

Abstract
Australia and New Guinea contain high levels of endemism and biodiversity, yet there have been few evaluations of population-level genetic diversity in fauna occurring throughout the Australo-Papuan region. Using extensive geographical sampling, we examined and compared the phylogenetic relationships, phylogeography and population structure of Anopheles farauti, An. hinesorum and An. irenicus throughout their ranges in the southwest Pacific using mitochondrial (mtDNA COI) and nuclear (ribosomal protein S9 and ribosomal DNA ITS2) loci. Phylogenetic analyses suggest that the ability to utilize humans as hosts has been lost repeatedly, coincident with independent colonizations of the Solomon Islands. As some of the species under investigation transmit malaria in the region, this is a medically important finding. Maximum likelihood and Bayesian phylogenetic analyses of nuclear loci also showed that the three species are monophyletic. However, putative introgression of An. hinesorum mtDNA onto a nuclear background of An. farauti was evident in populations from Queensland, Torres Strait and southern New Guinea. Haplotype networks and pairwise FST values show that there is significant genetic structure within New Guinea and Australia in both An. farauti and An. hinesorum, consistent with a long-term history of low gene flow among populations.
Keywords: Anopheles, introgression, malaria vector, New Guinea, parallel evolution, phylogeography
Introduction
Background
Until recently (about 8000 years ago), Australia and New Guinea were connected by a land bridge (Voris 2000) and the two landmasses are considered part of the same continent known as Sahul (O’Connell & Allen 2004). Now separated, both landmasses support exceptional biodiversity (Allison 2007; Marshall 2007) and there are close affinities between their biotas, reflecting their historical connection (Allison 2007). High levels of local animal endemism are present in parts of New Guinea (Taylor et al. 1982; Allison 2007), which may be due to the island’s topographical and geological complexity (Pigram & Davies 1987; Polhemus 2007). This complexity has been the focus of many biogeographical studies on the fauna of New Guinea (Beehler & Swaby 1991; de Boer & Duffels 1996; Polhemus & Polhemus 1998; Heads 2002).
However, despite the importance of the Australo-Papuan region as an evolutionary centre and a biodiversity hotspot (Allison 2007), few studies have investigated population-level patterns of genetic diversity in terrestrial organisms of the area (Rawlings & Donnellan 2003; Murphy et al. 2007; Kearns et al. 2011; Macqueen et al. 2011). Most genetic studies published from the region either contain little intraspecific sampling (phylogenetic studies; Keogh et al. 1998; Osborne & Christidis 2001; Rawlings et al. 2004; Wuster et al. 2005; Norman et al. 2007) and/or limited geographical sampling from within New Guinea (De Bruyn et al. 2004; Baker et al. 2008; Cook & Hughes 2010; Craft et al. 2010). This may be due to the fact that many species in New Guinea have narrow ranges (Taylor et al. 1982; Allison 2007), making the inference of broadscale patterns of intraspecific genetic structure in the Australo-Papuan region difficult.
Previous studies have identified areas of endemism in the southwest Pacific, based on the distribution of various taxa (Van Welzen 1989; de Boer & Duffels 1996). Within New Guinea, major areas of endemism identified include, southern New Guinea, the Papuan Peninsula and northern New Guinea. Additionally, the Bismarck Archipelago, the Solomon Islands and islands of Vanuatu each possess unique species (de Boer & Duffels 1996). Biogeographical features that have led to vicariance have also been identified based on genetic relationships. These include the following: (i) the Carpentaria Gap, an ecological–climatic barrier separating the Northern Territory and Queensland (Bowman et al. 2010), (ii) the Torres Strait, the body of water currently separating Australia and New Guinea (Kearns et al. 2011) and (iii) the Central cordillera of New Guinea (Rawlings & Donnellan 2003; Kearns et al. 2011).
Study system
In this study, we focus on three closely related mosquito species: Anopheles farauti (formerly An. farauti 1), An. hinesorum (formerly An. farauti 2) and An. irenicus (formerly An. farauti 7; Beebe et al. 2000c). Although morphologically indistinguishable, these species are identifiable using allozymes (Foley et al. 1993), PCR-based diagnostics and genomic DNA probes (Cooper et al. 1991; Beebe & Saul 1995) and are part of a larger species complex. Phylogenetic studies of the species complex suggest that the species under investigation are each other’s closest relatives (with the inclusion of the highland restricted species, An. farauti 5 and 6, which are most closely related to An. hinesorum and are only found in the Central Highlands of New Guinea; Foley et al. 1998; Beebe et al. 2000c; Beebe & Cooper 2002). Two of the species (An. farauti and An. hinesorum) are widespread malaria transmitting mosquitoes (Cooper et al. 2009), which occur east of the Webber line through New Guinea, the Bismarck Archipelago and the Solomon Islands with An. farauti extending to Vanuatu. The range of both species also extends southward into northern Australia, with An. farauti also present on the Torres Strait Islands between Australian and New Guinea (see Fig. 1a for a map of the region; Beebe & Cooper 2002; Cooper et al. 2002). In contrast An. irenicus has a very narrow range, limited to Guadalcanal in the Solomon Islands, and it does not bite humans (Beebe & Cooper 2002; Cooper et al. 2002). The species also differ in distribution and ecology; An. farauti is a coastally restricted mosquito (Beebe & Cooper 2002; Cooper et al. 2002) with larvae that are tolerant of brackish water (Sweeney 1987)—as are the larvae of An. irenicus (Foley & Bryan 2000). On the other hand, An. hinesorum is found both on the coast and inland, as well as in the cooler climate of New Guinea’s highlands (up to 1500 m; Cooper et al. 2002), and its larvae develop in freshwater. Despite this difference in larval salt tolerance, An. farauti and An. hinesorum are frequently found cohabiting freshwater coastal larval sites (Cooper et al. 2002). A better understanding of the population genetics and evolutionary relationships of these mosquitoes is of significant value in terms of malaria control, particularly with regards to the evolution and potential spread of insecticide-resistant (biochemical or behavioural) phenotypes.
Fig. 1.
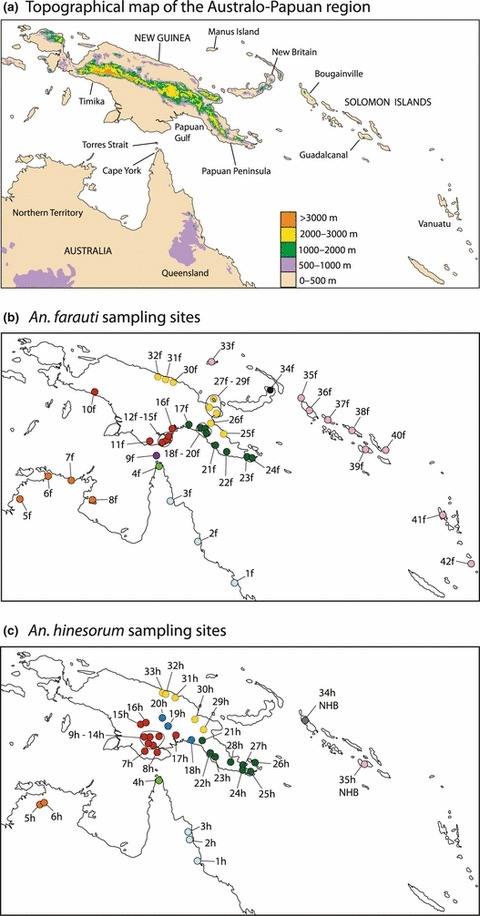
Maps of the southwest Pacific, including the topography of the region and approximate sampling locations for Anopheles farauti and An. hinesorum. Sites are colour coded by region in the same way throughout the study. See Fig 5 for key and Tables 1 and 2 for detailed sampling information.
Both An. hinesorum and An. farauti bite humans and mammals, as do most other species in the broader group of closely related cryptic species (the Punctulatus group; Cooper et al. 2009). Additionally, An. koliensis and An. punctulatus are positioned basally within the Punctulatus group (Beebe et al. 2000c) and are generalist feeders that bite humans and transmit malaria in the region (Cooper et al. 2009). Thus, the (generalist) ability to perceive humans as suitable hosts is most likely the ancestral character of the group. With this in mind, an interesting behavioural pattern has been observed—two populations belonging to different species co-occurr in the Solomon Islands and do not bite humans. These are An. irenicus (found only on the island of Guadalcanal) and the Solomon Island populations of An. hinesorum (Foley et al. 1994; Beebe et al. 2000a; Cooper & Frances 2002). Although limited to few individuals per species and mostly to single markers, previous phylogenetic studies suggest that An. irenicus is more closely related to An. farauti than to An. hinesorum (Foley et al. 1998; Beebe et al. 1999, 2000c, d; Bower et al. 2009). These relationships indicate that the ability to perceive humans as hosts may have been lost in co-occurring island populations of An. irenicus and An. hinesorum. If this is the case, there may be opportunities to investigate the genetic mechanisms involved in this loss, which would be of significant medical interest. Throughout the rest of manuscript, we refer to this loss of perception of humans as hosts as ‘non-human biting behaviour’. In contrast to An. hinesorum and An. irenicus, An. farauti bite humans and transmit malaria wherever there are infected hosts, including on the Solomon Islands.
The history of colonization of An. farauti into the Solomon Islands remains unclear and has only been investigated using ribosomal DNA ITS genotyping methods (Beebe et al. 2000b; Cooper et al. 2002) or limited geographical sampling (Hasan et al. 2008). Further, the history of colonization by An. hinesorum of the Solomon Islands has not yet been investigated. However, previous studies on An. farauti in the southwest Pacific have found significant regional population structure, both within and between landmasses, based on multiple ribosomal DNA internal transcribed spacer (rDNA ITS1 and ITS2) genotypes (Beebe et al. 2000b; Bower et al. 2008).
In this study, we aim to further examine evolutionary relationships between An. farauti, An. hinesorum and An. irenicus to explicitly assess whether human biting ability has been repeatedly lost in the Solomon Islands. We hypothesize that multiple gene trees will support the existence of discretely evolving lineages corresponding to recognized species and that An. irenicus will be most closely related to An. farauti, with An. hinesorum being more distantly related. Therefore, we also hypothesize that non-human biting behavior has evolved more than once in the species complex.
