

Background: The Escherichia coli O8 and O9a antigens are influential models for bacterial glycan assembly.
Results: O8 and O9a glycan biosynthesis requires three mannosyltransferases, whose activities were defined.
Conclusion: Two conserved mannosyltransferases (WbdCB) produce an adaptor trisaccharide on which each WbdA polymerizes a serotype-specific repeat unit polysaccharide.
Significance: These systems require multiple mannosyltransferase modules in a flexible arrangement.
Keywords: Cell Wall, Escherichia coli, Glycobiology, Glycoconjugate, Glycosyltransferases, Lipopolysaccharide (LPS), O-antigen, Bacterial Cell Envelope, Mannosyltransferases, Polysaccharides, O-polysaccharide, Glycan Synthesis
Abstract
The Escherichia coli O9a and O8 O-antigen serotypes represent model systems for the ABC transporter-dependent synthesis of bacterial polysaccharides. The O9a and O8 antigens are linear mannose homopolymers containing conserved reducing termini (the primer-adaptor), a serotype-specific repeat unit domain, and a terminator. Synthesis of these glycans occurs on the polyisoprenoid lipid-linked primer, undecaprenol pyrophosphoryl-GlcpNAc, by two conserved mannosyltransferases, WbdC and WbdB, and a serotype-specific mannosyltransferase, WbdA. The glycan structure and pattern of conservation in the O9a and O8 mannosyltransferases are not consistent with the existing model of O9a biosynthesis. Here we establish a revised pathway using a combination of in vivo (mutant complementation) experiments and in vitro strategies with purified enzymes and synthetic acceptors. WbdC and WbdB synthesize the adaptor region, where they transfer one and two α-(1→3)-linked mannose residues, respectively. The WbdA enzymes are solely responsible for forming the repeat unit domains of these O-antigens. WbdAO9a has two predicted active sites and polymerizes a tetrasaccharide repeat unit containing two α-(1→3)- and two α-(1→2)-linked mannopyranose residues. In contrast, WbdAO8 polymerizes trisaccharide repeat units containing single α-(1→3)-, α-(1→2)-, and β-(1→2)-mannopyranoses. These studies illustrate assembly systems exploiting several mannosyltransferases with flexible active sites, arranged in single- and multiple-domain formats.
Introduction
Lipopolysaccharide (LPS) is an essential constituent of the outer membrane of most Gram-negative bacteria. The prototypical LPS molecule consists of three structural regions: the lipid A, core oligosaccharide, and O-antigenic polysaccharide (O-PS)5 (1). The structure of lipid A is well conserved, as are some features of the core oligosaccharide. In contrast, O-PSs are hypervariable glycans of variable chain length that consist of repeat units containing one or more sugars and non-carbohydrate components. Unique O-PS structures define more than 180 different O-antigen serotypes in Escherichia coli (2, 3). The lipid A-core and O-PS components of LPS are assembled in separate pathways that converge in a ligation reaction at the periplasmic face of the inner membrane (1), and the completed molecule is then transported to the cell surface (4). Despite the structural diversity, all O-PS glycans are thought to be assembled by one of only three pathways, all of which involve biosynthetic intermediates built on the 55-carbon polyisoprenoid lipid acceptor, undecaprenol phosphate (reviewed in Ref. 1).
The O-PSs of E. coli O9, O9a, and O8 are a family of related structures comprising linear homopolymers of mannopyranose (Manp) (3) (Fig. 1A). Investigation of their biosynthesis began in 1971 (5), and they represent influential prototypes for O-PSs synthesized via the well distributed ATP-binding cassette (ABC) transporter-dependent pathway (6). The E. coli O9a and O8 O-PSs are identical to the Klebsiella pneumoniae O3 and O5 O-PSs, respectively, and the genetic loci encoding the corresponding O-PS biosynthesis enzymes are highly conserved (7). The close evolutionary relationships in these antigens are believed to reflect lateral gene transfer between the two species (8, 9). The E. coli O9a O-PS is a variant of the E. coli O9 structure. Seroconversion of O9a to O9 requires a single amino acid substitution in WbdA, one of three mannosyltransferases required for their synthesis (10). Detailed structural studies have identified four domains in the K. pneumoniae O3 and O5 O-PS molecules: a conserved β-GlcpNAc residue (the primer) at the reducing terminus, an adapter region containing two or more α-linked Manp residues, the variable serotype-specific repeat unit domain, and the terminator comprising an O-methyl group (O8) or a methyl phosphate modification (O9 and O9a) (Fig. 1A) (11–13).
FIGURE 1.

Structures of the E. coli polymannose O-PSs and organization of the corresponding biosynthetic gene clusters. A, structures of the O8, O9, and O9a O-PSs. Each polysaccharide contains four structural regions (the primer, adaptor, repeat unit domain, and terminal modification), and these are represented in the schematic in the context of the und-PP-linked biosynthetic intermediate. The gene products dedicated to the synthesis of each domain are identified in red. In the structures, GlcpNAc is represented by a blue square and Manp by a green circle following the nomenclature established by the Consortium for Functional Glycomics. B, organization of the corresponding biosynthetic gene clusters.
The gene clusters responsible for expression of these O-PSs encode proteins involved in synthesis of the nucleotide precursor, GDP-Man (ManBC), the transmembrane and nucleotide-binding domains of the ABC transporter (Wzm and Wzt, respectively), a chain terminator (WbdD), and three mannosyltransferases (WbdC, WbdB, and WbdA; formerly MtfCBA) (14) (Fig. 1B). WbdC and WbdB are highly conserved in these serotypes. The WbdAO8 and WbdAO9/O9a proteins differ in size and sequence, and experimental evidence supports the conclusion that WbdA is the serotype-defining mannosyltransferase (10). WbdDO9/O9a is a bifunctional kinase-methyl transferase that adds a phosphomethyl group to the O9/O9a non-reducing terminus, whereas WbdDO8 adds only a methyl group in O8 (12, 15). The terminated glycan is then recognized by a serotype-specific carbohydrate-binding module located at the C terminus of Wzt, prior to its export via the ABC transporter (16). Chain termination is essential for recognition and export.
The proposed biosynthesis pathway of these O-PS glycans is based primarily on data from E. coli O9a; this was initially reported as O9 in earlier studies and corrected later (14, 17). In the published model, WbdC is thought to add the first α-Manp residue to the β-GlcpNAc-PP-und acceptor that is synthesized by WecA (14). WecA is part of the machinery for biosynthesis of the enterobacterial common antigen glycan, and the wecA gene is not located in the O-PS locus (18, 19). The activity of WbdC is proposed to be followed by the addition of two α-(1→3)-linked Manp residues by WbdB. Further chain extension then occurs through the combined alternating activities of WbdB and WbdA (14). However, this pathway is difficult to rationalize with the O-PS structures and the pattern of conserved (WbdC/B) and variable (WbdA) mannosyltransferases.
Here, we establish a revised biosynthetic pathway using purified proteins and defined acceptors. We conclude that biosynthesis of these important model O-PSs involves a unique combination of two enzymes containing a single mannosyltransferase domain, one of which is capable of adding two Manp residues in succession, and multidomain-polymerizing mannosyltransferases.
EXPERIMENTAL PROCEDURES
Bacterial Strains, Plasmids, and Growth Conditions
The bacterial strains and plasmids used in this study are described in Table 1. Bacteria were routinely grown in Luria-Bertani (LB) medium (20) or M9 minimal medium (21). M9 minimal medium was supplemented, where appropriate, with glycerol (0.4%, w/v), thiamine HCl (0.5 μg/ml), niacinamide (0.5 μg/ml), l-histidine (20 μg/ml), l-tryptophan (20 μg/ml), d-glucose (0.4%, w/v), d-mannose (0.1%, w/v), l-arabinose (0.2%, w/v), isopropyl β-d-1-thiogalactopyranoside (0.5 mm), ampicillin (100 μg/ml), or chloramphenicol (34 μg/ml).
TABLE 1.
