mg/mL using ultrafiltration. Protein was set up for crystallization using protein to cocktail ratios 2:1 and 3:1. The protein was mixed with reservoir cocktails of 24–30% polyethylene glycol 4000 in 50 mM NaCl, 50 mM Tris, pH 8.5, and vapor diffused in sealed 24-well sitting drop plates against the corresponding reservoir solution. The inhibition of aromatase by C6β-2-alkynyloxy derivative 4/5 measured by the 3H-water method showed 93% and 96%, respectively, inhibition of the enzyme activity. Crystals appeared in about 2 weeks.
Refinement of the Androstenedione-Complex Structure at 2.75 Å
Details of crystallization and data collection have already been described.6 Briefly, the diffraction data set was collected on an ADSC Quantam 315 CCD detector at beamline 19-ID, Structural Biology Center, Advanced Photon Source, Argonne National Laboratory, Argonne, IL. The crystal was flash cooled in a stream of liquid nitrogen and maintained at ~100 K during data collection. The data set was recently reprocessed with HKL2000,41 and the resolution extended to 2.75 Å based on linearity of the Wilson plot. The crystal and data processing summary is provided in Table S3 (Supporting Information). Model building and refinement were performed with Coot42 and Refmac543 routines, respectively, on a MacPro workstation with OS × 10.5 operating system. The refinement was initiated with the previously published coordinates6 (PDB code 3EQM). The refinement results are summarized in Table S3 (Supporting Information). The new coordinates and structure factors have been deposited with the RCSB Protein Data Bank44 (PDB code 3S79).
Determination of Crystal Structures of the Complexes with Exemestane, 4, and 5
Diffraction data sets for the complexes with EXM and 5 were collected at beamline A-1, Cornell High Energy Synchrotron (CHESS) and Macromolecular Diffraction at CHESS (MacCHESS), Ithaca, NY. The data were recorded on an ADSC Quantum 210 CCD detector and processed with the HKL2000 software package.41 The data for the complex with 4 were gathered at beamline 19-ID, Structural Biology Center, Advanced Photon Source, Argonne National Laboratory, Argonne, IL, using an ADSC Quantum 315 CCD detector and processed as above. All complexes were isomorphous to the androstenedione complex, i.e., the same space group and cell parameters within the limits of errors. The androstenedione complex was refined at 2.75 Å without the solvent, and the androstenedione molecule was used as the starting model for the protein for each refinement. The difference electron density maps showed the unbiased electron density for the bound inhibitor molecules (Figure 4). These maps were used for the initial positioning of the ligand molecule at the active site and the starting model for each complex. A difference electron density calculation with androstenedione as the ligand (missing the C6 methylidene carbon) showed a peak at 2.0σ at the missing carbon position for EXM (Figure S1, Supporting Information). The entire model including the atoms of EXM was then refined using Refmac543 until convergence. Coot42 was used for model building purposes. The coordinates and structure factors for the EXM, 4 and 5 complexes have been deposited with the RCSB Protein Data Bank44 (PDB codes 3S7S, 4GL5 and 4GL7, respectively).
Docking of the New Series of Inhibitors into Other Cytochrome P450s and ERα
All the docking calculations were carried out using Molecular Operating Environment (MOE, version 2011.10), Chemical Computing Group, Montreal, Canada.45 The coordinate files for ERα (PDB code 1QKU), CYP3A4 (PDB code 3UA1), CYP2A6 (PDB code 3T3R), CYP2C9 (PDB code 1OG5), CYP2D6 (PDB code 3QM4), and the steroidogenic enzyme CYP17A1 (PDB code 3RUK) were obtained from the RCSB Protein Data Bank. Only single conformation of the active site and its ligand given by the X-ray structure for a given PDB code was used for docking, although some of the P450 active sites are known to have multiple conformations. Predocking procedure included deletion of ligand, water, and phosphate molecules from the 3D protein structure, addition of hydrogen to the protein, energy minimization, and identification of the binding site. Protonate 3D, a module in MOE platform, was used to assign protons to donor side chains.45,46 Protonation of Asp309 side chain in the aromatase structure6 was manually implemented. Following the identification of the binding site, the ligand from each crystal structure was first docked to its corresponding receptor. The 2-pentynyloxy derivative 5, the most potent of the new inhibitors, was then docked to the binding site of each receptor. Poses were generated using the default Triangle Matcher45 method that aligned ligand triplets of atoms on triplets of α spheres and scored by Affinity dG, London dG, and GBVI/WSA dG scoring functions.45 The score used for tabulation was the empirical free energy of ligand binding after refinement (Table S3, Supporting Information). Closer examination revealed that for most receptors the top-ranked pose listed in Table S3 was indeed the most likely binding mode of the 2-pentynyloxy derivative 5. The fifth pose was used for CYP2C9 and the 15th for CYP2A6 because they were the most similar to the ligand in the crystal structure.
