

Abstract
Renal proximal tubular damage and repair are hallmarks of acute kidney injury. Because glycogen synthase kinase-3β (GSK-3β) is an important cellular regulator of survival and proliferation, we determined its role during injury and recovery of proximal tubules in a mercuric chloride-induced nephrotoxic model of acute kidney injury. Renal proximal tubule-specific GSK-3β knockout mice exposed to mercuric chloride had improved survival and renal function compared to wild type mice. Apoptosis, measured by TUNEL staining, Bax activation, and caspase 3 cleavage were all reduced in the knockout mice. The restoration of renal structure, function, and cell proliferation was also accelerated in the GSK-3β knockout mice. This enhanced repair, evidenced by increased Ki-67 and BrdU staining, along with increased cyclin D1 and c-myc levels, was recapitulated by treatment of wild type mice with the small-molecule GSK-3 inhibitor TDZD-8 following injury. This confirmed that hastened repair in the knockout mice was not merely due to lower initial injury levels. Thus, inhibition of GSK-3β prior to nephrotoxic insult protects from renal injury. Such treatment after acute kidney injury may accelerate repair and regeneration.
Introduction
Acute kidney injury (AKI) is characterized by an abrupt loss of renal function caused by ischemia–reperfusion or nephrotoxic insult and increases risk of later chronic kidney disease1–4. AKI involves a complex series of events that leads to tissue injury including endothelial and epithelial cell death, intra-tubular obstruction, changes in local microvascular blood flow and inflammatory processes5. The epithelial cells of proximal tubules are most susceptible to injury as they have a high metabolic rate and have greater ability to take up and concentrate toxins from both the luminal and the basolateral sides6, 7. However, these cells also have an amazing capacity to regenerate1, 8. Glycogen synthase kinase-3 (GSK3) is a serine/threonine protein kinase that is well positioned to coordinate multiple signaling pathways that regulate various cellular processes including gene transcription, translation, cytoskeletal organization, cell cycle progression and survival9, 10. GSK3 exists in two isoforms encoded by distinct genes, α and β. Since GSK3α and GSK3β isoforms share 98% sequence homology in their kinase domains11, no truly isoform-specific GSK3β inhibitors have been developed yet12, 13.
GSK3β is widely expressed in the kidneys14–19 and recent studies have identified a possible role for GSK3β in renal tubular injury. Gene silencing of GSK3β in cultured proximal tubular cells reduced ATP-depletion induced apoptosis20. Further, inhibition using GSK3 isoform non-selective inhibitors reduced injury in endotoxemia and ischemia-reperfusion induced AKI20–22. Since these studies employed systemic inhibition of GSK3 to examine renal injury, the specific role of GSK3β in survival of the proximal tubules per se, has remained unclear. In addition, the role of GSK3β in repair and regeneration of proximal tubules in AKI has not been explored. GSK3 isoforms have a pivotal role in cell cycle progression in embryonic stem cells and other cultured cell types10. Although the relative importance of GSK3α and GSK3β isoforms in proliferation is not clear, targeted global KO of GSK3β in mice resulted in hyperproliferation of cardiomyocytes during embryonic development, while mice with global knockout of GSK3α appeared to be normal in this respect23. In a recent study, Peng et al reported that inhibition of GSK3 reduced migration of cultured proximal tubule cells in a scratch wound healing assay suggesting suppression of wound healing in renal tubular cells24. This result potentially contradicts the observations that inhibition of GSK3 is generally protective and reduces damage. Hence in the current study we used proximal tubule specific GSK3β knockout mice to examine the specific role of GSK3β in tubular injury and repair in AKI.
We chose the widely used HgCl2-induced model of AKI25–28 because mercury is a potent nephrotoxin, and its uptake via luminal γ-glutamyltranspeptidase (γ-GT) and the basolateral organic anion transporter system results in preferential accumulation and cytotoxicity of proximal tubules 25, 29–34. Here, we report on the impact of selective genetic and chemical inhibition of GSK3 on initial damage and subsequent repair of renal proximal tubules in AKI.
