

Abstract
The diagnosis of vascular dementia (VaD) describes a group of various vessel disorders with different types of vascular lesions that finally contribute to the development of dementia. Most common forms of VaD in the elderly brain are subcortical vascular encephalopathy, strategic infarct dementia, and the multi infarct encephalopathy. Hereditary forms of VaD are rare. Most common is the cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy (CADASIL). Sporadic forms of VaD are caused by degenerative vessel disorders such as atherosclerosis, small vessel disease (SVD) including small vessel arteriosclerosis, arteriolosclerosis, and lipohyalinosis, and cerebral amyloid angiopathy (CAA). Less frequently inflammatory vessel disorders and tumor-associated vessel lesions (e.g. angiocentric T-cell or angiotropic large cell lymphoma) can cause symptoms of dementia. Here, we review and discuss the impact of vessel disorders to distinct vascular brain tissue lesions and to the development of dementia in elderly individuals. The impact of coexisting neurodegenerative pathology in the elderly brain to VaD as well as the correlation between SVD and CAA expansion in the brain parenchyma with that of Alzheimer’s disease (AD)-related pathology is highlighted. We conclude that “pure” VaD is rare and most frequently caused by infarctions. However, there is a significant contribution of vascular lesions and vessel pathology to the development of dementia that may go beyond tissue damage due to vascular lesions. Insufficient blood blow and alterations of the perivascular drainage mechanisms of the brain may also lead to a reduced protein clearance from extracellular space and subsequent increase of proteins in the brain parenchyma, such as the amyloid β-protein, and foster, thereby, the development of AD-related neurodegeneration. As such, it seems to be important for clinical practice to consider treatment of potentially coexisting AD pathology in cognitively impaired patients with vascular lesions.
Keywords: Atherosclerosis, small vessel disease, cerebral amyloid angiopathy, dementia, neurodegeneration, perivascular drainage
Introduction
Vascular cognitive impairment (VCI) is a syndrome with evidence of clinical stroke or subclinical vascular brain injury and cognitive impairment affecting at least one cognitive domain. The most severe form of VCI is vascular dementia (VaD) (Gorelick et al., 2011; Roman et al., 1993). Most authors distinguish familial and sporadic forms of VaD (Ferrer 2010; Gorelick et al., 2011; Ince 2005; Kalimo and Kalaria 2005). Familial VaD is usually caused by gene mutations. The most frequent subtype of familial VaD is the “cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy” (CADASIL) (Ferrer 2010; Gorelick et al., 2011; Kalimo and Kalaria 2005). However, CADASIL that is caused by mutations in the Notch 3 gene (Joutel et al., 1996) and other types of familial VaD are not within the scope of this article. Here, we want to review the relationship of the three major types of sporadic VaD, i.e. 1. multi infarct dementia, 2. strategic infarct dementia, and 3. subcortical vascular encephalopathy (synonymous with Binswanger’s disease) (Ferrer 2010; Ince 2005), with their underlying vascular pathologies. In addition, we will discuss VaD overlapping with other age-related pathologies leading to dementia, e.g. Alzheimer’s disease (AD)-related changes.
Vessel disorders and their relationship to vascular lesions
Vessel disorders
The vessel disorders that are most frequently associated with VaD are atherosclerosis of cerebral arteries (AS), cerebral small vessel disease (SVD), and cerebral amyloid angiopathy (CAA) (Ferrer 2010; Gorelick et al., 2011; Hachinski et al., 2006; Ince 2005; Kalaria 2003; Kalaria and Erkinjuntti 2006; Roman et al., 1993). These vessel disorders frequently occur in the brains of elderly individuals and become more prevalent and severe with advancing age (Table 1) (Jellinger and Attems 2010).
Tab. 1A. Age group-related prevalence (%) of AS, SVD, CAA, brain infarction, cerebral hemorrhage, and vascular dementia.
Prevalence (in %) of AS, SVD, CAA, brain infarction, hemorrhage, and VaD in the brain in different age groups in a non-selected autopsy sample. Partial correlation analysis controlled for gender was performed for the prevalence of brain infarction and cerebral hemorrhage but did not exhibit a significant increase with age.
| −60 years | 61–70 years | 71–80 years | 81–90 years | 91–104 years | Correlation: p | |
|---|---|---|---|---|---|---|
| AS | 52.94 (n=9/17) | 79.17 (n=38/48) | 97.06 (n=66/68) | 97.30 (n=36/37) | 100.00 (n=3/3) | n.a. |
| SVD | 77.27 (n=17/22) | 80.41 (n=78/97) | 82.44 (n=108/131) | 87.50 (n=63/72) | 90.91 (n=10/11) | n.a. |
| CAA | 0.00 (n=0/26) | 42.34 (n=47/111) | 47.74 (n=74/155) | 66.67 (n=66/99) | 80.00 (n=20/25) | n.a. |
| Brain infarction | 20.51 (n=8/39) | 23.48 (n=27/115) | 38.78 (n=57/147) | 49.37 (n=39/79) | 18.18 (n=2/11) | 0.260 |
| Cerebral hemorrhage | 12.82 (n=5/39) | 8.70 (n=10/115) | 12.24 (n=18/147) | 10.13 (n=8/79) | 9.09 (n=1/11) | 0.906 |
| VaD | 0.00 (n=0/36) | 3.30 (n=3/91) | 2.17 (n=3/138) | 6.12 (n=6/98) | 0.00 (n=0/24) | n.a. |
Determinations: AS severity (Larionov et al., 2006): 0 = no AS, 1 = no more than three AS-plaques in an artery and no circular AS-plaques, 2 = more than three plaques in at least one artery but no concentric plaques, 3 = more than three plaques in at least one vessel and concentric plaques; SVD severity (Kolsch et al., 2007): 0 = no SVD, 1 = single (1–3) vessels exhibit SVD in one area of the brain, 2 = more than 3 vessels in at least one brain area exhibit SVD, 3 = in more than two brain regions more than 3 vessels are affected by SVD; CAA severity (Vonsattel et al., 1991): 0 = no CAA, 1 = Aβ deposits in the vessel wall without loss of smooth muscle cells in the vessel wall, 2 = Aβ deposits in the vessel wall accompanied by degeneration of the smooth muscle cell layer, 3 = extensive Aβ deposition and focal vessel wall fragmentations, microaneurysms, signs of hemorrhage, and fibrinoid necrosis.
Abbreviations: Aβ = amyloid β-protein, AS = Atherosclerosis, CAA = cerebral amyloid angiopathy, n = number of cases studied, p = p-value assessed by partial correlation analysis controlled for gender, r = correlation coefficient assessed by partial correlation analysis controlled for gender, SVD = small vessel disease, VaD = vascular dementia.
Atherosclerosis (AS)
AS is a degenerative disorder of large and medium sized arteries that leads to intima proliferation and accumulation of blood-derived lipids and proteins, especially cholesterol within the vessel wall (Kolsch et al., 2007; Larionov et al., 2007; Stary et al., 1995; Stary et al., 1994). These processes result in the generation and finally in the calcification of atherosclerotic plaques and in further degeneration and fibrosis of the vessel wall. Plaque rupture frequently induces local thrombosis due to endothelial damage (Stary 2000; Stary et al., 1995). AS-related thrombosis can cause large brain infarcts, whereas embolism of atherogenic thrombi can lead to a broad variety of infarcts (Grinberg and Thal 2010; Liberato et al., 2005; Marti-Vilalta and Arboix 1999).
Small vessel disease (SVD)
SVD comprises small vessel arteriosclerosis/atherosclerosis, lipohyalinosis, and arteriolosclerosis (Grinberg and Thal 2010). The vessel wall changes of small artery arteriosclerosis/atherosclerosis (diameter between 200 to 800 μm) are similar to that seen in AS of larger blood vessels except for calcifications not seen in small arteries (Fisher 1991; Hachinski et al., 2006; Lammie 2005). Lipohyalinosis occurs in arteries with a diameter between 40 and 300 μm. Asymmetric areas of fibrosis, hyalinosis associated with foam cells, and accumulation of blood-derived lipids and proteins characterize this type of SVD (Grinberg and Thal 2010; Lammie 2002; Nag 1996; Nag and Robertson 2005; Utter et al., 2008). Arteriolosclerosis describes the concentric hyaline thickening of the vessel wall with stenosis of the arterioles with a caliber of 40 and 150 μm (Lammie 2005). The hyaline lesions often exhibit blood-derived proteins (Utter et al., 2008). SVD can result in lacunar infarcts, microinfarcts, which are smaller than lacunar infarcts, hemorrhages, and microbleeds (Challa et al., 1990; Grinberg and Thal 2010; Jeong et al., 2002; Vinters et al., 2000). SVD affects first arteries of the basal ganglia and then expands into the peripheral white matter and leptomeningeal arteries, as well as into thalamic and cerebellar white matter vessels. Finally, also brain stem arteries exhibit SVD. These three stages of the expansion of SVD have been previously described in detail (Thal et al., 2003). Cortical vessels are usually not involved in SVD (Thal et al., 2003).
Cerebral amyloid angiopathy (CAA)
Sporadic CAA is characterized by the deposition of the amyloid β-protein (Aβ) in the wall of leptomeningeal and cerebral blood vessels (Glenner and Wong 1984; Scholz 1938). Most frequently these deposits are located near the basement membrane or in the smooth muscle cell layer (Grinberg et al. 2012; Vinters 1992; Vonsattel et al., 1991; Wisniewski et al., 1992; Yamaguchi et al., 1992). CAA can lead to vessel wall rupture and hemorrhage (Vonsattel et al., 1991), microbleeds (Greenberg et al., 2009), capillary occlusion and blood flow disturbances (Thal et al., 2009) as well as to microinfarcts (Cadavid et al., 2000; Okamoto et al., 2012). Familial forms of CAA are not in the focus of this review. In these cases severe CAA can be caused by Aβ as well as by the deposition of other proteins, e.g. prion protein and cystatin C, etc. (for detailed review see (Revesz et al., 2009)). CAA most frequently involves leptomeningeal and neocortical arteries, veins, and/or capillaries. In the second stage of CAA, vessels in allocortical regions (hippocampus, entorhinal and cingulate cortex, amygdala), the hypothalamus and in the cerebellum exhibit Aβ deposits as well whereas blood vessels of the basal ganglia, thalamus, lower brain stem, and white matter are affected only in the third and final stage (Thal et al., 2003).