Lastly, pharmacophore query and filtering were performed by pharmacophore applications in MOE. A pharmacophore is a set of structural features in a ligand that are directly related to the ligand’s recognition at a receptor site and its biological activity.47 Pharmacophore filtering used pharmacophores derived from receptor–ligand interactions in the complex X-ray structures to probe whether the docked poses of the query ligand (compound 5) could replicate these interactions.
Supplementary Material
ACKNOWLEDGMENTS
This work is supported, in whole or in part, by Grant GM086893 from the National Institutes of Health (to D.G.). The authors thank Drs. Giovanna Di Nardo and Karen Haswell for assistance in the diffraction data collection, and Prof. James L. Thomas, Mercer University School of Medicine, Macon, GA, for the gift of the MCF7a cell line. Dr. Haswell is also thanked for critically reading the manuscript. Mary Erman is thanked for technical assistance in protein purification. The Structural Biology Center at the Advanced Photon Source, Argonne, IL, is operated by UChicago Argonne, LLC, for the U.S. Department of Energy, Office of Biological and Environmental Research under Contract DE-AC02-06CH11357. CHESS is supported by the NSF and NIH/NIGMS via NSF Award DMR-0936384, and the MacCHESS resource is supported by NIH/NIGMS Award GM103485.
ABBREVIATIONS USED
- BDM
β-d-dodecylmaltopyranoside
- HTST
16α-hydroxytestosterone
- E2
17β-estradiol
- E3
17β,16α-estriol
- ANZ
anastrozole
- ASD
androstenedione
- ASDD
androsta-1,4-diene-3,17-dione
- AI
aromatase inhibitor
- CYP
cytochrome P450
- CYP19A1
cytochrome P450 aromatase
- CPR
cytochrome P450 reductase
- ER
estrogen receptor
- E1
estrone
- EXM
exemestane
- LTZ
letrozole
- MCF-7
Michigan Cancer Foundation 7
- MOE
molecular operating environment
- TST
testosterone
Footnotes
ASSOCIATED CONTENT Supporting Information Tables of detailed inhibition and antiproliferation data, figures showing unbiased electron density maps, and tables and figures giving accounts of docking results. This material is available free of charge via the Internet at http://pubs.acs.org.
Notes The authors declare no competing financial interest.