RESULTS
1) Generating renal proximal tubule specific GSK3β KO mice
To obtain renal proximal tubule specific gene deletion of GSK3β, we bred GSK3βloxp/loxp mice35 with γ-GT-Cre+/+ mice36. Mice progeny exhibited the expected Mendelian ratio and histo-pathological examination revealed no renal abnormalities in the knockout (KO) mice (GSK3βloxp/loxp, γ-GT-Cre+/+) when compared to wild type (WT) (GSK3βloxp/loxp, γ-GT-Cre−/−). Western blot analysis of lysate from whole renal cortex (Fig 1a) and isolated proximal tubules (Fig 1b) showed significantly reduced levels of GSK3β expression in the KO mice compared to WT mice. Immunohistochemical staining with antibodies selective for GSK3β showed significantly reduced levels of GSK3β in the proximal tubules but not in other nephron segments of KO mice (Fig 1c).
Fig. 1. Reduced expression of GSK3β in renal proximal tubules of KO mice.
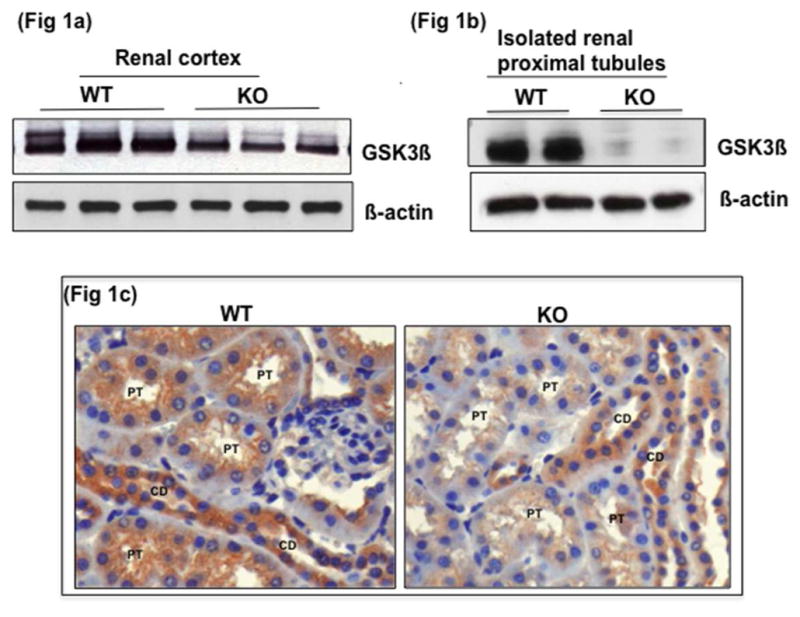
Western blot analysis shows reduced GSK3β protein levels in tissue lysate of GSK3βloxp/loxp, γ-GT-Cre+/+ (KO) compared to GSK3βloxp/loxp, γ-GT-Cre−/− (WT) in a) whole renal cortex and b) acutely isolated proximal tubules from renal cortex. c) Immunohistochemical staining shows expression of GSK3β reduced in proximal tubules (PT) but not in the collecting ducts (CD) of KO mice (anti GSK3β antibody, x200 original magnification).
2) Proximal tubule-specific KO of GSK3β reduces HgCl2-induced mortality and tubular injury
When treated with 8.14 mg/kg body weight of HgCl2, a 10% mortality rate was observed in both WT and KO groups within 24 hours (Fig 2). By days 2–3, the mortality rate further increased to 40% in the WT group, while it remained unchanged in the KO cohort, indicating that GSK3β gene deletion in the proximal tubule reduced HgCl2-induced mortality (Fig 2). Kidneys showed tubular dilatation, cellular necrosis and loss of brush border in both WT and KO mice by day 2 after HgCl2 treatment (Fig 3a). Semiquantitative injury score showed that on day 2, injury score in the KO mice was only half of that in WT mice (Fig 3b). The injury score did not change significantly in WT mice by day 4 and remained 16-fold higher than baseline values on day 6. In the KO mice, the injury score decreased by 1.8-fold on day 4 and attained baseline levels by day 6. Blood urea nitrogen (BUN) levels increased by 7.8-fold in WT and 3.5-fold in the KO group by day 2 after HgCl2 treatment, compared to baseline (20±6 vs 155±15 mg/dl in WT and 25±7 vs 87±18 mg/dl in KO). By days 4 and 6, BUN levels in the KO mice were reduced by 42% and 60%, respectively, returning to close to baseline levels. In contrast, BUN levels remained essentially unchanged in the WT group on day 4, followed by a 40% decrease on day 6 but failed to reach baseline levels even by day 8 (Fig 3c). Plasma creatinine levels showed a similar pattern (Fig 3d).