Bacterial strains and plasmids
| Strain/plasmid | Description or genotype | Reference or source |
|---|---|---|
| Strains | ||
| Top10 | E. coli F−, mcrA, Δ (mrr-hsdRMS-mcrBC), φ80, lacZΔM15, ΔlacX74, deoR, nupG, recA1, araD139, Δ(ara-leu)7697, galU, galK, repsL(Strr), endA1 | Invitrogen |
| BL21 | E. coli B F− dcm ompT hsdS(rB− mB−) gal [malB+]K-12(λS) | Novagen |
| CWG634 | E. coli O9a:K−; trp his lac rpsL cpsK30; manA; Smr; Tcr | Ref. 15 |
| CWG636 | E. coli O8:K−; ugd::aacC1 manA; Gmr; Tcr | Ref. 15 |
| CWG1005 | K-12 lacZ trp Δ(sbcB-rfb) upp rel rpsL manA | Ref. 12 |
| CWG1006 | CWG1005 derivative; waaL::cat; Cmr | This study |
| CWG1007 | CWG636 derivative; ΔwbdB; Gmr; Tcr | This study |
| CWG1008 | CWG636 derivative; ΔwbdC; Gmr; Tcr | This study |
| CWG1009 | CWG634 derivative; ΔwbdB; Smr; Tcr | This study |
| CWG1010 | CWG634 derivative; ΔwbdC; Smr; Tcr | This study |
| CWG1104 | CWG636 derivative; ΔwbdA; Gmr; Tcr | This study |
| CWG1105 | CWG634 derivative; ΔwbdA; Smr; Tcr | This study |
| Plasmids | ||
| pMAL-c2X | IPTG-inducible plasmid for expressing cytoplasmic maltose-binding protein fusions; Apr | New England Biolabs |
| pBADHisA | l-Arabinose-inducible plasmid to express N-terminal hexahistidine-tagged constructs; Apr | Invitrogen |
| pBAD24 | l-Arabinose-inducible plasmid; Apr | Ref. 23 |
| pKD3 | Source of Cmr resistance cassette | Ref. 22 |
| pKD46 | Helper plasmid encoding the Red recombinase genes, γ, β, and exo | Ref. 22 |
| pCP20 | Helper plasmid encoding the FLP recombinase | Ref. 22 |
| pWQ492 | pBAD24 derivative containing an EcoRI/HindIII fragment encoding WbdAO9a-His10; Apr | Ref. 58 |
| pWQ575 | pMAL-c2X derivative containing an XmnI/HindIII fragment encoding WbdCO9a; Apr | This study |
| pWQ576 | pMALc-2X derivative containing an EcoRI/HindIII fragment encoding WbdBO9a; Apr | This study |
| pWQ577 | pMALc-2X derivative containing an EcoRI/HindIII fragment encoding WbdCO8; Apr | This study |
| pWQ578 | pMALc-2X derivative containing an EcoRI/HindIII fragment encoding WbdBO8; Apr | This study |
| pWQ579 | pBAD24 derivative containing an EcoRI/HindIII fragment encoding WbdCO9a; Apr | This study |
| pWQ580 | pBAD24 derivative containing an EcoRI/HindIII fragment encoding WbdCO9a-WbdBO9a; Apr | This study |
| pWQ581 | pBAD24 derivative containing an EcoRI/HindIII fragment encoding WbdCO8; Apr | This study |
| pWQ582 | pBAD24 derivative containing an EcoRI/HindIII fragment encoding WbdCO8-WbdBO8; Apr | This study |
| pWQ587 | pBADHisA derivative containing an XhoI and PvuII fragment encoding WbdAO8; Apr | This study |
| pWQ588 | pBAD24 derivative containing an NcoI/XbaI fragment encoding His10-WbdAO8; Apr | This study |
DNA Methods
InstaGene Matrix (Bio-Rad) or DNAzol reagent (Invitrogen) was used to purify chromosomal DNA. DNA fragments were PCR-amplified using Pwo DNA polymerase (Roche Applied Science) or Pfu Ultra high fidelity DNA polymerase (Stratagene), using custom oligonucleotide primers (Sigma) containing restriction sites to facilitate cloning. The sequences and features of the oligonucleotide primers are described in supplemental Table S1. The PureLink PCR purification kit (Invitrogen) was used to purify DNA fragments from PCRs or restriction enzyme digests, and DNA fragments from agarose gels were purified using the PureLink quick gel extraction kit (Invitrogen). Plasmid DNA was purified using either the PureLink quick plasmid miniprep kit (Invitrogen) or the Qiagen HiSpeed Midi kit (Qiagen). Restriction endonucleases (Invitrogen and New England Biolabs) and T4 DNA ligase (New England Biolabs) were used according to the manufacturer's instructions. DNA sequencing was performed by the AAC Genomics Facility (University of Guelph, Guelph, Canada).
Construction of Chromosomal Mutants
The λred-recombinase system (22) was used to generate CWG1006 (waaL::cat), CWG1105 (ΔwbdAO9a), CWG1104 (ΔwbdAO8), CWG1009 (ΔwbdBO9a), CWG1007 (ΔwbdBO8), CWG1010 (ΔwbdCO9a), and CWG1008 (ΔwbdCO8). The chloramphenicol resistance cassette was amplified by PCR from pKD3 using oligonucleotide primers containing 50 nucleotide homology extensions upstream and downstream of the target gene. The oligonucleotide primers were designed to generate deletions of the entire open reading frame. After recombination to replace the targeted gene with an antibiotic resistance marker, the FLP-recombinase plasmid (pCP20) was used to remove the marker from the chromosomes of mutants. The exception was CWG1006 (waaL::cat), where removal of the marker was unnecessary because any potential polar effects would not influence experiments conducted in this study.
Cloning of wbdC, wbdB, and wbdA Mannosyltransferase Genes from E. coli O9a and O8
The wbdC and wbdB genes were amplified from E. coli CWG634 (O9a) and CWG636 (O8) chromosomal DNA and cloned into the isopropyl β-d-1-thiogalactopyranoside-inducible pMAL-c2X vector (New England Biolabs) to generate N-terminal maltose-binding protein (MalE) fusions. The wbdC and wbdBC genes from both serotypes were also cloned into the arabinose-inducible pBAD24 vector (23). The wbdA genes from serotypes O8 and O9a were cloned into the pBAD24 or pBADHisA (Invitrogen) vector to produce proteins with polyhistidine tags. All constructs were confirmed by restriction endonuclease digestion or by sequence and by their ability to complement their corresponding chromosomal mutation.
Purification of MalE-WbdB and MalE-WbdC Fusion Proteins
A 250-ml culture of E. coli Top10 transformed with the appropriate pMAL-c2X-based plasmid was grown in LB medium at 37 °C until an A600 of ∼0.3 was reached. Cultures were then transferred to an incubator set at 20 °C and grown until midexponential phase (A600 = 0.6). 0.5 mm isopropyl β-d-1-thiogalactopyranoside was then added to induce expression of the MalE fusion protein, and incubation was continued overnight at 20 °C. Cells were then collected by centrifugation and resuspended in 25 ml of buffer A (20 mm Tris, 100 mm NaCl, 1 mm EDTA, pH 7.5), prior to lysis by sonication with intermittent cooling on ice. The cell lysate was cleared by sequential centrifugation steps at 3000 × g for 10 min and 27,000 × g for 30 min. The resulting supernatant was loaded onto a column containing amylose resin (New England Biolabs) and washed extensively with buffer A. The bound MalE fusion protein was eluted using buffer A containing 10 mm maltose. The protein concentrations were determined using the A280, based on theoretical extinction coefficients of MalE-WbdCO8/O9a (105,910 m−1 cm−1) and MalE-WbdBO8/O9a (140,985 m−1 cm−1), predicted by the ProtParam program (24). The elution fraction containing the most protein typically contained 2–4 mg/ml protein and was stored in buffer A containing 10 mm maltose at −80 °C.
Purification of WbdAO9a-His10 and His10-WbdAO8
A 500-ml culture of E. coli BL21 transformed with the appropriate pBAD24-based plasmid was grown in LB medium at 37 °C until an A600 of ∼0.3 was reached. Gene expression was induced as described above, with the exception that 0.2% (w/v) l-arabinose was used as inducer. Cells were then collected by centrifugation and resuspended in 25 ml of buffer B (20 mm BisTris, 250 mm NaCl, 5% (w/v) glycerol, pH 7.0) prior to lysis by sonication with intermittent cooling on ice. The cell lysate was cleared by sequential centrifugation steps at 5000 × g for 10 min and 12,000 × g for 20 min. The resulting supernatant was centrifuged at 100,000 × g for 60 min, and the soluble fraction was loaded onto a 5-ml HiTrap chelating (Ni2+) column (GE Healthcare). For His10-WbdAO9a, the column was washed sequentially with buffer B containing 50 and 75 mm imidazole, and the bound protein was eluted in buffer B containing 125 mm imidazole. The column loaded with His10-WbdAO8 was washed with buffer B and then eluted in the same buffer containing 50 mm imidazole. A PD-10 column (GE Healthcare) was used to exchange the purified proteins into buffer C (20 mm BisTris, 50 mm NaCl, pH 7.0), and aliquots were frozen at −80 °C. The protein concentrations were determined using the A280, based on theoretical extinction coefficients of His10-WbdAO9a (123,230 m−1 cm−1) and His10-WbdAO8 (160,420 m−1 cm−1) predicted by the ProtParam program (24). The protein concentration in frozen aliquots of His10-WbdAO9a and His10-WbdAO8 was typically 2 mg/ml.
Preparation of Membrane Fractions
A 200-ml culture of CWG1006 containing the desired pBAD24-derived plasmid was grown in M9 minimal medium at 37 °C until an A600 of ∼0.4 was achieved. Recombinant protein expression was induced by the addition of 0.2% (w/v) l-arabinose, and the culture was grown for a further 3.5 h. Cells were collected by centrifugation and resuspended in 20 ml of buffer D (20 mm HEPES, pH 7.5) prior to lysis by sonication. The lysate was cleared by sequential centrifugation steps at 4000 × g for 10 min and 12,000 × g for 20 min. The resulting supernatant was centrifuged at 100,000 × g for 60 min, and the membrane pellet was resuspended in 0.2 ml of buffer E (50 mm HEPES, 20 mm MgCl2, 1 mm dithiothreitol, pH 7.5). The total membrane protein concentration was typically 5–10 mg/ml, based on the DC Protein Assay (Bio-Rad), and membrane preparations were stored in buffer E at −80 °C.
In Vitro WbdC and WbdB Mannosyltransferase Reactions Using a Synthetic Acceptor
A synthetic acceptor lipid analog β-GlcpNAc-pyrophosphoryl-C13 (GlcpNAc-PP-C13) (25) was generously provided by Dr. E. D. Brown (McMaster University, Hamilton, Canada) and used as a substrate for MalE-WbdC and/or MalE-WbdB. Radiolabeled mannosyltransferase assays were carried out for 1 h at 25 °C in 10-μl reaction volumes of buffer E containing 0.5 mm GlcpNAc-PP-C13, 0.31 μm GDP-[14C(U)]Man (262 mCi/mmol; PerkinElmer Life Sciences) and 10 μg of enzyme. Reactions were terminated by the addition of an equal volume of stop solution (50% (v/v) aqueous acetonitrile, 1% (w/v) SDS, 10 mm EDTA). Radiolabeled reaction products were analyzed by thin layer chromatography (TLC). Aliquots (2–4 μl) of stopped reaction mixtures were spotted on AL SIL G TLC plates (Whatman) and developed in solvent A (ethyl acetate/methanol/water/acetic acid, 4:2:1:0.1). Dried TLC plates were exposed to film (Kodak Biomax MR film, Amersham Biosciences) for 1 week.
To prepare non-radiolabeled products for mass spectrometry (MS), reactions were carried out for 3 h at 25 °C in 50-μl reaction volumes consisting of buffer E containing 1 mm GlcpNAc-PP-C13, 1 mm GDP-Man, and 50 μg of enzyme. After termination in stop solution, reaction products were diluted in 1 ml of H2O and loaded onto a C18 Sep-Pak cartridge (Waters). After washing extensively with water, the products were eluted in 60% acetonitrile and concentrated using a SpeedVac concentrator. Capillary electrophoresis-mass spectrometry (CE-MS) of these products was conducted on a Prince CE system (Prince Technologies, Netherlands) coupled to a 4000Qtrap mass spectrometer (AB/Sciex, Concord, Canada), via a micro-ion spray interface. Survey scan spectra of the products were obtained in negative ion mode. Sheath solution (isopropyl alcohol/methanol, 2:1) was delivered at a flow rate of 2.0 μl/min. Separations were obtained on a ∼90-cm length bare fused silica capillary, using 10 mm ammonium acetate in chloroform/methanol (2:1). Separations were performed by applying a voltage of 30 kV together with 500 millibars of pressure. MS/MS experiments were performed in positive ion mode. The instrument was operated with low (0.7 atomic mass unit peak width at half height) resolution on quadrupole 1, and typical collision energy of 50 eV was applied to induce fragmentation of ions selected for collision-induced dissociation. Nitrogen was used as the collision gas with peak width at half-height.
