

ABSTRACT
The cellular autophagy response induced by herpes simplex virus 1 (HSV-1) is countered by the viral γ34.5 protein. γ34.5 modulates autophagy by binding to the host autophagy protein Beclin-1 and through this binding inhibits the formation of autophagosomes in fibroblasts and neurons. In contrast, in this study dendritic cells (DCs) infected with HSV-1 showed an accumulation of autophagosomes and of the long-lived protein p62. No such accumulations were observed in DCs infected with a γ34.5-null virus or a virus lacking the Beclin-binding domain (BBD) of γ34.5. To explore this further, we established stably transduced DC lines to show that γ34.5 expression alone induced autophagosome accumulation yet prevented p62 degradation. In contrast, DCs expressing a BBD-deleted mutant of γ34.5 were unable to modulate autophagy. DCs expressing γ34.5 were less capable of stimulating T-cell activation and proliferation in response to intracellular antigens, demonstrating an immunological consequence of inhibiting autophagy. Taken together, these data show that in DCs, γ34.5 antagonizes the maturation of autophagosomes and T cell activation in a BBD-dependent manner, illustrating a unique interface between HSV and autophagy in antigen-presenting cells.
IMPORTANCE
Herpes simplex virus 1 (HSV-1) is a highly prevalent pathogen causing widespread morbidity and some mortality. HSV infections are lifelong, and there are no vaccines or antivirals to cure HSV infections. The ability of HSV to modulate host immunity is critical for its virulence. HSV inhibits host autophagy, a pathway with importance in many areas of health and disease. Autophagy is triggered by many microbes, some of which harness autophagy for replication; others evade autophagy or prevent it from occurring. Autophagy is critical for host defense, either by directly degrading the invading pathogen (“xenophagy”) or by facilitating antigen presentation to T cells. In this study, we show that HSV manipulates autophagy through an unsuspected mechanism with a functional consequence of reducing T cell stimulation. These data further our understanding of how HSV evades host immunity to persist for the lifetime of its host, facilitating its spread in the human population.
Introduction
Herpes simplex virus 1 (HSV-1) is a common and significant pathogen with two distinct phases of infection (1). Acute infection occurs at peripheral mucocutaneous sites with widespread expression of viral genes. Infection of innervating neurons is followed by retrograde transport of virus to cell bodies within sensory ganglia and establishment of a latent infection therein. During latency, viral gene expression is limited until the viral genome reactivates to form progeny virions. Following anterograde transport to the periphery, the reactivated virus may form new lesions and be shed to infect other susceptible individuals. The ability of HSV to repeatedly reactivate from infected individuals underscores both the power and importance of its immune-modulating activities which allow HSV to replicate in, and be shed from, a primed and immunocompetent host. One such immunomodulatory factor, γ34.5, the focus of this study, is now emerging as a multifunctional viral protein that is effective at manipulating both the innate and adaptive immune responses.
Host cell translational shutdown is a key antiviral defense pathway mediated by double-stranded RNA-dependent protein kinase (PKR), which phosphorylates the alpha subunit of the translation initiation factor eIF2 (2, 3). γ34.5, expressed by HSV at approximately 3 h postinfection, serves to reverse this translational shutdown by bridging protein phosphatase 1 (PP1) and eIF2α, thereby dephosphorylating eIF2α (4–8). Another target for γ34.5 is Tank-binding kinase 1 (TBK1), which is responsible for signaling to interferon regulatory factors 3 and 7 (IRF3/7) (9, 10). γ34.5 thereby inhibits IRF3/7 activation, repressing the induction of many antiviral genes within infected cells. In addition to these roles in modulating the innate immune response, γ34.5 also regulates autophagy (11). Autophagy is a catabolic homeostatic process involving the breakdown of cellular components in cytosolic vacuoles (12–14). It is induced by starvation, heat shock, hypoxia, hormones, immune signaling, and other triggers (15–19). Among its myriad roles, autophagy is involved in survival and apoptosis, organelle maintenance, removal of protein aggregates, and via a process called xenophagy, direct clearance of intracellular pathogens (20, 21). Mechanistically, autophagy progresses through the formation of an isolation membrane in the cytosol, which surrounds and segregates cytosolic material (22, 23). This matures to a double-membrane structure, the autophagosome, which in turn fuses with the lysosome leading to the enzymatic breakdown of its contents (24, 25). Although autophagy is constitutive, the rate of autophagosome formation and autophagic flux is tightly controlled, with Beclin-1 as a major regulator (26, 27). Autophagy also plays a key role in antigen processing for major histocompatibility complex (MHC) presentation, especially to CD4+ T cells (28), and this activity is critical in vivo for protection against HSV-2 and other pathogens (29).
Modulation of autophagy is important for the virulence of many viruses, including HIV, hepatitis B and C, and Coxsackie B (30–36), underscoring the importance of understanding the interplay between viruses and autophagy. HSV-1 mutants lacking γ34.5 demonstrate a PKR- and eIF2α phosphorylation-dependent reduction of long-lived proteins and reduced volume of autophagic vacuoles in infected fibroblasts and neurons (11, 37). Control of autophagy by γ34.5 is mediated not only by its manipulation of eIF2α phosphorylation but also by its capacity to bind Beclin-1 through a 20-amino-acid Beclin-binding domain (BBD) in both mouse and human cells (38). Mutants lacking BBD (ΔBBD) induce increased numbers of autophagosomes in epithelial cells and are neuro-attenuated in vivo (38). This attenuation of the ΔBBD mutant is dependent upon a functional adaptive immune response, and ΔBBD mutant-infected mice display higher CD4+ T cell responsiveness than mice infected with wild-type virus (39). These observations suggested a possible role for γ34.5 in modulation of the immune response via preclusion of autophagy in antigen-presenting cells (APCs). Emerging data, however, has suggested that the effect of γ34.5 on autophagy in professional APCs may differ from that observed in fibroblasts or neurons. In infected macrophages, γ34.5 leads to the formation of morphologically distinct autophagosomes that are associated with the nuclear envelope, and infected cells retain the ability to prime CD8+ T cells (40). Also, in contrast to neurons and fibroblasts, γ34.5 does not inhibit the induction of autophagy in dendritic cells (DCs) (41). Finally, the maturation of infected DCs is inhibited by γ34.5 expression, further illustrating that γ34.5 manipulates immune surveillance through multiple mechanisms (42–44).
In this study, we wished to address the role of the BBD of γ34.5 in alteration of the autophagy pathway in DCs. We also sought to study the functional consequences of γ34.5 expression in these cells and to test whether γ34.5 alone was sufficient for alteration of autophagy and T cell responses to presented antigen. To address this, we infected DCs with wild-type or mutant viruses and examined their capacity to form autophagosomes and process long-lived proteins. We also constructed DC lines stably expressing γ34.5 or its ΔBBD mutant to examine their roles in modulation of autophagy in the absence of expression of other HSV genes. Our results showed that γ34.5 does not prevent the induction of autophagy in DCs but rather prevents the maturation of autophagosomes. Significantly, we also illustrated a γ34.5- and BBD-dependent interference with the ability of DCs to stimulate antigen-specific T cells. These data suggest an important role for the BBD of γ34.5 in the modulation of autophagy in DCs and genesis of the adaptive immune response.
RESULTS
HSV-1 infection induces autophagosome accumulation in infected DC2.4 cells.