Fig. 2. KO mice show higher survival rate following HgCl2 treatment.
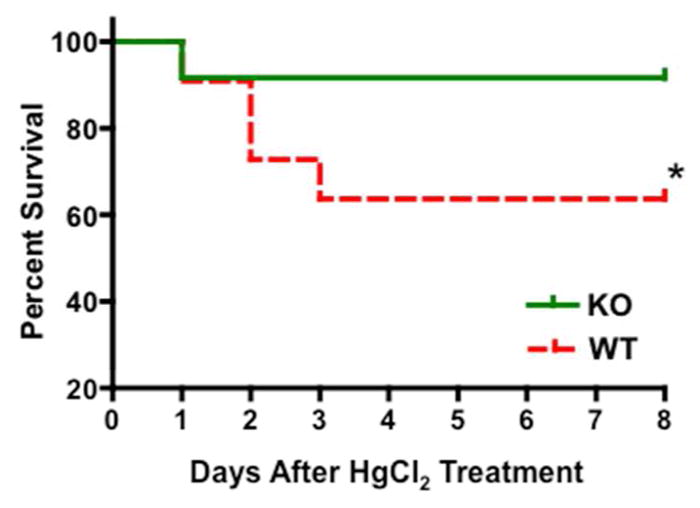
Kaplan-Meier survival curve of WT and KO mice injected with a single subcutaneous injection of 8.14 mg/kg body weight of HgCl2. *, P<0.01, n=20 per group
Fig. 3. Proximal tubule specific GSK3β gene deletion reduced HgCl2-induced renal injury.

a) Tubular injury is manifest as necrotic tubules (*), flattened epithelium (arrows) and loss of nuclei (arrows) and was less severe in KO compared to WT at all time points (hematoxylin and eosin, x200 original magnification). b) Injury score of renal tissue sections. **, P<0.001, n=5 per group c) Blood urea nitrogen and d) Serum creatinine levels are lower in KO mice. **, P<0.001, n=21 per group
3) Reduced apoptosis in the renal cortex of GSK3β KO mice following HgCl2 treatment
To examine if the reduced levels of injury in the KO mice could be attributed to a decreased rate of apoptosis, TUNEL assay was carried out. Within 24h after HgCl2 treatment, 12% of nuclei were TUNEL positive in the WT kidney compared to only 4% in KO (Fig 4a, 4b). These apoptotic cells were mostly in proximal tubules as shown by an immunoperoxidase- labeled TUNEL assay (Supplemental Fig. 1). This result indicated that apoptosis was associated with tissue injury early in HgCl2-induced AKI and the rate of apoptosis was reduced in KO mice.
Fig. 4. HgCl2 induced apoptosis is reduced in KO mice.

a) TUNEL nuclear staining in renal cortex, 24h after HgCl2 treatment and b) quantitation which shows reduced TUNEL positive nuclei in KO group. c) Cleaved caspase 3 and activated Bax levels are lower by Western blot in KO kidney compared to WT (d & e). **, P<0.001, ***, P<0.0001. Fig 4d & e based on two time-course studies. Fig 4b n=5 per group.