REFERENCES
- (1).Thompson EA, Jr., Siiteri PK. The involvement of human placental microsomal cytochrome P-450 in aromatization. J. Biol. Chem. 1974;249:5373–5378. [PubMed] [Google Scholar]
- (2).Simpson ER, Mahendroo MS, Means GD, Kilgore MW, Hinshelwood MM, Graham-Lorence S, Amarneh B, Ito Y, Fisher CR, Michael MD. Aromatase cytochrome P450, the enzyme responsible for estrogen biosynthesis. Endocr. Rev. 1994;15:342–355. doi: 10.1210/edrv-15-3-342. [DOI] [PubMed] [Google Scholar]
- (3).Johnston JO. Aromatase inhibitors. Crit. Rev. Biochem. Mol. Biol. 1998;33:375–405. [PubMed] [Google Scholar]
- (4).Shimozawa O, Sakaguchi M, Ogawa H, Harada N, Mihara K, Omura T. Core glycosylation of cytochrome P-450(arom) Evidence for localization of N terminus of microsomal cytochrome P-450 in the lumen. J. Biol. Chem. 1993;268:21399–21402. [PubMed] [Google Scholar]
- (5).Amarneh B, Corbin CJ, Peterson JA, Simpson ER, Graham-Lorence S. Functional domains of human aromatase cytochrome P450 characterized by linear alignment and site-directed mutagenesis. Mol. Endocrinol. 1993;7:1617–1624. doi: 10.1210/mend.7.12.8145767. [DOI] [PubMed] [Google Scholar]
- (6).Ghosh D, Griswold J, Erman M, Pangborn W. Structural basis for androgen specificity and oestrogen synthesis in human aromatase. Nature. 2009;457:219–223. doi: 10.1038/nature07614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Jiang W, Ghosh D. Motion and flexibility in human cytochrome p450 aromatase. PLoS One. 2012;7:e32565. doi: 10.1371/journal.pone.0032565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Brueggemeier RW. Update on the use of aromatase inhibitors in breast cancer. Expert Opin. Pharmacother. 2006;7:1919–1930. doi: 10.1517/14656566.7.14.1919. [DOI] [PubMed] [Google Scholar]
- (9).Brueggemeier RW, Hackett JC, Diaz-Cruz ES. Aromatase inhibitors in the treatment of breast cancer. Endocr. Rev. 2005;26:331–345. doi: 10.1210/er.2004-0015. [DOI] [PubMed] [Google Scholar]
- (10).Bulun SE, Attar E, Gurates B, Chen Y-H, Tokunaga H, Monsivais D, Pavone ME. Medical Therapies: Aromatase Inhibitors. In: Giudice LC, Evers JLH, Healy DL, editors. Endometriosis: Science and Practice. Wiley-Blackwell; Oxford, U.K.: 2012. DOI: 10.1002/9781444398519.ch35. [Google Scholar]
- (11).AlHilli M, Long H, Podratz K, Bakkum-Gamez J. Aromatase inhibitors in the treatment of recurrent ovarian granulosa cell tumors: brief report and review of the literature. J. Obstet. Gynaecol. Res. 2012;38:340–344. doi: 10.1111/j.1447-0756.2011.01698.x. [DOI] [PubMed] [Google Scholar]
- (12).Miki Y, Abe K, Suzuki S, Suzuki T, Sasano H. Suppression of estrogen actions in human lung cancer. Mol. Cell. Endocrinol. 2011;340:168–174. doi: 10.1016/j.mce.2011.02.018. [DOI] [PubMed] [Google Scholar]
- (13).Kellis JT, Jr., Vickery LE. Purification and characterization of human placental aromatase cytochrome P-450. J. Biol. Chem. 1987;262:4413–4420. [PubMed] [Google Scholar]
- (14).Yoshida N, Osawa Y. Purification of human placental aromatase cytochrome P-450 with monoclonal antibody and its characterization. Biochemistry. 1991;30:3003–3010. doi: 10.1021/bi00226a004. [DOI] [PubMed] [Google Scholar]
- (15).Zhou D, Cam LL, Laughton CA, Korzekwa KR, Chen S. Mutagenesis study at a postulated hydrophobic region near the active site of aromatase cytochrome P450. J. Biol. Chem. 1994;269:19501–19508. [PubMed] [Google Scholar]