In Vitro WbdC and WbdB Mannosyltransferase Reactions Using Endogenous Acceptors in Membranes
The use of membranes from CWG1006 (manA waaL::cat) in these experiments ensured that the reaction products in these experiments remained as und-PP-linked intermediates rather than being transferred to lipid A core by the WaaL ligase enzyme (1). The manA mutation prevented synthesis of GDP-Man in the absence of mannose in the growth medium (15), allowing de novo synthesis in membranes following the addition of the radiolabeled sugar donor. Mannosyltransferase assays were carried out for 1.5 h at 25 °C in 100-μl reaction volumes of buffer E containing 0.31 μm GDP-[14C(U)]Man and 100 μg of membrane protein. The und-PP-linked oligosaccharides were extracted from membranes in an equal volume of 1-butanol (26). The organic (upper phase) was kept, and the extractions were repeated. The combined organic phases were washed once with an equal volume of water and then dried to completeness. 100 μl of 1-propanol, 2 m trifluoroacetic acid (1:1) was added to each of the dried lipid extracts, and the samples were heated to 50 °C for 15 min to liberate the oligosaccharide phosphates from the und-PP-linked products. The samples were dried and then treated with 10 units of calf intestinal alkaline phosphatase (2000 units/mg; Roche Applied Science) in dephosphorylation buffer (0.5 m Tris-HCl, 1 mm EDTA, pH 8.5) for ∼2 h at 37 °C.
The dephosphorylation reaction mixtures were diluted to 1 ml with water and then loaded onto a column (75 × 2.5 cm) containing Toyopearl HW-40(S) resin (Tosoh Bioscience LLC). 100 μl of radiolabeled WbdC (∼5500 cpm) or WbdCB (∼10,000 cpm) products were mixed with 25 μl each of Manp, maltose, maltotriose, and maltotetraose (10 mg/ml) standards. The mixture was separated, and eluates were collected in 1-ml fractions using 0.1 m acetic acid as the eluant. The radiolabeled oligosaccharides were detected by scintillation counting of 0.4 ml of each fraction in Ecolite scintillation fluid (ICN Biomedicals). The non-radiolabeled standards were detected using the colorimetric Dubois assay for total sugars (27).
In Vitro WbdA Mannosyltransferase Reactions Using Synthetic Substrates
Two synthetic acceptors were used. Acceptor A (α-Manp-(1→2)-α-Manp-(1→2)-α-Manp-(1→3)-α-Manp) represents the repeat unit of the O9a antigen, whereas Acceptor B (α-Manp-(1→3)-α-Manp-(1→3)-β-GlcpNAc) is the conserved reducing terminal trisaccharide of the O8 and O9a antigens (Fig. 1A). Their synthesis as 8-azidooctyl glycoside derivatives has been described elsewhere (28, 29), and both were used as fluorescein-tagged derivatives. Preparation of a fluorescein-tagged derivative of Acceptor A and its use in characterizing the WbdDO9a-mediated chain-terminating activity has been described previously (12). The synthesis of the fluorescein-tagged derivative of Acceptor B followed the same protocol.
Initial assay conditions were established using WbdAO9a. The enzyme's activity was determined to be optimal at room temperature (∼25 °C) and declined rapidly at temperatures above 30 °C (data not shown). Approximate Km values for the acceptor and GDP-Man were determined using a CE-based assay. The percentage conversion of Acceptor A into products was determined by integration of the product peak areas on electropherograms. The percentage conversion was used to establish reaction conditions at which the rate of product formation was linear with time (i.e. the initial velocity). Keeping within the linear range, apparent Km values for Acceptor A and GDP-Man were determined in two separate trials. The percentage conversion was used to calculate the enzyme activity for each substrate concentration, and the data were fit to the Michaelis-Menten equation in GraphPad Prism version 4.0. The Km(app) values for Acceptor A in the independent experiments were 1.5 ± 0.2 and 1.2 ± 0.3 mm. The Km(app) values for GDP-Man were 0.5 ± 0.05 and 0.6 ± 0.08 mm (data not shown). Insufficient acceptor was available for a complete kinetic analysis, but these experiments defined conditions of excess substrate used for standard reactions. These reactions were carried out for 30 min at 25 °C in 10-μl volumes of buffer E containing 0.5 mm acceptor, 5 mm GDP-mannose, and 2 μm purified His10-WbdAO9a or 10 μm purified His10-WbdAO8. Reactions were terminated by the addition of an equal volume of stop solution, diluted 1:4 with 50% (v/v) aqueous acetonitrile, and 2-μl aliquots were spotted on AL SIL G TLC plates (Whatman). The plates were developed in solvent B (ethyl acetate/water/1-butnaol/acetic acid, 5:4:4:2.5) and fluorescent reaction products were detected with a hand-held UV lamp.
CE analysis of the WbdAO9a products synthesized using Acceptor A was conducted according to published methods (30). The stopped reactions were centrifuged at 13,000 rpm for 5 min and then diluted 1:250 in water (to 1 μm acceptor concentration). The samples were pressure-injected for 10 s into a bare silica capillary (50 μm × 50 cm) and separated in CE buffer (89 mm Tris, 89 mm boric acid, 2 mm EDTA, 20 mm SDS, pH 8.27) over 15 min. The samples were resolved at 30 kV using a Beckman-Coulter P/ACE MDQ CE system equipped with an argon ion laser-induced fluorescence detector (λEx = 488 nm, with emission filter at 520 nm).
Structural Analyses of the Products of WbdA Proteins Using Synthetic Acceptors
To generate sufficient products for NMR analyses, 250-μl reaction volumes were used. After 30 min, the reactions were diluted in 1 ml of H2O and loaded onto a C18 Sep-Pak cartridge (Waters), and the cartridge was washed extensively with water. The products were eluted in 2 ml of 60% (v/v) aqueous acetonitrile and concentrated using a SpeedVac concentrator.
MALDI-TOF mass spectra of the reaction products were obtained on a Bruker Ultraflextreme MALDI TOF/TOF. The MALDI spectra of the reaction products of WbdAO9a with Acceptor B were obtained in positive ion mode, whereas the reaction products of WbdAO8 or WbdAO9a with Acceptor A were obtained in negative ion mode.
NMR spectra for the reaction products were acquired in D2O at 27 °C on a 700-MHz spectrometer equipped with a cryoprobe. The spectra were referenced to an external standard of acetone (2.22 ppm for 1H, 31.07 for 13C). One-dimensional, gCOSY, 1H-tROESY, and 1H-13C gHSQC spectra were obtained. For all of the 1H spectra, the intensity of the residual HOD peak was decreased using a presaturation pulse sequence, irradiating at 4.76 ppm. The spectral window for the one-dimensional 1H spectra was 8446 Hz (from 10.8 to −1.3 ppm), and a line broadening function of 0.5 Hz was applied to improve the signal/noise ratio. For all of the two-dimensional spectra, sine-bell functions were applied interactively to improve signal/noise, but no line-broadening was used. For the 1H-13C gHSQC spectra, proton signals were decoupled during acquisition, and the 1JC,H value was set to 140 Hz to determine the appropriate delays. Additional gradient-enhanced total correlation spectroscopy spectra were also acquired to facilitate interpretation.
For the O9a WbdA products, the spectral window for the gCOSY was 8446 Hz (from 10.8 to −1.3 ppm) in both dimensions, with 512 increments in F1 and 64 transients in F2. The tROESY was acquired with a spectral window of 5605 Hz (8.0 to 0.0 ppm) in both dimensions, 299 increments in the F1 dimension, 64 transients in the F2 dimension, and a mixing time of 0.4 s. The spectral window for the 1H-13C gHSQC was 4223 Hz (from 6.0 to 0.0 ppm) in F2 (1H dimension, 64 transients) and 24.6 kHz (from 130 to −10 ppm) in F1 (13C dimension, 128 increments).
To investigate the structure of the O8 WbdA products, the spectral windows for the gCOSY and tROESY were 5605 Hz (from 9.0 to 1.0 ppm) in both dimensions. The gCOSY was acquired with 600 increments in F1 and 16 transients in F2, and the tROESY was acquired with 550 increments in F1, 16 transients in F2, and a mixing time of 0.4 s The spectral window for the 1H-13C gHSQC was 5605 Hz (from 9.0 to 1.0 ppm) in F2 (1H dimension, 16 transients) and 28.2 kHz (from 160 to 0 ppm) in F1 (13C dimension, 432 increments).
Protein and LPS PAGE
Protein and LPS samples were analyzed by SDS-PAGE in Tris/glycine buffer (31). Protein was visualized using Simply Blue stain (Invitrogen). LPS samples were prepared by proteinase K digestion of whole-cell lysates according to the method of Hitchcock and Brown (32). LPS was visualized by silver staining (33). Western immunoblots of LPS were prepared by transferring samples to PROTRAN nitrocellulose membranes (PerkinElmer Life Science). Samples were probed at a 1:500 dilution with O9a-specific or O8-specific antiserum (15). Alkaline phosphatase-conjugated goat anti-rabbit secondary antibody (Cedar Lane Laboratories) was used at a dilution of 1:3000, and nitro blue terazolium and 5-bromo-4-chloro-3-indolyl phosphate (Roche Applied Science) were used as substrates for detection.