The conversion of LC3-I to its phosphatidylethanolamine-conjugated form, LC3-II, is requisite for the formation of the autophagosome and is a widely used measurement of autophagy (45). When DC2.4 cells were infected with the wild type (WT), the LC3-I isoform (top band) was reduced and LC3-II concentrations were ~6-fold higher than in uninfected cells and significantly higher than observed in cells infected with Δγ34.5 and ΔBBD mutants (Fig. 1A and B). We hypothesized that this unexpected increased conversion of LC3 was indicative of increased autophagy and sought to assess this further by immunofluorescence microscopy. DC2.4 cells were infected with the WT, Δγ34.5, or ΔBBD strain and analyzed 8 h postinfection. As a positive control, cells were treated with bafilomycin, which causes an accumulation of autophagosomes by preventing lysosome acidification (46, 47). We observed a significant increase of LC3 puncta in WT-infected cells that was similar to levels seen following bafilomycin treatment (Fig. 1C and D). In contrast, infection with the Δγ34.5 or ΔBBD mutant did not result in an increase in LC3 puncta. In these experiments, cells scored as positive for autophagy contained from 20 to 60 puncta, whereas negative cells contained 0 to 3 puncta. Uninfected bystander cells, unstained for ICP8, did not accumulate LC3-specific puncta, indicating that the effect is dependent upon direct infection. Based on these results, we conclude that infection with wild-type virus, but not the Δγ34.5 or ΔBBD mutant, results in an increased number of LC3-II-positive autophagosomes in DCs.
FIG 1.
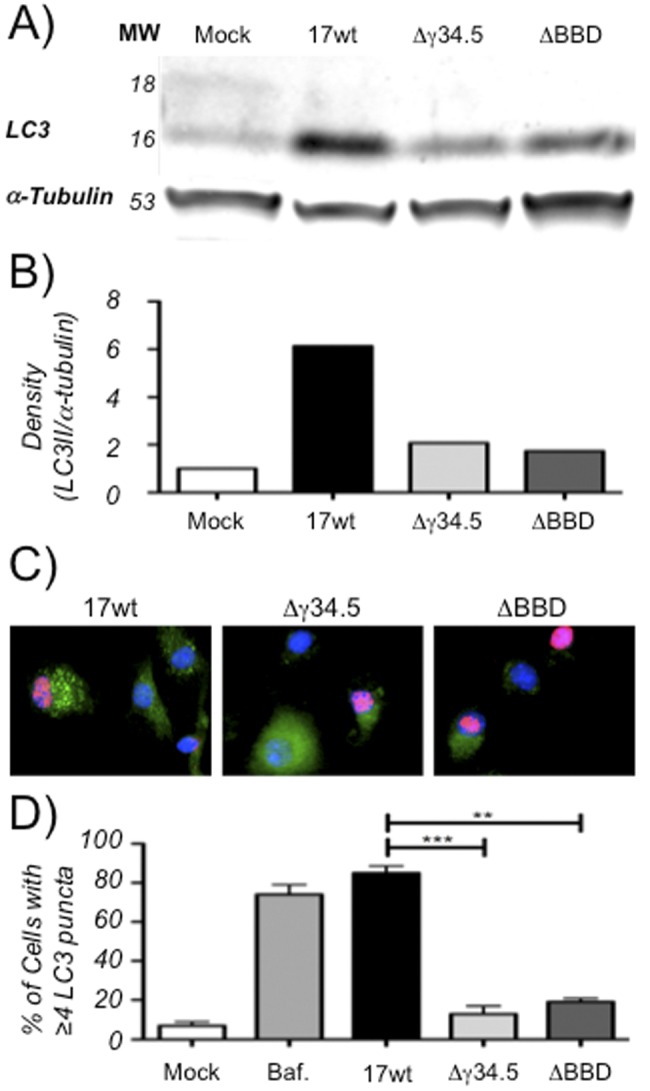
Infection of DCs with HSV leads to autophagosome accumulation. (A) Representative Western blot (of two experiments) of lysates from DC2.4 cells that were mock infected or infected at an MOI of 8.0 with WT, Δγ34.5, or ΔBBD virus for 8 h. Blots were probed with anti-LC3 or anti-α-tubulin. (B) Quantification of representative Western blot showing relative band density for LC3-II (the 16-kDa band) normalized to α-tubulin. (C) Immunofluorescence micrographs of cells infected at an MOI of 2.0 with WT, Δγ34.5, or ΔBBD virus for 8 h and probed for autophagosomes with anti-LC3 and for viral infection with anti-ICP8. Bystander cells without ICP8 stain serve as internal, uninfected controls. (D) Quantification of cells displaying 4 or more bright LC3 puncta for various treatments following analysis of at least three experiments involving 100 cells in four or more fields for each treatment. Positive cells are graphed as a function of total cells using DAPI for mock-infected cultures or as a function of ICP8-positive stain for infected cultures. Error bars indicate standard deviations between visualized fields. **, P < 0.005; ***, P < 0.001 (t test).
Long-lived proteins accumulate in HSV-infected dendritic cells.
While an increase in LC3 lipidation often results from increased initiation of autophagy, LC3-II can also accumulate due to interference with the maturation phase of autophagy. To differentiate between these possibilities, we examined the stability of the long-lived protein p62. p62 is primarily involved in clearing ubiquitinated proteins from the cell, and its degradation by autophagy renders it a suitable marker for autophagic flux (48, 49). Western blots for p62 were performed on lysates from DC2.4 cells infected for 8 h with WT, Δγ34.5-null, or ΔBBD strains (Fig. 2A and B). DC2.4 is a cell line derived from mouse bone marrow, and in the inactive form of the cells, like the epidermal myeloid-derived DCs infected by HSV in vivo, they are poorly inflammatory and express little MHC class II (MHC-II), CD40, and CD80 (44, 50, 51). They are phagocytosis-competent and mature when activated, upregulating MHC-II, CD40, and CD80 and secreting a wide variety of proinflammatory cytokines and chemokines. Cells infected with the WT had greater concentrations of p62 than uninfected cells or cells infected with the Δγ34.5 or ΔBBD mutant, indicating that HSV interferes with catabolic breakdown of p62 in a BBD-dependent manner. These data are consistent with the hypothesis that the increased number of autophagosomes in HSV-infected dendritic cells is due to a BBD-dependent interference with the maturation phase of autophagy, analogous to the activities of influenza A M2 and HIV-1 Nef (30, 31). To test this hypothesis further, we performed immunofluorescence microscopy for p62, with the prediction that we would see a greater number of p62-containing puncta in WT-infected cells than in cells infected with the Δγ34.5 and ΔBBD mutants (Fig. 2C and D). Untreated cells showed p62 in a diffuse cytoplasmic pattern, in contrast to cells treated with bafilomycin, where distinct multiple cytoplasmic puncta were observed in the majority of cells. Consistent with our prediction, infection of DC2.4 with WT caused a significant accumulation of cytoplasmic puncta compared to that of mock-infected cells. In contrast Δγ34.5 or ΔBBD mutant-infected cells did not significantly differ from mock-infected cells. Taken together, these data suggest that γ34.5, through its BBD, prevents the maturation of autophagosomes, leading to the intracellular accumulation of p62.
FIG 2.