- (16).Graham-Lorence S, Amarneh B, White RE, Peterson JA, Simpson ER. A three-dimensional model of aromatase cytochrome P450. Protein Sci. 1995;4:1065–1080. doi: 10.1002/pro.5560040605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Hong Y, Yu B, Sherman M, Yuan YC, Zhou D, Chen S. Molecular basis for the aromatization reaction and exemestanemediated irreversible inhibition of human aromatase. Mol. Endocrinol. 2007;21:401–414. doi: 10.1210/me.2006-0281. [DOI] [PubMed] [Google Scholar]
- (18).Hong Y, Cho M, Yuan YC, Chen S. Molecular basis for the interaction of four different classes of substrates and inhibitors with human aromatase. Biochem. Pharmacol. 2008;75:1161–1169. doi: 10.1016/j.bcp.2007.11.010. [DOI] [PubMed] [Google Scholar]
- (19).Lala P, Higashiyama T, Erman M, Griswold J, Wagner T, Osawa Y, Ghosh D. Suppression of human cytochrome P450 aromatase activity by monoclonal and recombinant antibody fragments and identification of a stable antigenic complex. J. Steroid Biochem. Mol. Biol. 2004;88:235–245. doi: 10.1016/j.jsbmb.2003.12.010. [DOI] [PubMed] [Google Scholar]
- (20).Ghosh D, Griswold J, Erman M, Pangborn W. X-ray structure of human aromatase reveals an androgen-specific active site. J. Steroid Biochem. Mol. Biol. 2010;118:197–202. doi: 10.1016/j.jsbmb.2009.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Ghosh D, Jiang W, Lo J, Egbuta C. Higher order organization of human placental aromatase. Steroids. 2011;76:753–758. doi: 10.1016/j.steroids.2011.02.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Varela C, Tavares da Silva EJ, Amaral C, Correia da Silva G, Baptista T, Alcaro S, Costa G, Carvalho RA, Teixeira NA, Roleira FM. New structure–activity relationships of A- and D-ring modified steroidal aromatase inhibitors: design, synthesis, and biochemical evaluation. J. Med. Chem. 2012;55:3992–4002. doi: 10.1021/jm300262w. [DOI] [PubMed] [Google Scholar]
- (23).Wood PM, Woo LW, Thomas MP, Mahon MF, Purohit A, Potter BV. Aromatase and dual aromatase-steroid sulfatase inhibitors from the letrozole and vorozole templates. ChemMedChem. 2011;6:1423–1438. doi: 10.1002/cmdc.201100145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Caporuscio F, Rastelli G, Imbriano C, Del Rio A. Structure-based design of potent aromatase inhibitors by high-throughput docking. J. Med. Chem. 2011;54:4006–4017. doi: 10.1021/jm2000689. [DOI] [PubMed] [Google Scholar]
- (25).Bahar I, Atilgan AR, Erman B. Direct evaluation of thermal fluctuations in proteins using a single-parameter harmonic potential. Folding Des. 1997;2:173–181. doi: 10.1016/S1359-0278(97)00024-2. [DOI] [PubMed] [Google Scholar]
- (26).Jeong SW,M, Flockhart D, Zerusenav D. Inhibition of drug metabolizing cytochrome P450s by the aromatase inhibitor drug letrozole and its major oxidative metabolite 4,4′-methanol-bisbenzonitrile in vitro. Cancer Chemother. Pharmacol. 2009;64:867–875. doi: 10.1007/s00280-009-0935-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Wirz B, Valle B, Parkinson A. CYP3A4 and CYP2A6 Are Involved in the Biotransformation of Letrozole (Femara). Proceedings of 7th North American ISSX Meeting; Washington, DC: ISSX; 1996. p. 359. [Google Scholar]
- (28).Miller WL. Minireview: regulation of steroidogenesis by electron transfer. Endocrinology. 2005;146:2544–2550. doi: 10.1210/en.2005-0096. [DOI] [PubMed] [Google Scholar]
- (29).Brueggemeier RW. Aromatase, aromatase inhibitors, and breast cancer. Am. J. Ther. 2001;8:333–344. doi: 10.1097/00045391-200109000-00007. [DOI] [PubMed] [Google Scholar]
- (30).Budzar AU, Roberston JF, Eiermann W, Nabholtz JM. An overview of the pharmacology and pharmacokinetics of the newer generation aromatase inhibitors anastrozole, letrozole, and exemestane. Cancer. 2002;95:2006–2016. doi: 10.1002/cncr.10908. [DOI] [PubMed] [Google Scholar]
