

Abstract
The first total synthesis of triantennary, fully sialylated N-glycan of complex type is described. Two strategies for installation of sialylated antennae are explored and both approaches converge on a global glycosylation step that delivers the desired tetradecasaccharide in good yields.
Introduction
It is well established that the post-translational attachment of glycan domains can play a central role in mediating protein stability, function, and structure.1 Interestingly, changes in protein glycosylation patterns may signal the onset of aberrant biochemical processes. This phenomenon has been exploited as a means by which to monitor disease progression.2 Of particular interest to our laboratory is the finding that transformed tumor cells often exhibit characteristic levels and types of cell surface carbohydrate expression. Theoretically, it should be possible to design vaccine constructs bearing these tumor-associated carbohydrate antigens, which, upon presentation to the immune system, would induce a selective immune response, leading to the eradication of tumor cells expressing these carbohydrates. A major research program underway in our laboratory is devoted to the de novo synthesis and immunological evaluation of complex carbohydrate-based anticancer vaccine candidates.3 A number of our most promising constructs have been advanced to clinical trials.
In the context of this research program, we identified Prostate Specific Membrane Antigen (PSMA) as an attractive target for a potential vaccine against prostate cancer.4 PSMA, a membrane-bound glycoprotein possessing carboxypeptidase activity, is apparently localized in the prostate sac and is heavily over-expressed in prostate cancer.5 Additionally, PSMA is upregulated in an androgen-independent stage.6 Moreover, this glycoprotein is found on the neovasculature of a panel of solid tumors, including renal cells, breast, and colonic carcinomas.7 The immunogenic properties of the peptidic domain of PSMA, as well as the enzymatic activity of the glycoprotein,8 have been harnessed for the development of anti-tumor therapies.4 However, less well understood is the role of the oligosaccharide domain of PSMA, and its potential application for the development of prostate cancer vaccines.9 PSMA possesses ten sites of N-linked glycosylation, and the glycans comprise about 20-25% of the total molecular weight of the glycoprotein.10 Based on the crystal structure of truncated PSMA, eight asparagine sites are exposed to the extracellular environment11 and certain glycosylation positions (e.g. Asn638) can play a critical role in maintaining enzymatic function of this homodimeric glycoprotein.12 A consensus structure of PSMA glycan, isolated from prostate tumor cells, is depicted in Figure 1. This complex-type oligosaccharide is characterized by the presence of sialic acid residues (Neu5Ac) at each of its three antennae. The sialic moieties play an important role in recognition processes, through mediation of glycoprotein conformation and binding of positively charged compounds.13 In addition, resident sialic acid motifs can complicate immune surveillance of potential antigenic sites.14 Synthetic access to homogeneous 1 would enable studies on the biological role of the glycan expression in PSMA and its potential role in the etiology and pathogenesis of prostate cancer. We previously accomplished the synthesis of a related core biantennary sialylated glycan structure often found in the mammalian glycoproteins.15 Since the highly branched tetradecasaccharide 1 has also been identified in other glycoproteins of biological interest,16 studies on the role of glycosylation in these systems could deepen our understanding of how the phenomenon of altered carbohydrate expression can be harnessed for therapeutic purposes.
Figure 1.