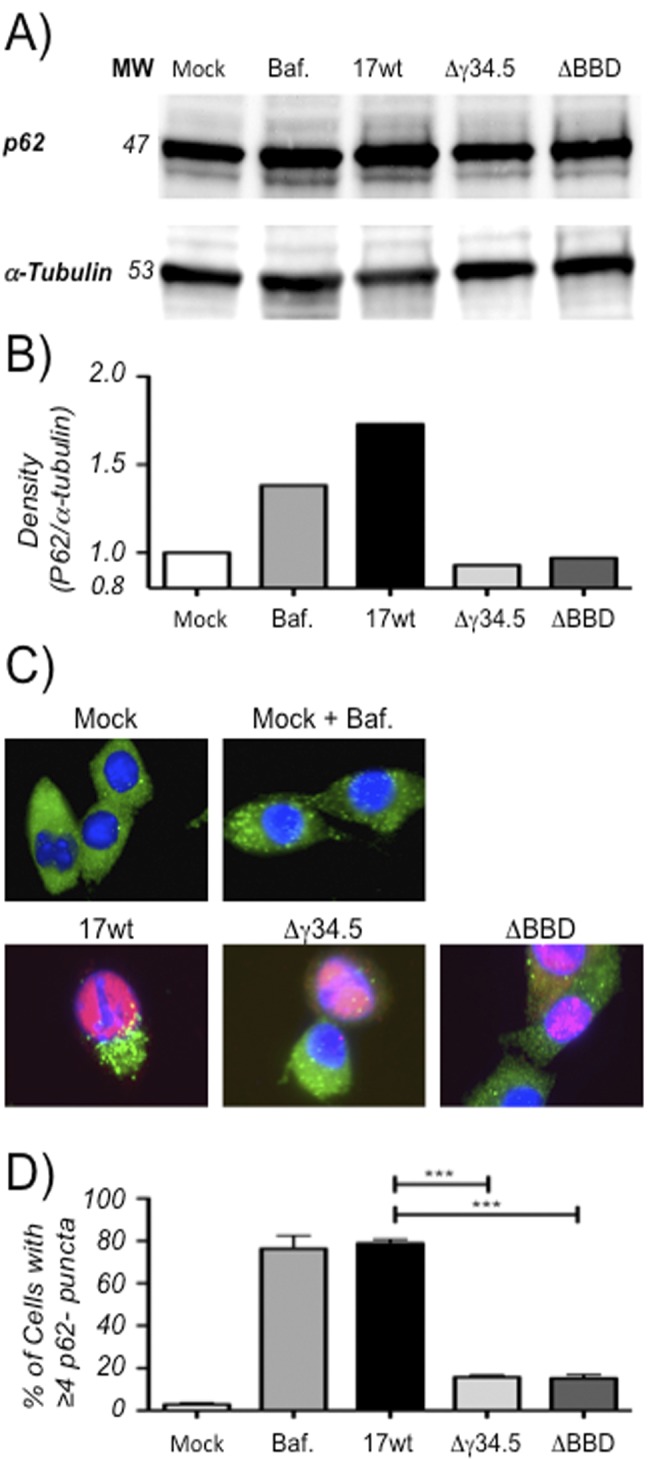
p62 accumulates in DC2.4 cells infected with HSV-1. (A) Representative Western blots (from two experiments) from lysates of DC2.4 cells that were mock infected or infected at an MOI of 8.0 with WT, Δγ34.5, or ΔBBD virus for 8 h and probed with anti-p62 or anti-α-tubulin antibodies. (B) Graph from representative Western blot showing relative band density for p62 normalized to α-tubulin. (C) Immunofluorescence micrographs of cells infected at an MOI of 2.0 with WT, Δγ34.5, or ΔBBD virus for 16 h and probed for p62 and HSV ICP0 expression. (D) Quantification of cells from fluorescence micrographs. Cells displaying 4 or more p62 puncta were counted as positive and graphed as a function of total cells using DAPI for mock-infected cells or as a function of ICP0-positive stain for infected cells. Analysis included at least 60 cells in three or more fields for each treatment in two experiments. Error bars indicate standard deviations between visualized fields. ***, P < 0.001 (t test).
Stably transduced DC2.4 lines express functional γ34.5.
Having shown that the BBD of γ34.5 was necessary for impacting autophagy maturation in DCs, we next sought to determine whether γ34.5 is sufficient for this function and whether the BBD is necessary. To examine this, we used a lentivirus expression vector to create γ34.5- and γ34.5ΔBBD mutant-expressing DC2.4 cell lines. Probing cell lysates from the stably transduced DC2.4 lines by Western blotting showed γ34.5- and γ34.5ΔBBD mutant-reactive bands with the expected migration patterns (data not shown). To determine if the expressed proteins were functional in an autophagy-independent manner, we used Western blotting to test the levels of phosphorylated eIF2α induced by HSV-1 or poly(I:C) treatment (Fig. 3A). Δγ34.5 infection was used to avoid the interfering effects of γ34.5 being expressed by both the infected cells and the incoming virus. Both Δγ34.5 infection and poly(I:C) treatment strongly induced a band detectable by an eIF2α serine-51 phosphorylation-specific antibody in lysates from control cells but not in cells expressing full-length γ34.5 or the γ34.5ΔBBD mutant. These results demonstrated that the stably transduced DCs were expressing γ34.5 and γ34.5ΔBBD proteins that were equally capable of mediating the dephosphorylation of eIF2α.
FIG 3.
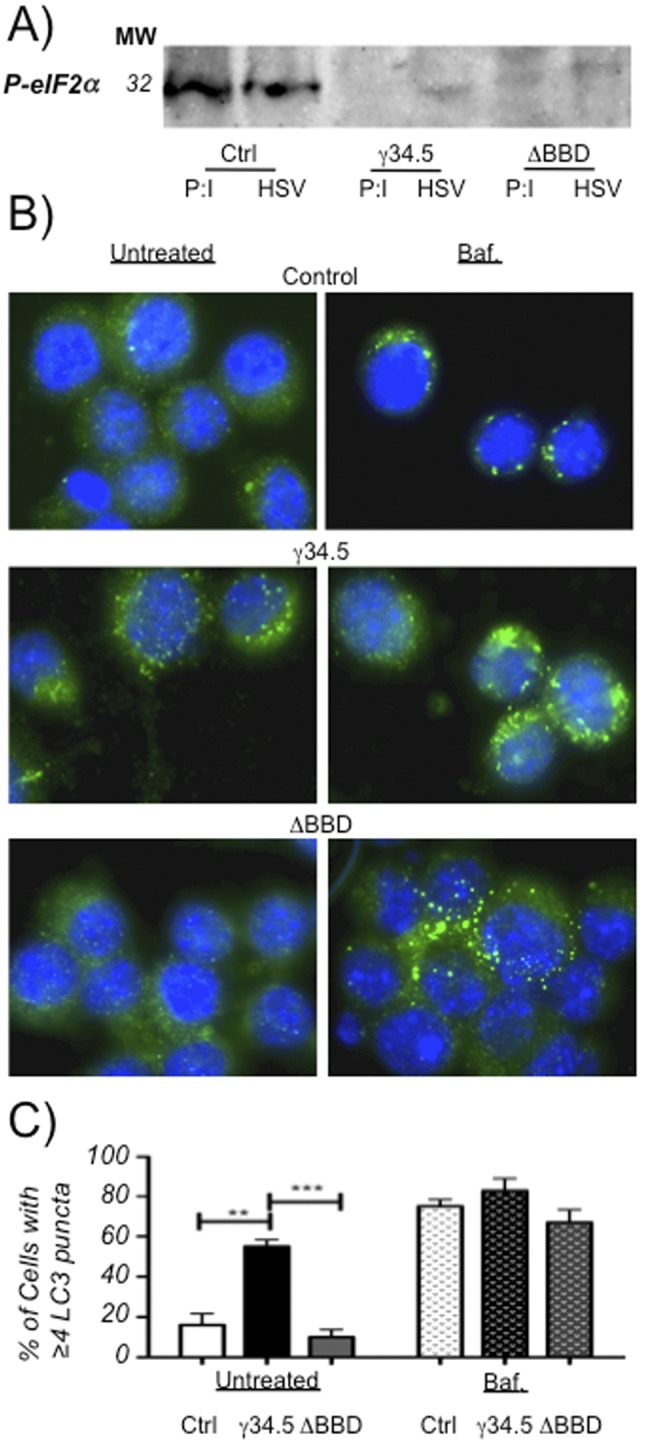
Stably transduced DC2.4 cells express functional γ34.5 and accumulate autophagosomes. (A) Western blot (representative of two experiments) of cell lysates from γ34.5 or ΔBBD mutant-expressing DC2.4 cells or control DC2.4 cells transfected with poly(I:C) at 20 µg/ml or infected with Δγ34.5 HSV-1 at an MOI of 8.0 for 12 h. Western blots were probed with an antibody for phospho-eIF2α. (B) Fluorescence micrographs of γ34.5- or ΔBBD mutant-expressing or control cells probed with an anti-LC3 antibody. Cells were untreated or treated with 100 µM bafilomycin for 6 h. (C) Quantification of cells from panel B displaying 4 or more LC3 puncta. At least 100 cells in 4 or more fields were counted for each cell type and each treatment in 2 experiments. Positive cells are graphed as a function of total cells using DAPI stain, and the error bars indicate standard deviations between visualized fields. **, P < 0.005; ***, P < 0.001 (t test).