Additionally, we aim to compare geographical patterns of genetic differentiation between populations of An. farauti and An. hinesorum occurring in the Australo-Papuan region and islands of the southwest Pacific. We hypothesize that mitochondrial and nuclear sequence data will reveal population structure within New Guinea and Australia, as well as between the landmasses. As An. hinesorum is most commonly found inland, it is likely to encounter more barriers to dispersal than An. farauti, which inhabits a comparatively uniform coastal habitat. Thus, we predict that within New Guinea, An. hinesorum will contain more population structure than An. farauti. Sequence data for the mitochondrial cytochrome oxidase 1 (COI) gene, the single copy nuclear locus ribosomal protein S9 (rpS9) and cloned ITS2 sequence data were used to address these aims. This study design has allowed us to make comparisons between species, as well as between unlinked markers with very different modes of evolution.
Materials and methods
Sampling and laboratory procedures
Most mosquitoes were field collected from northern Australia, New Guinea, the Solomon Islands and Vanuatu by RD Cooper from the Australian Army Malaria Unit between 1991 and 2009. A few collection sites in Queensland were obtained through colleagues at Queensland Health. Significantly, this sampling encompasses almost the entire known geographical range of all species included in this study. Adult mosquitoes were caught either in CO2 light traps or by night human biting catches. Some specimens were also collected as larvae and reared to adulthood. Mosquitoes were identified by species-specific DNA probe and/or PCR–RFLP analysis (Cooper et al. 1991; Beebe et al. 1994; Beebe & Saul 1995), and those identified as Anopheles farauti, An. hinesorum and An. irenicus were used in this study. It should be noted that we do not have a complete (multilocus) set of data for all individuals. Sampling information on mosquito material (An. farauti and An. hinesorum) used is summarized in Tables 1–3 and sampling locations are shown in Fig. 1. For An. irenicus, all individuals used were sampled from Guadalcanal Island (one individual for ITS2, four individuals for COI and three individuals for rpS9).
Table 1.
Sampling data and genetic diversity indices for Anopheles farauti
| COI | rpS9 | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| Region | Site ID* | Co-ordinates† | n (COI, rpS9)‡ | Hd§ | π¶ | TD** | Hd§ | π¶ | TD** |
| Queensland | 1f | −21.133, 149.183 | 2, 0 | 1 | 0.57 | n/a | — | — | — |
| 2f | −17.691, 146.107 | 10, 0 | 0.956 | 0.61 | 0.03 | — | — | — | |
| 3f | −14.086, 143.676 | 5, 0 | 0.9 | 0.38 | 0.27 | — | — | — | |
| Cape York | 4f | −10.778, 142.415 | 5, 0 | 0.8 | 0.99 | 1.45 | — | — | — |
| Northern Territory | 5f | −13.926, 130.482 | 3, 0 | 0 | 0 | n/a | — | — | — |
| 6f | −11.8, 132.633 | 5, 0 | 0.9 | 0.49 | −0.67 | — | — | — | |
| 7f | −12.125, 134.746 | 9, 0 | 0.417 | 0.21 | −1.68 | — | — | — | |
| 8f | −13.841, 136.493 | 3, 0 | 0.667 | 0.13 | n/a | — | — | — | |
| Torres Strait | 9f | −10.167, 142.2 | 10, 0 | 0.8 | 0.46 | −0.09 | — | — | — |
| Timika | 10f | −4.835, 136.758†† | 5, 5 | 0.6 | 0.11 | 1.22 | 0.867 | 0.50 | −1.16 |
| Southern New Guinea | 11f | −9.2, 141.583 | 10, 5 | 0.511 | 0.34 | 0.07 | 0.867 | 0.30 | −0.4 |
| 12f | −9.25, 142.768 | 0, 1 | — | — | — | — | — | n/a | |
| 13f | −9.07, 143.211 | 0, 10 | — | — | — | 0.632 | 0.20 | −0.77 | |
| 14f | −8.938, 143.409 | 0, 3 | — | — | — | 0.733 | 0.21 | n/a | |
| 15f | −8.6, 143.557 | 0, 5 | — | — | — | 0.778 | 0.44 | −0.58 | |
| 16f | −8.043, 143.723 | 10, 0 | 0.844 | 0.43 | −0.36 | — | — | — | |
| 17f | −7.777, 144.889 | 13, 6 | 0.872 | 1.01 | −1.36 | 0.939 | 1.42 | 1.2 | |
| Southern Papuan Peninsula | 18f | −8.072, 146.04 | 9, 6 | 0.944 | 0.69 | −0.09 | 0.909 | 1.25 | 0.54 |
| 19f | −8.339, 146.549 | 6, 0 | 0.933 | 0.44 | −1.39 | — | — | — | |
| 20f | −8.724, 146.569 | 0, 4 | — | — | — | 0.786 | 1.20 | n/a | |
| 21f | −9.491, 147.293 | 9, 0 | 0.944 | 0.84 | 0.46 | — | — | — | |
| 22f | −10.15, 148.368 | 0, 5 | — | — | — | 0.867 | 1.32 | 0.16 | |
| 23f | −10.438, 149.891 | 9, 7 | 0.833 | 0.36 | −1.19 | 0.813 | 0.53 | −1.43 | |
| 24f | −10.347, 150.564 | 9, 0 | 0.889 | 0.27 | −0.91 | — | — | — | |
| Northern Papuan Peninsula | 25f | −8.148, 148.137 | 5, 8 | 0.700 | 0.27 | −0.17 | 0.742 | 0.26 | −0.34 |
| 26f | −7.123, 147.041 | 9, 9 | 0.806 | 0.45 | −0.32 | 0.660 | 0.24 | −0.47 | |
| 27f | −6.741, 147.535 | 0, 5 | — | — | — | 0.733 | 0.34 | 1.15 | |
| 28f | −5.959, 147.079 | 0, 3 | — | — | — | 0.8 | 0.32 | n/a | |
| 29f | −5.268, 147.178 | 0, 8 | — | — | — | 0.242 | 0.06 | −1.5 | |
| Northern New Guinea | 30f | −4.056, 144.741 | 9, 12 | 0.972 | 0.54 | −0.17 | 0.663 | 0.22 | −1.29 |
| 31f | −3.865, 144.064 | 9, 6 | 0.944 | 0.47 | −0.68 | 0.758 | 0.32 | −0.72 | |
| 32f | −3.531, 143.589 | 3, 8 | — | — | — | 0.783 | 0.30 | 0.14 | |
| Manus | 33f | Manus Island | 5, 5 | 0.800 | 0.26 | −0.17 | 0 | 0 | 0 |
| New Britain | 34f | −4.237, 152.15†† | 1, 0 | n/a | n/a | n/a | — | — | — |
| Solomon Islands | 35f | Buka Island | 3, 0 | 0 | 0 | n/a | — | — | — |
| 36f | Bougainville Island | 14, 10 | 0.538 | 0.31 | 2.18 | 0.279 | 0.07 | −1.14 | |
| 37f | Choiseul Island | 10, 4 | 0.756 | 0.31 | −0.33 | 0.464 | 0.12 | n/a | |
| 38f | Santa Isabel Island | 0, 20 | — | — | — | 0.677 | 0.22 | −0.6 | |
| 39f | Guadalcanal | 9, 0 | 0.639 | 0.15 | 0.19 | — | — | — | |
| 40f | Ulawa Island | 10, 5 | 0.733 | 0.19 | −0.13 | 0.467 | 0.11 | 0.82 | |
| Vanuatu Islands | 41f | Espiritu Santo | 4, 0 | 0.833 | 0.41 | n/a | — | — | — |
| 42f | Tanna Island | 10, 0 | 0 | 0 | 0 | — | — | — | |
| Total Data | n/a | n/a | 233, 160 | 0.976 | 2.20 | 0.44 | 0.766 | 0.98 | −0.978 |
Data shown include sampling information (*site ID and †geographic co-ordinates, ‡number of individuals (n) sequenced per locus), as well as genetic diversity indices for each locus [§haplotype diversity (Hd), ¶nucleotide diversity (π) and **neutrality tests (Tajima’s D)] per site and for total species data. Locations for islands are given as island names. ††Approximate co-ordinates. GenBank Accession numbers (COI: JN384217–JN384661; rpS9: JN384662–JN385275; ITS2: JN564766–JN564794, AF033218, HM584426, AF1404314–AF104326).
Table 3.
Summary of An. hinesorum ITS2 sequencing
| Region | Site ID | n | ITS2 sequences* |
|---|---|---|---|
| Queensland | 3h | 3† | 2–4 |
| Cape York | 4h | 3† | 5–7 |
| Northern Territory | 5h | 3† | 1, 2 |
| 6h | 3† | 1, 2 | |
| Southern New Guinea | 8h | 3† | 8–9, 12–14 |
| 9h | 3† | 2, 11–15 | |
| Central New Guinea | 18h | 4 | 16, 20 |
| 19h | 5 | 20 | |
| 20h | 5 | 20 | |
| Papuan Peninsula | 21h | 3† | 17–19 |
| 22h | 2† | 17, 18 | |
| 25h | 3† | 17–19 | |
| 28h | 3† | 17, 18 | |
| Northern New Guinea | 29h | 4 | 21 |
| 31h | 1 | 21 | |
| 32h | 1 | 21 | |
| 33h | 3 | 21 | |
| Bougainville Island | 34h | 3‡ | 22–26 |
| Guadalcanal Island | 35h | 3‡ | 27–29 |
ITS2 sequences 1–29 correspond to GenBank Accession nos JN564766–JN564794.
Three individuals per site were cloned with 3–5 random clones sequenced.
Mosquito isolates from three separate larval sites.