RESULTS
WbdCO8/O9a Is a Monofunctional Mannosyltransferase
Previous experimental data from E. coli O9a support the conclusion that WbdCO9a performs the first committed step in biosynthesis of this polymannose O-PS, by transferring a single Manp residue to the multifunctional acceptor, β-GlcpNAc-PP-und (14). This is consistent with both of the O-PS structures (Fig. 1A), which contain an α-Manp-(1→3)-β-GlcpNAc disaccharide at their reducing termini (11). The predicted WbdCO8 and WbdCO9a proteins share 96% identity, and, as expected, each gene could rescue O-PS synthesis in the ΔwbdC mutants constructed in the O8 and O9a serotypes (Fig. 2, A–D), suggesting an identical function in vivo. O-PS was even synthesized in the absence of inducer, due to the “leaky” promoter (34). Induced cultures generated reduced amounts of O-PS, which may be a consequence of high levels of protein expression. This may reflect increased formation of inactive enzyme aggregates or altered stoichiometry of components in an enzyme complex. However, this does not influence interpretation of the current data and was not pursued further.
FIGURE 2.

The O8 and O9a wbdC and wbdB genes are functionally exchangeable. The results show gene complementation experiments with CWG1008 (ΔwbdCO8) (A and B), CWG1010 (ΔwbdCO9a) (C and D), CWG1007 (wbdBO8) (E and F), and CWG1009 (ΔwbdBO9a) (G and H). The top panels in each pair show the silver-stained SDS-polyacrylamide gel of LPS samples from whole-cell lysates, and the bottom panels show the corresponding confirmatory Western immunoblots using O8- or O9a-specific antisera. In each case, native O-PS biosynthesis was restored by the introduction of plasmids carrying the relevant gene from either serotype O8 or O9a. The plasmids used were as follows: pWQ577 (malE-wbdCO8), pWQ575 (malE-wbdCO9a), pWQ578 (malE-wbdBO8), and pWQ576 (malE-wbdBO9a).
To confirm the function of WbdC, in vitro synthesis was performed in reactions containing membranes from cells expressing WbdCO8/O9a, GDP-[14C]Man, and endogenous acceptor. TLC and autoradiography revealed a single major co-migrating oligosaccharide product from each enzyme (data not shown). The products from WbdCO9a were subjected to gel filtration chromatography, revealing a product co-migrating with a disaccharide standard, consistent with an α-Manp-(1→3)-β-GlcpNAc disaccharide (Fig. 3A).
FIGURE 3.

Analysis of the in vitro products from WbdC and WbdCB mannosyltransferase activities using an endogenous acceptor (membrane fraction) and a synthetic acceptor. A, gel filtration chromatography of the WbdCO9a and WbdCBO9a oligosaccharide products generated using the endogenous (β-GlcpNAc-PP-und) acceptor in membranes. The standards were mannose (1), maltose (2), maltotriose (3), and maltotetraose (4). B, autoradiograms of TLC-separated reactions synthesized by WbdCO8, WbdCO9a, WbdCBO8, and WbdCBO9a using the synthetic GlcpNAc-PP-C13 acceptor. The CE-MS spectra of the reaction products generated by WbdCO9a and the WbdCBO9a pair using the synthetic GlcpNAc-PP-C13 acceptor and the unmodified acceptor are shown in C.
To unequivocally establish the activity of WbdC, in vitro reactions were performed using purified MalE-WbdC fusion proteins and GlcpNAc-PP-C13 acceptor. When GDP-[14C]Man was used as the donor, a single product with identical TLC migration was observed for both WbdC proteins (Fig. 3B). CE-MS of the reaction products from WbdCO9a showed a novel peak at m/z 724.8 corresponding to the expected molecular weight for α-Manp-(1→3)-β-GlcpNAc-PP-C13 (Fig. 3C), and this assignment was confirmed by MS-MS (supplemental Table S2). A signal corresponding to unmodified β-GlcpNAc-PP-C13 (562 Da) was not evident in the reaction mixture, suggesting its complete conversion into product under these reaction conditions. Thus, the activity seen in membranes can be entirely attributed to WbdC.
Collectively, these analyses unequivocally confirmed the earlier predicted activity of WbdC as a GDP-Manp:GlcpNAc-PP-und α-(1→3)-mannosyltransferase (14) and showed that the enzymes from serotypes O8 and O9a possessed identical activities in vivo and in vitro.
WbdB Extends the WbdC Product by the Addition of a Manp2 Disaccharide
In the published model for O9a O-PS synthesis, WbdB is thought to act after WbdC, adding two Manp residues to the growing polymer. Furthermore, it is proposed that WbdB and WbdA act in an alternating fashion to form the repeat unit (14). The existence of a conserved adaptor region in the native O-PS structure was unknown at the time. However, the predicted WbdBO8/O9a proteins are almost identical (96% identity), and the O-PS repeat unit structures cannot accommodate the addition of the same disaccharide in each serotype, suggesting that the enzyme does not act in tandem with WbdA. Therefore, the activity of WbdBO8/O9a was reinvestigated.
Cross-complementation of ΔwbdB mutants with the corresponding genes from each serotype provided evidence that the proteins had identical activities in vivo (Fig. 2, E–H). Membranes from cells expressing WbdCB from either serotype produced a single major product that co-migrated on TLC, and this activity was dependent on WbdC (data not shown). On gel filtration chromatography, the WbdCBO9a oligosaccharide product migrated as a tetrasaccharide (Fig. 3A), consistent with the predicted α-Manp3-β-GlcpNAc product from previous studies (14).
To confirm this result and to rule out any unexpected contributions from host enzymes, an in vitro reaction was performed using β-GlcpNAc-PP-C13 acceptor and purified enzymes. The WbdCB enzymes from each serotype generated the same major and minor products, whose slower migrations in TLC were consistent with oligosaccharides larger than the WbdC product (Fig. 3B). MS analysis of the WbdCO9a reaction products revealed two product peaks (Fig. 3C), and both were identified by MS-MS analysis (supplemental Table S2). One product possessed an m/z of 1048.8 (α-Manp3-β-GlcpNAc-PP-C13), consistent with the product synthesized using the endogenous acceptor in membranes and presumably reflecting the major product seen by TLC. The second product had an m/z of 1210.8 and comprised α-Manp4-β-GlcpNAc-PP-C13. This is consistent with the appearance of a minor larger product on TLC. No intermediate product reflecting a single Manp transfer by WbdC was seen in these reactions, nor was any unmodified acceptor detected (Fig. 3C), suggesting efficient transfer by WbdB in these reactions. The collective data indicate that WbdB is an α-(1→3), mannosyltransferase that installs, in succession, two Manp residues onto the disaccharide α-Manp-(1→3)-β-GlcpNAc-PP-und intermediate, which is the WbdC product. In vivo, the activity is limited to the addition of two Manp residues, whereas in vitro conditions with the surrogate soluble acceptor can result in further extension of the glycan chain.
WbdA Enzymes Are Polymerizing Multidomain Mannosyltransferases
The activity assigned above to WbdB is not consistent with an identical involvement in synthesizing both the O8 and O9a repeat units. However, it has been reported that WbdAO9a possesses a duplicated sequence EX7E motif (14, 35), which is now known to be present in other retaining glycosyltransferases (36). These include members, like WbdA, of the GT4 family in the CAZy database (37–39). The WbdA proteins from serotypes O8 and O9a share only 16% identity (35% similarity) and differ in size. Moreover, unlike WbdCB, the WbdA enzymes are not functionally exchangeable between serotypes (data not shown). One interpretation of these observations is that the WbdA proteins contain multiple glycosyltransferase modules and are sufficient by themselves for repeat unit biosynthesis. This hypothesis was tested with purified enzymes and defined synthetic acceptors.
WbdAO9a transferred multiple Manp residues to both Acceptor A and Acceptor B, yielding reaction products with a range of sizes on TLC (Fig. 4A). At high GDP-Man/acceptor ratios, the product was confined to the origin of a TLC plate, whereas 3–4 faster migrating products were observed when the molar concentrations of donor and acceptor were 1:1. A CE trace of samples taken at various time points from a reaction containing Acceptor A demonstrated the accumulation of larger products over time (data not shown). These observations are consistent with a reaction mechanism that is distributive rather than processive.
FIGURE 4.

Analysis of the in vitro products from WbdAO9a mannosyltransferase activity using synthetic acceptors. The reaction products were separated by TLC (A), using the fluorescein tag on the acceptors for detection. The products generated with Acceptor B were examined by MALDI MS (B), revealing a series of incrementally sized products that differ by the addition of one Manp residue. C, anomeric region from the 1H NMR spectrum of the reaction products generated by WbdAO9a using Acceptor B.
The reaction products obtained with Acceptor A and WbdAO9a were examined by MALDI MS, and a range of products was detected. These consist of Acceptor A alone (m/z 1182) and acceptor plus increasing numbers of Manp residues (data not shown). The largest product (m/z 4911) corresponds to the mass of the acceptor plus 23 Manp residues. Each peak was separated by the mass of one Manp residue (162.1). MALDI MS of the products made by WbdAO9a with Acceptor B also revealed products of increasing size. The largest (m/z 4487.5) corresponds to the acceptor plus 21 Manp residues (Fig. 4B). The smallest product (m/z 1246.5) is Acceptor B plus one added Manp; no unmodified acceptor was detected, indicating that all of the starting material was converted into product by the enzyme.
WbdAO9a is capable of polymerizing sufficient Manp residues to encompass multiple repeat units of the O-PS, but MS offers no insight into the linkage sequence in the product. To resolve this question, reaction products generated using Acceptor B were examined by 1H NMR spectroscopy. The one-dimensional 1H spectrum revealed that at least two compounds were produced in the reaction (Fig. 4C). Integration of the anomeric signals in the 1H spectrum showed that that these compounds were present in an approximate ratio of 3:1. Four signals were obtained in the anomeric region for the major product, indicating that the H1 protons are present in four distinct chemical environments. This is consistent with the O9a repeat unit that contains blocks of two α-(1→2)- and two α-(1→3)-linked mannose residues. The chemical shifts for the anomeric protons and carbons of the major species closely match those published for the O9a antigen (Table 2) (40).
TABLE 2.
Comparison of the chemical shifts for the anomeric protons and carbons of the major WbdAO9a product generated using Acceptor B with those previously published for the O9a O-PS
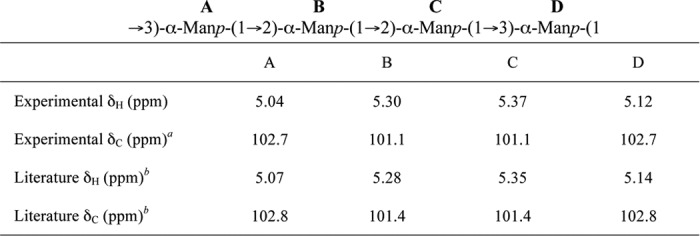
a The chemical shift values were determined from the 1H-13C gHSQC spectrum (supplemental Fig. S1).
b Values from Ref. 40.