Vascular lesions in the brain
As already mentioned these vessel disorders can cause vascular lesions. The most frequent vascular lesions in the brains of elderly individuals are: brain infarcts, white matter lesions, and hemorrhages.
Brain infarcts
Brain infarcts represent areas of brain parenchyma necrosis caused by insufficient blood flow. The prevalence of morphologically detectable brain infarcts regardless of size, quantity, and clinical impact increases with age up to 90 years of age (Table 1A). In our autopsy cases the prevalence of infarcts decreased in the age group of 91–104 years of age. Apparently, this finding is in line with the previously reported reduction of multi infarct dementia in this age group (Jellinger and Attems 2010) although most studies analyzed people older than 80 years of age as a single group or took in consideration only clinically relevant strokes (Ovbiagele 2010; Wieberdink et al., 2012). Brain infarcts are subclassified by its size into large infarcts (infarcts bigger than 1,5 cm3 or 1 cm in diameter) (Grinberg and Thal 2010; Hachinski et al., 2006), lacunar infarcts (infarcts of 0,5–1,5 cm3 in volume or 0,5–1,0 cm in diameter) (Petito 2005) and microinfarcts (less than 0,5 cm in diameter) (Ince 2005) (Fig. 1A–E). The distinction between large and small infarcts by 1cm in diameter is recommended in the National Institute of Neurological Disorders and Stroke-Canadian Stroke Network vascular cognitive impairment harmonization standards (Hachinski et al., 2006) for imaging. The other measures given here were taken from opinion and textbook articles as indicated, but are to our knowledge not published as consensus guidelines.
Fig. 1.
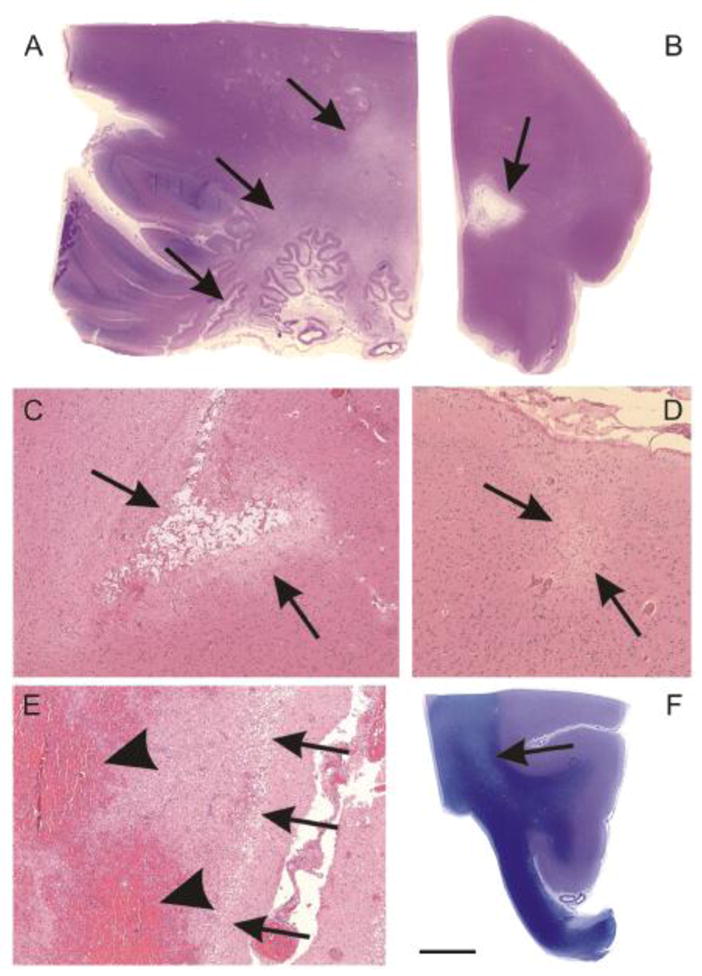
Infarcts and white matter lesions. A: Large ischemic infarct in the cerebellum (stage of necrosis; arrows). B, C: Lacunar infarcts in the pons (B) and in the basal ganglia (C) (stage of pseudocystic gliosis; arrows). D: Microinfarct in the parietal cortex (stage of gliosis) The arrows indicate the demarcation line of the infarct. E: Large hemorrhagic infarct in the cortex (stage of necrosis). The arrows indicate the infarct demarcation line at the border between the cortical layers I and II, the arrowheads point to the areas of blood extravasation in the hemorrhagic (red) infarct. F: White matter lesion in the frontocentral white matter near the cingulate gyrus (arrow). Note the U-fibers directly underneath the cortical layer VI are intact.
Calibration bar in F corresponds to A = 6900 μm, B, F = 5000μm, C, E = 450μm, D = 360 μm. A–E: Hematoxylin & Eosin staining; F: PAS-Luxol fast blue staining.
Large infarcts can be caused by thrombotic or embolic vessel occlusion. AS frequently induces thrombosis after rupture of an atherosclerotic plaque. Such a thrombus either causes local occlusion or embolism into smaller downstream blood vessels. Cardiogenic embolism is an alternative explanation for the embolic nature of a large infarct (Liberato et al., 2005; Marti-Vilalta and Arboix 1999). Lacunar infarcts are either caused by SVD-related vessel occlusion or by embolic events. Microinfarcts are often related to SVD (white matter microinfarcts) and CAA (cortical microinfarcts) but can also be caused by thromboembolism (Cadavid et al., 2000; Gerraty et al., 2002; Jellinger 2008; Kimberly et al., 2009; Okamoto et al., 2009; Okamoto et al., 2012). Hypoperfusion is another possible cause for “watershed” microinfarcts occurring in the border zone between two vessel territories (Suter et al., 2002). The number of microinfarcts was associated with cognitive impairment in the Honolulu Asia study (Launer et al., 2011). Thrombosis of atherosclerotic lesions has not been reported to cause lacunar and microinfarcts.
Brain infarcts can be further subclassified as ischemic (synonymous with anemic or white infarcts (Fig. 1A–D)) and hemorrhagic (synonymous with red) infarcts (Fig. 1E). Ischemic infarcts are caused by vessel occlusion in the absence of collateral or remaining blood flow or reperfusion. In contrast, hemorrhagic infarcts occur after reperfusion of an ischemic infarct area in the stage of necrosis or when remaining or collateral blood flow is not sufficient to keep the infarcted area viable but causes blood extravasation into the damaged area. Infarct mechanisms leading to hemorrhagic infarcts are: 1. embolism due to AS or cardiogenic embolism, 2. reperfusion of an ischemic infarct (e.g. lysis of a thrombus/embolus), 3. fragile vessel walls of already SVD- or CAA-affected vessels, 4. collateral perfusion, and 5. venous occlusion and subsequent congestion of blood into the respective drainage area (Grinberg and Thal 2010). Ischemic damage of the vessel walls in the infarct area and impaired coagulation mechanisms (e.g. lysis therapy) may facilitate blood leakage under the above mentioned conditions leading the hemorrhagic infarcts. In the elderly brain the presence of brain infarction correlates best with SVD (Table 3) suggesting that SVD-related infarcts are most frequent in this age group.
Tab. 1B. Age group-related severity of AS, SVD, and CAA (mean values).
Mean severity values of AS (determined according to (Larionov et al., 2006)), SVD (determined according to (Kolsch et al., 2007)), and CAA (determined according to (Vonsattel et al., 1991)) in different age groups in an autopsy sample that included non-demented, AD, and VaD cases. Cases with other neurological disorders where excluded from this analysis. Partial correlation analysis was controlled for gender showed a significant increase in AS severity and CAA severity with age whereas SVD was already seen in the age groups −60 and 61–70 with out a significant increase with further aging.
| −60 years | 61–70 years | 71–80 years | 81–90 years | 91–104 years | Correlation: p | |
|---|---|---|---|---|---|---|
| AS | 0.71 (n=9/17) | 1.50 (n=38/48) | 1.91 (n=66/68) | 2.11 (n=36/37) | 2.67 (n=3/3) | 0.025 (r = 0.228) |
| SVD | 1.18 (n=17/22) | 1.30 (n=78/97) | 1.45 (n=108/131) | 1.49 (n=63/72) | 1.64 (n=10/11) | 0.629 |
| CAA | 0.00 (n=0/26) | 0.57 (n=47/111) | 0.66 (n=74/155) | 1.06 (n=66/99) | 1.12 (n=20/25) | 0.043 (r = 0.207) |
Determinations: AS severity (Larionov et al., 2006): 0 = no AS, 1 = no more than three AS-plaques in an artery and no circular AS-plaques, 2 = more than three plaques in at least one artery but no concentric plaques, 3 = more than three plaques in at least one vessel and concentric plaques; SVD severity (Kolsch et al., 2007): 0 = no SVD, 1 = single (1–3) vessels exhibit SVD in one area of the brain, 2 = more than 3 vessels in at least one brain area exhibit SVD, 3 = in more than two brain regions more than 3 vessels are affected by SVD; CAA severity (Vonsattel et al., 1991): 0 = no CAA, 1 = Aβ deposits in the vessel wall without loss of smooth muscle cells in the vessel wall, 2 = Aβ deposits in the vessel wall accompanied by degeneration of the smooth muscle cell layer, 3 = extensive Aβ deposition and focal vessel wall fragmentations, microaneurysms, signs of hemorrhage, and fibrinoid necrosis.
Abbreviations: Aβ = amyloid β-protein, AS = Atherosclerosis, CAA = cerebral amyloid angiopathy, n = number of cases studied, p = p-value assessed by partial correlation analysis controlled for gender, r = correlation coefficient assessed by partial correlation analysis controlled for gender, SVD = small vessel disease, VaD = vascular dementia.
White matter lesions
White matter lesions (synonymous with leukoaraiosis or vascular leukoencephalopathy) represent non-necrotic changes in the white matter that are characterized by demyelination, axon loss, astrogliosis, and microglia activation in the white matter. These changes occur in the deep white matter and spare U-fibers (Fig. 1F). White matter lesions are associated with SVD (Fazekas et al., 1993; Hachinski et al., 2006; Ince 2005; Schmidt et al., 2011; Sze et al., 1986).