DC2.4 cells expressing γ34.5 exhibit altered autophagy patterns.
We next sought to determine if the stably transduced DC2.4 lines expressing γ34.5 or the γ34.5ΔBBD mutant exhibit altered patterns of autophagy (Fig. 3B and C). We stained stably transduced DC2.4 with an LC3-specific antibody and quantified cells per field with 4 or more puncta by immunofluorescence microscopy. Relative to control cells, we observed a significant increase in the number of cells with ≥4 LC3-positive puncta in the line expressing γ34.5 but not in the control or γ34.5ΔBBD mutant lines. All cell lines treated with bafilomycin displayed an expected increase in the number of LC3-specific puncta, showing that the pathways for induction of LC3-II were intact in all 3 cell lines. Based on these results, we conclude that γ34.5 is sufficient to cause an increase in accumulation of LC3 puncta and that the changes in autophagy observed during infection of DCs with HSV are due largely to the activities of γ34.5 and the BBD.
γ34.5 reduces cell survival following starvation and interferes with autophagic flux.
Autophagy is induced in cells in response to starvation, thereby making biosynthetic precursors available to starving cells to preclude nutritional crisis and apoptosis. Cells with functional autophagy can therefore survive brief starvation stress, while cells deficient in autophagy are less resistant (52–54). We utilized this classic observation to ask whether cells expressing wild-type γ34.5 are less able to survive starvation relative to cells expressing the γ34.5ΔBBD strain, thereby demonstrating a block to functional autophagy and survival. To assess this, cells were stained for the apoptosis marker annexin V during normal culture conditions and following starvation (Fig. 4A). All cell lines stained equivalently for annexin V under normal culture conditions, but following a 2-h starvation, DC2.4 lines expressing γ34.5, but not control cells or cells expressing the γ34.5ΔBBD strain, had significantly increased annexin V staining. Cell viability, as measured by propidium iodide incorporation, was also reduced in starved cells expressing γ34.5 (data not shown). These results indicate that despite γ34.5-induced LC3-II positive puncta, autophagy-derived catabolites are not available for de novo biosynthesis under nutritional stress conditions, consistent with the idea that γ34.5 inhibits the maturation phase of autophagy. To examine this further, we measured autophagic flux by examining p62 concentrations in each of the 3 cell lines in the presence and absence of bafilomycin (Fig. 4B and C). Untreated γ34.5-expresssing cells exhibit higher concentrations of p62 than control cells or cells expressing the γ34.5ΔBBD strain, relative to expression of α-tubulin. Bafilomycin treatment leads to an accumulation of p62 in all cells, showing that the pathways that promote p62 accumulation are intact in all cells. We infer from these data that autophagic flux is reduced by γ34.5 and that this activity is dependent upon binding of Beclin-1.
FIG 4.
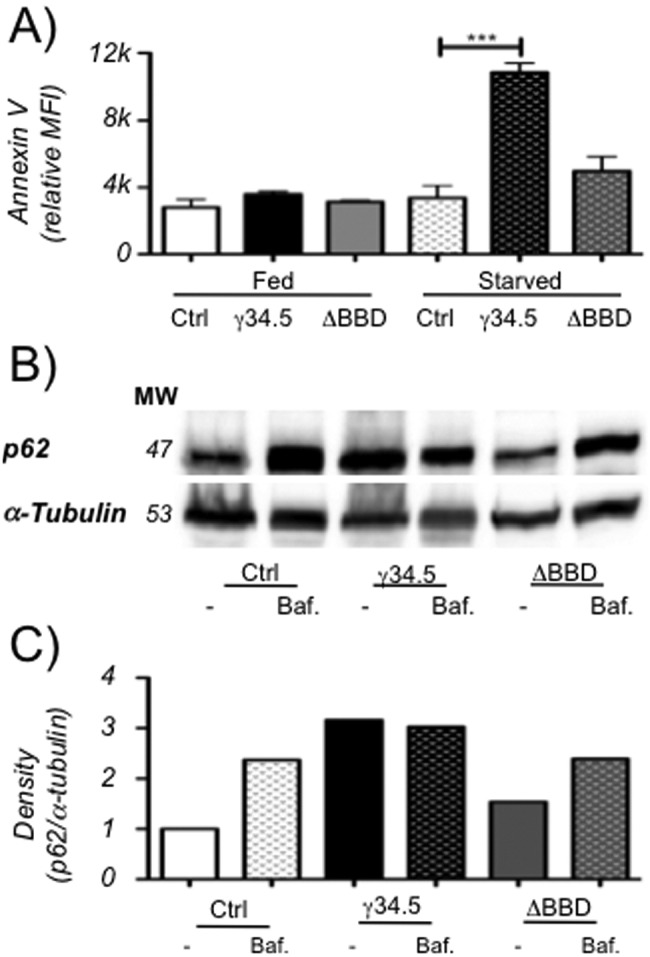
Stably transduced DC2.4 cells expressing γ34.5 are sensitive to starvation and accumulate long-lived proteins. (A) Annexin V staining of DC2.4 cells stably expressing γ34.5 or the ΔBBD mutant or control cells. Cells were untreated or starved and analyzed by flow cytometry, and mean fluorescence intensity was recorded. Graph shows the mean from two experiments, each consisting of at least 2 biological duplicates, and error bars indicate standard deviations. ***, P < 0.001 (t test). (B) Representative Western blot (of three experiments) of p62 or α-tubulin on cell lysates of control cells or cells expressing γ34.5 or the ΔBBD mutant. Cells were untreated or incubated with 100 µM bafilomycin for 6 h. (C) Quantification of a representative Western blot showing band density of p62 normalized to α-tubulin band density.
Autophagy modulation by γ34.5 in DCs interferes with T cell stimulation.
Autophagy is involved in the presentation of intracellular antigens on MHC-II via its delivery of cytoplasmic components to the lysosome and multivesicular MHC-II-loading compartment (28). We hypothesized that γ34.5, by virtue of interfering with autophagic delivery of intracellular antigens to the lysosome, would prevent the presentation of intracellular antigens when expressed in DCs. To this end, we engineered a γ34.5-deficient virus that expressed a truncated form of ovalbumin (OVA) lacking its amino-terminal signal sequence (virus termed Δγ34.5tOVA). Truncated OVA is not secreted and accumulates in the cytoplasm (55, 56). This makes it an attractive tool for immunological analysis of intracellular antigen processing by autophagy. We used Δγ34.5tOVA in conjunction with OT-II T cell receptor transgenic mice. OT-II mice have CD4+ T cells specific to the MHC-II immunodominant chicken ovalbumin peptide (residues 323 to 339) (57, 58). We infected control DC2.4 lines or the stably transduced lines expressing γ34.5 or the γ34.5ΔBBD mutant with the Δγ34.5tOVA strain and examined their ability to present intracellular antigen by coculturing them with splenocytes from OT-II mice. We measured the percentage of responding CD4+ T cells with a high degree of CD44 surface expression to quantify antigen exposure (Fig. 5). High-CD44 populations were robustly induced in OT-II CD4+ cells responding to the infected control DC line and to the cells expressing the γ34.5ΔBBD strain. In marked contrast, the γ34.5-expressing cell line infected with the Δγ34.5tOVA strain did not stimulate OT-II cells. These differences were due to the presence or absence of the BBD and independent of the remainder of the γ34.5 protein. The phenotype can therefore be primarily attributed to γ34.5’s effects upon the autophagic pathway. This T cell stimulation was antigen specific since CD44 surface expression was not upregulated in response to mock infection or infection with Δγ34.5 HSV-1 that lacked ovalbumin (Fig. 5B). In addition, no stimulation was observed when infected DCs were cocultured with splenocytes from nontransgenic C57BL/6 mice (data not shown). With all cocultures, treatment with phorbol myristate acetate (PMA) and ionomycin increased CD44 surface expression in CD4+ OT-II splenocytes, demonstrating that the T cells were responsive regardless of the DC lines in the coculture. We therefore conclude that γ34.5 manipulates autophagosome maturation in DCs, via Beclin-1 interaction, resulting in functional interference with immune surveillance of intracellular antigens.