- (31).Ariazi EA, Leitao A, Oprea TI, Chen B, Louis T, Bertucci AM, Sharma CG, Gill SD, Kim HR, Shupp HA, Pyle JR, Madrack A, Donato AL, Cheng D, Paige JR, Jordan VC. Exemestane’s 17-hydroxylated metabolite exerts biological effects as an androgen. Mol. Cancer Ther. 2007;6:2817–2827. doi: 10.1158/1535-7163.MCT-07-0312. [DOI] [PubMed] [Google Scholar]
- (32).Morton D, Dick AR, Ghosh D, Davies HML. Convenient method for the functionalization of the 4- and 6-positions of the androgen skeleton. Chem. Commun. 2012;48:5838–5840. doi: 10.1039/c2cc31973j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Anzenbacherova E, Bec N, Anzenbacher P, Hudecek J, Soucek P, Jung C, Munro AW, Lange R. Flexibility and stability of the structure of cytochromes P450 3A4 and BM-3. Eur. J. Biochem. 2000;267:2916–2920. doi: 10.1046/j.1432-1327.2000.01305.x. [DOI] [PubMed] [Google Scholar]
- (34).Negishi M, Uno T, Darden TA, Sueyoshi T, Pedersen LG. Structural flexibility and functional versatility of mammalian P450 enzymes. FASEB J. 1996;10:683–689. doi: 10.1096/fasebj.10.7.8635685. [DOI] [PubMed] [Google Scholar]
- (35).Xu LH, Fushinobu S, Ikeda H, Wakagi T, Shoun H. Crystal structures of cytochrome P450 105P1 from Streptomyces avermitilis: conformational flexibility and histidine ligation state. J. Bacteriol. 2009;191:1211–1219. doi: 10.1128/JB.01276-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Giudici D, Ornati G, Briatico G, Buzzetti F, Lombardi P, di Salle E. 6-Methylenandrosta-1,4-diene-3,17-dione (FCE 24304): a new irreversible aromatase inhibitor. J. Steroid Biochem. 1988;30:391–394. doi: 10.1016/0022-4731(88)90129-x. [DOI] [PubMed] [Google Scholar]
- (37).Moreau RJ, Sorensen EJ. Classical carbonyl reactivity enables a short synthesis of the core structure of acutumine. Tetrahedron. 2007;63:6446–6453. [Google Scholar]
- (38).Kittigowittana K, Yang CT, Cheah WC, Chuang KH, Tuang CY, Chang YT, Golay X, Bates RW. Development of intravascular contrast agents for MRI using gadolinium chelates. ChemMedChem. 2011;6:781–787. doi: 10.1002/cmdc.201100066. [DOI] [PubMed] [Google Scholar]
- (39).Motulsky H. GraphPad Prism. version 5.0 GraphPad Software; San Diego, CA, U.S.: 2003. [Google Scholar]
- (40).Thomas JL, Bucholtz KM, Sun J, Mack VL, Kacsoh B. Structural basis for the selective inhibition of human 3β-hydroxysteroid dehydrogenase 1 in human breast tumor MCF-7 cells. Mol. Cell. Endocrinol. 2009;301:174–182. doi: 10.1016/j.mce.2008.09.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Otninowski Z, Minor W. HKL Manual. Yale University; New Haven, CT, U.S.: 1995. [Google Scholar]
- (42).Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- (43).Murshudov GN, Vagin AA, Dodson EJ. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr., Sect. D: Biol. Crystallogr. 1997;53:240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- (44).Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, Shindyalov IN, Bourne PE. The Protein Data Bank. Nucleic Acids Res. 2000;28:235–242. doi: 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Molecular Operating Environment. version 2011.10 Chemical Computing Group; Montreal, Canada: 2011. [Google Scholar]
- (46).Labute P. Protonate3D: assignment of ionization states and hydrogen coordinates to macromolecular structures. Proteins. 2009;75:187–205. doi: 10.1002/prot.22234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Lin A. Overview of Pharmacophore Applications in MOE. Chemical Computing Group Inc.; Montreal, Canada: 2012. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.