Cerebral hemorrhage
In contrast to hemorrhagic infarction, cerebral hemorrhage is characterized by the influx of blood into the intact brain parenchyma because of vessel wall rupture. In our autopsy sample the prevalence of morphologically detectable hemorrhage regardless of its size and clinical significance (except for microbleeds) varied between 8.7 and 12.82% of our autopsy cases without significant increase by age (Table 1A). Large hemorrhages displace brain tissue and can be fatal when vital brain areas are destroyed, e.g., brain stem hemorrhage (Fig. 2A, B), or when hemorrhage is accompanied by brain edema, that subsequently leads to increased intracranial pressure and transtentorial herniation (Ferrer et al., 2008). The most frequent cause of these hemorrhages is SVD associated with arterial hypertension (Takebayashi and Kaneko 1983; Yamori et al., 1976). CAA-related hemorrhages frequently show a lobar pattern of occurrence and represent the second most frequent cause of hemorrhage in the brain of elderly individuals (Greenberg 1997; Mandybur 1986; Thal et al., 2008b). Aneurysms and vascular malformations rarely cause hemorrhage in the elderly brain. Microbleeds are blood extravasations into the perivascular or Virchow-Robin space without further tissue damage and usually measure less than 10 mm in diameter (Henry-Feugeas 2007) (Fig. 2C). Siderin-laden macrophages within the perivascular space indicate previous microbleeds (Winkler et al., 2001). They frequently occur in the presence of SVD and CAA (Greenberg et al., 2009; Jeong et al., 2002) while AS is not involved in the pathogenesis of microbleeds. Their exact pathogenesis and cognitive effects remain to be clarified, as these may be a surrogate for microvascular disease (De Reuck et al., 2011). Radiologically, microbleeds can be easily detected by magnetic resonance imaging as areas of signal loss (Greenberg et al., 2009). As such, radiologically detected microbleeds in the cortex are indicative for CAA; those seen in the white matter point to SVD (Greenberg et al., 2009; Hommet et al., 2011; van Es et al., 2011). However, De Reuck et al. (De Reuck et al., 2011) reported recently that there is significant overestimation of striatal microbleeds even using 7.0T MRI compared to its post-mortem histopathological confirmation.
Fig. 2.
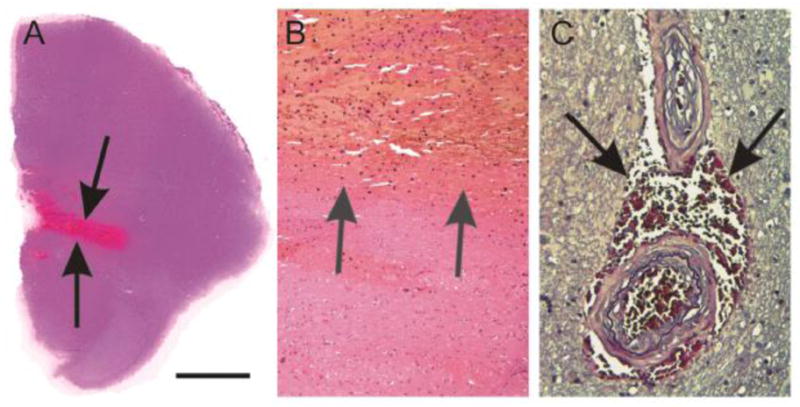
Hemorrhage and microbleeds. A, B: Cerebral hemorrhage in the pons (arrows in A). At higher magnification level (B) it becomes evident that blood clots (arrows) displace the vital brain parenchyma. C Microbleed in the basal ganglia. The microbleed, thereby, represents a hemorrhage restricted to blood extravasation into the perivascular space (arrows) without tissue damage and displacement. Note the two arteries associated with the microbleed exhibit pattern of SVD with a concentric intima proliferation and partial fibrosis of the vessel wall.
Calibration bar in A corresponds to A = 4500μm, B = 226μm, C = 85 μm. A–B: Hematoxylin & Eosin staining; C: Elastica van Gieson staining.
Types of vascular dementia (VaD) and their relation to vessel disorders and vascular lesions
VaD is the net result of vascular lesions that lead to an impairment of brain function (Ferrer 2010; Ince 2005; Roman et al., 1993). Hypoperfusion is thereby, considered to contribute significantly to cognitive decline (Kitamura et al., 2012; Suter et al., 2002). Pure VaD (extensive vascular lesions without widespread AD pathology, i.e., no AD or low degree of AD pathology with Braak NFT stage III or less, CERAD score for neuritic plaque pathology 1 or less, and Aβ phase 2 or less according to the National Institute of Aging – Alzheimer Association (NIA-AA) guidelines (Braak and Braak 1991; Hyman et al., 2012; Mirra et al., 1991; Thal et al., 2002)) was found in only ~5% in our sample of demented and non-demented autopsy cases from university and municipal hospitals in Germany with an age spectrum shown in Table 1. All other cases with dementia and vascular changes also exhibited significant levels of AD-related pathology (i.e., low – severe AD pathology with Braak NFT stage III or higher, CERAD score 1 or higher, and Aβ phase 2 or higher according to the NIA-AA guidelines (Hyman et al., 2012)) and were classified as AD (Table 3A). Different types of vascular lesions, thereby, contribute to the development of VaD: brain infarcts, hemorrhage, and leukoencephalopathy (Ferrer 2010; Ince 2005; Roman et al., 1993). The patterns of the vascular brain lesions leading to dementia distinguish three major forms of VaD (Ferrer 2010; Ince 2005) (Fig. 3): 1. multi infarct dementia, 2. strategic infarct dementia, and 3. subcortical vascular encephalopathy.
Tab. 3A. Prevalence (%) of symptomatic AD, VaD, and cognitively normal individuals in an autopsy sample.
Prevalence (in %) of VaD, AD, and cognitively normal individuals in an autopsy sample that included non-demented, AD, and VaD cases. Cases with other neurological disorders where excluded from this analysis. AD was diagnosed in the presence of high-intermediate degrees of AD-related pathology in the absence of severe vascular and non-AD neurodegenerative changes (Hyman et al., 2012). VaD was considered only when severe and widespread or strategic vascular lesions were observed in demented individuals while no or only a low degree of AD-related lesions and no other neurodegenerative disorders were found in the brain (Gorelick et al., 2011; Roman et al., 1993). AD and VaD cases exhibited clinical signs of dementia fulfilling the DSM IV criteria for dementia (American Psychiatric Association 1994).
| VaD | AD | Cognitively Normals | |
|---|---|---|---|
| 5.08 (n=12/236) | 29.24 (n=69/236) | 65.68 (n=155/236) |
Abbreviations: AD = Alzheimer’s disease, AS = Atherosclerosis, CAA = cerebral amyloid angiopathy, CERAD = consortium to establish a registry for Alzheimer’s disease, CI = 95% confidence interval determined by logistic regression analysis, MTL = medial temporal lobe, n = number of cases studied, NIA-AA = National Institute on Aging – Alzheimer Association, NFT = neurofibrillary tangle, OR = Odds-ratio determined by logistic regression analysis, p = p-value determined by logistic regression analysis and corrected for multiple testing, SVD = small vessel disease, VaD = vascular dementia.
Fig. 3.

Schematic representation of multi infarct dementia, strategic infarct dementia, and subcortical vascular encephalopathy. The gray areas mark the regions where infarcts and white matter lesions are located. In multi infarct dementia multiple microinfarcts, lacunar infarcts, and small large infarcts are distributed all over the gray matter. Strategic infarct dementia is characterized by infarcts in strategic regions that alone explain dementia, i.e., in the hippocampal formation and in the paramedian nuclei of the thalamus. Subcortical vascular encephalopathy is characterized by confluent white matter lesions in the central and peripheral white matter. Small infarcts in subcortical brain regions may also co-occur with this type of VaD.
Abbreviations: Amy = amygdala, Bgl. = basal ganglia, CA1 = Ammon’s horn sector CA1, Cing. = cingulate gyrus, ER = entorhinal cortex, F = frontal neocortex, Hypoth = hypothalamus, NBM = basal nucleus of Meynert, T = temporal neocortex, Thal = thalamus.
Hippocampal sclerosis, i.e. neuron loss and astrogliosis in the CA1 sector of the hippocampus, has also been considered to be related to vascular changes (Ferrer 2010; Hachinski et al., 2006). However, this type of lesion is associated with transactive response desoxyribonucleic acid (TAR DNA)-binding protein TDP43-positive pathology (Amador-Ortiz et al., 2007). As such, it is not fully clear whether hippocampal sclerosis in the aged brain is indeed of vascular origin or a TDP43-proteinopathy or may result from different origin (Hyman et al., 2012).
Multi infarct dementia
Multi infarct dementia is characterized by multiple lacunar and microinfarcts as well as small or large infarcts in the cortex and in subcortical areas (Fig. 3). The total amount of the damaged brain tissue passes the threshold for cognitive impairment by reducing the functional brain capacity significantly. The infarcts can be caused by atherosclerosis in the circle of Willis, embolic events, but also by CAA and SVD. CAA- or SVD-related hemorrhages may contribute to the overall brain damage as well (Ferrer 2010; Hachinski et al., 1974; Ince 2005; Jellinger 2008; Kalaria and Erkinjuntti 2006; Launer et al., 2011; Smith et al., 2012). In so doing, multi infarct dementia cannot be linked to a specific vessel disorder. Moreover, its relation to age-related vessel disorders varies and a combination of vascular lesions due to different vessel disorders is frequently seen. In our sample brain infarction was observed in 66 % of our cases with “pure” VaD (Table 3B). In comparison to CAA and SVD, AS showed a trend to more severe lesions in the circle of Willis in VaD cases than in cognitively normal controls (Table 3C) suggesting that AS-related thrombosis and embolic events are important for this type of VaD. This trend is supported by the finding that the likelihood of dementia is increased in the presence of high-grade internal carotid artery atherosclerosis (Suemoto et al., 2011).
Tab. 3B. Prevalence (%) of AS, SVD, CAA, brain infarction, and cerebral hemorrhage in symptomatic AD, VaD, and cognitively normal individuals.