FIG 5.
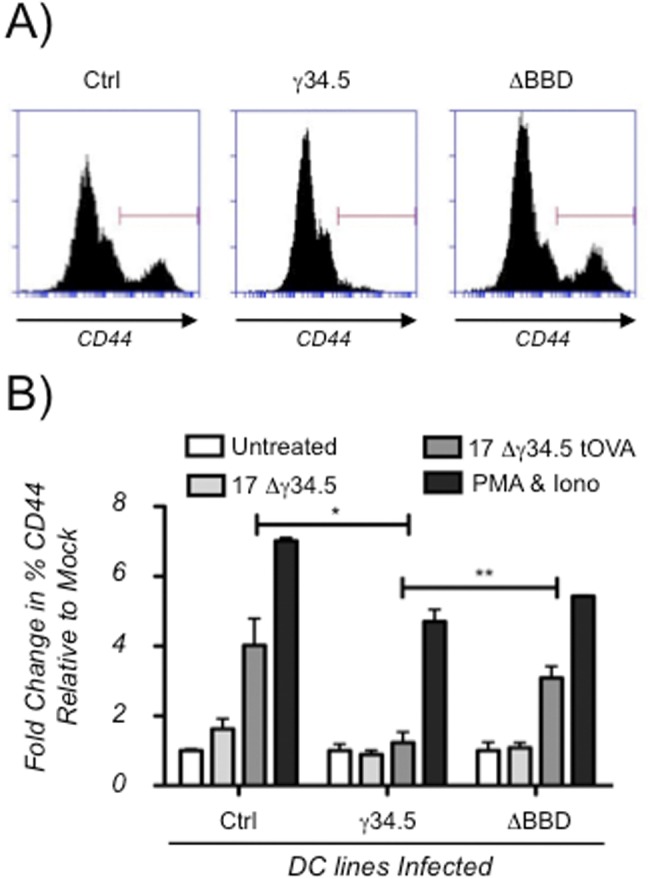
γ-34.5 expression in dendritic cells antagonizes activation of CD4+ T cells via Beclin-1 binding. (A) Representative flow cytometry histograms from DC2.4 cell lines infected with the 17Δ34.5tOva strain. Histograms represent one of two experiments analyzing 2 biological replicates. The γ34.5 or ΔBBD strains expressing DC2.4 cells or control cells were mock infected or infected with the Δ34.5 or Δ34.5tOva strain. Cells were cocultured with whole splenocytes from OT-II mice, stained cells were gated for CD4+ T cells, and CD44 staining was measured. (B) Graph showing fold changes in percentages of CD4+ T cell populations staining high for CD44 relative to mock-infected controls. Results are means from two experiments, each consisting of biological duplicates. Error bars indicate standard deviations. *, P < 0.05; **, P < 0.005 (t test).
DISCUSSION
Data from a number of laboratories have suggested that γ34.5 modulates autophagy during infection of fibroblasts and neurons via two activities, binding to Beclin-1 and dephosphorylation of eIF2α (11, 37, 38). These data were consistent with the idea that autophagy is dependent on Beclin-1 and on the activities of PKR, whose principle target is eIF2α. Two recent studies, however, have suggested that this model may be too simplistic, since the effect of γ34.5 on autophagy appears to be cell type specific. First, and consistent with the data in this study, induction of autophagosomes in murine myeloid cells is not antagonized by γ34.5 (41). Second, a recent study showed that HSV-1 induces incomplete/abortive autophagy in infected neuroblastoma cells (59). These latter results differ from those which examined infected primary neurons in culture (37, 38). This discordance may result from differences in autophagy pathways in immortalized SK-N-SH cells relative to postmitotic neurons or may simply reflect differences between human and mouse cells. The key question, however, is why do HSV-induced autophagy patterns differ so markedly in different cell types? Cell permissivity has been invoked as a possible explanation for the differences between DCs and fibroblasts (41), although this cannot be the entire explanation, since primary DCs and neurons are both poorly permissive for productive HSV-1 infection yet markedly differ in their autophagy responses. Another key question is do different autophagy patterns confer any advantage for HSV as a pathogen? It is possible that following infection of certain cells (e.g., fibroblasts), HSV has evolved to evade xenophagy, whereas in others (e.g., APCs), HSV has evolved to minimize antigen presentation, to sequester pathogen-associated molecular patterns (PAMPs) in autophagosomes, or possibly to regulate the inflammasome (60). It is also likely that other effects of γ34.5 on DCs, such as the prevention of DC maturation (42–44), impact the manner in which autophagy is inhibited by γ34.5 in these unique cells. Cell-specific differential use of autophagy-related proteins, due to their shared use in phagocytosis, antigen presentation, or immune modulation (61–63), may cause invading pathogens to evolve alternative strategies to subvert autophagy in different cell types. While evidence from our laboratory suggests that γ34.5 does not modulate phagocytosis in DCs (P. A. M. Gobeil and D. A. Leib, unpublished data), this is clearly an important area that warrants further investigation.
The initial observation of a role for γ34.5 in regulating MHC-II-restricted antigen presentation was described 10 years ago (64). Work from our lab and others has served to show a role for γ34.5 in controlling immunity through modulation of autophagy and APC maturation which underscores the multifunctional nature of γ34.5’s role in controlling immunity (42, 43, 65). The maturation of DCs is dependent upon NF-κB activation (66), which can be suppressed through γ34.5’s regulation of IKKβ activity (43). Intriguingly, the posttranslational modification of IKKβ is also important for the control of autophagy, so it is possible that this is yet another way in which γ34.5 modulates the formation or maturation of autophagosomes (67). Abortive autophagy, as observed in this study, occurs following infection with other viruses (e.g., hepatitis B and C, coxsackievirus B, and influenza) in a wide variety of cell types (31–36, 68). A close functional analog to γ34.5 is HIV Nef, which can bind Beclin-1 and blocks virus-induced autophagosome maturation, causing accumulation of long-lived proteins, lipidated LC3, and autophagosomes in infected macrophages (30). The lack of sequence similarity between γ34.5 and HIV Nef, as well as between other viral modulators of autophagy, suggests that these genes and their functions have evolved independently. This mode of evolution highlights the common importance of this pathway in controlling virulence and development of innate and adaptive immunity.
During the revision of this report, TBK-1 was shown to be necessary for the maturation of autophagosomes (69). TBK-1 inhibition leads to the accumulation of both LC3-II and p62 in a manner comparable to bafilomycin treatment and to the effects of γ34.5 described herein. This is especially of interest given that γ34.5 possesses a TBK-1 binding domain which partially overlaps the BBD (10). It could be argued, therefore, that mutagenesis of the BBD may have caused a defect in the ability of γ34.5 to modulate TBK-1 in addition to ablation of Beclin binding. While this is a possibility, the virulence of a virus lacking BBD remained attenuated in mice lacking IRF-3 (39). Given that TBK is critical for activation of IRF-3, we would have expected significantly increased virulence in these mice if the BBD-deleted virus was completely incapable of modulating TBK-1. This is an aspect of γ34.5 function that clearly warrants closer scrutiny.
In conclusion, we have shown in this study that γ34.5 is sufficient for control of autophagy in DCs and that the BBD of γ34.5 is necessary for this control. In contrast to its activities in other cells, γ34.5 allows autophagosomes to accumulate but interrupts autophagic degradation of long-lived proteins in DCs, significantly affecting presentation of intracellular antigens. This finding is important for enhancing our understanding of how adaptive immunity develops to HSV and how γ34.5 can facilitate evasion of both innate and adaptive immunity. Currently, we are addressing the role of autophagy control in APCs during HSV-1 infection in vivo and investigating the DC-specific mechanisms that result in this atypical mode of manipulation of autophagy by γ34.5. This work will serve to further elucidate the immune-modulatory activities of HSV-1 with impact on vaccine development and antitumor therapies using HSV as an oncolytic vector.