To determine the mechanism underlying the anti-apoptotic effect of GSK3β gene deletion, we examined protein levels of pro-apoptotic- activated Bax, cleaved caspase 3 and an anti-apoptotic factor, Bcl 2. Although a time dependent increase in activated Bax levels was observed in both WT and KO mice, the levels were significantly lower in KO mice (Fig 4c). Activated Bax levels in the KO mice were lower by 2 and 2.8-fold respectively on days 1 and 2 when compared to WT mice (Fig 4d). Total Bax levels showed no change. Cleaved caspase 3 levels peaked on day 1, increasing by 5-fold compared to day 0 levels. Compared to WT mice, in KO mice, cleaved caspase 3 levels were 5 and 2-fold lower on days 1 and 2, respectively (Fig 4c, 4e). In the WT group, Bcl 2 levels were reduced by 20% and 40% respectively by days 1 and 2, compared to day 0. In the KO mice, no significant difference was observed (Fig 4c, 4f). These data demonstrate that the KO mice were protected from the apoptosis caused by HgCl2.
3) Accelerated cell proliferation in renal proximal tubules of KO mice
We next determined whether GSK3β gene deletion in proximal tubule cells alters their proliferative response after injury. Ki-67 nuclear staining, a proliferative marker, was higher by 2-fold on day 2 and 3.5-fold on day 4 in the KO group compared to the WT group (500-fold more than baseline) (Fig 5a & 5b). BrdU positive nuclei were also 2.5-fold higher in the KO kidney on day 4 compared to WT (Fig 5c & 5d). Both Ki-67 as well as BrdU staining decreased in KO kidneys after day 4, consistent with the improved renal function and decreased histopathology injury data. Triple immunofluorescence staining of kidneys on day 4 showed that the dividing cells, indicated by PCNA staining, were more abundant in the KO mice, and were primarily located in the proximal tubule (stained with Lotus tetragonolobus agglutinin (LTA)) (Fig 5e).
Fig. 5. Proliferation is accelerated in KO kidneys.

Immunohistochemical staining and quantitation for a) & b) Ki-67 and c & d) BrdU show more proliferating cells in KO kidneys. **, P<0.001. ***, P<0.0001, n=5 per group, x200 original magnification). immunofluorescence staining for PCNA and LTA shows more dividing cells in proximal tubule cells of KO mice on day 4 after HgCl2 treatment, x400 original magnification.
To examine the mechanism for the higher rate of proliferation in the KO mice, protein levels of the pro-proliferative factors, cyclin D1, c-myc and β-catenin, known to be regulated by GSK3β were measured. Cyclin D1 and c-myc levels in the kidney cortex were markedly upregulated in KO mice compared to WT mice, especially on days 1–4 (Fig 6a). Interestingly, cyclin D1 levels decreased by day 6 in the KO mice, while in the WT mice, the highest cyclin D1 levels were observed on days 6 and 8 (Fig 6b). c-myc levels were significantly higher in KO mice on day 3, 4 and 6 compared to that in WT mice (Fig 6c). β-catenin levels did not differ significantly between the WT and KO groups. These results indicated an accelerated rate of proliferation in KO mice when compared to WT mice. Examination of renal fibrosis by sirius red staining, a marker of collagen, revealed no significant difference between the WT and KO mice 8 days after HgCl2 treatment, though it is possible that fibrosis occurs after a longer period of time (supplemental data).
Fig. 6. Cyclin D1 and c-myc levels are higher in KO Kidneys.