Prevalence (in %) of AS, SVD, CAA, brain infarction, and hemorrhage in the brain in VaD, AD, and cognitively normal individuals.
| VaD | AD | Cognitively Normals | OR and CI | p | |
|---|---|---|---|---|---|
| AS | 100.00 (n=6/6) | 100.00 (n=14/14) | 84.52 (n=71/84) | n.a. | n.a. |
| SVD | 75.00 (n=6/8) | 82.98 (n=39/47) | 73.68 (n=98/133) | n.a. | n.a. |
| CAA | 33.33 (n=4/12) | 95.52 (n=64/67) | 30.67 (n=46/150) | n.a. | n.a. |
| Brain infarction | 66.67 (n=6/9) | 22.45 (n=11/49) | 22.97 (n=34/148) | VaD 6.7 (1.6–28.5) | 0.01 |
| Cerebral hemorrhage | 22.22 (n=2/9) | 8.16 (n=4/49) | 10.81 (n=16/148) | VaD n.s. | 0,229 |
Abbreviations: AD = Alzheimer’s disease, AS = Atherosclerosis, CAA = cerebral amyloid angiopathy, CERAD = consortium to establish a registry for Alzheimer’s disease, CI = 95% confidence interval determined by logistic regression analysis, MTL = medial temporal lobe, n = number of cases studied, NIA-AA = National Institute on Aging – Alzheimer Association, NFT = neurofibrillary tangle, OR = Odds-ratio determined by logistic regression analysis, p = p-value determined by logistic regression analysis and corrected for multiple testing, SVD = small vessel disease, VaD = vascular dementia.
Tab. 3C. Severity of AS, SVD, and CAA (mean values) in the brains of symptomatic AD, VaD, and cognitively normal individuals.
Mean severity values of AS (determined according to (Larionov et al., 2006)), SVD (determined according to (Kolsch et al., 2007)), and CAA (determined according to (Vonsattel et al., 1991)) in VaD, AD, and cognitively normal individuals.
| VaD | AD | Cognitively Normals | OR and CI | p | |
|---|---|---|---|---|---|
| AS | 2.00 (n=6/104) | 1.86 (n=14/104) | 1.57 (n=84/104) | AD and VaD n.s. | 0,305 |
| SVD | 1.13 (n=8/188) | 1.43 (n=47/188) | 1.10 (n=133/188) | AD n.s. | 0,113 |
| CAA | 0.50 (n=12/229) | 1.51 (n=67/229) | 0.41 (n=150/229) | AD 4.9 (2.9–8.1) | < 0.002 |
Abbreviations: AD = Alzheimer’s disease, AS = Atherosclerosis, CAA = cerebral amyloid angiopathy, CERAD = consortium to establish a registry for Alzheimer’s disease, CI = 95% confidence interval determined by logistic regression analysis, MTL = medial temporal lobe, n = number of cases studied, NIA-AA = National Institute on Aging – Alzheimer Association, NFT = neurofibrillary tangle, OR = Odds-ratio determined by logistic regression analysis, p = p-value determined by logistic regression analysis and corrected for multiple testing, SVD = small vessel disease, VaD = vascular dementia.
Strategic infarct dementia
Strategic infarct dementia, in contrast, is caused by single infarcts in strategic regions, i.e. in regions that cause significant cognitive deficits when destroyed by an infarct. Large infarcts, lacunar infarcts, but also microinfarcts in the hippocampus or the paramedian nuclei of the thalamus can contribute to strategic infarct dementia (Ferrer 2010; Jellinger 2008; Kalaria and Erkinjuntti 2006; Szirmai et al., 2002; Tomlinson et al., 1970) (Fig. 3). These infarcts can be caused by SVD and embolic events. CAA (except for familial CAA cases) is usually not associated with thalamic or hippocampal infarcts (Thal et al., 2008b) although capillary CAA and capillary CAA-associated vessel occlusion has been reported in the CA1-subiculum region and the thalamus (Thal et al., 2009; Thal et al., 2008b).
Subcortical vascular encephalopathy (Binswanger’s disease)
Subcortical vascular encephalopathy describes a widespread demyelination and axon loss in the white matter sparing U-fibers, i.e. confluent white matter lesions (Fig. 3). These changes are caused by severe SVD, predominantly SVD of the arteriolosclerosis and lipohyalinosis type (Ferrer 2010; Hachinski et al., 2006; Ince 2005; Jellinger 2008; Kalaria and Erkinjuntti 2006). CADASIL can cause a similar pattern of lesions (Kalimo and Kalaria 2005).
Dementia as a result of reduced brain capacity by vascular types of dementia and neurodegeneration
VaD is usually a disease of elderly individuals (Ferrer 2010; Gorelick et al., 2011; Hachinski et al., 2006; Ince 2005; Jellinger 2008; Kalaria and Erkinjuntti 2006; Roman et al., 1993). In the elderly brain, Alzheimer’s disease (AD)-related pathological changes and argyrophilic grain disease (AGD) frequently occur (Braak and Braak 1997; Braak et al., 2011; Jellinger and Attems 2012; Thal et al., 2008a; Togo et al., 2002; Tolnay et al., 2001) in addition to CAA, AS, and SVD (Table 1, 3) (Deramecourt et al., 2012; Grinberg and Thal 2010; Jellinger and Attems 2012; Thal et al., 2003; Thal et al., 2008b). As such, pure AD, pure AGD, and pure VaD are rare (Korczyn 2002). Usually, AD is associated with CAA (Attems 2005; Joachim et al., 1988; Thal et al., 2008b; Vinters 1992), SVD (Brun and Englund 1986; Thal et al., 2003) (Table 3C), AS (Roher et al., 2003), as well as with white matter lesions (Gootjes et al., 2004; Meier et al., 2012) indicative for blood-brain barrier alterations due to SVD (Wardlaw et al., 2009). Vascular lesions, similar to neurodegenerative disorders such as AGD (Thal et al., 2005), are capable of reducing the threshold of AD pathology required for the development of dementia (Esiri et al., 1999; Schneider et al., 2009). Thus, the development of dementia may be the net result of all lesions in the brain, including vascular lesions, neurodegenerative lesions, but also traumatic or inflammatory lesions in any given brain. As soon as a given threshold is reached, the cognition gets impaired, regardless of the cause of the lesion, i.e, vascular or neurodegenerative or both (Table 3).
For the vascular component of dementia, as discussed in detail above, all kinds of lesions and related vessel disorders can contribute to the development of dementia. However, in our series of elderly autopsy cases VaD was associated with brain infarction in 66% of the VaD cases. SVD with its associated leukoencephalopathy occurred more frequently in cases categorized as AD (83%) whereas these lesions were less frequently observed in cases categorized as VaD (75%) and cognitively normal (74%). As such SVD-related subcortical vascular encephalopathy changes frequently coexist in AD patients (Table 3C) although they are also common in cognitively normal elderly individuals which frequently exhibit low levels of AD-related pathology as well (Table 3D).
Tab. 3D. Stages and degrees of AD pathology (mean values) in the brains of symptomatic AD, VaD, and cognitively normal individuals.
Stages of AD pathology (mean values) in VaD, AD, and cognitively normal individuals. To describe AD-related pathology in the observed cases Aβ phases, Braak-NFT stages and CERAD neuritic plaque scores were determined as recommended and previously described and recommended in the NIA-AA guidelines for the diagnosis of AD (Braak and Braak 1991; Hyman et al., 2012; Mirra et al., 1991; Thal et al., 2000). The NIA-AA degree of AD pathology was determined according to recently published guidelines (Hyman et al., 2012): no AD = Aβ phase = 0; low AD pathology = Aβ phase 1–2 and CERAD 0–1 or any Aβ phase ≥ 1 or CERAD score ≥ 0 combined with Braak-NFT stage 0–2; intermediate AD pathology = Braak-NFT stage III–VI and Aβ phase 3–5 except for cases fulfilling the criteria of high degree of AD pathology or Braak-NFT stage III–VI, Aβ phase 1–2 and CERAD 2–3; high AD pathology = Braak-NFT stage V–VI, Aβ phase 4–5, and CERAD 2–3. A detailed description of the NIA-AA guidelines is given by (Hyman et al., 2012) and (Montine et al., 2012).
| VaD | AD | Cognitively Normals | OR and CI | p | |
|---|---|---|---|---|---|
| Aβ-Phase (MTL) | 1.50 (n=12/233) | 3.75 (n=68/233) | 1.37 (n=153/233) | AD 10.2 (5 – 20.5) | < 0.002 |
| Braak-NFT-Stage | 1.83 (n=12/236) | 4.84 (n=69/236) | 1.58 (n=155/236) | AD 101.7 (14.8 – 696.7) | < 0.002 |
| CERAD-neuritic plaque score | 0.17 (n=12/234) | 2.41 (n=69/234) | 0.10 (n=153/234) | AD 30.7 (10.1 – 93.2) | < 0.002 |
| NIA-AA degree of AD pathology | 0.73 (n=11/229) | 249 (n=65/229) | 0.73 (n=153/229) | AD 195.1 (26.5 – 1436.5) | < 0.002 |
Abbreviations: AD = Alzheimer’s disease, AS = Atherosclerosis, CAA = cerebral amyloid angiopathy, CERAD = consortium to establish a registry for Alzheimer’s disease, CI = 95% confidence interval determined by logistic regression analysis, MTL = medial temporal lobe, n = number of cases studied, NIA-AA = National Institute on Aging – Alzheimer Association, NFT = neurofibrillary tangle, OR = Odds-ratio determined by logistic regression analysis, p = p-value determined by logistic regression analysis and corrected for multiple testing, SVD = small vessel disease, VaD = vascular dementia.