MATERIALS AND METHODS
Cells, viruses, and mice.
BMDCs were generated and infected as previously described (70). DC2.4 cells are BMDC-derived immortalized cell lines made available by Kenneth Rock (University of Massachusetts, Worcester, MA) and kindly provided by Ed Usherwood (Dartmouth) (50). All infection protocols for DC2.4 cells were as previously described for BMDCs (71). The DC2.4-derived stably transduced cells were made using the Clontech Lenti-X system. Briefly, γ34.5 or Δ34.5ΔBBD gene sequences were cloned into pLVX-IRES-Hyg vectors by digestion of pcγ34.5 and pcγ34.5ΔBBD expression vectors (72) with Sau3AI and XbaI (New England Biolabs) and of the lentiviral vector with BamHI and XbaI, thus placing the chicken beta-actin promoter upstream of γ34.5 in the vector. New pLVX-γ34.5 and pLVX-γ34.5dBBD or control pLVX-IRES-Hyg vectors were then transfected into 293 Lenti-X cells along with helper plasmid according to the manufacturer’s protocol. The lentiviruses produced were then used to transduce DC2.4 cells. Stable clones were isolated using 250 µg/ml hygromycin. Expressing clones were identified by Western blotting and routinely propagated in hygromycin at a concentration of 100 to 300 µg/ml to ensure stability. HSV-1 strain 17 (WT) was the background for all viruses in this study. The γ34.5-null mutant, Δγ34.5, and the Beclin-binding domain mutant, ΔBBD, were made as previously described (40, 72). HSV-1 strain 17 Δ34.5 tOVA was made using a truncated cytoplasmic-retained chicken ovalbumin (OVA)-encoding plasmid generously supplied by Charles Sentman (Dartmouth) (73). The sequence was cloned into pCI using EcoRI and XhoI/SalI digests. The promoter and gene sequences from the resulting plasmid, pCI-tOVA, were transferred into the pUIC17 vector using BamHI/BglII cutting. This vector possesses a sequence from the UL49 and UL50 genes of strain 17 separated by a BglII restriction site (74). The pUIC17-tOVA vector was cotransfected into Vero cells with Δγ34.5 infectious DNA to make strain 17 Δγ34.5 tOVA by homologous recombination as previously described (75). Viruses were screened by PCR and Western blotting for expression of truncated OVA (data not shown). OT-II mice, originally developed by Francis Carbone (57), were generously provided by Ed Usherwood (Dartmouth).
Fluorescence microscopy.
Cells were plated on glass coverslips and infected at the indicated multiplicities of infection (MOIs) for 8 or 16 h. Where applied, 100 µM bafilomycin A1 was added for 6 h. All samples were fixed in 4% paraformaldehyde in phosphate-buffered saline (PBS) for 20 min at room temperature, blocked, and permeabilized in 2% goat serum (Vector Laboratories), 1% bovine serum albumen (BSA) (Sigma), 0.1% cold fish gelatin (Sigma), 0.1% Triton X-100 (Sigma) in PBS, pH 7.2, for 20 min. Primary and secondary antibodies were sequentially added, diluted in 5% goat serum in PBS. Coverslips were mounted using Vectashield mounting medium (Vector Laboratories). Antibodies used were specific to LC3 (1:400) (MBL PD014), p62 (1:1,000) (Novus NBP1-48320), ICP0 (1:1,500) (Virusys, HA027), or ICP8 (1:700) (kindly provided by David Knipe, Harvard Medical School). Microscopy was performed on a Zeiss AX10 microscope fitted with a QImaging cooled mono 14-bit camera. Images were captured at either ×66 magnification for puncta quantification or ×100 magnification for the images shown in this study. Equivalent contrast enhancement was applied to all images using Q-Capture Pro software.
When quantifying populations accumulating p62 or LC3 puncta, antibody-positive puncta were counted, and those with a minimum of 4 (LC3) or 5 (p62) puncta per cell were scored positive (76). Total cell population, for determining ratios, were derived by counting DAPI (4′,6-diamidino-2-phenylindole)-positive nuclei, and infected cell populations were determined by counting nuclei costaining for ICP8 or ICP0. All fluorescence ratios were determined by imaging four or more fields and counting a minimum of 60 cells.
Western blotting.
LC3 conversion and p62 accumulation assays were performed on DCs and BMDCs at an MOI of 8. Cells were infected for 8 or 16 h and treated with bafilomycin as described for fluorescence microscopy. Stable cells were induced to phosphorylate eIF2α using poly(I:C) at a concentration of 20 µg/ml or infected with the Δγ34.5 strain, at an MOI of 8 for 12 h. Cell lysate was prepared by rinsing cells in PBS and resuspending them in sample buffer (62.5 mM Tris [lsqb]pH 6.8[rsqb], 4.65% SDS, 20% glycerol, 10% β-mercaptoethanol, 0.025% bromophenol blue) (Sigma). Membranes were probed with rabbit polyclonal anti LC3 (MBL PD014) or p62 (Novus NBP1-48320) using antibody concentrations of 1:1,000 and anti-α-tubulin antibody (Novus NB100-690) as a loading control at 1:2,000. Anti-phospho-eIF2α (BioSource, AH01182) antibodies were used at 1:800, and anti-γ34.5 antibodies (a generous gift from Ian Mohr, New York University) were used at 1:1,000. Secondary goat-anti-rabbit or mouse horseradish peroxidase antibodies (Bio-Rad) were used in conjunction with ECL Western blotting substrate (Thermo) and detected on an Alpha Innotech Fluor Chem Q multi-imager. The molecular weights (MW) of bands of interest were determined by interpolation from MW 15.3 to 101.4 (Bio-Rad). Images were captured, molecular weights were determined, contrast was optimized, and band density was quantified using AlphaImager software. All blots are representative of a minimum of two experiments.
Starvation/survival assay.
Cells were starved by replacing media with Earl’s balanced salt solution for 2 h. Cells were then analyzed for cell viability by propidium iodide and Alexa Fluor 647-annexin V (BioLegend) staining according to the manufacturer’s protocol. Flow cytometry and data analysis were performed on an Accuri flow cytometer (BD Biosciences).
T cell response assay and flow cytometry.
Stably transduced DC2.4 cells were mock infected or infected with the 17 Δ34.5 or 17 Δ34.5 tOva strains at an MOI of 8.0, incubated for 1 h, and rinsed three times in Hank’s balanced salt solution (HBSS). Whole OT-II splenocytes were then added at a ratio of 4 splenocytes to 1 DC. For positive controls, PMA and ionomycin were added at concentrations of 10 ng/ml and 1 µg/ml, respectively. The coculture was incubated for 44 h, and cells were harvested for analysis by flow cytometry using anti-CD4-PercP and anti-CD44-FITC antibodies (Biolegends). Cells were gated based on the high surface expression of CD4, and gated cells were analyzed for high surface expression of CD44. All flow cytometry and analysis were performed on a BD Accuri C6 using CFlow software. Relative high-CD44 staining was expressed as a ratio relative to mock-infected cells.
ACKNOWLEDGMENTS
We thank members of the Leib Laboratory for helpful discussions throughout the development of this work and Ed Usherwood, Brent Berwin, and their labs for helpful advice.
This work was supported by NIH RO1 EY09083 (to D.A.L.) and by P20RR016437 from the National Center for Research Resources (to Dartmouth).
Footnotes
Citation Gobeil PAM, Leib DA. 2012. Herpes simplex virus γ34.5 interferes with autophagosome maturation and antigen presentation in dendritic cells. mBio 3(5):e00267-12. doi:10.1128/mBio.00267-12.