DNA extraction, amplification, cloning and sequencing
Genomic DNA was extracted from all specimens using a standard salt extraction method (Beebe et al. 1999). Final PCR mixtures for all loci amplified contained 2.5 mm MgCl, 0.125 mm of each dNTP, 0.4 μm of each primer, 0.5–1.0 unit of Taq polymerase and 5.0–10.0 ng of extracted genomic DNA (1 μL of extraction). The cycling for all loci involved an initial denaturation step of 95 °C for 3 min, followed by 35 cycles of 95 °C for 1 min, 50 °C for 1 min (57 °C for rpS9) and 72 °C for 1 min. Part of the mtDNA COI gene was amplified using primers from Walton et al. (2000) and a novel reverse primer (5′-TAA TAT AGC ATA AAT TAT TCC-3′) yielding a 527 base pair (bp) product after editing. An exon priming intron crossing (EPIC) fragment of the nuclear-ribosomal protein S9 (rpS9) was amplified using the forward primer (5′-GAA AAG CCR CGT CTC GAT GCG G-3′) and the reverse primer (5′-GCC AAT CCC AGC TTG AAS ACC-3′), designed by aligning an Aedes aegypti mRNA sequence (GenBank: XM_001653919) to the An. gambiae genome, yielding a final length of 431 bp after editing. The ITS2 locus was amplified and cloned following procedures outlined in a previous study (Beebe et al. 1999). Amplified products were purified using the QIAquick PCR purification kit (Qiagen, Hilden, Germany) or using an enzyme digestion containing 1 unit of Exonuclease I, 1 unit of Antarctic Phosphatase and a final concentration of 1× Antarctic Phosphatase buffer per reaction, with an incubation period of 37 °C for 30 min followed by 80 °C for 5 min. Sequencing was performed by the Australian Genome Research Facility and by Macrogen (Korea) and sequences have been submitted to GenBank (refer to Table 1 for accession numbers).
The ITS2 is part of the rDNA repetitive gene family, and intragenomic copy variants of ITS2 frequently occur in Anopheles mosquitoes, preventing direct DNA sequencing. The ITS2 sequence data of An. farauti (GenBank numbers AF104314–AF104326) and An. irenicus (GenBank number AF033218) used in this study was generated in previous studies (Beebe et al. 1999), with geographically selected individuals of An. farauti PCR amplified, cloned and sequenced. New ITS2 sequences for An. hinesorum were generated for this study via PCR and cloning, from a subset of individuals representing as best as possible the distribution of this species. Approximately three An. hinesorum individuals per site were cloned and 3–5 randomly selected colonies sequenced following methods of (Beebe et al. 1999). Table 3 provides a summary of ITS2 sequencing of An. hinesorum individuals.
Sequence quality and editing
Prior to analysis, sequences were verified as being of mosquito origin by performing a blast search against the NCBI GenBank nucleotide database. For quality assurance, we translated all mtDNA COI nucleotide sequences into amino acid sequences and found no stop codons in the reading frame of any sequence. Sequences were aligned and edited in the program Geneious (Drummond et al. 2010), and pseudo-haplotypes of the nuclear locus rpS9 were inferred using the program PHASE, implemented in DNAsp v5 (Rozas et al. 2003). We tested for recombination within the nuclear loci using the program RPD3 (Martin et al. 2010).
Data analysis
Genetic diversity and tests of neutrality
Tajima’s D (TD) test (Tajima 1989) was used to test for selective neutrality and equilibrium expectations and was computed for sites with five or more individuals sampled, as well as for the total data of each species in Arlequin v 3.5 (Excoffier & Lischer 2010). Bonferroni corrections were applied to assess the significance of TD tests. To estimate levels of genetic diversity, Arlequin was used to compute π (nucleotide diversity), and haplotype diversity (Hd) was calculated in DNAsp.
Phylogenetic and coalescent analyses
To infer evolutionary relationships between the species at mtDNA and nuclear DNA (nDNA) loci, phylogenies were constructed using Bayesian (beast; Drummond & Rambaut 2007) and maximum likelihood (ML) methods (PhyML; Guindon et al. 2009). For ML phylogenies, duplicate sequences (individuals with identical sequences) were removed from alignments prior to analysis, whereas for beast (coalescent) analyses, all sampled haplotypes were included. jModelTest version 0.1.1 (Posada 2008) was run using three substitution schemes to determine the most appropriate model of evolution to use in subsequent analyses. An HKY model (Hasegawa et al. 1985) with a gamma distribution and a proportion of invariable sites was used in analyses of the COI locus. For the rpS9 locus, a K80 (K2P) model (Kimura 1980) with a gamma distribution was used, and for the ITS2 locus, a GTR model with a gamma distribution was used. For all ML analyses, PhyML was run using the ML method with 1000 bootstraps. ITS2 data were only used for phylogenetic analysis because it is part of the multicopy rDNA gene family that evolves through a poorly described non-Mendelian process (Beebe et al. 1999; Alquezar et al. 2010). Additionally, Anopheles mosquitoes often carry multiple ITS2 sequence variants in the rDNA array, thus cloning is required prior to sequencing.
Analyses were also run in *beast (Heled & Drummond 2010) to estimate species/population relationships based on multilocus data and to estimate dates of divergence between An. farauti and An. hinesorum. Individuals were assigned to populations and species (as traits) in *beast analyses based on their geographic origin. Groups assigned for An. farauti include: southern New Guinea (sites 10–16f), Papuan Peninsula (sites 17–24f), northern New Guinea (sites 25–32f), Manus Island (site 33f) and Solomon Islands (sites 35–40f); groups for An. hinesorum include: southern New Guinea (sites 8–20h), Papuan Peninsula (sites 21–26h), northern New Guinea (sites 29–33h), north Solomon Islands (site 34h) and south Solomon Islands (site 35h). An. irenicus and the outgroup An. koliensis were also included in the *beast analysis. We specified a rate of 0.0115 mutations (2.3%) per million years for the COI locus, based on the widely used mitochondrial insect molecular clock (Brower 1994) and used the relaxed lognormal clock as recommended by the developers of beast as it allows the mutation rate to vary between branches (Drummond et al. 2006). We allowed the program to estimate the substitution rate of the rpS9 locus. beast was also run using the simpler strict clock model, but we found that the relaxed lognormal clock model outperformed the strict clock model. This was based on an estimation of Bayes factors between models: the log10 BF of the relaxed clock relative to the strict clock model = 0.667, which is taken as evidence in favour of a relaxed clock model (Kass & Raftery 1995). Bayes Factors were computed in Tracerv.1.5 (Rambaut & Drummond 2009) from the mean likelihoods of each model.
Two independent analyses of 50 million generations were run in BEAST and *BEAST, with trees sampled every 1000 generations. Log files and tree files from the two runs were then combined in LogCombiner v1.6.1 and a burnin of 10 million generations was applied. We assessed the performance of the combined runs in Tracer v1.5, and all ESS values were 200 or above for beast analyses, as recommended by the developers, however, for the *beast analyses, a few ESS values failed to reach 200 despite the long run time. Tree files generated from the programs were run in Tree Annotator version v1.6.1 and viewed in FigTree v1.3.1 (Rambaut 2009). We also performed parsimony based ancestral state reconstruction on the mitochondrial ML tree using the program Mesquite (Maddison & Maddison 2011). Where citations are not included, the above-mentioned programs are codistributed with beast.
Phylogeography and population structure
To examine phylogeographic relationships in An. farauti and An. hinesorum, we constructed maximum parsimony haplotype networks (using all sampled haplotypes) in tcs (Clement et al. 2000). Levels of differentiation between sites for COI (mtDNA) and rpS9 (nDNA) were estimated by generating pairwise FST values in Arlequin version 3.5 (distance method). The significance levels of FST comparisons were assessed using permutation tests (1023 permutations per comparison), also implemented in Arlequin. We only performed pairwise FST comparisons for populations with five or more individuals sampled. Due to the large number of sites sampled, we summarized pairwise FST relationships using multidimensional scaling plots generated in the program R version 1.4 (R Development Core Team 2011). Multidimensional scaling plots are similar to principle component analysis plots but relative distances between points represent relationships between pairwise comparisons. This allows a visually intuitive and compact representation of FST-based relationships between sites (and the regions they were sampled from). Detailed tables summarizing FST relationships and significance of comparisons are available as supplementary material (Tables S1–S4, Supporting information).
Results
Genetic diversity and tests of neutrality/recombination
Tables 1 and 2 show genetic diversity indices and results of TD tests. Our data for Anopheles farauti contained a total of 82 COI haplotypes (Hd = 0.976, π = 2.20) and 49 (inferred) rpS9 haplotypes (Hd = 0.766, π = 0.98; Table 1). For An. hinesorum, we found a total of 94 COI haplotypes (Hd = 0.969, π = 2.54) and 45 (inferred) rpS9 haplotypes (Hd = 0.872, π = 1.09; Table 2). Most tests of selection gave nonsignificant results, including tests performed with the total data of each species at both loci. This suggests that populations approximate the expectations of mutation-drift equilibrium. The only site that gave a significant TD test value following Bonferroni correction was the An. hinesorum site 34h (TD = −2.45, P < 0.001). We found no evidence of recombination at either of the nuclear loci.
Table 2.