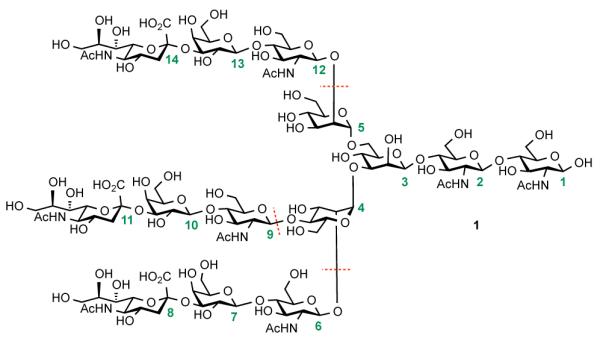
As depicted in Figure 1, glycan 1 is composed of a pentasaccharide core (saccharides 1-5), equipped with three trisaccharide antennae. Our general synthetic strategy envisioned the three-fold glycosylation of the partially protected pentasaccharide with excess sialylated trisaccharide donor. The pentasaccharide precursor itself would be accessed through a sequence featuring coupling of the “reducing” chitobiose end (saccharides 1 and 2) with a mannose donor (saccharide 3). The resultant core system would then be further elaborated via α-mannosylation, with building blocks orthogonally protected at the 2′ and 4′-OH (saccharide 4) and at the 2′-OH (saccharide 5). At the outset of these studies, we were mindful of the difficulties typically associated with glycosylation of sterically hindered 4′-OH mannose acceptors, which could well pose a significant challenge to the successful execution of the plan.15
Results and Discussion
We first sought to gain access to core pentasaccharide 14, bearing three free hydroxyl groups, in order to examine the feasibility of installing all three sialylated antennae in a single step. The synthesis of key intermediate 14 commenced with the coupling of two protected glucosamine monosaccharides, 217 and 3,18 to generate disaccharide 4 in nearly quantitative yield (Scheme 1). Removal of the acetyl group, under Zemplén conditions, afforded the chitobiose acceptor, 5, properly functionalized for the requisite β-mannosylation. Thus, under Crich β-mannosylation conditions,19 5 and 620 were efficiently coupled with high stereoselectivity (dr>10:1). The crude mixture was then treated with a buffered solution of DDQ to provide 8 in 56% yield over two steps. Coupling of 8 and 9, with subsequent reductive cleavage of the benzylidene linkage, proceeded in 88% yield to afford only the desired isomer, 11. Next, attachment of the mannose thioethyl donor 12 to tetrasaccharide 11 provided pentasaccharide 13 in 90% yield. Finally, liberation of the acyl functionalities under basic conditions afforded triol 14 in 83% yield. Use of the 2,5-difluorobenzoyl (dFBz)21 protecting group was very helpful in allowing for a high yield in this step. Similar attempts to perform removal of unsubstituted benzoyl groups led to incomplete conversion and eventual cleavage of the phthalimidyl functionalities.22
Scheme 1. Reagents and Conditions.
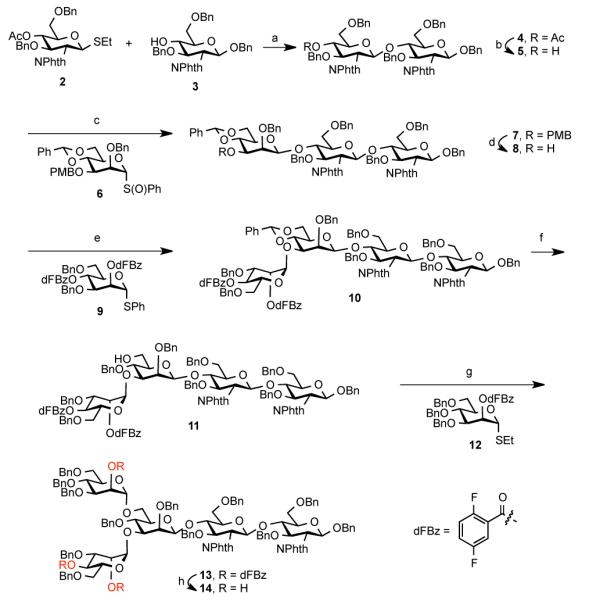
(a) NIS, AgOTf, CH2Cl2, 3Å MS, −25 °C, quant.; (b) NaOMe, MeOH, THF, rt, 1 h, 91%; (c) 6, Tf2O, DTBMP, CH2Cl2, 4A MS; (d) DDQ, CH2Cl2, phosphate buffer pH 7, 56% (over 2 steps); (e) 9, NIS, AgOTf, CH2Cl2, 3Å MS, 90%; (f) BH3/THF, n-Bu2BOTf, CH2Cl2, 0 °C, 4 h, 88%; (g) 12, NIS, AgOTf, CH2Cl2, −50 to −25 °C, h, 90%; (h) NaOMe, MeOH, THF, rt, 1.5 h, 83%.
We next prepared a series of trisaccharide derivatives (Scheme 2). Thioether trisaccharide 15 (itself prepared in 25 steps from commercially available materials),23 was converted to anomeric fluoride 16 through exposure to HF/pyridine. Alternatively, the Schmidt type donor, 19, could be prepared through coupling of 15 with 2-(trimethylsilyl)ethanol, followed by acidic cleavage of the anomeric protecting group and formation of the corresponding trichloroacetimidate. With a range of glycosyl donors in hand, we were now in a position to evaluate the feasibility of the proposed global three-fold glycosylation of pentasaccharide 14.