REFERENCES
- 1. Nicoll MP, Proença JT, Efstathiou S. 2012. The molecular basis of herpes simplex virus latency. FEMS Microbiol. Rev. 36:684–705 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Kaufman RJ, Murtha P. 1987. Translational control mediated by eucaryotic initiation factor-2 is restricted to specific mRNAs in transfected cells. Mol. Cell. Biol. 7:1568–1571 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Samuel CE, Brody MS. 1990. Biosynthesis of reovirus-specified polypeptides. 2-Aminopurine increases the efficiency of translation of reovirus s1 mRNA but not s4 mRNA in transfected cells. Virology 176:106–113 [DOI] [PubMed] [Google Scholar]
- 4. Chou J, Roizman B. 1992. The gamma 1(34.5) gene of herpes simplex virus 1 precludes neuroblastoma cells from triggering total shutoff of protein synthesis characteristic of programed cell death in neuronal cells. Proc. Natl. Acad. Sci. U. S. A. 89:3266–3270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Chou J, Chen JJ, Gross M, Roizman B. 1995. Association of a M(r) 90,000 phosphoprotein with protein kinase PKR in cells exhibiting enhanced phosphorylation of translation initiation factor eIF-2 alpha and premature shutoff of protein synthesis after infection with gamma 134.5- mutants of herpes simplex virus 1. Proc. Natl. Acad. Sci. U. S. A. 92:10516–10520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. He B, et al. 1997. Suppression of the phenotype of gamma(1)34.5- herpes simplex virus 1: failure of activated RNA-dependent protein kinase to shut off protein synthesis is associated with a deletion in the domain of the alpha47 gene. J. Virol. 71:6049–6054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Zhang C, et al. 2008. A conserved domain of herpes simplex virus ICP34.5 regulates protein phosphatase complex in mammalian cells. FEBS Lett. 582:171–176 [DOI] [PubMed] [Google Scholar]
- 8. Li Y, et al. 2011. ICP34.5 protein of herpes simplex virus facilitates the initiation of protein translation by bridging eukaryotic initiation factor 2alpha (eIF2alpha) and protein phosphatase 1. J. Biol. Chem. 286:24785–24792 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Verpooten D, Ma Y, Hou S, Yan Z, He B. 2009. Control of TANK-binding kinase 1-mediated signaling by the gamma(1)34.5 protein of herpes simplex virus 1. J. Biol. Chem. 284:1097–1105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Ma Y, et al. 2012. Inhibition of TANK binding kinase 1 by herpes simplex virus 1 facilitates productive infection. J. Virol. 86:2188–2196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Tallóczy Z, et al. 2002. Regulation of starvation- and virus-induced autophagy by the eIF2alpha kinase signaling pathway. Proc. Natl. Acad. Sci. U. S. A. 99:190–195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Ashford TP, Porter KR. 1962. Cytoplasmic components in hepatic cell lysosomes. J. Cell Biol. 12:198–202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Hruban Z, Spargo B, Swift H, Wissler RW, Kleinfeld RG. 1963. Focal cytoplasmic degradation. Am. J. Pathol. 42:657–683 [PMC free article] [PubMed] [Google Scholar]
- 14. Kovács AL, Reith A, Seglen PO. 1982. Accumulation of autophagosomes after inhibition of hepatocytic protein degradation by vinblastine, leupeptin or a lysosomotropic amine. Exp. Cell Res. 137:191–201 [DOI] [PubMed] [Google Scholar]
- 15. Mortimore GE, Pösö AR. 1988. Amino acid control of intracellular protein degradation. Methods Enzymol. 166:461–476 [DOI] [PubMed] [Google Scholar]
- 16. Arico S, et al. 2001. The tumor suppressor PTEN positively regulates macroautophagy by inhibiting the phosphatidylinositol 3-kinase/protein kinase B pathway. J. Biol. Chem. 276:35243–35246 [DOI] [PubMed] [Google Scholar]
- 17. Liu Y, et al. 2005. Autophagy regulates programmed cell death during the plant innate immune response. Cell 121:567–577 [DOI] [PubMed] [Google Scholar]
- 18. Azad MB, et al. 2008. Hypoxia induces autophagic cell death in apoptosis-competent cells through a mechanism involving BNIP3. Autophagy 4:195–204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. He C, Klionsky DJ. 2009. Regulation mechanisms and signaling pathways of autophagy. Annu. Rev. Genet. 43:67–93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Mizushima N, Klionsky DJ. 2007. Protein turnover via autophagy: implications for metabolism. Annu. Rev. Nutr. 27:19–40 [DOI] [PubMed] [Google Scholar]
- 21. Deretic V, Levine B. 2009. Autophagy, immunity, and microbial adaptations. Cell Host Microbe 5:527–549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Mizushima N, et al. 2001. Dissection of autophagosome formation using Apg5-deficient mouse embryonic stem cells. J. Cell Biol. 152:657–668 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Mizushima N, Ohsumi Y, Yoshimori T. 2002. Autophagosome formation in mammalian cells. Cell Struct. Funct. 27:421–429 [DOI] [PubMed] [Google Scholar]
- 24. Sou YS, et al. 2008. The Atg8 conjugation system is indispensable for proper development of autophagic isolation membranes in mice. Mol. Biol. Cell 19:4762–4775 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Tong J, Yan X, Yu L. 2010. The late stage of autophagy: cellular events and molecular regulation. Protein Cell 1:907–915 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Liang XH, et al. 1999. Induction of autophagy and inhibition of tumorigenesis by beclin 1. Nature 402:672–676 [DOI] [PubMed] [Google Scholar]
- 27. Kihara A, Kabeya Y, Ohsumi Y, Yoshimori T. 2001. Beclin-phosphatidylinositol 3-kinase complex functions at the trans-Golgi network. EMBO Rep. 2:330–335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Schmid D, Pypaert M, Münz C. 2007. Antigen-loading compartments for major histocompatibility complex class II molecules continuously receive input from autophagosomes. Immunity 26:79–92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Lee HK, et al. 2010. In vivo requirement for Atg5 in antigen presentation by dendritic cells. Immunity 32:227–239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Kyei GB, et al. 2009. Autophagy pathway intersects with HIV-1 biosynthesis and regulates viral yields in macrophages. J. Cell Biol. 186:255–268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Gannagé M, et al. 2009. Matrix protein 2 of influenza A virus blocks autophagosome fusion with lysosomes. Cell Host Microbe 6:367–380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Sir D, et al. 2008. Induction of incomplete autophagic response by hepatitis C virus via the unfolded protein response. Hepatology 48:1054–1061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Sir D, et al. 2010. The early autophagic pathway is activated by hepatitis B virus and required for viral DNA replication. Proc. Natl. Acad. Sci. U. S. A. 107:4383–4388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Wong J, et al. 2008. Autophagosome supports coxsackievirus B3 replication in host cells. J. Virol. 82:9143–9153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Comber JD, Robinson TM, Siciliano NA, Snook AE, Eisenlohr LC. 2011. Functional macroautophagy induction by influenza A virus without a contribution to major histocompatibility complex class II-restricted presentation. J. Virol. 85:6453–6463 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Dumit VI, Dengjel J. 2012. Autophagosomal protein dynamics and influenza virus infection. Front. Immunol. 3:43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Tallóczy Z, Virgin HW, IV, Levine B. 2006. PKR-dependent autophagic degradation of herpes simplex virus type 1. Autophagy 2:24–29 [DOI] [PubMed] [Google Scholar]
- 38. Orvedahl A, et al. 2007. HSV-1 ICP34.5 confers neurovirulence by targeting the Beclin 1 autophagy protein. Cell Host Microbe 1:23–35 [DOI] [PubMed] [Google Scholar]