In addition to the impact of classical vascular lesions on dementia, the association between AD and SVD (Brun and Englund 1986; Thal et al., 2003), AS (Roher et al., 2003), and white matter lesions (Gootjes et al., 2004; Grimmer et al., 2012; Meier et al., 2012) points to a possible pathogenic link between AD and the above mentioned vessel disorders. However, another group found no relationship between AD and AS (Luoto et al., 2009). In our sample, AD cases exhibited slightly higher degrees of AS severity than cognitively normal individuals without reaching significance (Table 3C). The correlation analysis presented in Table 2 shows a correlation between SVD and CAA suggesting a pathogenic link between these vessel disorders and to AD which is closely correlated with CAA. A possible explanation for such a pathogenic link between AD and/ or CAA, on the one hand, and SVD with its associated white matter lesions, on the other, is the alteration of perivascular clearance mechanisms. Perivascular clearance describes the drainage of extracellular fluid and its proteins along the perivascular channels and the basement membranes into the cerebrospinal fluid and into the cervical lymph nodes (Carare et al., 2008; Weller et al., 2009). SVD leads to a fibrosis of the vessel wall (Lin et al., 2000) and to a leakage of blood-derived proteins into the vessel wall, perivascular space, and into the perivascular brain parenchyma (Alafuzoff et al., 1985; Utter et al., 2008). Accordingly, drainage along the blood vessels is impaired by fibrosis (Weller et al., 2009) and by the influx of blood-derived proteins and Aβ accumulates in the brain (Grinberg and Thal 2010; Utter et al., 2008). Since white matter lesions are associated with SVD (Pantoni and Garcia 1995; Schmidt et al., 2011; Urbach et al., 2007) and since they are related to defects in the blood-brain barrier (Wardlaw et al., 2009) and AD (Gootjes et al., 2004; Grimmer et al., 2012; Meier et al., 2012) it is tempting to speculate that an impaired perivascular clearance of Aβ and apolipoprotein E (apoE) due to SVD contributes to the development of AD (Attems et al., 2011; Grimmer et al., 2012; Grinberg and Thal 2010; Thal 2009). Moreover, the link between AD and CAA has been widely accepted because most AD cases exhibit CAA (Joachim et al., 1988; Thal et al., 2008b), CAA is caused by Aβ deposits in the vessel wall similar to those in AD-related Aβ plaques (Glenner and Wong 1984; Masters et al., 1985). Mouse models, which are made to produce Aβ plaques by neuronal Aβ production, exhibit CAA as well (Calhoun et al., 1999). CAA in these animals is, thereby, the result of Aβ drained through the perivascular space that aggregates at the vascular basement membranes. This supports the hypothesis of altered perivascular drainage of parenchymal Aβ as a cause of CAA in the human brain (Weller et al., 2009; Weller et al., 1998). Furthermore, evidence was provided that Aβ protein, especially Aβ1–40 may damage brain vessels via vasoactive mechanisms which disrupt the brain’s blood supply and render the brain more vulnerable to injury (Iadecola 2004).
Tab. 2. Correlations between AS, SVD, and CAA severity with the presence of brain infarction and cerebral hemorrhage.
Partial correlation analysis between AS, SVD, CAA, brain infarction and cerebral hemorrhage controlled for age and gender. The severity of SVD correlated with the occurrence of brain infarction and CAA severity whereas no significant correlations were observed between AS and SVD, CAA, brain infarction, or cerebral hemorrhage, between CAA and AS, SVD, brain infarction of cerebral hemorrhage, between SVD and AS or cerebral hemorrhage, and between brain infarction and AS, CAA, or cerebral hemorrhage.
| AS | SVD | CAA | |
|---|---|---|---|
| Brain infarction | 0.384 | 0.022 (r= 0.235) | 0.530 |
| Cerebral hemorrhage | 0.664 | 0.993 | 0.370 |
| CAA | 0.061 | 0.002 (r = 0.237) | |
| SVD | 0.258 | 0.020 (r = 0.237) |
Abbreviations: AS = Atherosclerosis, CAA = cerebral amyloid angiopathy, n = number of cases studied, p = p-value assessed by partial correlation analysis controlled for age and gender, r = correlation coefficient assessed by partial correlation analysis controlled for age and gender, SVD = small vessel disease.
In light of these considerations, dementia develops in the elderly brain as the result of an accumulation of neurodegenerative, vascular and other lesions whereby the diagnosis of VaD describes the development of dementia only due to vascular lesions. Since pure VaD or pure AD are quite rare the NIA-AA consensus guidelines for the diagnosis of AD recommend to report the degree each AD pathology, i.e. neurofibrillary tangles, Aβ plaques and neuritic plaques, separately as well as vascular pathology by category (Hyman et al., 2012). These guidelines are a first effort to unify the description of vascular lesions, although until today there is no widely accepted set of criteria for VaD classification and the assessment of these lesions by different observers varies strikingly (Alafuzoff et al. 2012; Grinberg and Heinsen 2010).
Conclusions
This review of the current literature and of our case collection revealed that vascular lesions and vessel disorders are prevalent in most demented elderly individuals often combined with concurrent AD-related lesions (Table 2). The frequent coexistence of AD pathology with SVD and its associated white matter lesions suggests that SVD is a risk factor for AD. As such, it seems to be important for clinical practice to consider the prevalence of AD-related brain lesions in patients with vascular, especially white matter lesions and to examine and treat the patients accordingly.
VaD with only negligible AD-related pathology is rare and when diagnosed most frequently caused by infarcts due to AS and embolic events. SVD and CAA did not play a major role in these cases.
Highlights.
Vascular dementia is the net results of vascular tissue lesions impairing cognitive function. Coexisting vascular and neurodegenerative lesions frequently co-occur in the elderly brain and the development of dementia is often due to both pathologies. Thus, clinical diagnosis and treatment of Alzheimer disease-related lesions should be considered in cases exhibiting dementia and vascular lesions.
Acknowledgments
Dietmar R. Thal received research grants from the Deutsche Forschungsgemeinschaft (DFG: Grant Nos. TH624/4-1,2; TH624/6-1) and the Alzheimer Forschung Initiative (AFI: Grant No. #10810). Johannes Attems is grateful for the support of the Dunhill Medical Trust (R173/1110). Lea Tenenholz Grinberg receives research grants from the National Institute of Health (1R01AG040311-01A1 and 2 P50 AG023501-06), John Douglas French Alzheimer Foundation and Albert Einstein Research Institute – São Paulo. The studies on human brain tissue reviewed here have been approved by the respective local ethic committees.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Alafuzoff I, Adolfsson R, Grundke-Iqbal I, Winblad B. Perivascular deposits of serum proteins in cerebral cortex in vascular dementia. Acta Neuropathol. 1985;66:292–298. doi: 10.1007/BF00690961. [DOI] [PubMed] [Google Scholar]
- Alafuzoff I, Al-Sarraj S, Arzberger T, Attems J, Bodi I, Bogdanovic N, Budka H, Bugiani O, Englund E, Ferrer I, Gelpi E, Gentleman S, Giaccone G, Graeber M, Hortobagyi T, Hoftberger R, Ironside J, Jellinger K, Kavantzas M, King A, Korkolopoulou P, Kovacs GG, Meyronet D, Monoranu C, Parchi P, Patsouris E, Roggendorf W, Rozemuller A, Seilhean D, Streichenberger N, Thal DR, Wharton S, Kretzschmar H. Diversity of neuropathological assessments of vascular alterations in the ageing brain. Multicenter survey by the BrainNet Europe consortium. Exp Gerontol. 2012 doi: 10.1016/j.exger.2012.06.001. in consideration. [DOI] [PubMed] [Google Scholar]
- Amador-Ortiz C, Lin WL, Ahmed Z, Personett D, Davies P, Duara R, Graff-Radford NR, Hutton ML, Dickson DW. TDP-43 immunoreactivity in hippocampal sclerosis and Alzheimer’s disease. Ann Neurol. 2007;61:435–445. doi: 10.1002/ana.21154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- American Psychiatric Association. Diagnostic and statistical manual of mental disorders. Washington DC: 1994. [Google Scholar]
- Attems J. Sporadic cerebral amyloid angiopathy: pathology, clinical implications, and possible pathomechanisms. Acta Neuropathol. 2005;110:345–359. doi: 10.1007/s00401-005-1074-9. [DOI] [PubMed] [Google Scholar]
- Attems J, Jellinger K, Thal DR, Van Nostrand W. Review: sporadic cerebral amyloid angiopathy. Neuropathol Appl Neurobiol. 2011;37:75–93. doi: 10.1111/j.1365-2990.2010.01137.x. [DOI] [PubMed] [Google Scholar]
- Braak H, Braak E. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol. 1991;82:239–259. doi: 10.1007/BF00308809. [DOI] [PubMed] [Google Scholar]
- Braak H, Braak E. Frequency of stages of Alzheimer-related lesions in different age categories. Neurobiol Aging. 1997;18:351–357. doi: 10.1016/s0197-4580(97)00056-0. [DOI] [PubMed] [Google Scholar]