Scheme 2. Reagents and Conditions.
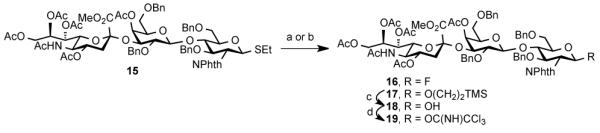
(a) NBS, HF/pyridine, CH2Cl2, rt, 91%; (b) TMSCH2CH2OH, NIS, AgOTf, CH2Cl2, 3Å MS, −25 °C, 94%; (c) TFA, CH2Cl2, H2O, rt, 5 h, 67%; (d) CCl3CN, DBU, CH2Cl2, 0 °C, 87%.
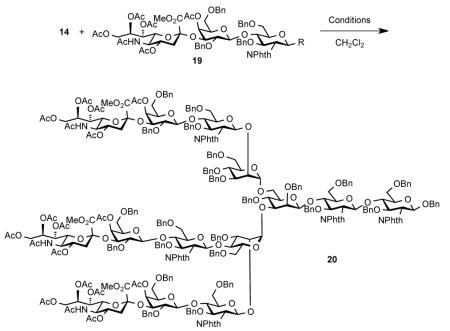
The results of the attempted global glycosylation with pentasaccharide 14 are summarized in Table 1. Thus, when a five-fold excess of donor 14 was exposed to Sinaÿ radical conditions,24 only 10% of the desired product, 20, was observed, along with unidentified side-products (entry 1). A survey of coupling protocols revealed thioether 15 to be an efficient donor under NIS/TfOH-mediated conditions (entry 3). The yield of this transformation was further improved when the reaction was conducted with AgOTf as a stoichiometric additive (entry 4). It is also of note that, when the reaction was carried out under more dilute conditions (<0.025M), no triglycosylated product was observed (MS-ESI) and only partially substituted intermediates were detected. Thus, under the optimized conditions (0.04 M), the desired saccharide 20 was formed in 21% yield, which translates to an average yield of 60% per glycosyl acceptor coupling. In a related glycosylation event with a disaccharide donor, we observed an improved yield of triantennary oligosaccharide, indicating a detrimental steric impact of the sialic acid moiety on global glycosylation reactions.22 Saccharide 20 was isolated following extensive chromatographic purifications on silica gel and gel filtrations (Sephadex LH-20 and Bio-Gel S-X1). We were also able to isolate 20% of biantennary products from this reaction, which, upon re-exposure to the above glycosylation conditions, afforded 20 in 10% yield. Attempts to achieve coupling with fluoride donor 16 (under Cp2HfCl2/AgClO4 or BF3*Et2O conditions), or through sulfoxide-mediated glycosylation protocols,25 led to clean activation of the donor, but only decomposition of pentasaccharide 14 (entries 5 and 6). Interestingly, Schmidt donor 19 was found to be a suitable reagent for this transformation, delivering 20 in 19% yield, with only trace amounts of diglycosylated intermediates (entry 7).
Table 1.
| Entry | R | Conditions | Yield [%] |
|---|---|---|---|
| 1 | SEt (15) | (4-BrC6H4)3NSbCl6, 0 °C → rt | 10% |
| 2 | SEt (15) | BSP, Tf2O, TTBP, −60 °C | ND |
| 3 | SEt (15) | NIS, TfOH, −20 °C | 16% |
| 4 | SEt (15) | NIS, AgOTf, −20 °C | 21% |
| 5 | F (16) | Cp2HfCl2, AgClO4, 0 °C | <5% |
| 6 | OH (18) | Ph2SO, Tf2O, −60 °C | ND |
| 7 | OC(NH)CCl3 (19) | TMSOTf, −30 °C | 19% |
Key: ND=None Detected
Based on the findings summarized in Table 1, we hypothesized that the efficiency of the synthetic route toward 1 could be greatly improved if the sialylated antenna at the most sterically hindered position (ring 4, 4′-OH) were installed first. To this end, trisaccharide 8 was coupled with orthogonally protected mannose donor 21 and the resultant tetrasaccharide was then exposed to reductive conditions to provide 22 in 53% yield (Scheme 3). The top mannose branching was introduced via reaction with donor 23 and removal of the silyl functionality. When 25 was coupled with 15 under the optimized conditions (Table 1, entry 4), octasaccharide 26 was obtained in 60% yield. The subsequent reductive removal of the 2′-OH protective groups proceeded uneventfully. We were pleased to find that installation of the remaining two trisaccharide antennae proceeded in 50% yield (>70% per glycosyl acceptor), which represents a significant improvement in overall efficiency, in comparison to the global glycosylation strategy.