Sampling data and genetic diversity indices for Anopheles hinesorum
| COI | rpS9 | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| Region | Site ID* | Co-ordinates† | n (COI, S9)‡ | Hd§ | π¶ | TD** | Hd§ | π¶ | TD** |
| Queensland | 1h | −18.216, 145.854 | 8, 0 | 0.929 | 0.50 | 0.69 | — | — | — |
| 2h | −16.673, 145.328 | 6, 0 | 0.867 | 0.56 | −0.25 | — | — | — | |
| 3h | −16.252, 145.302 | 7,0 | 0.952 | 0.49 | −1.59 | — | — | — | |
| Cape York | 4h | −10.954, 142.462 | 19, 0 | 0.965 | 1.19 | −0.74 | — | — | — |
| Northern Territory | 5h | −12.96, 132.56 | 7, 0 | 0.810 | 0.23 | 0.05 | — | — | — |
| 6h | −12.76, 132.66 | 4, 0 | 1 | 1.55 | n/a | — | — | — | |
| Southern New Guinea | 7h | −8.634, 142.218 | 1, 2 | — | — | — | 1 | 1.51 | n/a |
| 8h | −8.621, 141.137 | 3, 0 | 0 | 0 | n/a | — | — | — | |
| 9h | −8.074, 141.750 | 5, 2 | 0.900 | 0.91 | −0.34 | 0 | 0 | n/a | |
| 10h | −7.728, 141.490 | 9, 6 | 0.889 | 1.22 | 0.46 | 0.939 | 1.31 | 0.58 | |
| 11h | −6.978, 141.503 | 0, 2 | — | — | — | 0.833 | 0.35 | n/a | |
| 12h | −7.182, 141.247 | 5, 9 | 0.900 | 0.64 | −0.81 | 0.961 | 0.90 | −1.01 | |
| 13h | −7.079, 141.989 | 6, 7 | 1 | 1.15 | −0.06 | 0.571 | 0.83 | n/a | |
| 14h | −6.823, 141.371 | 10, 8 | 0.844 | 0.25 | −1.04 | 0.575 | 0.43 | 0.73 | |
| 15h | −6.317, 141.024 | 0, 1 | — | — | — | 1 | 0.70 | n/a | |
| 16h | −6.121, 141.294 | 0, 2 | — | — | — | 0.833 | 0.70 | n/a | |
| 17h | −7.319, 144.178 | 0, 2 | — | — | — | 0.500 | 0.46 | n/a | |
| Central New Guinea | 18h | −7.864, 145.669 | 11, 8 | 0.182 | 0.45 | −2.05 | 0.342 | 0.09 | −1.04 |
| 19h | −6.78, 143.469 | 11, 12 | 0.618 | 0.23 | −1.57 | 0.471 | 0.13 | 0.06 | |
| 20h | −6.205, 143.01 | 1, 10 | — | — | — | 0.584 | 0.18 | 0.87 | |
| Papuan Peninsula | 21h | −7.966, 146.214 | 8, 9 | 0.964 | 0.47 | −0.35 | 0.562 | 0.17 | −1.53 |
| 22h | −8.999, 146.801 | 9, 12 | 0.972 | 0.50 | −0.97 | 0.308 | 0.08 | −1.49 | |
| 23h | −9.145, 147.166 | 0, 1 | — | — | — | 0 | 0 | n/a | |
| 24h | −10.203, 149.695 | 0, 1 | — | — | — | 0 | 0 | n/a | |
| 25h | −10.329, 150.271 | 4, 3 | 1 | 0.69 | n/a | 0.533 | 0.12 | n/a | |
| 26h | −9.668, 150.787 | 12, 12 | 0.894 | 0.51 | −0.37 | 0.163 | 0.04 | −1.51 | |
| 27h | −9.704, 149.652 | 0, 2 | — | — | — | 0 | 0 | 0 | |
| 28h | −9.505, 148.462 | 4, 0 | 1 | 0.51 | n/a | — | — | — | |
| Northern New Guinea | 29h | −6.86, 146.351 | 4, 3 | 0.833 | 0.38 | −0.78 | 0.600 | 0.45 | n/a |
| 30h | −5.769, 145.592 | 1, 0 | n/a | n/a | n/a | — | — | — | |
| 31h | −3.956, 143.927 | 1, 0 | n/a | n/a | n/a | — | — | — | |
| 32h | −3.782, 143.336 | 3, 3 | 0.667 | 0.38 | 0 | 0 | 0 | 0 | |
| 33h | −3.8, 143.066 | 5, 4 | 0.400 | 0.23 | −1.05 | 0.786 | 0.39 | −1.28 | |
| Solomon Islands | 34h | Bougainville Island | 21, 8 | 0.271 | 0.43 | −2.45* | 0 | 0 | 0 |
| 35h | Guadalcanal | 21, 11 | 0.348 | 0.80 | −1.29 | 0.416 | 0.39 | 1.44 | |
| Total Data | n/a | n/a | 206, 140 | 0.969 | 2.54 | −0.343 | 0.872 | 1.09 | −1.166 |
Data shown include sampling information (*site ID and †geographic co-ordinates, ‡number of individuals (n) sequenced per locus), as well as genetic diversity indices for each locus [§haplotype diversity (Hd), ¶nucleotide diversity (π) and **neutrality tests (Tajima’s D)] per site and for total species data. Locations for islands are given as island names. Statistically significant results following Bonferroni correction with an asterisk. GenBank Accession numbers (COI: JN384217–JN384661; rpS9: JN384662–JN385275; ITS2: JN564766–JN564794, AF033218, HM584426, AF1404314–AF104326).
Phylogenetic and coalescent analysis
Phylogenies of mitochondrial and nuclear markers (Figs 2a and 3) suggest that the non-human biting species, An. irenicus, is monophyletic and most closely related to the human biting An. farauti. On the basis of mitochondrial genealogies, we infer that four independent and strongly supported clades from the three species under investigation occur on the Solomon Islands (two An. hinesorum, and one of each of An. farauti and An. irenicus; Fig. 2a). Interestingly, the two (non-human biting) An. hinesorum Solomon Islands mtDNA lineages are phylogenetically distinct from each other. Ancestral state reconstruction supported human biting behaviour as the ancestral trait, and given the close relationship between An. irenicus and An. farauti, this supports the hypothesis that non-human biting behaviour has evolved at least twice on the Solomon Islands. Although monophyletic and distinct (with the exception of one Solomon Islands haplotype that is nested within a closely related clade sampled from the Papuan Peninsula), the less divergent lineage is most closely related to populations found on the Papuan Peninsula.
Fig. 2.
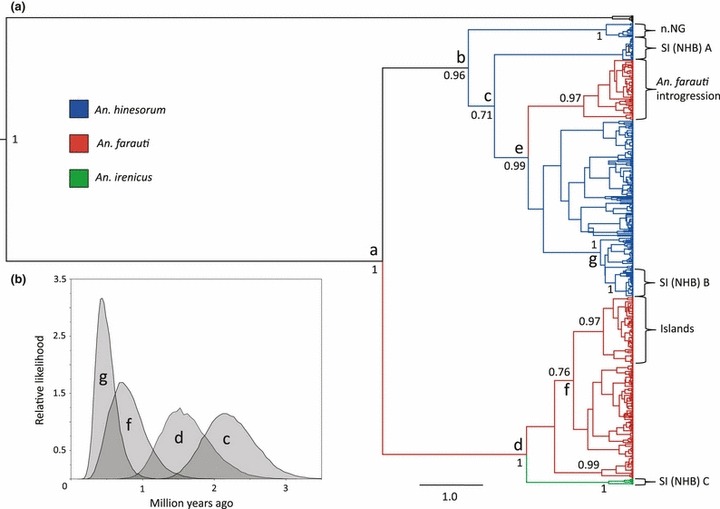
(a) Bayesian (beast) phylogeny (maximum clade credibility) of the COI locus containing all haplotypes sampled. Branches are coloured by species [red = Anopheles farauti, blue = An. hinesorum, green = An. irenicus, black = An. koliensis (outgroup)]. Support values are posterior probabilities, and scale bar shows time in millions of years. A standard insect molecular clock of 2.3% divergence per million years was used to scale the tree. Abbreviations used on labels include: NHB, non-human biting; SI, Solomon Islands. Further details on node and clade labels are described in the results section. (b) Graph generated from the combined beast log files showing the most likely dates of divergence of nodes on the beast phylogeny (a), as well as the range of dates divergence for nodes.
Fig. 3.
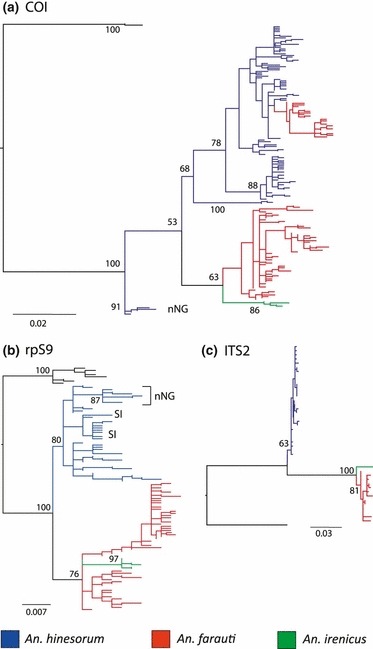
Maximum likelihood phylogenies of: (a) COI, (b) rpS9 and (c) ITS2. Duplicate sequences were removed prior to the generation of phylogenies. Branches are coloured by species [red = Anopheles farauti, blue = An. hinesorum, green = An. irenicus, black = An. koliensis (outgroup)]. Support values on nodes are percentages out of 1000 bootstraps; scale bar is in units of number of nucleotide substitutions per site, nNG, northern New Guinea.
We found that An. farauti and An. hinesorum are polyphyletic for mtDNA, with a monophyletic lineage of An. farauti (found throughout Queensland, the Torres Strait Islands and southern New Guinea) nested within An. hinesorum (Figs 2a and 3a). In contrast to the mitochondrial trees, phylogenies of the two nuclear markers show that An. farauti and An. hinesorum are reciprocally monophyletic (Fig. 3b,c). This conflict between gene trees may be due to either ancient mitochondrial introgression, or incomplete lineage sorting at the mitochondrial locus.
In the mtDNA phylogenies, a basal lineage within An. hinesorum clearly stands out (∼3% divergence) and was sampled exclusively from northern New Guinea. However, there is conflict in the position of this clade between trees generated using different phylogenetic models. Maximum likelihood phylogenies suggest that it diverged earlier than the An. hinesorum—An. farauti split (Fig. 3a), whereas Bayesian methods suggest that it diverged after the two species split. In the rpS9 phylogeny, An. hinesorum from northern New Guinea also form a distinct clade, although unlike the mtDNA phylogenies populations from this region are nested within the rest of An. hinesorum (Fig. 3b). These populations are currently identified as An. hinesorum but their high level of divergence and genetic distinctness suggest that they may be a separate species.
The multilocus tree generated using *beast (Fig. 4) reveals that An. hinesorum (with the exception of northern New Guinea) and An. farauti are reciprocally monophyletic. On the basis of the molecular clock used, it appears that the two species diverged about 2 Ma (95% HPD height: 3.04–1.15 Mya, Fig. 4). This confidence interval is overlapping with the estimated timing of the putative mitochondrial introgression (95% HPD height: 2.19–1.11 Mya, Fig. 3 node e and Fig. 4). Thus, the null hypothesis of ancestral polymorphism cannot be rejected as the cause of the mitochondrial polyphyly of An. farauti. The sister taxa relationship between An. farauti and An. irenicus is also supported by the multilocus tree, as is the early divergence (pre the split of An. farauti and An. hinesorum) of northern New Guinean populations of An. hinesorum. A close relationship can be seen between northern and southern Solomon Island populations in the multilocus tree. This can be explained by both mitochondrial lineages being found in the northern and the southern Solomon Islands, as well as there being heterozygotes between populations for the nuclear (rpS9) locus.
Fig. 4.