To confirm the linkages of the major product, gCOSY and tROESY spectra were acquired (supplemental Fig. S2). The gCOSY spectrum was used to establish through-bond correlations between the protons of each ring system. The H2 and H3 resonances of each mannose residue were correlated to the anomeric signals in the spectrum. This information was used to interpret the tROESY spectrum, which established the through space correlations between protons of adjacent ring systems. tROESY showed a correlation between the anomeric signal at 5.37 ppm and a signal at 3.99 ppm, which was assigned as the H3 proton in the same Manp residue as the anomeric signal at 5.12 ppm. Thus, the Manp residues corresponding to the H1 at 5.37 and 5.12 ppm are linked (1→3). Similarly, there is a correlation between the anomeric signal at 5.12 ppm and the peak at 3.93 ppm, which was assigned as the H3 in the same residue as the anomeric signal at 5.04 ppm. Therefore, the Manp residues corresponding to the H1 at 5.12 ppm and 5.04 ppm are also linked (1→3). The anomeric signal at 5.04 ppm was correlated to 4.11 ppm, which was assigned as H2 in the residue corresponding to the remaining anomeric peak at 5.30 ppm. This indicated that the Manp residues of the H1 at 5.04 and 5.30 ppm are linked (1→2). The final important correlation was between the anomeric signal at 5.30 ppm and a resonance at 4.09 ppm, which was assigned as the H2 in the mannose with an anomeric peak at 5.37 ppm. Thus, the last linkage between the residues of the H1 at 5.30 and 5.37 ppm is also (1→2). Key correlations from the tROESY spectrum are labeled in supplemental Fig. S2B. From these data, the structure of the major product produced by WbdAO9a with the Acceptor B was identified as a repeating chain of the following tetrasaccharide: →3)-α-d-Manp-(1→2)-α-d-Manp-(1→2)-α-d-Manp-(1→3)-α-d-Manp-(1). This structure is identical to the repeat unit of the O9a antigen (40).
The structure of the minor reaction product was more difficult to determine. Resonances for five anomeric protons were visible in the one-dimensional 1H spectrum (Fig. 4C), but many of the correlations in the two-dimensional spectra were similar in intensity to the noise. Therefore, to aid in the interpretation of the data, the computer program CASPER (41, 42) was used. This software predicts 1H and 13C chemical shifts from a database built from mono-, di-, and trisaccharides. Sequences were entered into the program for several penta- and hexasaccharides of various linkages with the Acceptor B trisaccharide (α-d-Manp-(1→3)-α-d-Manp-(1→3)-β-d-GlcpNAc) at the reducing end. The best match between the anomeric shifts of the minor reaction product and the chemical shifts predicted by CASPER occurred for the following hexasaccharide: α-d-Manp-(1→3)-α-d-Manp-(1→2)-α-d-Manp-(1→2)-α-d-Manp-(1→3)-α-d-Manp-(1→3)-β-d-GlcpNAc (Table 3). For comparison, the sequence of a dodecasaccharide containing three repeating units of the major structure was entered into CASPER. The experimental and predicted chemical shifts agreed remarkably well (Table 3). Therefore, we propose that the minor reaction product is a truncated version of the major product with the structure of the hexasaccharide above. Its synthesis may result from relaxed fidelity in the in vitro reaction conditions because there is no evidence for such a product in the native O-PS.
TABLE 3.
Comparison of experimental 1H chemical shifts (ppm) with those predicted by CASPER for the anomeric protons of the major WbdAO9a product (1) and predicted minor WbdAO9a product (2) generated using Acceptor B

a The HOD peak at 4.76 ppm is too large to determine if the anomeric signal for the β-G1cNAc residue is present.
Similar experiments were performed using WbdAO8. This enzyme could not extend Acceptor B under the conditions tested but, perhaps surprisingly, yielded a range of different sized products with Acceptor A (Fig. 5, A and B). The reaction products obtained with Acceptor A and WbdAO8 were examined by MALDI MS, and a range of products were detected consisting of Acceptor A alone (m/z 1183) and acceptor plus increasing numbers of Manp residues (Fig. 5B). The largest product (m/z 4427) corresponds to the mass of the acceptor plus 20 Manp residues. There is a non-statistical distribution of intensities in the MALDI spectrum, indicating varied substrate activity for the different length oligosaccharides. For example, the intensity corresponding to the addition of seven Manp residues (m/z 2318) is much lower than that of nine additional residues (m/z 2643).
FIGURE 5.

Analysis of the in vitro products from WbdAO8 mannosyltransferase activity using synthetic acceptors. The reaction products were separated by TLC (A), using the fluorescein tag on the acceptors for detection. The reaction products generated with Acceptor A were examined by MALDI MS (B), revealing a series of incrementally sized products that differ by the addition of one Manp residue. Some of the product masses are 1 or 2 mass units higher than expected because the MS analysis was performed after their NMR spectra were acquired, and there were some residual deuterons remaining in the sample. C, the anomeric region from the 1H NMR spectrum of the reaction products generated by WbdAO8 using Acceptor A.
The anomeric signals of the one-dimensional 1H spectrum showed that at least two compounds were present in the reaction mixture generated by WbdAO8 using Acceptor A (Fig. 5C). The signals for the three anomeric protons of the major product integrate to one proton each, and the chemical shifts for the corresponding anomeric protons and carbons closely match those published for the O8 antigen (Table 4) (43). The signals for the minor compound are consistent with unmodified acceptor.
TABLE 4.
Comparison of the chemical shifts for the anomeric protons and carbons of the WbdAO8 product generated using Acceptor A with those previously published for the O8 O-PS
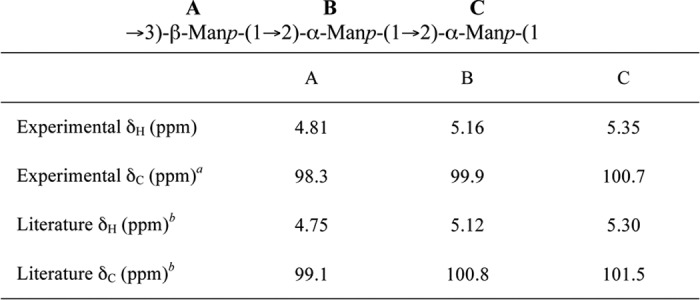
a The chemical shift values were determined from the 1H-13C gHSQC spectrum (supplemental Fig. S3).
b Values from Ref. 43.
Further analysis of the gCOSY and tROESY spectra confirmed the linkages between each of the Manp residues. As with the major WbdAO9a product, the resonances for H2 and H3 for each of the residues in O8 product could be correlated to each anomeric signal from the gCOSY spectrum (supplemental Fig. S4A). This information was used to interpret the tROESY spectrum and assign the linkages. Key correlations from the tROESY are labeled supplemental Fig. S4B. The anomeric signal at 5.35 ppm correlated to a signal at 3.75 ppm, which was assigned as the H3 proton in the same residue as the signal at 4.81 ppm. Thus, the resonance at 5.35 ppm is indicative of a (1→3)-linked moiety. The anomeric signal at 5.16 ppm had correlations to protons on different residues, one to the anomeric proton at 4.81 ppm and a second to the signal at 4.13 ppm. This signal was assigned to H2 on the same residue as the anomeric proton at 5.35 ppm, and the correlation is consistent with a (1→2)-linked residue. The anomeric signal at 4.81 correlated to a signal at 4.30 ppm, which is proton H2 on the same ring as the signal at 5.16 ppm. This resonance indicates another (1→2) linkage. From these data, we conclude that the major compound in the NMR spectrum is the O8 antigen.
DISCUSSION
The O8, O9, and O9a antigens were the first representatives of O-PS synthesized via an ABC transporter-dependent system to be studied biochemically. These systems have been influential in the development of an understanding of the shared features of this biosynthetic strategy in O-PS and in other cell surface glycans from a wide range of bacteria with different lifestyles (44). Biosynthesis of the E. coli O9a and O8 polymannose O-PSs occurs through the sequential transfer of Manp residues to β-GlcpNAc-PP-und, and this is catalyzed by three mannosyltransferases, WbdC, WbdB, and WbdA (14). WbdC and WbdB are highly conserved in serotypes O8 and O9a, and all experimental evidence indicates that they perform the same reaction in each serotype. Although the published model correctly assigned the catalytic activity of the WbdC mannosyltransferase, the context of the activity of WbdB was misinterpreted as being involved in repeat unit synthesis, because the adaptor region at the reducing terminus of the O-PS was unknown at the time. Furthermore, definitive assignments can only be made when purified enzymes are used with defined acceptors. Our data confirm that, operating together, WbdC and WbdB are responsible for completing the und-PP-linked tetrasaccharide (α-Manp-(1→3)-α-Manp-(1→3)-α-Manp-(1→3)-β-GlcpNAc), to form a conserved reducing terminal structure (the adaptor-primer) in the serotype O8 and O9a O-PSs. In our earlier report of the O-PS structure (11), we suggested that the conserved region may be confined to a trisaccharide (α-Manp-(1→3)-α-Manp-(1→3)-β-GlcpNAc). Both possibilities are accommodated by the reported structure, but they result in a different position of the first Manp residue in the repeat unit domain, essentially shifting the register of the repeat unit. However, only the conserved reducing terminal tetrasaccharide reconciles all of the structural, genetic, and biochemical data.