- Braak H, Thal DR, Ghebremedhin E, Del Tredici K. Stages of the pathological process in Alzheimer’s disease: Age categories 1 year to 100 years. J Neuropathol Exp Neurol. 2011;70:960–969. doi: 10.1097/NEN.0b013e318232a379. [DOI] [PubMed] [Google Scholar]
- Brun A, Englund E. A white matter disorder in dementia of the Alzheimer type: a pathoanatomical study. Ann Neurol. 1986;19:253–262. doi: 10.1002/ana.410190306. [DOI] [PubMed] [Google Scholar]
- Cadavid D, Mena H, Koeller K, Frommelt RA. Cerebral beta amyloid angiopathy is a risk factor for cerebral ischemic infarction, A case control study in human brain biopsies. J Neuropathol Exp Neurol. 2000;59:768–773. doi: 10.1093/jnen/59.9.768. [DOI] [PubMed] [Google Scholar]
- Calhoun ME, Burgermeister P, Phinney AL, Stalder M, Tolnay M, Wiederhold KH, Abramowski D, Sturchler-Pierrat C, Sommer B, Staufenbiel M, Jucker M. Neuronal overexpression of mutant amyloid precursor protein results in prominent deposition of cerebrovascular amyloid. Proc Natl Acad Sci U S A. 1999;96:14088–14093. doi: 10.1073/pnas.96.24.14088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carare RO, Bernardes-Silva M, Newman TA, Page AM, Nicoll JA, Perry VH, Weller RO. Solutes, but not cells, drain from the brain parenchyma along basement membranes of capillaries and arteries: significance for cerebral amyloid angiopathy and neuroimmunology. Neuropathol Appl Neurobiol. 2008;34:131–144. doi: 10.1111/j.1365-2990.2007.00926.x. [DOI] [PubMed] [Google Scholar]
- Challa VR, Bell MA, Moody DM. A combined hematoxylin-eosin, alkaline phosphatase and high-resolution microradiographic study of lacunes. Clin Neuropathol. 1990;9:196–204. [PubMed] [Google Scholar]
- De Reuck J, Auger F, Cordonnier C, Deramecourt V, Durieux N, Pasquier F, Bordet R, Maurage CA, Leys D. Comparison of 7.0-T T*-magnetic resonance imaging of cerebral bleeds in post-mortem brain sections of Alzheimer patients with their neuropathological correlates. Cerebrovasc Dis. 2011;31:511–517. doi: 10.1159/000324391. [DOI] [PubMed] [Google Scholar]
- Deramecourt V, Slade JY, Oakley AE, Perry RH, Ince PG, Maurage CA, Kalaria RN. Staging and natural history of cerebrovascular pathology in dementia. Neurology. 2012;78:1043–1050. doi: 10.1212/WNL.0b013e31824e8e7f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esiri MM, Nagy Z, Smith MZ, Barnetson L, Smith AD. Cerebrovascular disease and threshold for dementia in the early stages of Alzheimer’s disease. Lancet. 1999;354:919–920. doi: 10.1016/S0140-6736(99)02355-7. [DOI] [PubMed] [Google Scholar]
- Fazekas F, Kleinert R, Offenbacher H, Schmidt R, Kleinert G, Payer F, Radner H, Lechner H. Pathologic correlates of incidental MRI white matter signal hyperintensities. Neurology. 1993;43:1683–1689. doi: 10.1212/wnl.43.9.1683. [DOI] [PubMed] [Google Scholar]
- Ferrer I. Cognitive impairment of vascular origin: neuropathology of cognitive impairment of vascular origin. J Neurol Sci. 2010;299:139–149. doi: 10.1016/j.jns.2010.08.039. [DOI] [PubMed] [Google Scholar]
- Ferrer I, Kaste M, Kalimo H. Vascular Diseases. In: Love S, Louis DN, Ellison DW, editors. Greenfield’s Neuropathology. 8. Arnold; London: 2008. pp. 121–240. [Google Scholar]
- Fisher CM. Lacunar infarcts - A review. Cerebrovasc Dis. 1991;1:311–320. [Google Scholar]
- Gerraty RP, Parsons MW, Barber PA, Darby DG, Desmond PM, Tress BM, Davis SM. Examining the lacunar hypothesis with diffusion and perfusion magnetic resonance imaging. Stroke. 2002;33:2019–2024. doi: 10.1161/01.str.0000020841.74704.5b. [DOI] [PubMed] [Google Scholar]
- Glenner GG, Wong CW. Alzheimer’s disease: initial report of the purification and characterization of a novel cerebrovascular amyloid protein. Biochem Biophys Res Commun. 1984;120:885–890. doi: 10.1016/s0006-291x(84)80190-4. [DOI] [PubMed] [Google Scholar]
- Gootjes L, Teipel SJ, Zebuhr Y, Schwarz R, Leinsinger G, Scheltens P, Moller HJ, Hampel H. Regional distribution of white matter hyperintensities in vascular dementia, Alzheimer’s disease and healthy aging. Dement Geriatr Cogn Disord. 2004;18:180–188. doi: 10.1159/000079199. [DOI] [PubMed] [Google Scholar]
- Gorelick PB, Scuteri A, Black SE, Decarli C, Greenberg SM, Iadecola C, Launer LJ, Laurent S, Lopez OL, Nyenhuis D, Petersen RC, Schneider JA, Tzourio C, Arnett DK, Bennett DA, Chui HC, Higashida RT, Lindquist R, Nilsson PM, Roman GC, Sellke FW, Seshadri S. Vascular contributions to cognitive impairment and dementia: a statement for healthcare professionals from the american heart association/american stroke association. Stroke. 2011;42:2672–2713. doi: 10.1161/STR.0b013e3182299496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenberg SM. Handbook of Neurosurgery. Greenberg Graphics; Lakeland, Florida: 1997. [Google Scholar]
- Greenberg SM, Vernooij MW, Cordonnier C, Viswanathan A, Al-Shahi Salman R, Warach S, Launer LJ, Van Buchem MA, Breteler MM. Cerebral microbleeds: a guide to detection and interpretation. Lancet Neurol. 2009;8:165–174. doi: 10.1016/S1474-4422(09)70013-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grimmer T, Faust M, Auer F, Alexopoulos P, Forstl H, Henriksen G, Perneczky R, Sorg C, Yousefi BH, Drzezga A, Kurz A. White matter hyperintensities predict amyloid increase in Alzheimer’s disease. Neurobiol Aging. 2012 doi: 10.1016/j.neurobiolaging.2012.01.016. [DOI] [PubMed] [Google Scholar]
- Grinberg LT, Heinsen H. Toward a pathological definition of vascular dementia. J Neurol Sci. 2010;299:136–138. doi: 10.1016/j.jns.2010.08.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grinberg LT, Korczyn AD, Heinsen H. Cerebral amyloid angiopathy impact on endothelium - new hopes for therapeutic interventions. Exp Gerontol. 2012 doi: 10.1016/j.exger.2012.08.005. in consideration. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grinberg LT, Thal DR. Vascular pathology in the aged human brain. Acta Neuropathol. 2010;119:277–290. doi: 10.1007/s00401-010-0652-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hachinski V, Iadecola C, Petersen RC, Breteler MM, Nyenhuis DL, Black SE, Powers WJ, DeCarli C, Merino JG, Kalaria RN, Vinters HV, Holtzman DM, Rosenberg GA, Wallin A, Dichgans M, Marler JR, Leblanc GG. National Institute of Neurological Disorders and Stroke-Canadian Stroke Network vascular cognitive impairment harmonization standards. Stroke. 2006;37:2220–2241. doi: 10.1161/01.STR.0000237236.88823.47. [DOI] [PubMed] [Google Scholar]
- Hachinski VC, Lassen NA, Marshall J. Multi-infarct dementia, A cause of mental deterioration in the elderly. Lancet. 1974;2:207–210. doi: 10.1016/s0140-6736(74)91496-2. [DOI] [PubMed] [Google Scholar]
- Henry-Feugeas MC. MRI of the ‘Alzheimer syndrome’. J Neuroradiol. 2007;34:220–227. doi: 10.1016/j.neurad.2007.07.002. [DOI] [PubMed] [Google Scholar]
- Hommet C, Mondon K, Constans T, Beaufils E, Desmidt T, Camus V, Cottier JP. Review of cerebral microangiopathy and Alzheimer’s disease: relation between white matter hyperintensities and microbleeds. Dement Geriatr Cogn Disord. 2011;32:367–378. doi: 10.1159/000335568. [DOI] [PubMed] [Google Scholar]
- Hyman BT, Phelps CH, Beach TG, Bigio EH, Cairns NJ, Carrillo MC, Dickson DW, Duyckaerts C, Frosch MP, Masliah E, Mirra SS, Nelson PT, Schneider JA, Thal DR, Thies B, Trojanowski JQ, Vinters HV, Montine TJ. National Institute on Aging Alzheimer’s Association guidelines for the neuropathologic assessment of Alzheimer’s disease. Alzheimer Dementia. 2012;8:1–13. doi: 10.1016/j.jalz.2011.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iadecola C. Neurovascular regulation in the normal brain and in Alzheimer’s disease. Nat Rev Neurosci. 2004;5:347–360. doi: 10.1038/nrn1387. [DOI] [PubMed] [Google Scholar]
- Ince PG. Acquired forms of vascular dementia. In: Kalimo H, editor. Cerebrovascular diseases. ISN Neuropath Press; Basel: 2005. pp. 316–323. [Google Scholar]
- Jellinger KA. The pathology of “vascular dementia”: a critical update. J Alzheimers Dis. 2008;14:107–123. doi: 10.3233/jad-2008-14110. [DOI] [PubMed] [Google Scholar]
- Jellinger KA, Attems J. Prevalence of dementia disorders in the oldest-old: an autopsy study. Acta Neuropathol. 2010;119:421–433. doi: 10.1007/s00401-010-0654-5. [DOI] [PubMed] [Google Scholar]
- Jellinger KA, Attems J. Neuropathology and general autopsy findings in nondemented aged subjects. Clin Neuropathol. 2012;31:87–98. doi: 10.5414/np300418. [DOI] [PubMed] [Google Scholar]
- Jeong JH, Yoon SJ, Kang SJ, Choi KG, Na DL. Hypertensive pontine microhemorrhage. Stroke. 2002;33:925–929. doi: 10.1161/01.str.0000013563.73522.cb. [DOI] [PubMed] [Google Scholar]
- Joachim CL, Morris JH, Selkoe DJ. Clinically diagnosed Alzheimer’s disease: autopsy results in 150 cases. Ann Neurol. 1988;24:50–56. doi: 10.1002/ana.410240110. [DOI] [PubMed] [Google Scholar]
- Joutel A, Corpechot C, Ducros A, Vahedi K, Chabriat H, Mouton P, Alamowitch S, Domenga V, Cecillion M, Marechal E, Maciazek J, Vayssiere C, Cruaud C, Cabanis EA, Ruchoux MM, Weissenbach J, Bach JF, Bousser MG, Tournier-Lasserve E. Notch3 mutations in CADASIL, a hereditary adult-onset condition causing stroke and dementia. Nature. 1996;383:707–710. doi: 10.1038/383707a0. [DOI] [PubMed] [Google Scholar]