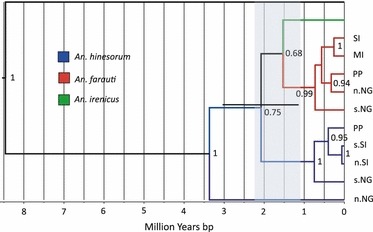
Phylogenetic relationships between species and populations estimated by jointly analysing COI and rpS9 loci in *beast. Branches are coloured by species as shown in the key and the outgroup is Anopheles koliensis (black). Support values are posterior probabilities, and the scale bar shows time in millions of years (based on the molecular clock used—2.3% per million years for mtDNA; nuclear DNA estimated by the program). The error bar on the node at which An. farauti and An. hinesorum split is the 95% HPD (height) and shows the error associated with the estimate of the timing of divergence between the species. The shaded area shows the 95% HPD (height) of the putative introgression event based on the mtDNA beast phylogeny (node e). Abbreviations include: SI, Solomon Islands; MI, Manus Island; PP, Papuan Peninsula; n.NG, northern New Guinea; s.NG, southern New Guinea; s.SI, southern Solomon Islands; and n.SI, northern Solomon Islands.
Phylogeography and population structure
Anopheles farauti
The An. farauti mtDNA COI haplotype network (Fig. 5b), as well as MDS plots of pairwise FST comparisons (Fig. 6a), reveal well-defined geographical groups. Groups identified include the following: (1) Queensland—including Cape York; (2) Torres Strait, southern New Guinea and Timika (both of which are part of the An. farauti lineage containing mtDNA introgressed from An. hinesorum and are therefore shown on the An. hinesorum haplotype network: Fig. 5e, Group 1); (3) Northern Territory and (4) the Solomon and Vanuatu islands; (Figs 5b and 6a). Some structure is also evident between An. farauti populations from the southern Papuan Peninsula and northern New Guinea (Fig. 6a). Island populations of An. farauti form a monophyletic lineage (Fig. 5b). The most closely related mitochondrial relatives to the lineage with putatively introgressed mtDNA appear to be An. hinesorum from the Cape York Peninsula, which differ from the introgressed lineage by at least four mutational steps (Fig. 5e).
Fig. 5.
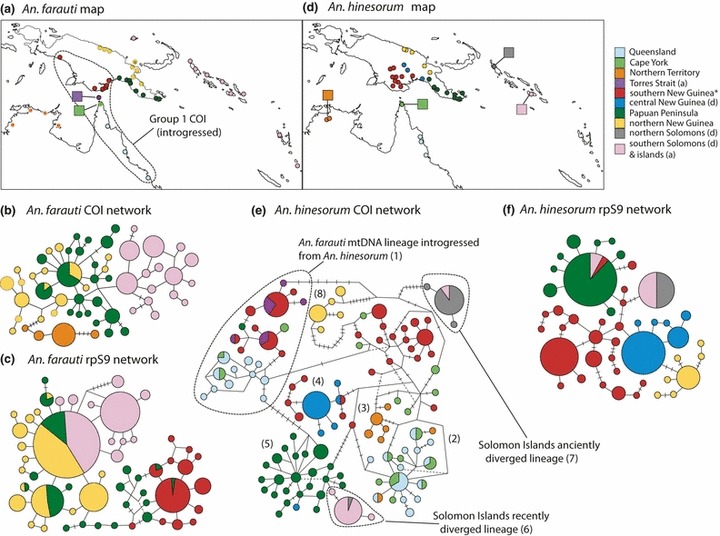
Maximum parsimony haplotype networks of Anopheles farauti and An. hinesorum at COI and rpS9 loci. Circles represent haplotypes and the size of circles is relative to the number of individuals per haplotype. Lines between haplotypes represent a single mutational change, as do dashes across lines, bends and intersections. Colours indicate sampling locality of haplotypes (i.e. geographic region), see maps and legend for details. The An. farauti lineage containing mtDNA introgressed from An. hinesorum is shown in the An. hinesorum network (Group 1 in e).
Fig. 6.
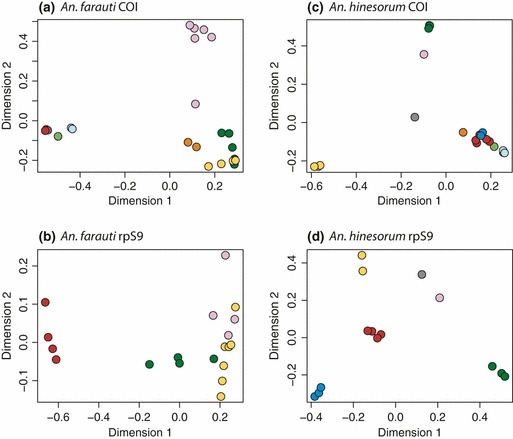
Multidimensional scaling plots of pairwise FST values. Each point represents a sampling site and distances between points are relative to FST values. Sites are coloured by geographical region, consistent with maps presented throughout.
Though less informative, the rpS9 locus is largely concordant with the COI locus. The strongest genetic break at the rpS9 locus is between southern New Guinea and the other sites (Figs 5c and 6b). However, some specimens from the Papuan Peninsula have close affinities to the southern New Guinea populations, with two individuals (17f-15 and 17f-13) possessing a haplotype more commonly found in southern New Guinea (Fig. 5c). Although there are high fixation indices between some sites (Fig. 6b, Table S2, Supporting information), patterns of structure throughout the rest of New Guinea are difficult to distinguish.
Anopheles hinesorum
Both the haplotype network (Fig. 5e) and MDS plots of pairwise FST values (Fig. 6c) for the COI locus of An. hinesorum show that there is strong genetic structure between geographically distinct groups of this species. The haplotype network reveals that the Queensland and Cape York populations form a group (Fig. 5e: Group 2) that is closely related to a Northern Territory group (Fig. 5e: Group 3) and also to haplotypes from southern New Guinea. Within Group 2 are two haplotypes shared with individuals from the Northern Territory 3, possibly suggesting recent dispersal events from Queensland to the Northern Territory. Additionally, a number of individuals from Cape York do not fall within Group 2, but are more closely related to individuals from southern New Guinea with which they make up the highly reticulated centre of the network (Fig. 5e), although no haplotypes are shared between Cape York and southern New Guinea. Also closely related to the southern New Guinean populations are populations from central New Guinea (Fig. 5e: Group 4), and there are shared haplotypes between these regions. The MDS plot of pairwise FST values suggests a close affinity between all regions outlined previously (Fig. 6c), although sites from different regions are significantly differentiated from each other (Table S3, Supporting information).
Mitochondrial haplotypes of all An. hinesorum individuals from the Papuan Peninsula form a distinct group (Group 5) and MDS plots of pairwise FST values show that this region is very different from other regions sampled (Fig. 6c). Group 6 was sampled from the Solomon Islands (mostly Guadalcanal) and is nested within Group 5, suggesting a founder event from the Papuan Peninsula to Guadalcanal. Group 7 is another Solomon Island lineage (mostly from Bougainville) that is located at the opposite end of the network to Group 6 and is highly diverged from all groups. MDS plots show that sites from the Solomon Islands are highly distinct from each other and from all other regions. Finally, Group 8 is a highly differentiated lineage from northern New Guinea that differs from its closest relatives by at least 15 mutational steps (∼3%).
The An. hinesorum rpS9 locus revealed strong genetic structure, supported both by the haplotype network (Fig. 5f) as well as by MDS plots and pairwise FST values (Fig. 6d, Table S4, Supporting information). Distinct groups identified include the following: (i) southern New Guinea—forming the centre of the network; (ii) central New Guinea; and (iii) the Papuan Peninsula (Fig. 5f)—with one haplotype of seven found on the Papuan Peninsula being the most common. This common haplotype was also found on Guadalcanal (in the Solomon Islands) and there is a second more divergent Solomon Island haplotype found on both Bougainville and Guadalcanal. This finding supports the evidence from the mitochondrial data; that there are two genetically distinct An. hinesorum lineages inhabiting the Solomon Islands, with the less divergent lineage being most similar to populations from the Papuan Peninsula. Interestingly, some individuals from the Solomon Islands were found to be heterozygous for the two rpS9 alleles sampled from this region, suggesting that despite being highly diverged at the mitochondrial locus, individuals from the two putative founder events have remained reproductively compatible.
Discussion
In this study, we examined evolutionary relationships of three closely related mosquito species at mtDNA and nDNA loci. We inferred that the non-human biting species Anopheles irenicus is most closely related to An. farauti and more distantly related to the non-human biting populations of An. hinesorum. Therefore, we conclude that non-human biting behaviour has evolved more than once in this group of cryptic species. Given the role of these mosquitoes in the transmission of malaria in this region, this is a medically significant finding.
Further, we provided the first genetic data on the colonization history of An. hinesorum to the Solomon Islands and found two distinct mitochondrial lineages of An. hinesorum on the Solomon Islands. We also found nonmonophyly between An. hinesorum and An. farauti at the mtDNA COI locus. Given that the species are monophyletic at nuclear loci, mtDNA introgression of An. hinesorum onto a nuclear background of An. farauti may have occurred; however, we cannot rule out incomplete lineage sorting as an alternative explanation. Finally, we showed that there are highly distinct populations of both An. farauti and An. hinesorum within and between New Guinea and Australia, with more strongly defined groups in An. hinesorum.
Colonization of the Solomon Islands and the repeated evolution of non-human biting behaviour
We have presented evidence that four separate mtDNA lineages from the three species studied occur on the Solomon Islands (Fig. 2a): two lineages of An. hinesorum (one highly diverged and one less diverged), one lineage of An. irenicus and one lineage of An. farauti. The less divergent lineage of An. hinesorum on the Solomon Islands appears to be descended from a population on the Papuan Peninsula (Fig. 5e, group 6) that is known to bite humans and carry malaria and probably arrived on the Solomon Islands within the last 500 000 years. Due to the greater separation and recent tMRCA of the more anciently diverged Solomon Islands lineage of An. hinesorum, it is difficult to tell where it originated or when it diverged from the rest of the species. According to the haplotype networks, it is most closely related to populations from southern New Guinea (Fig. 5e, group 7) that bite humans and transmit malaria.