WbdC and WbdB are single active site glycosyltransferases belonging to the GT4 family (39). Several monospecific GT4 representatives from cell surface glycoconjugate assembly systems have been characterized, and solved structures are available for some. Selected examples include WaaG (E. coli LPS biosynthesis (45)), WasF (Geobacillus stearothermophilus NRS 2004/3A S-layer glycan (46)), and PimA (mycobacterial phosphoinositol mannoside (47)). In contrast, WbdB transfers two Manp residues to the native acceptor in vivo. Although less common, this is not without precedent. One well characterized example of this phenomenon is seen in PglH (another GT4 family member), which is involved in the synthesis of an und-PP-linked glycan that is used for N-glycosylation of proteins in Campylobacter jejuni (48). In a situation that is reminiscent of the activity of WbdB, PglH transfers three GalpNAc residues to a terminal GalpNAc in the growing glycan (49). Other examples of this type of activity are provided by GlfT1, an enzyme that transfers two galactofuranosyl residues to a decaprenol-P-linked acceptor in the biosynthesis of cell wall arabinogalactan in Mycobacterium tuberculosis (50, 51) and KdtA in E. coli, which adds two 2-deoxy-d-manno-octulosonic acid residues to lipid A in LPS biosynthesis (52). Examples are not confined to prokaryotes, because Alg11 of Saccharomyces cervisiae adds two Manp residues to the dolichol-PP-linked intermediate used for N-glycosylation (53, 54). These enzymes are distinguished from single-site processive polymerases by the unique ability to limit the number of sequential glycosyl transfers to 2–3 residues. PglH offers insight into how this may occur. It has been proposed that the transfer of a discrete number of residues is controlled by the relative binding affinities for the growing acceptor (49). These increase with size, resulting in a lower kcat. A counting mechanism for PglH is effectively provided by inhibition of enzymatic activity after the acceptor has been elongated by three GalpNAc residues. Whether this applies to other examples remains to be established.
WbdB adds only two Manp residues to the endogenous acceptor in vivo. In contrast, using a purified enzyme and soluble acceptor WbdB also generates a minor product with three Manp residues transferred. This relaxed activity presumably results from the artificial (soluble) reaction conditions and/or the absence of other enzymes in the pathway. A similar phenomenon has been observed with the bifunctional α-(2→3), α-(2→8) sialyltransferase, Cst-II. This enzyme normally adds one or two sialic acid residues to lipooligosaccharide of C. jejuni (55), but the purified enzyme can add up to four sialyl residues to a synthetic acceptor (56).
The studies described here indicate that the earlier proposal that WbdB participates in repeat unit synthesis (14) is incorrect. Instead, our data unequivocally demonstrate that WbdAO9a is a bifunctional α-(1→2), α-(1→3)-mannosyltransferase and that WbdAO8 is a trifunctional enzyme with α-(1→2), α-(1→3), and β-(1→2) mannosyltransferase activities. These enzymes are sufficient for formation of the repeat unit domain of the cognate O-PS. WbdAO9a can polymerize the authentic O9a mannan using either Acceptor A or Acceptor B, indicating that it can elongate acceptors containing either α-(1→3)- or α-(1→2)-linked terminal Manp residues. This might be expected from the repeat unit structure. The accumulation of larger products over time and the altered size distribution with varying acceptor/donor ratios are consistent with an enzyme that is non-processive, or distributive. In this mechanism, the enzyme dissociates from the growing polymer after each Manp transfer. Surprisingly, WbdAO8 could also use Acceptor A (the O9a repeat unit), although this enzyme does see an α-(1→2)-linked terminal Manp in the biosynthesis of the native glycan. In principle, the enzyme should also be able to add to an α-(1→3)-linked terminal Manp, but no activity was seen with Acceptor B. Because neither of the available synthetic oligosaccharides represented a true physiological acceptor for WbdAO8, the difference in the capacity to use these acceptors cannot be readily interpreted. Nevertheless, the extension of Acceptor A to generate an O8 glycan does provide clear evidence that WbdAO8, like its counterpart from serotype O9a, is sufficient for assembly of the corresponding repeat unit domain.
Unlike WbdC and WbdB, the WbdA mannosyltransferases are not functionally exchangeable in vivo (data not shown). This result is expected, given that the ability of the ABC transporter to recognize the nascent O-PS (and form completed, O-PS-substituted LPS molecules) requires that the O-PS be first capped by the action of the cognate WbdD enzyme (15, 57). These enzymes differ in specificity and create different terminal structures (11, 13, 15, 43) (Fig. 1A). Furthermore, in O9a, a C-terminal region of WbdD is essential for recruiting the cognate WbdA to the membrane (58), and specificity in these interactions might be anticipated based on sequence differences between the two serotypes.
In silico predictions using the NCBI Conserved Domain Database (59) identify two putative glycosyltransferase domains in WbdAO9a and three in WbdAO8. These predictions correlate with sizes of the WbdAO9a (95.5 kDa) and WbdAO8 (137 kDa) proteins. WbdAO9a is approximately twice the size of a typical monofunctional mannosyltransferase, and WbdAO8 is roughly 3 times as large. For reference, WbdC and WbdB have predicted sizes of 42.5 and 43.9 kDa, respectively. Previous studies have shown that the domains in WbdAO9a can be separated; however, both are required for O9a biosynthesis (35), and it is currently unknown whether a specific mannosyltransferase activity is associated with each domain.
In light of the observations reported here, other glycan biosynthesis systems following the same general theme can be proposed. One example is the E. coli O99 antigen, which has the following repeat unit backbone: →3)-α-d-Rhap-(1→2)-α-d-Rhap-(1→2)-α-d-Rhap-(1→3)-α-d-Rhap-(1 (60). The structure carries additional α-(1→2)-linked Glcp substituents on the first and fourth Rhap residues in the repeat unit. Genetic analyses suggest that like other glycans containing d-Rhap, the backbone is synthesized using GDP-d-Rhap as the donor and is therefore very similar to the O9a antigen. The fine structure of the reducing terminus of the O99 glycan has not been reported, but its biosynthesis involves three glycosyltransferases, WejKJI, which are similar to WbdCBA, respectively (60). Catalytic assignments have been proposed based on the earlier model of O9a biosynthesis (60), but WejI contains two putative glycosyltransferase domains like WbdA and may be solely responsible for synthesis of the repeat unit domain. Another multidomain glycosyltransferase is predicted in synthesis of the E. coli O52 antigen (61), but further investigation is needed to confirm the activities of the enzymes involved.
Some Gram-positive S-layer glycoproteins provide non-O-PS systems that share striking similarities with the E. coli O9a and O8 antigens in both structure and biosynthesis. For example, the O-linked S-layer glycan of G. stearothermophilus NRS 2004/3a contains an adaptor and a repeat unit domain. The glycan repeat unit is composed of the trisaccharide →2)-α-l-Rhap-(1→3)-β-l-Rhap-(1→2)-α-l-Rhap-(1 and is terminated with a methyl group (62). Biosynthesis requires a multidomain glycosyltransferase that also contains the chain-terminating methyltransferase (63).
Despite these examples, it is not possible to make a reliable prediction of the synthesis strategy and the organization of glycosyltransferase modules based on the glycan structure alone. For example, the A-band O-PS of Pseudomonas aeruginosa possesses a trisaccharide, →2)-α-d-Rhap-(1→3)-α-d-Rhap-(1→3)-α-d-Rhap-(1 (64), reminiscent of both the O99 antigen and the G. steaothermophilus NRS 2004/3a glycan. However, the three glycosyltransferases involved, WbpZYX (65), all appear to be single-domain enzymes based on predictions using their primary sequence. In the biosynthesis of the E. coli K4 capsular polysaccharide, a two-domain enzyme polymerizes a chondroitin backbone composed of a disaccharide repeat unit, →4)-d-GlcpA-β-(1→3)-β-d-GalpNAc-(1 (66, 67). In contrast, in E. coli K5, the modified heparosan backbone, →4)-α-d-GlcpA-(1→4)-β-d-GlcpNAc-(1, requires the concerted activity of two glycosyltransferases, KfiC and KfiA (68–70). KfiC and KfiA are both single-domain glycosyltransferases, and they must associate with one another to polymerize the K5 capsule (70).
In summary, the E. coli O9a and O8 polymannose O-PSs represent important model systems for the ABC transporter-dependent assembly of O-PS. They provide examples where a combination of two enzymes containing a single mannosyltransferase domain, one of which is capable of adding two Manp residues in succession, and multidomain-polymerizing mannosyltransferases is exploited to build a single glycan. Although these features have been seen individually in other glycan biosynthesis systems, we are unaware of any others where all appear in the same dedicated assembly pathway.
Supplementary Material
Acknowledgments
We thank C. Bouwman for creation of CWG1009 and CWG1010 and Dr. B. R. Clarke for reagents and helpful discussions. We also thank M. Schur for technical support for CE methods. Dr. R. Whittal and J. Zheng (University of Alberta, Department of Chemistry Mass Spectrometry Facility) and J. Stupak (Institute for Biological Sciences, National Research Council of Canada) provided invaluable assistance in obtaining the mass spectra of enzymatically produced products. Drs. D. Hou and C. Liu prepared Acceptors A and B, and Dr. E. D. Brown (McMaster University) generously provided the β-GlcpNAc-PP-C13 acceptor prepared by E. W. Sewell.
This work was supported by grants from the Natural Sciences and Engineering Research Council (awarded to C. W. and T. L. L.) and by the Alberta Glycomics Centre (to T. L. L.).

This article contains supplemental Tables S1 and S2 and Figs. S1–S4.
- O-PS
- O-polysaccharide
- und-PP
- undecaprenol pyrophosphate
- ABC
- adenosine triphosphate-binding cassette
- Manp
- mannose
- GlcpNAc
- N-acetylglucosamine
- Rhap
- rhamnose
- Glcp
- glucose
- GlcpA
- glucuronic acid
- MalE
- maltose-binding protein
- CE
- capillary electrophoresis
- gCOSY
- gradient-enhanced correlation spectroscopy
- tROESY
- transverse rotating-frame Overhauser enhancement spectroscopy
- gHSQC
- gradient-enhanced heteronuclear single quantum correlation
- GlcpNAc-PP-C13
- β-GlcpNAc-pyrophosphoryl-C13
- BisTris
- 2-[bis(2-hydroxyethyl)amino]-2-(hydroxymethyl)propane-1,3-diol.