Scheme 3. Reagents and Conditions.
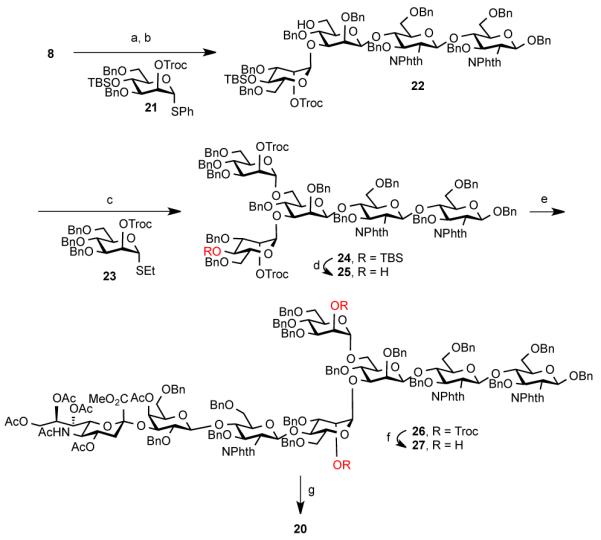
(a) 21, NIS, AgOTf, CH2Cl2, 3Å MS, 2 h; (b) BH3.THF, n-Bu2BOTf, CH2Cl2/THF, 0 °C, 3 h, 53% (over 2 steps) (c) 23, NIS, AgOTf, CH2Cl2, 3Å MS, −20 °C, 3 h, 78%; (d) HF/pyridine, THF, rt, 44 h, 58%; (e) 15, NIS, AgOTf, CH2Cl2, 3 Å MS, −20 °C, 36 h, 60%; (f) nano-Zn, AcOH, THF, rt, 20 min, 85%; (g) 15, NIS, AgOTf, CH2Cl2, 3 Å MS, −20 °C, 36 h, 50%.
With compound 20 in hand, we next proceeded to the stage of global deprotection. As shown in Scheme 4, the ester functionalities were cleaved through exposure to NaOMe/MeOH, and the resultant crude triacid was treated with 1,2-ethylenediamine in n-butanol and PhMe, to liberate the five amine functionalities.26 This mixture was then exposed to selective N-acetylation conditions to deliver triacid 28. The best yield for this sequence was obtained when the excess of diamine was removed following initial cleavage of the phthalimidyl ring and the deprotection was completed in refluxing toluene. Although we were unable to remove the benzyl groups through hydrogenolysis, exposure of 28 to dissolving metal conditions provided clean deprotection of the 24 benzyl functionalities in one step to deliver the target glycan 1, in moderate overall yield.
Scheme 4. Reagents and Conditions.

(a) NaOMe/NaOH, MeOH/H2O, rt, 20 h; (b) 1,2-ethylenediamine, n-BuOH, PhMe, 90 °C, 24 h; (c) Ac2O, Et3N, MeOH, rt; (d) Na/NH3, THF, −78 °C, 11-25% (over 4 steps).
Conclusion
In summary, we have disclosed the first total synthesis of fully sialylated triantennary N-linked glycan, 1. In toto, oligosaccharide 1 was reached in 71 steps from commercially available materials, through a synthetic route featuring an optimized convergent installation of the terminal sialylated antennae. While a large number of steps were required to reach this complex target, very little material is needed to allow for evaluation of the underlying biological question. Moreover, the synthetic program outlined in this paper will surely provide guidance for the synthesis of other highly branched members of this family of complex PSMA-related N-linked glycodomains. Studies on the biological and immunological properties of glycan 1 are currently underway.
Supplementary Material
ACKNOWLEDGMENT
Support for this research was provided by the National Institute of Health (CA103823 to S.J.D.). M.A.W. would like to acknowledge Terrie Brodeur Breast Cancer Foundation for postdoctoral fellowship. We also thank Dr. George Sukenick, Hui Fang, and Sylvi Rusli of SKI′s NMR core facility for mass spectral and NMR assistance.