As previously mentioned, neither An. hinesorum nor An. irenicus bite humans in the Solomon Islands. On the basis of the phylogenetic evidence available from both mitochondrial and nuclear data, we conclude that this medically important behaviour has evolved more than once in these mosquitoes, possibly in the same geographical region. However, it is difficult to tell whether this behavior evolved more than once in An. hinesorum, because the two divergent mitochondrial lineages from the Solomon Islands appear to be reproductively compatible and may have exchanged this trait. On the basis of the geographical distribution of this trait, we think it most likely that non-human biting behaviour evolved in the Solomon Islands. We cannot, however, rule out the possibility that it evolved in New Guinea (where it may still be present in a subset of the population) or elsewhere and subsequently became fixed in the Solomon Islands through a founder event.
The finding of parallel evolution of non-human biting behaviour raises important evolutionary questions about the driving force behind the repeated evolution of this behaviour. We hypothesize that a reduction in host availability may have driven the parallel evolution of non-human biting behavior. Olfactory receptors (ORs) are a highly diverse gene family and it has recently been suggested that the number of OR genes possessed by a mosquito species is positively correlated with diversity of host preference (Arensburger et al. 2010). Rodents, bats and birds would have constituted the only warm-blooded hosts on the Solomon Islands at the times of initial colonization by An. hinesorum and An. irenicus (Carvajal & Adler 2005). Thus, it is possible that the colonizations of the Solomon Islands by An. irenicus and An. hinesorum resulted in a reduction in their OR gene repertoires due to relaxation of (purifying) selection on some OR genes upon exposure to a less diverse array of hosts. By genomic comparison of closely related populations with different host preferences, it may be possible to identify genes that enable mosquitoes to cue to humans. This would potentially reveal the mechanistic basis of attraction of Anopheles mosquitoes to humans, which may lead to the development of improved mosquito attractants and repellents, or other novel technologies to reduce direct exposure to infectious mosquitoes.
Mitochondrial introgression and its potential role in range expansion
Mitochondrial introgression has been found by many studies across diverse groups (Ballard 2000; Funk & Omland 2003) and we suggest that mitochondrial introgression may have occurred between An. hinesorum and An. farauti. We base this on the finding that An. farauti is polyphyletic for mtDNA (COI), with a clade currently defined as An. farauti falling within An. hinesorum (Figs 2a and 3a), and the two species being monophyletic at nuclear loci (Fig. 3b,c). Although we cannot rule out incomplete lineage sorting as an explanation for this observation, we suggest that the observed polyphyly is probably to be the result of introgression. Because of its smaller effective population size, mtDNA tends to have a higher rate of genetic drift than nuclear DNA. Therefore, incomplete lineage sorting is more likely for nuclear than for mitochondrial loci. Additionally, all An. farauti individuals with introgressed mtDNA were sampled from an area geographically separate from the rest of the species, namely in Queensland, Torres Strait, southern New Guinea and Timika. These introgressed haplotypes are most closely related to populations of An. hinesorum in the same area (Fig. 5b, Queensland, Cape York and southern New Guinea). Overlapping distributions of species between which introgression is suspected is one of the keys to discriminating between mitochondrial introgression and incomplete lineage sorting (Funk & Omland 2003).
Mitochondria may frequently play an important role in thermal adaptation (Ballard & Whitlock 2004) and other studies have found evidence of temperature-based selection on mitochondria (Weinstein & Somero 1998; Doi et al. 1999). With this in mind, it is interesting to note that An. hinesorum occur at altitudes of >1000 m in the Central Ranges of New Guinea in colder conditions (avg. temp. 20 °C) than the coastally restricted An. farauti (avg. coastal temp. 26 °C; McAlpine et al. 1983). Additionally, the two sister species of An. hinesorum—An. farauti 5 and An. farauti 6—are only extant in the highlands of New Guinea (>1000 m; Cooper et al. 2002). Thus, a selective sweep may have driven an introgressed An. hinesorum mitotype, pre-adapted to cooler climates, through the southerly distribution of An. farauti, facilitating the spread of the species south (as this lineage occupies the species’ most southern latitude). Alternatively, rather than being favoured by selection, the introgressed mitochondrial lineage may have become fixed in populations during a range expansion. Fixation of introgressed genes and/or organelles following, or as a result of a range expansion, is an evolutionary phenomenon that has received recent support (Excoffier et al. 2009; Neiva et al. 2010).
Patterns of divergence and genetic structure in New Guinea and northern Australia
Many of the genetically distinct groups identified by this study correlate with areas of endemism identified by previous biogeographic studies of insects and other taxa (see Introduction for a brief description of these areas; Van Welzen 1989; de Boer & Duffels 1996). We found phylogeographic patterns that are in agreement with these studies. For example, we found that populations of An. farauti and An. hinesorum from southern New Guinea are genetically similar to populations from Northern Queensland (and, in the case of An. farauti, to populations from Torres Strait). These relationships may be due to the fact that Australia and New Guinea were connected during most of the late Pleistocene (Voris 2000), and are similar to patterns observed in other species (Rawlings et al. 2004; Bower et al. 2008), although others have found that there has been little or no recent gene flow between the regions (Kearns et al. 2011). Also in agreement with previous studies of non-mosquito species was the strong differentiation between Northern Territory and Queensland populations (Bowman et al. 2010; Kearns et al. 2011). This may be due to the low rainfall area in the Gulf of Carpentaria (the Carpentaria Gap) that is hypothesized to have posed a barrier to dispersal for a number of species (Bowman et al. 2010). On the basis of mtDNA, An. farauti from the Northern Territory is not part of the introgressed An. farauti lineage and is most closely related to northern New Guinea populations. This pattern is different to those shown in previous nuclear-ribosomal-based studies of An. farauti which have suggested that there is some sharing of rDNA copy variant sequences between Queensland and the Northern Territory populations (Beebe et al. 2000b; Bower et al. 2008). This difference suggests that there may have been greater nuclear than mitochondrial gene exchange between Queensland and Northern Territory populations.
A number of geographically coherent groups of An. hinesorum within New Guinea were found to be almost completely genetically separate from each other. The greater distinction between groups of An. hinesorum, versus between groups of An. farauti, may be due to differences in behaviour or ecology. For example, there may be less distinction between An. farauti populations because of the relative uniformity of their coastal habitats and the absence of physical barriers to dispersal. The most highly diverged group that we found in either species within New Guinea was a group of An. hinesorum sampled from the Sepik and Ramu River regions in northern New Guinea (29–33h: Table 2, Fig. 1c). It is unsurprising to find such high levels of divergence between the north and south of New Guinea, as the Central Cordillera (up to 4000 m altitude) runs the length of the New Guinea and provides an obvious barrier to gene flow. Other studies have also found high levels of divergence between the north and south of New Guinea (Rawlings & Donnellan 2003; Kearns et al. 2011), although others have found no genetic structure between the regions (Murphy et al. 2007). The absence of shared nuclear rpS9 alleles between the northern New Guinean and other lineages, as well as the level of COI divergence (approximately 3%), and the fact that genomic DNA probes developed to identify An. hinesorum do not hybridize with samples of the species from this region (Cooper et al. 2002), indicate that An. hinesorum occupying northern New Guinea may be a separate species.
Some phylogeographic patterns that are congruent between the species and loci examined are not as easily explained as the divergence between populations from the north and south of New Guinea. One such example is the major genetic break in New Guinea, separating the Papuan Peninsula and southern New Guinea. This break is present at both mitochondrial and nuclear loci of both species (Figs 5 and 6). In An. hinesorum, the discontinuity occurs in the Gulf region of New Guinea, at which point three distinct groups are found in close proximity. This genetic break may be due to a mountain ridge of the Central Ranges that extends to approximately 20 km from the coast and separates the Purari River Basin from the Papuan Peninsula. The Purari River flows from Mt. Hagen in the Central Ranges to the ocean near site 18h (Fig. 1c) in the Gulf Province of PNG, offering an explanation for the close affinities observed between the lowland site (18h) and the sites sampled in the highlands (19 and 20h: Fig. 1c). Site 20h sits above 600 m in the Central Ranges and An. hinesorum has been collected from above 1300 m in this region (Cooper et al. 2002).
A similar genetic break between the Papuan Peninsula and southern New Guinea was also observed in An. farauti at both loci, but it occurs further to the west. Interestingly, this break correlates with a climatic interchange between monsoonal to the south and continuous hot wet to the north, suggesting that differences in rainfall or habitat associated with rainfall may have maintained the distinction between the regions (despite their close geographic proximity). This climatic interchange also delimits the group containing introgressed mtDNA from the group containing native An. farauti mtDNA. An. farauti rpS9 data revealed evidence of hybridization between populations from either side of the genetic break at a single site (17f: Figs 1b and 5c) but there is no evidence of these alleles elsewhere in the Papuan Peninsula.
This study provides important insights into the population genetic connectivity of two important malaria transmitting species in the southwest Pacific region. We find substantial population structure consistent with low gene flow both within and between major landmasses. This result implies that the spread of any newly evolved alleles conferring advantages against mosquito control initiatives, including biochemical and behavioural insecticide resistance, would be somewhat restricted in moving among populations relative to a high gene flow scenario (although the spread would also depend on the specific selection coefficient of a new allele). Genetic structure that cannot be explained by obvious barriers to dispersal, such as oceans and mountains, is apparent for both species.
Acknowledgments
Work was funded through start-up funds from the University of Queensland and the CSIRO Climate Change Adaptation Flagship as well as the NHMRC Project Grant 117102. The authors thank Richard Fuller for assistance with the maps and James Bower for laboratory assistance.
Acknowledgments
Research Interests of authors (NWB, RDC KSL and WO) revolve around developing a better understanding of vector biology, specifically the evolution, distribution, ecology and biology of mosquitoes that transmit human pathogens. LA and CR research interests include using population genetics tools to address evolutionary and ecological questions.
Data accessibility
DNA sequences: GenBank Accession nos
COI: JN384217–JN384661
rpS9: JN384662–JN385275
ITS2: JN564766–JN564794, AF033218, HM584426, AF104314–AF104326 (also see Table 1 and the supplementary spreadsheet).
Supporting information
Additional supporting information may be found in the online version of this article.
Data S1. Spreadsheet of Genbank Accession numbers with sample information for individuals used in this study.
Data S2. Spreadsheet of Genbank Accession numbers with sample information for individuals used in this study.
Data S3. Spreadsheet of Genbank Accession numbers with sample information for individuals used in this study.
Table S1 Pair-wise FST values between sites for the Anopheles farauti COI locus.