- Kalaria RN. The Cerebral Amyloid Angiopathies, Microvascular Degeneration and Dementia. Brain Pathol Suppl. 2003;S83 [Google Scholar]
- Kalaria RN, Erkinjuntti T. Small vessel disease and subcortical vascular dementia. J Clin Neurol. 2006;2:1–11. doi: 10.3988/jcn.2006.2.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalimo H, Kalaria R. Hereditary forms of vascular dementia. In: Kalimo H, editor. Cerebrovascular diseases. ISN Neuropath Press; Basel: 2005. pp. 324–334. [Google Scholar]
- Kimberly WT, Gilson A, Rost NS, Rosand J, Viswanathan A, Smith EE, Greenberg SM. Silent ischemic infarcts are associated with hemorrhage burden in cerebral amyloid angiopathy. Neurology. 2009;72:1230–1235. doi: 10.1212/01.wnl.0000345666.83318.03. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitamura A, Fujita Y, Oishi N, Kalaria RN, Washida K, Maki T, Okamoto Y, Hase Y, Yamada M, Takahashi J, Ito H, Tomimoto H, Fukuyama H, Takahashi R, Ihara M. Selective white matter abnormalities in a novel rat model of vascular dementia. Neurobiol Aging. 2012;33:1012, e1025–1035. doi: 10.1016/j.neurobiolaging.2011.10.033. [DOI] [PubMed] [Google Scholar]
- Kolsch H, Larionov S, Dedeck O, Orantes M, Birkenmeier G, Griffin WS, Thal DR. Association of the glutathione S-transferase omega-1 Ala140Asp polymorphism with cerebrovascular atherosclerosis and plaque-associated interleukin-1 alpha expression. Stroke. 2007;38:2847–2850. doi: 10.1161/STROKEAHA.107.484162. [DOI] [PubMed] [Google Scholar]
- Korczyn AD. Mixed dementia--the most common cause of dementia. Ann N Y Acad Sci. 2002;977:129–134. doi: 10.1111/j.1749-6632.2002.tb04807.x. [DOI] [PubMed] [Google Scholar]
- Lammie AG. Small vessel disease. In: Kalimo H, editor. Cerebrovascular diseases. ISN Neuropath Press; Basel: 2005. pp. 85–91. [Google Scholar]
- Lammie GA. Hypertensive cerebral small vessel disease and stroke. Brain Pathol. 2002;12:358–370. doi: 10.1111/j.1750-3639.2002.tb00450.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larionov S, Dedeck O, Birkenmeier G, Orantes M, Ghebremedhin E, Thal DR. The intronic deletion polymorphism of the a2-macroglobulin gene modulates the severity and extent of atherosclerosis in the circle of Willis. Neuropathol Appl Neurobiol. 2006;32:451–454. doi: 10.1111/j.1365-2990.2006.00743.x. [DOI] [PubMed] [Google Scholar]
- Larionov S, Dedeck O, Birkenmeier G, Thal DR. Expression of alpha(2)-macroglobulin, neutrophil elastase, and interleukin-1alpha differs in early-stage and late-stage atherosclerotic lesions in the arteries of the circle of Willis. Acta Neuropathol (Berl) 2007;113:33–43. doi: 10.1007/s00401-006-0134-0. [DOI] [PubMed] [Google Scholar]
- Launer LJ, Hughes TM, White LR. Microinfarcts, brain atrophy, and cognitive function: the Honolulu Asia Aging Study Autopsy Study. Ann Neurol. 2011;70:774–780. doi: 10.1002/ana.22520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liberato B, Chong JY, Sacco RL. Focal brain ischemia. Clinical features, epidemiology, risk factors and outcome. In: Kalimo H, editor. Cerebrovascular diseases. ISN Neuropath Press; Basel: 2005. pp. 176–185. [Google Scholar]
- Lin JX, Tomimoto H, Akiguchi I, Matsuo A, Wakita H, Shibasaki H, Budka H. Vascular cell components of the medullary arteries in Binswanger’s disease brains: a morphometric and immunoelectron microscopic study. Stroke. 2000;31:1838–1842. doi: 10.1161/01.str.31.8.1838. [DOI] [PubMed] [Google Scholar]
- Luoto TM, Haikonen S, Haapasalo H, Goebeler S, Huhtala H, Erkinjuntti T, Karhunen PJ. Large vessel cerebral atherosclerosis is not in direct association with neuropathological lesions of Alzheimer’s disease. Eur Neurol. 2009;62:93–98. doi: 10.1159/000222779. [DOI] [PubMed] [Google Scholar]
- Mandybur TI. Cerebral amyloid angiopathy: the vascular pathology and complications. J Neuropathol Exp Neurol. 1986;45:79–90. [PubMed] [Google Scholar]
- Marti-Vilalta JL, Arboix A. The Barcelona Stroke Registry. Eur Neurol. 1999;41:135–142. doi: 10.1159/000008036. [DOI] [PubMed] [Google Scholar]
- Masters CL, Simms G, Weinman NA, Multhaup G, McDonald BL, Beyreuther K. Amyloid plaque core protein in Alzheimer disease and Down syndrome. Proc Natl Acad Sci U S A. 1985;82:4245–4249. doi: 10.1073/pnas.82.12.4245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meier IB, Manly JJ, Provenzano FA, Louie KS, Wasserman BT, Griffith EY, Hector JT, Allocco E, Brickman AM. White Matter Predictors of Cognitive Functioning in Older Adults. J Int Neuropsychol Soc. 2012:1–14. doi: 10.1017/S1355617712000227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mirra SS, Heyman A, McKeel D, Sumi SM, Crain BJ, Brownlee LM, Vogel FS, Hughes JP, van Belle G, Berg L. The Consortium to Establish a Registry for Alzheimer’s Disease (CERAD). Part II. Standardization of the neuropathologic assessment of Alzheimer’s disease. Neurology. 1991;41:479–486. doi: 10.1212/wnl.41.4.479. [DOI] [PubMed] [Google Scholar]
- Montine TJ, Phelps CH, Beach TG, Bigio EH, Cairns NJ, Carrillo MC, Dickson DW, Duyckaerts C, Frosch MP, Masliah E, Mirra SS, Nelson PT, Schneider JA, Thal DR, Thies B, Trojanowski JQ, Vinters HV, Hyman BT. National Institute on Aging–Alzheimer’s Association guidelines for the neuropathologic assessment of Alzheimer’s disease: a practical approach. Acta Neuropathol. 2012;123:1–11. doi: 10.1007/s00401-011-0910-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nag S. Immunohistochemical localization of extracellular matrix proteins in cerebral vessels in chronic hypertension. J Neuropathol Exp Neurol. 1996;55:381–388. doi: 10.1097/00005072-199603000-00014. [DOI] [PubMed] [Google Scholar]
- Nag S, Robertson DM. The brain in hypertension. In: Kalimo H, editor. Pathology & Genetics: Cerebrovascular diseases. ISN Neuropath Press; Basel: 2005. pp. 286–292. [Google Scholar]
- Okamoto Y, Ihara M, Fujita Y, Ito H, Takahashi R, Tomimoto H. Cortical microinfarcts in Alzheimer’s disease and subcortical vascular dementia. Neuroreport. 2009;20:990–996. doi: 10.1097/WNR.0b013e32832d2e6a. [DOI] [PubMed] [Google Scholar]
- Okamoto Y, Yamamoto T, Kalaria RN, Senzaki H, Maki T, Hase Y, Kitamura A, Washida K, Yamada M, Ito H, Tomimoto H, Takahashi R, Ihara M. Cerebral hypoperfusion accelerates cerebral amyloid angiopathy and promotes cortical microinfarcts. Acta Neuropathol. 2012;123:381–394. doi: 10.1007/s00401-011-0925-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ovbiagele B. Hospital-based stroke diagnoses among the oldest old in the United States: 1997 to 2006. Stroke. 2010;41:1820–1822. doi: 10.1161/STROKEAHA.110.587816. [DOI] [PubMed] [Google Scholar]
- Pantoni L, Garcia JH. The significance of cerebral white matter abnormalities 100 years after Binswanger’s report, A review. Stroke. 1995;26:1293–1301. doi: 10.1161/01.str.26.7.1293. [DOI] [PubMed] [Google Scholar]
- Petito CK. The neuropathology of focal brain ischemia. In: Kalimo H, editor. Cerebrovascular Diseases. ISN Neuropath Press; Basel: 2005. pp. 215–221. [Google Scholar]
- Revesz T, Holton JL, Lashley T, Plant G, Frangione B, Rostagno A, Ghiso J. Genetics and molecular pathogenesis of sporadic and hereditary cerebral amyloid angiopathies. Acta Neuropathol. 2009;118:115–130. doi: 10.1007/s00401-009-0501-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roher AE, Esh C, Kokjohn TA, Kalback W, Luehrs DC, Seward JD, Sue LI, Beach TG. Circle of willis atherosclerosis is a risk factor for sporadic Alzheimer’s disease. Arterioscler Thromb Vasc Biol. 2003;23:2055–2062. doi: 10.1161/01.ATV.0000095973.42032.44. [DOI] [PubMed] [Google Scholar]
- Roman GC, Tatemichi TK, Erkinjuntti T, Cummings JL, Masdeu JC, Garcia JH, Amaducci L, Orgogozo JM, Brun A, Hofman A, Moody DM, O’Brien MD, Yamaguchi T, Grafman J, Drayer BP, Bennett DA, Fisher M, Ogata J, Kokmen E, Bermejo F, Wolf PA, Gorelick PB, Bick KL, Pajeau AK, Bell MA, DeCarli C, Culebras A, Korczyn AD, Bogousslavsky J, Hartmann A, Scheinberg P. Vascular dementia: diagnostic criteria for research studies, Report of the NINDS-AIREN International Workshop. Neurology. 1993;43:250–260. doi: 10.1212/wnl.43.2.250. [DOI] [PubMed] [Google Scholar]
- Schmidt R, Schmidt H, Haybaeck J, Loitfelder M, Weis S, Cavalieri M, Seiler S, Enzinger C, Ropele S, Erkinjuntti T, Pantoni L, Scheltens P, Fazekas F, Jellinger K. Heterogeneity in age-related white matter changes. Acta Neuropathol. 2011;122:171–185. doi: 10.1007/s00401-011-0851-x. [DOI] [PubMed] [Google Scholar]
- Schneider JA, Aggarwal NT, Barnes L, Boyle P, Bennett DA. The neuropathology of older persons with and without dementia from community versus clinic cohorts. J Alzheimers Dis. 2009;18:691–701. doi: 10.3233/JAD-2009-1227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scholz W. Studien zur Pathologie der Hirngefäße. II. Die drusige Entartung der Hirnarterien und -capillaren (Eine Form seniler Gefäßerkrankung) Z ges Neurol Psychiatr. 1938;4:694–715. [Google Scholar]