REFERENCES
- 1. Raetz C. R., Whitfield C. (2002) Lipopolysaccharide endotoxins. Annu. Rev. Biochem. 71, 635–700 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Orskov I., Orskov F., Jann B., Jann K. (1977) Serology, chemistry, and genetics of O- and K-antigens of Escherichia coli. Bacteriol. Rev. 41, 667–710 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Stenutz R., Weintraub A., Widmalm G. (2006) The structures of Escherichia coli O-polysaccharide antigens. FEMS Microbiol. Rev. 30, 382–403 [DOI] [PubMed] [Google Scholar]
- 4. Sperandeo P., Dehò G., Polissi A. (2009) The lipopolysaccharide transport system of Gram-negative bacteria. Biochim. Biophys. Acta 1791, 594–602 [DOI] [PubMed] [Google Scholar]
- 5. Fitzgerald-Chandler D. K., Jann K. (1971) Studies of the biosynthesis of the O9 antigen from Escherichia coli O9:K30(A):H12. Eur. J. Biochem. 24, 222–231 [DOI] [PubMed] [Google Scholar]
- 6. Greenfield L. K., Whitfield C. (2012) Synthesis of lipopolysaccharide O-antigens by ABC transporter-dependent pathways. Carbohydr. Res. 356, 12–24 [DOI] [PubMed] [Google Scholar]
- 7. Saeki A., Kido N., Sugiyama T., Ohta M., Iwashita T., Uchiya K., Kato N. (1993) Isolation of rfb gene clusters directing the synthesis of O-polysaccharides consisting of mannose homopolymers and serological analysis of lipopolysaccharides. Microbiol. Immunol. 37, 601-606 [DOI] [PubMed] [Google Scholar]
- 8. Sugiyama T., Kido N., Kato Y., Koide N., Yoshida T., Yokochi T. (1997) Evolutionary relationship among rfb gene clusters synthesizing mannose homopolymer as O-specific polysaccharides in Escherichia coli and Klebsiella. Gene 198, 111–113 [DOI] [PubMed] [Google Scholar]
- 9. Sugiyama T., Kido N., Kato Y., Koide N., Yoshida T., Yokochi T. (1998) Generation of Escherichia coli O9a serotype, a subtype of E. coli O9, by transfer of the wb* gene cluster of Klebsiella O3 into E. coli via recombination. J. Bacteriol. 180, 2775–2778 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Kido N., Kobayashi H. (2000) A single amino acid substitution in a mannosyltransferase, WbdA, converts the Escherichia coli O9 polysaccharide into O9a. Generation of a new O-serotype group. J. Bacteriol. 182, 2567–2573 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Vinogradov E., Frirdich E., MacLean L. L., Perry M. B., Petersen B. O., Duus J. Ø., Whitfield C. (2002) Structures of lipopolysaccharides from Klebsiella pneumoniae. Eluicidation of the structure of the linkage region between core and polysaccharide O chain and identification of the residues at the non-reducing termini of the O chains. J. Biol. Chem. 277, 25070–25081 [DOI] [PubMed] [Google Scholar]
- 12. Clarke B. R., Richards M. R., Greenfield L. K., Hou D., Lowary T. L., Whitfield C. (2011) In vitro reconstruction of the chain termination reaction in biosynthesis of the Escherichia coli O9a O-polysaccharide. The chain-length regulator, WbdD, catalyzes the addition of methyl phosphate to the non-reducing terminus of the growing glycan. J. Biol. Chem. 286, 41391–41401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Kubler-Kielb J., Whitfield C., Katzenellenbogen E., Vinogradov E. (2012) Identification of the methyl phosphate substituent at the non-reducing terminal mannose residue of the O-specific polysaccharides of Klebsiella pneumoniae O3, Hafnia alvei PCM 1223, and Escherichia coli O9/O9a LPS. Carbohydr Res. 347, 186–188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kido N., Torgov V. I., Sugiyama T., Uchiya K., Sugihara H., Komatsu T., Kato N., Jann K. (1995) Expression of the O9 polysaccharide of Escherichia coli. Sequencing of the E. coli O9 rfb gene cluster, characterization of mannosyl transferases, and evidence for an ATP-binding cassette transport system. J. Bacteriol. 177, 2178–2187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Clarke B. R., Cuthbertson L., Whitfield C. (2004) Nonreducing terminal modifications determine the chain length of polymannose O-antigens of Escherichia coli and couple chain termination to polymer export via an ATP-binding cassette transporter. J. Biol. Chem. 279, 35709–35718 [DOI] [PubMed] [Google Scholar]
- 16. Cuthbertson L., Kimber M. S., Whitfield C. (2007) Substrate binding by a bacterial ABC transporter involved in polysaccharide export. Proc. Natl. Acad. Sci. U.S.A. 104, 19529–19534 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Kido N., Morooka N., Paeng N., Ohtani T., Kobayashi H., Shibata N., Okawa Y., Suzuki S., Sugiyama T., Yokochi T. (1997) Production of monoclonal antibody discriminating serological difference in Escherichia coli O9 and O9a polysaccharides. Microbiol. Immunol. 41, 519–525 [DOI] [PubMed] [Google Scholar]
- 18. Meier-Dieter U., Starman R., Barr K., Mayer H., Rick P. D. (1990) Biosynthesis of enterobacterial common antigen in Escherichia coli. Biochemical characterization of Tn10 insertion mutants defective in enterobacterial common antigen synthesis. J. Biol. Chem. 265, 13490–13497 [PubMed] [Google Scholar]
- 19. Meier-Dieter U., Barr K., Starman R., Hatch L., Rick P. D. (1992) Nucleotide sequence of the Escherichia coli rfe gene involved in the synthesis of enterobacterial common antigen. Molecular cloning of the rfe-rff gene cluster. J. Biol. Chem. 267, 746–753 [PubMed] [Google Scholar]
- 20. Miller J. H. (1972) Experiments in Molecular Genetics, pp. 432–433, Cold Spring Harbor Laboratory, Cold Spring Harbor, NY [Google Scholar]
- 21. Sambrook J., Fritsch E. F., Maniatis T. (1989) Molecular Cloning: A Laboratory Manual, 2nd Ed., pA3, Cold Spring Harbor Laboratory, Cold Spring Harbor, NY [Google Scholar]
- 22. Datsenko K. A., Wanner B. L. (2000) One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc. Natl. Acad. Sci. U.S.A. 97, 6640–6645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Guzman L. M., Belin D., Carson M. J., Beckwith J. (1995) Tight regulation, modulation, and high level expression by vectors containing the arabinose PBAD promoter. J. Bacteriol. 177, 4121–4130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Gasteiger E., Hoogland C., Gattiker A., Duvaud S., Wilkins M. R., Appel R. D., Bairoch A. (2005) in The Proteomics Protocols Handbook (Walker J. M., ed) pp. 571–607, Humana Press, Totowa, NJ [Google Scholar]
- 25. Ginsberg C., Zhang Y. H., Yuan Y., Walker S. (2006) In vitro reconstitution of two essential steps in wall teichoic acid biosynthesis. ACS Chem. Biol. 1, 25–28 [DOI] [PubMed] [Google Scholar]
- 26. Kanegasaki S., Jann K. (1979) Demonstration by membrane reconstitution of a butanol-soluble intermediate in the biosynthesis of the O9 antigen of Escherichia coli. Eur. J. Biochem. 95, 287–293 [DOI] [PubMed] [Google Scholar]
- 27. Dubois M., Gilles K. A., Hamilton J. K., Rebers P. A., Smith F. (1956) A colorimetric method for the determination of sugars substances. Nature 168, 167. [DOI] [PubMed] [Google Scholar]
- 28. Liu C., Skogman F., Cai Y., Lowary T. L. (2007) Synthesis of the “primer-adaptor” trisaccharide moiety of Escherichia coli O8, O9, and O9a lipopolysaccharide. Carbohydr. Res. 342, 2818–2825 [DOI] [PubMed] [Google Scholar]
- 29. Hou D., Skogman F., Lowary T. L. (2008) Synthesis of 8-azidooctyl glycoside derivatives of the O-chain repeating unit of Escherichia coli O9a lipopolysaccharide and a methylated analog. Carbohydr. Res. 343, 1778–1789 [DOI] [PubMed] [Google Scholar]
- 30. Wakarchuk W. W., Cunningham A. M. (2003) Capillary electrophoresis as an assay method for monitoring glycosyltransferase activity. Methods Mol. Biol. 213, 263–274 [DOI] [PubMed] [Google Scholar]
- 31. Laemmli U. K. (1970) Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227, 680–685 [DOI] [PubMed] [Google Scholar]
- 32. Hitchcock P. J., Brown T. M. (1983) Morphological heterogeneity among Salmonella lipopolysaccharide chemotypes in silver-stained polyacrylamide gels. J. Bacteriol. 154, 269–277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Tsai C. M., Frasch C. E. (1982) A sensitive silver stain for detecting lipopolysaccharides in polyacrylamide gels. Anal. Biochem. 119, 115–119 [DOI] [PubMed] [Google Scholar]
- 34. Shatzman A. R., Gross M. S., Rosenberg M. (2001) Expression using vectors with phage λ regulatory sequences. Current Protocols in Molecular Biology 11, chapter 16 unit 16.3 [DOI] [PubMed] [Google Scholar]
- 35. Kido N., Sugiyama T., Yokochi T., Kobayashi H., Okawa Y. (1998) Synthesis of Escherichia coli O9a polysaccharide requires the participation of two domains of WbdA, a mannosyltransferase encoded within the wb* gene cluster. Mol. Microbiol. 27, 1213–1221 [DOI] [PubMed] [Google Scholar]
- 36. Geremia R. A., Petroni E. A., Ielpi L., Henrissat B. (1996) Towards a classification of glycosyltransferases based on amino acid sequence similarities. Prokaryotic α-mannosyltransferases. Biochem. J. 318, 133–138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Campbell J. A., Davies G. J., Bulone V., Henrissat B. (1997) A classification of nucleotide-diphospho-sugar glycosyltransferases based on amino acid sequence similarities. Biochem. J. 326, 929–939 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Coutinho P. M., Deleury E., Davies G. J., Henrissat B. (2003) An evolving hierarchical family classification for glycosyltransferases. J. Mol. Biol. 328, 307–317 [DOI] [PubMed] [Google Scholar]