Table S2 Pair-wise FST values between sites for the Anopheles farauti rpS9 locus.
Table S3 Pair-wise FST values between sites for the Anopheles hinesorum COI locus.
Table S4 Pair-wise FST values between sites for the Anopheles hinesorum rpS9 locus.
Please note: Wiley-Blackwell are not responsible for the content or functionality of any supporting information supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
References
- Allison A. Introduction to the Fauna of Papua. The Ecology of Papua. EricOey, Singapore: A. McGuire; 2007. pp. 479–494. Part 1. [Google Scholar]
- Alquezar DE, Hemmerter S, Cooper RD, et al. Incomplete concerted evolution and reproductive isolation at the rDNA locus uncovers nine cryptic species within Anopheles longirostris from Papua New Guinea. BMC Evolutionary Biology. 2010;10:392. doi: 10.1186/1471-2148-10-392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arensburger P, Megy K, Waterhouse RM, et al. Sequencing of Culex quinquefasciatus Establishes a Platform for Mosquito Comparative Genomics. Science. 2010;330:86–88. doi: 10.1126/science.1191864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker N, De Bruyn M, Mather PB. Patterns of molecular diversity in wild stocks of the redclaw crayfish (Cherax quadricarinatus) from northern Australia and Papua New Guinea: Impacts of Plio-Pleistocene landscape evolution. Freshwater Biology. 2008;53:1592–1605. [Google Scholar]
- Ballard JWO. When one is not enough: introgression of mitochondrial DNA in Drosophila. Molecular Biology and Evolution. 2000;17:1126–1130. doi: 10.1093/oxfordjournals.molbev.a026394. [DOI] [PubMed] [Google Scholar]
- Ballard JWO, Whitlock MC. The incomplete natural histroy of mitochondria. Molecular Ecology. 2004;13:729–744. doi: 10.1046/j.1365-294x.2003.02063.x. [DOI] [PubMed] [Google Scholar]
- Beebe NW, Cooper RD. Distribution and evolution of the Anopheles punctulatus group (Diptera : Culicidae) in Australia and Papua New Guinea. International Journal for Parasitology. 2002;32:563–574. doi: 10.1016/s0020-7519(01)00359-9. [DOI] [PubMed] [Google Scholar]
- Beebe NW, Saul A. Discrimination of all members of the Anopheles punctulatus complex by polymerase chain reaction restriction fragment length polymorphism analysis. American Journal of Tropical Medicine and Hygiene. 1995;53:478–481. doi: 10.4269/ajtmh.1995.53.478. [DOI] [PubMed] [Google Scholar]
- Beebe NW, Foley DH, Saul A, et al. DNA probes for identifying the members of the Anopheles punctulatus complex in Papua New Guinea. American Journal of Tropical Medicine and Hygiene. 1994;50:229–234. doi: 10.4269/ajtmh.1994.50.229. [DOI] [PubMed] [Google Scholar]
- Beebe NW, Ellis JT, Cooper RD, et al. DNA sequence analysis of the ribosomal DNA ITS2 region for the Anopheles punctulatus group of mosquitoes. Insect Molecular Biology. 1999;8:381–390. doi: 10.1046/j.1365-2583.1999.83127.x. [DOI] [PubMed] [Google Scholar]
- Beebe NW, Bakote’e B, Ellis JT, et al. Differential ecology of Anopheles punctulatus and three members of the Anopheles farauti complex of mosquitoes on Guadalcanal, Solomon Islands, identified by PCR-RFLP analysis. Medical and Veterinary Entomology. 2000a;14:308–312. doi: 10.1046/j.1365-2915.2000.00248.x. [DOI] [PubMed] [Google Scholar]
- Beebe NW, Cooper RD, Foley DH, et al. Populations of the south-west Pacific malaria vector Anopheles farauti s.s. revealed by ribosomal DNA transcribed spacer polymorphisms. Heredity. 2000b;84:244–253. doi: 10.1046/j.1365-2540.2000.00665.x. [DOI] [PubMed] [Google Scholar]
- Beebe NW, Cooper RD, Morrison DA, et al. A phylogenetic study of the Anopheles punctulatus group of malaria vectors comparing rDNA sequence alignments derived from the mitochondrial and nuclear small ribosomal subunits. Molecular Phylogenetics and Evolution. 2000c;17:430–436. doi: 10.1006/mpev.2000.0853. [DOI] [PubMed] [Google Scholar]
- Beebe NW, Cooper RD, Morrison DA, et al. Subset partitioning of the ribosomal DNA small subunit and its effects on the phylogeny of the Anopheles punctulatus group. Insect Molecular Biology. 2000d;9:515–520. doi: 10.1046/j.1365-2583.2000.00211.x. [DOI] [PubMed] [Google Scholar]
- Beehler BM, Swaby RJ. Phylogeny and biogeography of the Ptiloris rifliebirds (Aves: Paradisaeidae) The Condor. 1991;93:738–745. [Google Scholar]
- de Boer AJ, Duffels JP. Historical biogegraphy of the cicadas of Wallacea, New Guinea and the West Pacific: a geotectonic explanation. Palaeogeography, Palaeoclimatology, Palaeoecology. 1996;124:153–177. [Google Scholar]
- Bower JE, Dowton M, Cooper RD, et al. Intraspecific concerted evolution of the rDNA ITS1 in Anopheles farauti sensu stricto (Diptera: Culicidae) reveals recent patterns of population structure. Journal of Molecular Evolution. 2008;67:397–411. doi: 10.1007/s00239-008-9161-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bower JE, Cooper RD, Beebe NW. Internal repetition and intraindividual variation in the rDNA ITS1 of the Anopheles punctulatus group (Diptera: Culicidae): multiple units and rates of turnover. Journal of Molecular Evolution. 2009;68:66–79. doi: 10.1007/s00239-008-9188-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowman D, Brown GK, Braby MF, et al. Biogeography of the Australian monsoon tropics. Journal of Biogeography. 2010;37:201–216. [Google Scholar]
- Brower AVZ. Rapid morphological radiation and convergence among races of the butterfly Heliconuserato inferred from patterns of mitochondrial DNA evolution. Proceedings of the National Academy of Sciences of the United States of America. 1994;91:6491–6495. doi: 10.1073/pnas.91.14.6491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carvajal A, Adler GH. Biogeography of mammals on tropical Pacific islands. Journal of Biogeography. 2005;32:1561–1569. [Google Scholar]
- Clement M, Posada D, Crandall KA. TCS: a computer program to estimate gene genealogies. Molecular Ecology. 2000;9:1657–1659. doi: 10.1046/j.1365-294x.2000.01020.x. [DOI] [PubMed] [Google Scholar]
- Cook BD, Hughes JM. Historical population connectivity and fragmentation in a tropical freshwater fish with a disjunct distribution (pennyfish, Denariusa bandata. Journal of the North American Benthological Society. 2010;29:1119–1131. [Google Scholar]
- Cooper RD, Frances SP. Malaria vectors on Buka and Bougainville islands, Papua New Guinea. Journal of The American Mosquito Control Association. 2002;18:100–106. [PubMed] [Google Scholar]
- Cooper L, Cooper RD, Burkot TR. The Anopheles punctulatus complex—DNA probes for identifying the Australian species using isotopic, chromogenic, and chemiluminescence detection systems. Experimental Parasitology. 1991;73:27–35. doi: 10.1016/0014-4894(91)90004-g. [DOI] [PubMed] [Google Scholar]
- Cooper RD, Waterson DG, Frances SP, et al. Speciation and distribution of the members of the Anopheles punctulatus (Diptera: Culicidae) group in Papua New Guinea. Journal of Medical Entomology. 2002;39:16–27. doi: 10.1603/0022-2585-39.1.16. [DOI] [PubMed] [Google Scholar]
- Cooper RD, Waterson DGE, Frances SP, et al. Malaria vectors of Papua New Guinea. International Journal for Parasitology. 2009;39:1495–1501. doi: 10.1016/j.ijpara.2009.05.009. [DOI] [PubMed] [Google Scholar]
- Craft KJ, Pauls SU, Darrow K, et al. Population genetics of ecological communities with DNA barcodes: An example from New Guinea Lepidoptera. Proceedings of the National Academy of Sciences of the United States of America. 2010;107:5041–5046. doi: 10.1073/pnas.0913084107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Bruyn M, Wilson JC, Mather PB. Reconciling geography and genealogy: phylogeography of giant freshwater prawns from the Lake Carpentaria region. Molecular Ecology. 2004;13:3515–3526. doi: 10.1111/j.1365-294X.2004.02348.x. [DOI] [PubMed] [Google Scholar]
- Doi A, Suzuki H, Matsuura ET. Genetic analysis of temperature-dependent transmission of mitochondrial DNA in Drosophila. Heredity. 1999;82:555–560. doi: 10.1038/sj.hdy.6885080. [DOI] [PubMed] [Google Scholar]
- Drummond AJ, Rambaut A. BEAST: Bayesian evolutionary analysis by sampling trees. Bmc Evolutionary Biology. 2007;7:214. doi: 10.1186/1471-2148-7-214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drummond AJ, Ho SYW, Phillips MJ, et al. Relaxed phylogenetics and dating with confidence. Plos Biology. 2006;4:e88. doi: 10.1371/journal.pbio.0040088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drummond AJ, Ashton B, Buxton S, et al. Geneious v5.1. 2010. [Google Scholar]
- Excoffier L, Lischer HEL. Arlequin suite ver 3.5: a new series of programs to perform population genetics analyses under Linux and Windows. Molecular Ecology Resources. 2010;10:564–567. doi: 10.1111/j.1755-0998.2010.02847.x. [DOI] [PubMed] [Google Scholar]
- Excoffier L, Foll M, Petit RJ. Genetic consequences of range expansions. Annual Review of Ecology Evolution and Systematics. 2009;40:481–501. [Google Scholar]
- Foley DH, Bryan JH. Shared salinity tolerance invalidates a test for the malaria vector Anopheles farauti s.s on Guadalcanal, Solomon Islands. Medical and Veterinary Entomology. 2000;14:450–452. doi: 10.1046/j.1365-2915.2000.00268.x. [DOI] [PubMed] [Google Scholar]