Footnotes
ASSOCIATED CONTENT
Supporting Information General experimental procedures, including spectroscopic and analytical data for new compounds. This material is available free of charge via the Internet at http://pubs.acs.org.
REFERENCES
- 1.Roth J. Chem. Rev. 2002;102:285. doi: 10.1021/cr000423j. [DOI] [PubMed] [Google Scholar]
- 2.Montreuil J, Vliegenthart JFG. In: Glycoproteins and Disease. H S, editor. Elsevier Science; 1996. [Google Scholar]
- 3.a) Danishefsky SJ, Allen JR. Angew. Chem. Int. Ed. 2000;39:836. doi: 10.1002/(sici)1521-3773(20000303)39:5<836::aid-anie836>3.0.co;2-i. [DOI] [PubMed] [Google Scholar]; b) Zhu J, Warren JD, Danishefsky SJ. Expert Rev. Vacc. 2009;8:1399. doi: 10.1586/erv.09.95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Schülke N, Varlamova OA, Donovan GP, Ma D, Gardner JP, Morrissey DM, Arrigale RR, Zhan C, Chodera AJ, Surowitz KG, Maddon PJ, Heston WDW, Olson WC. Proc. Natl. Acad. Sci. USA. 2003;100:12590. doi: 10.1073/pnas.1735443100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ghosh A, Heston WDW. J. Cell. Biochem. 2004;91:528. doi: 10.1002/jcb.10661. [DOI] [PubMed] [Google Scholar]
- 6.Kiessling A, Füssel S, Wehner R, Bachmann M, Wirth MP, Rieber EP, Schmitz M. Eur. Urol. 2008;53:694. doi: 10.1016/j.eururo.2007.11.043. [DOI] [PubMed] [Google Scholar]
- 7.Chang SS, Reuter VE, Heston WDW, Bander NH, Grauer LS, Gaudin PB. Cancer Res. 1999;59:3192. [PubMed] [Google Scholar]
- 8.(a) Dubrovska A, Kim C, Elliott J, Shen W, Kuo T-H, Koo D-I, Li C, Tuntland T, Chang J, Groessl T, Wu X, Gorney V, Ramirez-Montagut T, Spiegel DA, Cho CY, Schultz PG. ACS Chem. Biol. 2011;6:1223. doi: 10.1021/cb200222s. [DOI] [PubMed] [Google Scholar]; (b) Murelli RP, Zhang AX, Michel J, Jorgensen WL, Spiegel DA. J. Am. Chem. Soc. 2009;131:17090. doi: 10.1021/ja906844e. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Kozikowski AP, Zhang J, Nan F, Petukhov PA, Grajkowska E, Wroblewski JT, Yamamoto T, Bzdega T, Wroblewska B, Neale JH. J. Med. Chem. 2004;47:1729. doi: 10.1021/jm0306226. [DOI] [PubMed] [Google Scholar]; (d) Kularatne SA, Wang K, Santhapuram H-KR, Low PS. Mol. Pharmaceutics. 2009;6:780. doi: 10.1021/mp900069d. [DOI] [PubMed] [Google Scholar]
- 9.(a) Kuduk SD, Schwarz JB, Chen X-T, Glunz PW, Sames D, Ragupathi G, Livingston PO, Danishefsky SJ. J. Am. Chem. Soc. 1998;120:12474. [Google Scholar]; (b) Ragupathi G, Koide F, Livingston PO, Cho YS, Endo A, Wan Q, Spassova MK, Keding SJ, Allen J, Ouerfelli O, Wilson RM, Danishefsky SJ. J. Am. Chem. Soc. 2006;128:2715. doi: 10.1021/ja057244+. [DOI] [PubMed] [Google Scholar]; (c) Zhu J, Wan Q, Lee D, Yang G, Spassova MK, Ouerfelli O, Ragupathi G, Damani P, Livingston PO, Danishefsky SJ. J. Am. Chem. Soc. 2009;131:9298. doi: 10.1021/ja901415s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Holmes EH, Greene TG, Tino WT, Boynton AL, Aldape HC, Misrock SL, Murphy GP. Prostate Suppl. 1996;7:25. [PubMed] [Google Scholar]