- 39. Cantarel B. L., Coutinho P. M., Rancurel C., Bernard T., Lombard V., Henrissat B. (2009) The Carbohydrate-Active EnZymes database (CAZy). An expert resource for glycogenomics. Nucleic Acids Res. 37, D233–D238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Parolis L. A., Parolis H., Dutton G. G. (1986) Structural studies of the O-antigen polysaccharide of Escherichia coli 09a. Carbohydr Res. 155, 272–276 [DOI] [PubMed] [Google Scholar]
- 41. Jansson P. E., Stenutz R., Widmalm G. (2006) Sequence determination of oligosaccharides and regular polysaccharides using NMR spectroscopy and a novel Web-based version of the computer program CASPER. Carbohydr. Res. 341, 1003–1010 [DOI] [PubMed] [Google Scholar]
- 42. Lundborg M., Widmalm G. (2011) Structure analysis of glycans by NMR chemical shift prediction. Anal. Chem. 83, 1514–1517 [DOI] [PubMed] [Google Scholar]
- 43. Jansson P. E., Lönngren J., Widmalm G., Leontein K., Slettengren K., Svenson S. B., Wrangsell G., Dell A., Tiller P. R. (1985) Structural studies of the O-antigen polysaccharides of Klebsiella O5 and Escherichia coli O8. Carbohydr. Res. 145, 59–66 [DOI] [PubMed] [Google Scholar]
- 44. Cuthbertson L., Kos V., Whitfield C. (2010) ABC transporters involved in export of cell surface glycoconjugates. Microbiol. Mol. Biol. Rev. 74, 341–362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Martinez-Fleites C., Proctor M., Roberts S., Bolam D. N., Gilbert H. J., Davies G. J. (2006) Insights into the synthesis of lipopolysaccharide and antibiotics through the structures of two retaining glycosyltransferases from family GT4. Chem. Biol. 13, 1143–1152 [DOI] [PubMed] [Google Scholar]
- 46. Steiner K., Hagelueken G., Messner P., Schäffer C., Naismith J. H. (2010) Structural basis of substrate binding in WsaF, a rhamnosyltransferase from Geobacillus stearothermophilus. J. Mol. Biol. 397, 436–447 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Guerin M. E., Kordulakova J., Schaeffer F., Svetlikova Z., Buschiazzo A., Giganti D., Gicquel B., Mikusova K., Jackson M., Alzari P. M. (2007) Molecular recognition and interfacial catalysis by the essential phosphatidylinositol mannosyltransferase PimA from mycobacteria. J. Biol. Chem. 282, 20705–20714 [DOI] [PubMed] [Google Scholar]
- 48. Glover K. J., Weerapana E., Imperiali B. (2005) In vitro assembly of the undecaprenylpyrophosphate-linked heptasaccharide for prokaryotic N-linked glycosylation. Proc. Natl. Acad. Sci. U.S.A. 102, 14255–14259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Troutman J. M., Imperiali B. (2009) Campylobacter jejuni PglH is a single active site processive polymerase that utilizes product inhibition to limit sequential glycosyl transfer reactions. Biochemistry 48, 2807–2816 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Kremer L., Dover L. G., Morehouse C., Hitchin P., Everett M., Morris H. R., Dell A., Brennan P. J., McNeil M. R., Flaherty C., Duncan K., Besra G. S. (2001) Galactan biosynthesis in Mycobacterium tuberculosis. Identification of a bifunctional UDP-galactofuranosyltransferase. J. Biol. Chem. 276, 26430–26440 [DOI] [PubMed] [Google Scholar]
- 51. Belánová M., Dianisková P., Brennan P. J., Completo G. C., Rose N. L., Lowary T. L., Mikusová K. (2008) Galactosyltransferases in mycobacterial cell wall synthesis. J. Bacteriol. 190, 1141–1145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Brozek K. A., Hosaka K., Robertson A. D., Raetz C. R. (1989) Biosynthesis of lipopolysaccharide in Escherichia coli. Cytoplasmic enzymes that attach 3-deoxy-d-manno-octulosonic acid to lipid A. J. Biol. Chem. 264, 6956–6966 [PubMed] [Google Scholar]
- 53. O'Reilly M. K., Zhang G., Imperiali B. (2006) In vitro evidence for the dual function of Alg2 and Alg11. Essential mannosyltransferases in N-linked glycoprotein biosynthesis. Biochemistry 45, 9593–9603 [DOI] [PubMed] [Google Scholar]
- 54. Absmanner B., Schmeiser V., Kämpf M., Lehle L. (2010) Biochemical characterization, membrane association, and identification of amino acids essential for the function of Alg11 from Saccharomyces cerevisiae, an α1,2-mannosyltransferase catalyzing two sequential glycosylation steps in the formation of the lipid-linked core oligosaccharide. Biochem. J. 426, 205–217 [DOI] [PubMed] [Google Scholar]
- 55. Gilbert M., Brisson J. R., Karwaski M. F., Michniewicz J., Cunningham A. M., Wu Y., Young N. M., Wakarchuk W. W. (2000) Biosynthesis of ganglioside mimics in Campylobacter jejuni OH4384. Identification of the glycosyltransferase genes, enzymatic synthesis of model compounds, and characterization of nanomole amounts by 600-mHz 1H and 13C NMR analysis. J. Biol. Chem. 275, 3896–3906 [DOI] [PubMed] [Google Scholar]
- 56. Blixt O., Vasiliu D., Allin K., Jacobsen N., Warnock D., Razi N., Paulson J. C., Bernatchez S., Gilbert M., Wakarchuk W. (2005) Chemoenzymatic synthesis of 2-azidoethyl-ganglio-oligosaccharides GD3, GT3, GM2, GD2, GT2, GM1, and GD1a. Carbohydr. Res. 340, 1963–1972 [DOI] [PubMed] [Google Scholar]
- 57. Cuthbertson L., Powers J., Whitfield C. (2005) The C-terminal domain of the nucleotide-binding domain protein Wzt determines substrate specificity in the ATP-binding cassette transporter for the lipopolysaccharide O-antigens in Escherichia coli serotypes O8 and O9a. J. Biol. Chem. 280, 30310–30319 [DOI] [PubMed] [Google Scholar]
- 58. Clarke B. R., Greenfield L. K., Bouwman C., Whitfield C. (2009) Coordination of polymerization, chain termination, and export in assembly of the Escherichia coli lipopolysaccharide O9a antigen in an ATP-binding cassette transporter-dependent pathway. J. Biol. Chem. 284, 30662–30672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Marchler-Bauer A., Lu S., Anderson J. B., Chitsaz F., Derbyshire M. K., DeWeese-Scott C., Fong J. H., Geer L. Y., Geer R. C., Gonzales N. R. (2011) CDD. A conserved domain database for the functional annotation of proteins. Nucleic Acids Res. 39, D225–D229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Perepelov A. V., Li D., Liu B., Senchenkova S. N., Guo D., Shevelev S. D., Shashkov A. S., Guo X., Feng L., Knirel Y. A., Wang L. (2009) Structural and genetic characterization of Escherichia coli O99 antigen. FEMS Immunol. Med. Microbiol. 57, 80–87 [DOI] [PubMed] [Google Scholar]
- 61. Feng L., Senchenkova S. N., Yang J., Shashkov A. S., Tao J., Guo H., Cheng J., Ren Y., Knirel Y. A., Reeves P. R., Wang L. (2004) Synthesis of the heteropolysaccharide O-antigen of Escherichia coli O52 requires an ABC transporter. Structural and genetic evidence. J. Bacteriol. 186, 4510–4519 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Schäffer C., Wugeditsch T., Kählig H., Scheberl A., Zayni S., Messner P. (2002) The surface layer (S-layer) glycoprotein of Geobacillus stearothermophilus NRS 2004/3a. Analysis of its glycosylation. J. Biol. Chem. 277, 6230–6239 [DOI] [PubMed] [Google Scholar]
- 63. Steiner K., Novotny R., Werz D. B., Zarschler K., Seeberger P. H., Hofinger A., Kosma P., Schäffer C., Messner P. (2008) Molecular basis of S-layer glycoprotein glycan biosynthesis in Geobacillus stearothermophilus. J. Biol. Chem. 283, 21120–21133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Arsenault T. L., Hughes D. W., MacLean D. B., Szarek W. A., Kropinski A. M. B., Lam J. S. (1991) Structural studies on the polysaccharide portion of “A-band” lipopolysaccharide from a mutant (AK1401) of Pseudomonas aeruginosa strain PAO1. Can. J. Chem. 69, 1273–1280 [Google Scholar]
- 65. Rocchetta H. L., Burrows L. L., Pacan J. C., Lam J. S. (1998) Three rhamnosyltransferases responsible for assembly of the A-band d-rhamnan polysaccharide in Pseudomonas aeruginosa. A fourth transferase, WbpL, is required for the initiation of both A-band and B-band lipopolysaccharide synthesis. Mol. Microbiol. 28, 1103–1119 [DOI] [PubMed] [Google Scholar]
- 66. Ninomiya T., Sugiura N., Tawada A., Sugimoto K., Watanabe H., Kimata K. (2002) Molecular cloning and characterization of chondroitin polymerase from Escherichia coli strain K4. J. Biol. Chem. 277, 21567–21575 [DOI] [PubMed] [Google Scholar]
- 67. Sobhany M., Kakuta Y., Sugiura N., Kimata K., Negishi M. (2008) The chondroitin polymerase K4CP and the molecular mechanism of selective bindings of donor substrates to two active sites. J. Biol. Chem. 283, 32328–32333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Griffiths G., Cook N. J., Gottfridson E., Lind T., Lidholt K., Roberts I. S. (1998) Characterization of the glycosyltransferase enzyme from the Escherichia coli K5 capsule gene cluster and identification and characterization of the glucuronyl active site. J. Biol. Chem. 273, 11752–11757 [DOI] [PubMed] [Google Scholar]
- 69. Hodson N., Griffiths G., Cook N., Pourhossein M., Gottfridson E., Lind T., Lidholt K., Roberts I. S. (2000) Identification that KfiA, a protein essential for the biosynthesis of the Escherichia coli K5 capsular polysaccharide, is an α-UDP-GlcNAc glycosyltransferase. The formation of a membrane-associated K5 biosynthetic complex requires KfiA, KfiB, and KfiC. J. Biol. Chem. 275, 27311–27315 [DOI] [PubMed] [Google Scholar]
- 70. Sugiura N., Baba Y., Kawaguchi Y., Iwatani T., Suzuki K., Kusakabe T., Yamagishi K., Kimata K., Kakuta Y., Watanabe H. (2010) Glucuronyltransferase activity of KfiC from Escherichia coli strain K5 requires association of KfiA. KfiC and KfiA are essential enzymes for production of K5 polysaccharide, N-acetylheparosan. J. Biol. Chem. 285, 1597–1606 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.