- Foley DH, Paru R, Dagoro H, et al. Allozyme analysis reveals six species within the Anopheles punctulatus complex of mosquitoes in Papua New Guinea. Medical and Veterinary Entomology. 1993;7:37–48. doi: 10.1111/j.1365-2915.1993.tb00649.x. [DOI] [PubMed] [Google Scholar]
- Foley DH, Meek SR, Bryan JH. The Anopheles punctulatus group of mosquitoes in the Solomon Islands and Vanuatu surveyed by allozyme electrophoresis. Medical and Vetinary Entomology. 1994;8:340–350. doi: 10.1111/j.1365-2915.1994.tb00098.x. [DOI] [PubMed] [Google Scholar]
- Foley DH, Bryan JH, Yeates D, et al. Evolution and systematics of Anopheles: insights from a molecular phylogeny of Australasian mosquitoes. Molecular Phylogenetics and Evolution. 1998;9:262–275. doi: 10.1006/mpev.1997.0457. [DOI] [PubMed] [Google Scholar]
- Funk DJ, Omland KE. Species-level paraphyly and polyphyly: frequency, causes, and consequences, with insights from animal mitochondrial DNA. Annual Review of Ecology Evolution and Systematics. 2003;34:397–423. [Google Scholar]
- Guindon S, Dufayard JF, Hordijk W, et al. PhyML: fast and accurate phylogeny reconstruction by maximum likelihood. Infection Genetics and Evolution. 2009;9:384–385. [Google Scholar]
- Hasan AU, Suguri S, Fujimoto C, et al. Phylogeography and dispersion pattern of Anopheles farauti senso stricto mosquitoes in Melanesia. Molecular Phylogenetics and Evolution. 2008;46:792–800. doi: 10.1016/j.ympev.2007.09.018. [DOI] [PubMed] [Google Scholar]
- Hasegawa M, Kishino H, Yano TA. Dating of the human ape splitting by a molecular clock of mitochindrial DNA. Journal of Molecular Evolution. 1985;22:160–174. doi: 10.1007/BF02101694. [DOI] [PubMed] [Google Scholar]
- Heads M. Birds of paradise, vicariance biogeography and terrane tectonics in New Guinea. Journal of Biogeography. 2002;29:261–283. [Google Scholar]
- Heled J, Drummond AJ. Bayesian inference of species trees from multilocus data. Molecular Biology and Evolution. 2010;27:570–580. doi: 10.1093/molbev/msp274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kass RE, Raftery AE. Bayes Factors. Journal of the American Statistical Association. 1995;90:773–795. [Google Scholar]
- Kearns AM, Joseph L, Omland KE, et al. Testing the effect of transient Plio-Pleistocene barriers in monsoonal Australo-Papua: did mangrove habitats maintain genetic connectivity in the Black Butcherbird? Molecular Ecology. 2011;20:5042–5059. doi: 10.1111/j.1365-294X.2011.05330.x. [DOI] [PubMed] [Google Scholar]
- Keogh JS, Shine R, Donnellan S. Phylogenetic relationships of terrestrial Australo-Papuan elapid snakes (subfamily Hydrophiinae) based on cytochrome b and 16S rRNA sequences. Molecular Phylogenetics and Evolution. 1998;10:67–81. doi: 10.1006/mpev.1997.0471. [DOI] [PubMed] [Google Scholar]
- Kimura M. A simple method for estimating evolutionary rates of base substitutions through comparative studies of nucleotide sequences. Journal of Molecular Evolution. 1980;16:111–120. doi: 10.1007/BF01731581. [DOI] [PubMed] [Google Scholar]
- Macqueen P, Goldizen AW, Austin JJ, et al. Phylogeography of the pademelons (Marsupialia: Macropodidae: Thylogale) in New Guinea reflects both geological and climatic events during the Plio-Pleistocene. Journal of Biogeography. 2011;38:1732–1747. [Google Scholar]
- Maddison WP, Maddison DR. Mesquite: A Modular System for Evolutionary Analysis, Version 2.75. 2011. [Google Scholar]
- Marshall AJ. In: The Diversity and Conservation of Papua’s Ecosystems. In: The Ecology of Papua. Marshall AJ, Beehler BM, editors. Singapore: Periplus Editions; 2007. pp. 753–770. Part 2. [Google Scholar]
- Martin DP, Lemey P, Lott M, et al. RPD3: a flexible and fast computer program for analyzing recombination. Bioinformatics. 2010;26:2462–2463. doi: 10.1093/bioinformatics/btq467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McAlpine JR, Keig G, Falls R. Climate of Papua New Guinea. Canberra: CSIRO Publishing, Australian National University Press; 1983. [Google Scholar]
- Murphy SA, Double MC, Legge SM. The phylogeography of palm cockatoos, Probosciger aterrimus, in the dynamic Australo-Papuan region. Journal of Biogeography. 2007;34:1534–1545. [Google Scholar]
- Neiva J, Pearson GA, Valero M, et al. Surfing the wave on a borrowed board: range expansion and spread of introgressed organellar genomes in the seaweed Fucus ceranoides L. Molecular Ecology. 2010;19:4812–4822. doi: 10.1111/j.1365-294X.2010.04853.x. [DOI] [PubMed] [Google Scholar]
- Norman JA, Rheindt FE, Rowe DL, et al. Speciation dynamics in the Australo-Papuan Meliphaga honeyeaters. Molecular Phylogenetics and Evolution. 2007;42:80–91. doi: 10.1016/j.ympev.2006.05.032. [DOI] [PubMed] [Google Scholar]
- O’Connell JF, Allen J. Dating the colonization of Sahul (Pleistocene Australia–New Guinea): a review of recent research. Journal of Archaeological Science. 2004;31:835–853. [Google Scholar]
- Osborne MJ, Christidis L. Molecular phylogenetics of Australo-Papuan possums and gliders (Family Petauridae) Molecular Phylogenetics and Evolution. 2001;20:211–224. doi: 10.1006/mpev.2001.0960. [DOI] [PubMed] [Google Scholar]
- Pigram CJ, Davies HL. Terranes and the accretion history of the New Guinea Orogen. BMR Journal of Australian Geology and Geophysics. 1987;10:193–211. [Google Scholar]
- Polhemus DA. In: Tectonic Geology of Papua. In: The Ecology of Papua, Part 1. Marshall AJ, Beehler BM, editors. Singapore: Periplus Editions; 2007. pp. 137–164. [Google Scholar]
- Polhemus DA, Polhemus JT. In: Assembling New Guinea: 40 Million Years of Island arc Accretion as Indicated by the Distributions of Aquatic Heteroptera (Insecta). In: Biogeography and Geological Evolution of SE Asia. Hall R, Holloway JD, editors. Leiden: Backhuys Publishers; 1998. pp. 327–340. [Google Scholar]
- Posada D. jModelTest: phylogenetic model averaging. Molecular Biology and Evolution. 2008;25:1253–1256. doi: 10.1093/molbev/msn083. [DOI] [PubMed] [Google Scholar]
- R Development Core Team. R: A Language and Environment for Statistical Computing. Vienna: R Foundation for Statistical Computing; 2011. 3-900051-07-0, Available from http://www.R-project.org/ [Google Scholar]
- Rambaut A. 2009. FigTree v1.3.1. Available from http://tree.bio.ed.ac.uk/software/figtree/ (accessed 10 May 2012)
- Rambaut A, Drummond AJ. 2009. Tracer v1.5. Available from http://beast.bio.ed.ac.uk/Tracer (accessed 20 January 2010)
- Rawlings LH, Donnellan SC. Phylogeographic analysis of the green python, Morelia viridis, reveals cryptic diversity. Molecular Phylogenetics and Evolution. 2003;27:36–44. doi: 10.1016/s1055-7903(02)00396-2. [DOI] [PubMed] [Google Scholar]
- Rawlings LH, Barker D, Donnellan SC. Phylogenetic relationships of the Australo-Papuan Liasis pythons (Reptilia : Macrostomata), based on mitochondrial DNA. Australian Journal of Zoology. 2004;52:215–227. [Google Scholar]
- Rozas J, Sanchez-DelBarrio JC, Messeguer X, et al. DnaSP, DNA polymorphism analyses by the coalescent and other methods. Bioinformatics. 2003;19:2496–2497. doi: 10.1093/bioinformatics/btg359. [DOI] [PubMed] [Google Scholar]
- Sweeney AW. Larval salinity tolerances of the sibling species of Anopheles farauti. Journal of the American Mosquito Control Association. 1987;3:589–592. [PubMed] [Google Scholar]
- Tajima F. Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics. 1989;123:585–595. doi: 10.1093/genetics/123.3.585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor MJ, Calaby JH, van Deusen HM. A Revision of the Genus Rattus (Rodentia, Muridae) in the New Guinean Region. Bulletin of the American Museum of Natural History. 1982;173:177–336. [Google Scholar]
- Van Welzen PC. Guioa Cav (Sapindaceae): Taxonomy, Phylogeny, and Historical Biogeography. Rijksherbarium/Hortus Botanicus; 1989. ISBN: 9789071236044, Published in Netherlands. [Google Scholar]
- Voris HK. Maps of Pleistocene sea levels in Southeast Asia: shorelines, river systems and time durations. Journal of Biogeography. 2000;27:1153–1167. [Google Scholar]
- Walton C, Handley JM, Tun-Lin W, et al. Population structure and population history of Anopheles dirus mosquitoes in Southeast Asia. Molecular Biology and Evolution. 2000;17:962–974. doi: 10.1093/oxfordjournals.molbev.a026377. [DOI] [PubMed] [Google Scholar]
- Weinstein RB, Somero GN. Effects of temperature on mitochondrial function in the Antarctic fish Trematomus bernacchii. Journal of Comparative Physiology. 1998;168:190–196. [Google Scholar]
- Wuster W, Dumbrell AJ, Hay C, et al. Snakes across the Strait: trans-Torresian phylogeographic relationships in three genera of Australasian snakes (Serpentes : Elapidae : AcanthophisOxyuranus, and Pseudechis. Molecular Phylogenetics and Evolution. 2005;34:1–14. doi: 10.1016/j.ympev.2004.08.018. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
DNA sequences: GenBank Accession nos
COI: JN384217–JN384661
rpS9: JN384662–JN385275
ITS2: JN564766–JN564794, AF033218, HM584426, AF104314–AF104326 (also see Table 1 and the supplementary spreadsheet).