- 11.Davis MI, Bennett MJ, Thomas LM, Bjorkman PJ. Proc. Natl. Acad. Sci. USA. 2005;102:5981. doi: 10.1073/pnas.0502101102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Barinka C, Šácha P, Sklenář J, Man P, Bezouška K, Slusher BS, Konvalinka J. Protein Sci. 2004;13:1627. doi: 10.1110/ps.04622104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Schauer R. Curr. Opin. Struct. Biol. 2009;19:507. doi: 10.1016/j.sbi.2009.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Courtney AH, Puffer EB, Pontrello JK, Yang Z-Q, Kiessling LL. Proc. Natl. Acad. Sci. 2009;106:2500. doi: 10.1073/pnas.0807207106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wang P, Zhu J, Yuan Y, Danishefsky SJ. J. Am. Chem. Soc. 2009;131:16669. doi: 10.1021/ja907136d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.(a) Hård K, Mekking A, Damm JBL, Kamerling JP, De Boer W, Wijnands RA, Vliegenthart JFG. Eur. J. Biochem. 1990;193:263. doi: 10.1111/j.1432-1033.1990.tb19332.x. [DOI] [PubMed] [Google Scholar]; (b) Sakai H, Yamagishi F, Miura M, Hata K, Koyama I, Sakagishi Y, Komoda T. Tumor Biol. 1995;15:230. doi: 10.1159/000217896. [DOI] [PubMed] [Google Scholar]; (c) Ishii K, Iwasaki M, Inoue S, Kenny PT, Komura H, Inoue Y. J. Biol. Chem. 1989;264:1623. [PubMed] [Google Scholar]
- 17.(a) Unverzagt C. Chem. Eur. J. 2003;9:1369. doi: 10.1002/chem.200390156. [DOI] [PubMed] [Google Scholar]; (b) Nagorny P, Fasching B, Li X, Chen G, Aussedat B, Danishefsky SJ. J. Am. Chem. Soc. 2009;131:5792. doi: 10.1021/ja809554x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dinkelaar J, Duivenvoorden BA, Wennekes T, Overkleeft HS, Boot RG, Aerts JMFG, Codée JDC, van der Marel GA. Eur. J. Org. Chem. 2010;2010:2565. [Google Scholar]
- 19.(a) Crich D. Acc. Chem. Res. 2010;43:1144. doi: 10.1021/ar100035r. [DOI] [PubMed] [Google Scholar]; (b) Crich D, Sun S. J. Org. Chem. 1996;61:4506. doi: 10.1021/jo9606517. [DOI] [PubMed] [Google Scholar]
- 20.Crich D, Li H, Yao Q, Wink DJ, Sommer RD, Rheingold AL. J. Am. Chem. Soc. 2001;123:5826. doi: 10.1021/ja015985e. [DOI] [PubMed] [Google Scholar]
- 21.Sjölin P, Kihlberg J. J. Org. Chem. 2001;66:2957. doi: 10.1021/jo001584q. [DOI] [PubMed] [Google Scholar]
- 22.(a) Dudkin VY, Miller JS, Dudkina AS, Antczak C, Scheinberg DA, Danishefsky SJ. J. Am. Chem. Soc. 2008;130:13598. doi: 10.1021/ja8028137. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Dudkin VY, Miller JS, Danishefsky SJ. J. Am. Chem. Soc. 2003;126:736. doi: 10.1021/ja037988s. [DOI] [PubMed] [Google Scholar]
- 23.Wu B, Hua Z, Warren JD, Ranganathan K, Wan Q, Chen G, Tan Z, Chen J, Endo A, Danishefsky SJ. Tetrahedron Lett. 2006;47:5577. doi: 10.1016/j.tetlet.2006.09.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.(a) Marra A, Mallet J-M, Amatore C, Sinaÿ P. Synlett. 1990;1990:572. [Google Scholar]; (b) Zhang Y-M, Mallet J-M, Sinaÿ P. Carbohydr. Res. 1992;236:73. doi: 10.1016/0008-6215(92)85007-m. [DOI] [PubMed] [Google Scholar]
- 25.Garcia BA, Gin DY. J. Am. Chem. Soc. 2000;122:4269. [Google Scholar]
- 26.Kanie O, Crawley SC, Palcic MM, Hindsgaul O. Carbohydr. Res. 1993;243:139. doi: 10.1016/0008-6215(93)84087-m. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.