- 39. Leib DA, Alexander DE, Cox D, Yin J, Ferguson TA. 2009. Interaction of ICP34.5 with Beclin 1 modulates herpes simplex virus type 1 pathogenesis through control of CD4+ T-cell responses. J. Virol. 83:12164–12171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. English L, et al. 2009. Autophagy enhances the presentation of endogenous viral antigens on MHC class I molecules during HSV-1 infection. Nat. Immunol. 10:480–487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Rasmussen SB, et al. 2011. Activation of autophagy by α-herpesviruses in myeloid cells is mediated by cytoplasmic viral DNA through a mechanism dependent on stimulator of IFN genes. J. Immunol. 187:5268–5276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Jin H, et al. 2009. The gamma 1 34.5 protein of herpes simplex virus 1 is required to interfere with dendritic cell maturation during productive infection. J. Virol. 83:4984–4994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Jin H, Yan Z, Ma Y, Cao Y, He B. 2011. A herpesvirus virulence factor inhibits dendritic cell maturation through protein phosphatase 1 and Ikappa B kinase. J. Virol. 85:3397–3407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Eidsmo L, et al. 2009. Differential migration of epidermal and dermal dendritic cells during skin infection. J. Immunol. 182:3165–3172 [DOI] [PubMed] [Google Scholar]
- 45. Mizushima N, Yoshimori T. 2007. How to interpret LC3 immunoblotting. Autophagy 3:542–545 [DOI] [PubMed] [Google Scholar]
- 46. Yoshimori T, Yamamoto A, Moriyama Y, Futai M, Tashiro Y. 1991. Bafilomycin A1, a specific inhibitor of vacuolar-type H(+)-ATPase, inhibits acidification and protein degradation in lysosomes of cultured cells. J. Biol. Chem. 266:17707–17712 [PubMed] [Google Scholar]
- 47. Yamamoto A, et al. 1998. Bafilomycin A1 prevents maturation of autophagic vacuoles by inhibiting fusion between autophagosomes and lysosomes in rat hepatoma cell line, H-4-II-E cells. Cell Struct. Funct. 23:33–42 [DOI] [PubMed] [Google Scholar]
- 48. Bjørkøy G, et al. 2005. p62/SQSTM1 forms protein aggregates degraded by autophagy and has a protective effect on huntingtin-induced cell death. J. Cell Biol. 171:603–614 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Bjørkøy G, et al. 2009. Monitoring autophagic degradation of p62/SQSTM1. Methods Enzymol. 452:181–197 [DOI] [PubMed] [Google Scholar]
- 50. Shen Z, Reznikoff G, Dranoff G, Rock KL. 1997. Cloned dendritic cells can present exogenous antigens on both MHC class I and class II molecules. J. Immunol. 158:2723–2730 [PubMed] [Google Scholar]
- 51. Hargadon KM, Forrest OA, Reddy PR. 2012. Suppression of the maturation and activation of the dendritic cell line DC2.4 by melanoma-derived factors. Cell. Immunol. 272:275–282 [DOI] [PubMed] [Google Scholar]
- 52. Tsukada M, Ohsumi Y. 1993. Isolation and characterization of autophagy-defective mutants of Saccharomyces cerevisiae. FEBS Lett. 333:169–174 [DOI] [PubMed] [Google Scholar]
- 53. Onodera J, Ohsumi Y. 2005. Autophagy is required for maintenance of amino acid levels and protein synthesis under nitrogen starvation. J. Biol. Chem. 280:31582–31586 [DOI] [PubMed] [Google Scholar]
- 54. Boya P, et al. 2005. Inhibition of macroautophagy triggers apoptosis. Mol. Cell. Biol. 25:1025–1040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Tabe L, et al. 1984. Segregation of mutant ovalbumins and ovalbumin-globin fusion proteins in Xenopus oocytes. Identification of an ovalbumin signal sequence. J. Mol. Biol. 180:645–666 [DOI] [PubMed] [Google Scholar]
- 56. Boyle JS, Koniaras C, Lew AM. 1997. Influence of cellular location of expressed antigen on the efficacy of DNA vaccination: cytotoxic T lymphocyte and antibody responses are suboptimal when antigen is cytoplasmic after intramuscular DNA immunization. Int. Immunol. 9:1897–1906 [DOI] [PubMed] [Google Scholar]
- 57. Barnden MJ, Allison J, Heath WR, Carbone FR. 1998. Defective TCR expression in transgenic mice constructed using cDNA-based alpha- and beta-chain genes under the control of heterologous regulatory elements. Immunol. Cell Biol. 76:34–40 [DOI] [PubMed] [Google Scholar]
- 58. Robertson JM, Jensen PE, Evavold BD. 2000. DO11.10 and OT-II T cells recognize a C-terminal ovalbumin 323-339 epitope. J. Immunol. 164:4706–4712 [DOI] [PubMed] [Google Scholar]
- 59. Santana S, Bullido MJ, Recuero M, Valdivieso F, Aldudo J. 2012. Herpes simplex virus type I induces an incomplete autophagic response in human neuroblastoma cells. J. Alzheimers Dis. 30:815–831 [DOI] [PubMed] [Google Scholar]
- 60. Shi CS, et al. 2012. Activation of autophagy by inflammatory signals limits IL-1β production by targeting ubiquitinated inflammasomes for destruction. Nat. Immunol. 13:255–263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Ushio H, et al. 2011. Crucial role for autophagy in degranulation of mast cells. J. Allergy Clin. Immunol. 127:1267–1276.e6 PubMed; [DOI] [PubMed] [Google Scholar]
- 62. Florey O, Kim SE, Sandoval CP, Haynes CM, Overholtzer M. 2011. Autophagy machinery mediates macroendocytic processing and entotic cell death by targeting single membranes. Nat. Cell Biol. 13:1335–1343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Martinez J, et al. 2011. Microtubule-associated protein 1 light chain 3 alpha (LC3)-associated phagocytosis is required for the efficient clearance of dead cells. Proc. Natl. Acad. Sci. U. S. A. 108:17396–17401 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 64. Trgovcich J, Johnson D, Roizman B. 2002. Cell surface major histocompatibility complex class II proteins are regulated by the products of the gamma(1)34.5 and U(L)41 genes of herpes simplex virus 1. J. Virol. 76:6974–6986 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Salio M, Cella M, Suter M, Lanzavecchia A. 1999. Inhibition of dendritic cell maturation by herpes simplex virus. Eur. J. Immunol. 29:3245–3253 [DOI] [PubMed] [Google Scholar]
- 66. Reis e Sousa C. 2004. Toll-like receptors and dendritic cells: for whom the bug tolls. Semin. Immunol. 16:27–34 [DOI] [PubMed] [Google Scholar]
- 67. Criollo A, et al. 2010. The IKK complex contributes to the induction of autophagy. EMBO J. 29:619–631 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Li J, et al. 2011. Subversion of cellular autophagy machinery by hepatitis B virus for viral envelopment. J. Virol. 85:6319–6333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Pilli M, et al. 2012. TBK-1 Promotes autophagy-mediated antimicrobial defense by controlling autophagosome maturation. Immunity 37:223–234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Menachery VD, Pasieka TJ, Leib DA. 2010. Interferon regulatory factor 3-dependent pathways are critical for control of herpes simplex virus type 1 central nervous system infection. J. Virol. 84:9685–9694 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Menachery VD, Leib DA. 2009. Control of herpes simplex virus replication is mediated through an interferon regulatory factor 3-dependent pathway. J. Virol. 83:12399–12406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Alexander DE, Ward SL, Mizushima N, Levine B, Leib DA. 2007. Analysis of the role of autophagy in replication of herpes simplex virus in cell culture. J. Virol. 81:12128–12134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Barber MA, Zhang T, Gagne BA, Sentman CL. 2007. NK cells negatively regulate antigen presentation and tumor-specific CTLs in a syngeneic lymphoma model. J. Immunol. 178:6140–6147 [DOI] [PubMed] [Google Scholar]
- 74. Davido DJ, Leib DA, Schaffer PA. 2002. The cyclin-dependent kinase inhibitor roscovitine inhibits the transactivating activity and alters the posttranslational modification of herpes simplex virus type 1 ICP0. J. Virol. 76:1077–1088 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Cai WZ, Schaffer PA. 1989. Herpes simplex virus type 1 ICP0 plays a critical role in the de novo synthesis of infectious virus following transfection of viral DNA. J. Virol. 63:4579–4589 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Klionsky DJ, et al. 2008. Guidelines for the use and interpretation of assays for monitoring autophagy in higher eukaryotes. Autophagy 4:151–175 [DOI] [PMC free article] [PubMed] [Google Scholar]