- Smith EE, Schneider JA, Wardlaw JM, Greenberg SM. Cerebral microinfarcts: the invisible lesions. Lancet Neurol. 2012;11:272–282. doi: 10.1016/S1474-4422(11)70307-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stary HC. Natural history and histological classification of atherosclerotic lesions: an update. Arterioscler Thromb Vasc Biol. 2000;20:1177–1178. doi: 10.1161/01.atv.20.5.1177. [DOI] [PubMed] [Google Scholar]
- Stary HC, Chandler AB, Dinsmore RE, Fuster V, Glagov S, Insull W, Jr, Rosenfeld ME, Schwartz CJ, Wagner WD, Wissler RW. A definition of advanced types of atherosclerotic lesions and a histological classification of atherosclerosis, A report from the Committee on Vascular Lesions of the Council on Arteriosclerosis, American Heart Association. Arterioscler Thromb Vasc Biol. 1995;15:1512–1531. doi: 10.1161/01.atv.15.9.1512. [DOI] [PubMed] [Google Scholar]
- Stary HC, Chandler AB, Glagov S, Guyton JR, Insull W, Jr, Rosenfeld ME, Schaffer SA, Schwartz CJ, Wagner WD, Wissler RW. A definition of initial, fatty streak, and intermediate lesions of atherosclerosis, A report from the Committee on Vascular Lesions of the Council on Arteriosclerosis, American Heart Association. Circulation. 1994;89:2462–2478. doi: 10.1161/01.cir.89.5.2462. [DOI] [PubMed] [Google Scholar]
- Suemoto CK, Nitrini R, Grinberg LT, Ferretti RE, Farfel JM, Leite RE, Menezes PR, Fregni F, Jacob-Filho W, Pasqualucci CA. Atherosclerosis and dementia: a cross-sectional study with pathological analysis of the carotid arteries. Stroke. 2011;42:3614–3615. doi: 10.1161/STROKEAHA.111.628156. [DOI] [PubMed] [Google Scholar]
- Suter OC, Sunthorn T, Kraftsik R, Straubel J, Darekar P, Khalili K, Miklossy J. Cerebral hypoperfusion generates cortical watershed microinfarcts in Alzheimer disease. Stroke. 2002;33:1986–1992. doi: 10.1161/01.str.0000024523.82311.77. [DOI] [PubMed] [Google Scholar]
- Sze G, De Armond SJ, Brant-Zawadzki M, Davis RL, Norman D, Newton TH. Foci of MRI signal (pseudo lesions) anterior to the frontal horns: histologic correlations of a normal finding. AJR Am J Roentgenol. 1986;147:331–337. doi: 10.2214/ajr.147.2.331. [DOI] [PubMed] [Google Scholar]
- Szirmai I, Vastagh I, Szombathelyi E, Kamondi A. Strategic infarcts of the thalamus in vascular dementia. J Neurol Sci. 2002:203–204. 91–97. doi: 10.1016/s0022-510x(02)00273-3. [DOI] [PubMed] [Google Scholar]
- Takebayashi S, Kaneko M. Electron microscopic studies of ruptured arteries in hypertensive intracerebral hemorrhage. Stroke. 1983;14:28–36. doi: 10.1161/01.str.14.1.28. [DOI] [PubMed] [Google Scholar]
- Thal DR. The pre-capillary segment of the blood-brain barrier and its relation to perivascular drainage in Alzheimer’s disease and small vessel disease. ScientificWorldJournal. 2009;9:557–563. doi: 10.1100/tsw.2009.72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thal DR, Capetillo-Zarate E, Larionov S, Staufenbiel M, Zurbruegg S, Beckmann N. Capillary cerebral amyloid angiopathy is associated with vessel occlusion and cerebral blood flow disturbances. Neurobiol Aging. 2009;30:1936–1948. doi: 10.1016/j.neurobiolaging.2008.01.017. [DOI] [PubMed] [Google Scholar]
- Thal DR, Ghebremedhin E, Orantes M, Wiestler OD. Vascular pathology in Alzheimer’s disease: Correlation of cerebral amyloid angiopathy and arteriosclerosis / lipohyalinosis with cognitive decline. J Neuropathol Exp Neurol. 2003;62:1287–1301. doi: 10.1093/jnen/62.12.1287. [DOI] [PubMed] [Google Scholar]
- Thal DR, Griffin WS, Braak H. Parenchymal and vascular Abeta-deposition and its effects on the degeneration of neurons and cognition in Alzheimer’s disease. J Cell Mol Med. 2008a;12:1848–1862. doi: 10.1111/j.1582-4934.2008.00411.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thal DR, Griffin WST, De Vos RAI, Ghebremedhin E. Cerebral amyloid angiopathy and its relationship to Alzheimer’s disease. Acta Neuropathol. 2008b;115:599–609. doi: 10.1007/s00401-008-0366-2. [DOI] [PubMed] [Google Scholar]
- Thal DR, Rüb U, Orantes M, Braak H. Phases of Abeta-deposition in the human brain and its relevance for the development of AD. Neurology. 2002;58:1791–1800. doi: 10.1212/wnl.58.12.1791. [DOI] [PubMed] [Google Scholar]
- Thal DR, Rüb U, Schultz C, Sassin I, Ghebremedhin E, Del Tredici K, Braak E, Braak H. Sequence of Abeta-protein deposition in the human medial temporal lobe. J Neuropathol Exp Neurol. 2000;59:733–748. doi: 10.1093/jnen/59.8.733. [DOI] [PubMed] [Google Scholar]
- Thal DR, Schultz C, Botez G, Del Tredici K, Mrak RE, Griffin WS, Wiestler OD, Braak H, Ghebremedhin E. The impact of argyrophilic grain disease on the development of dementia and its relationship to concurrent Alzheimer’s disease-related pathology. Neuropathol Appl Neurobiol. 2005;31:270–279. doi: 10.1111/j.1365-2990.2005.00635.x. [DOI] [PubMed] [Google Scholar]
- Togo T, Cookson N, Dickson DW. Argyrophilic grain disease: neuropathology, frequency in a dementia brain bank and lack of relationship with apolipoprotein E. Brain Pathol. 2002;12:45–52. doi: 10.1111/j.1750-3639.2002.tb00421.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tolnay M, Monsch AU, Probst A. Argyrophilic grain disease, A frequent dementing disorder in aged patients. Adv Exp Med Biol. 2001;487:39–58. [PubMed] [Google Scholar]
- Tomlinson BE, Blessed G, Roth M. Observations on the brains of demented old people. J Neurol Sci. 1970;11:205–242. doi: 10.1016/0022-510x(70)90063-8. [DOI] [PubMed] [Google Scholar]
- Urbach H, Tschampa H, Flacke S, Thal DR. MRI of vascular dementia and differential diagnoses or is it really dementia? Clin Neuroradiol. 2007;17:88–97. [Google Scholar]
- Utter S, Tamboli IY, Walter J, Rijal Upadhaya A, Birkenmeier G, Pietrzik CU, Ghebremedhin E, Thal DR. Cerebral small vessel disease-induced apolipoprotein E leakage is associated with Alzheimer disease and the accumulation of amyloid beta-protein in perivascular astrocytes. J Neuropathol Exp Neurol. 2008;67:842–856. doi: 10.1097/NEN.0b013e3181836a71. [DOI] [PubMed] [Google Scholar]
- van Es AC, van der Grond J, de Craen AJ, Westendorp RG, Bollen EL, Blauw GJ, Greenberg SM, van Buchem MA. Cerebral microbleeds and cognitive functioning in the PROSPER study. Neurology. 2011;77:1446–1452. doi: 10.1212/WNL.0b013e318232ab1d. [DOI] [PubMed] [Google Scholar]
- Vinters HV. Cerebral Amyloid Angiopathy. In: Barnett HJM, Mohr JP, Stein BM, Yatsu FM, editors. Stroke Pathophysiology, diagnosis and management. Churchill Livingstone; New York: 1992. pp. 821–858. [Google Scholar]
- Vinters HV, Ellis WG, Zarow C, Zaias BW, Jagust WJ, Mack WJ, Chui HC. Neuropathologic substrates of ischemic vascular dementia. J Neuropathol Exp Neurol. 2000;59:931–945. doi: 10.1093/jnen/59.11.931. [DOI] [PubMed] [Google Scholar]
- Vonsattel JP, Myers RH, Hedley-Whyte ET, Ropper AH, Bird ED, Richardson EP., Jr Cerebral amyloid angiopathy without and with cerebral hemorrhages: a comparative histological study. Ann Neurol. 1991;30:637–649. doi: 10.1002/ana.410300503. [DOI] [PubMed] [Google Scholar]
- Wardlaw JM, Doubal F, Armitage P, Chappell F, Carpenter T, Munoz Maniega S, Farrall A, Sudlow C, Dennis M, Dhillon B. Lacunar stroke is associated with diffuse blood-brain barrier dysfunction. Ann Neurol. 2009;65:194–202. doi: 10.1002/ana.21549. [DOI] [PubMed] [Google Scholar]
- Weller RO, Djuanda E, Yow HY, Carare RO. Lymphatic drainage of the brain and the pathophysiology of neurological disease. Acta Neuropathol. 2009;117:1–14. doi: 10.1007/s00401-008-0457-0. [DOI] [PubMed] [Google Scholar]
- Weller RO, Massey A, Newman TA, Hutchings M, Kuo YM, Roher AE. Cerebral amyloid angiopathy: amyloid beta accumulates in putative interstitial fluid drainage pathways in Alzheimer’s disease. Am J Pathol. 1998;153:725–733. doi: 10.1016/s0002-9440(10)65616-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wieberdink RG, Ikram MA, Hofman A, Koudstaal PJ, Breteler MM. Trends in stroke incidence rates and stroke risk factors in Rotterdam, the Netherlands from 1990 to 2008. Eur J Epidemiol. 2012 doi: 10.1007/s10654-012-9673-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winkler DT, Bondolfi L, Herzig MC, Jann L, Calhoun ME, Wiederhold KH, Tolnay M, Staufenbiel M, Jucker M. Spontaneous hemorrhagic stroke in a mouse model of cerebral amyloid angiopathy. J Neurosci. 2001;21:1619–1627. doi: 10.1523/JNEUROSCI.21-05-01619.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wisniewski HM, Wegiel J, Wang KC, Lach B. Ultrastructural studies of the cells forming amyloid in the cortical vessel wall in Alzheimer’s disease. Acta Neuropathol (Berl) 1992;84:117–127. doi: 10.1007/BF00311383. [DOI] [PubMed] [Google Scholar]
- Yamaguchi H, Yamazaki T, Lemere CA, Frosch MP, Selkoe DJ. Beta amyloid is focally deposited within the outer basement membrane in the amyloid angiopathy of Alzheimer’s disease, An immunoelectron microscopic study. Am J Pathol. 1992;141:249–259. [PMC free article] [PubMed] [Google Scholar]
- Yamori Y, Horie R, Handa H, Sato M, Fukase M. Pathogenetic similarity of strokes in stroke-prone spontaneously hypertensive rats and humans. Stroke. 1976;7:46–53. doi: 10.1161/01.str.7.1.46. [DOI] [PubMed] [Google Scholar]