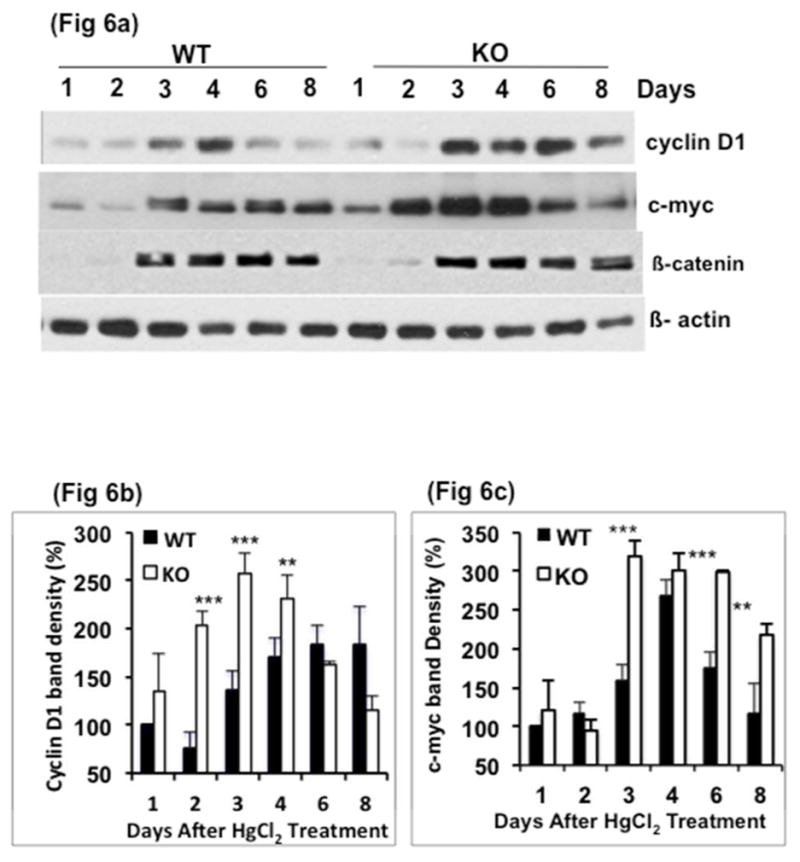
a) Western blot analysis and b, c) quantitation of band density for protein levels of cyclin D1, c-myc and β-catenin. Fig 6d & 6c based on two time course studies**, P<0.001. ***, P<0.0001. Data= mean ± SD
4) Paired study shows that GSK3β inhibition accelerates proliferation
To probe further the role of GSK3β in regeneration following AKI we tested the effect of TDZD-8, a small-molecule inhibitor of GSK337, 38. In this study, TDZD-8 treatment was carried out after onset of injury in WT mice. WT mice were first administered 6.3 mg /kg of HgCl2. On day 2 (48 hours after HgCl2), BUN levels were measured and mice with of approximately 82±2 mg/dl were separated into two groups. One group was administered vehicle and the other, TDZD-8 (1 and 0.5 mg/kg respectively on day 2 and 3 after HgCl2 treatment). In the TDZD-8 treated group, BUN levels decreased significantly compared to vehicle treated group (vehicle, 72±9 vs TDZD-8, 51±6 mg/dl on day 4, n=9 and vehicle, 51±8 vs TDZD-8, 30±7 mg/dl on day 6, n=6) (Fig 7a). TDZD-8 treated mice also showed 40% and 36% lower kidney injury scores compared to vehicle-treated mice on days 4 and 6, respectively (Fig 7b, 7c), indicating that inhibition of GSK3 accelerated regeneration. Further, protein levels of cyclin D1, c-myc and β-catenin were significantly higher on days 3 and 4 in the TDZD-8 treated compared to vehicle treated mice (Fig 8a). By day 4, the number of BrdU positive nuclei in TDZD-8 treated mice was almost double when compared to vehicle treated mice (Fig 8b and 8c). Similarly, Ki-67 staining was more than double in the TDZD-8 treated mice, compared to the vehicle treated mice on day 4 (Fig 8d and 8e). Sirius red staining, on tissue sections, 8 days after HgCl2 treatment revealed no significant change in fibrosis between the two groups at this time point (supplemental data).
Fig. 7. Recovery is faster in GSK3 inhibitor treated mice compared to similarly injured, but vehicle treated mice.

WT mice were injected once with 6.3 mg/kg HgCl2. After 48h, mice with comparable levels of BUN were administered vehicle or GSK3 inhibitor, TDZD-8 (1mg/kg BWt on day 2 and 0.5mg/kg BWt on day 3. a) Blood urea nitrogen levels decreased at a faster rate in TDZD-8 treated mice. b) injury score shows reduced injury in TDZD-8 treated mice, *, P<0.01. **, P<0.001, n=12 /group c) Representative image of renal cortex of WT and TDZD-8 treated mice, 4 days after HgCl2 treatment (hematoxylin and eosin, x100 original magnification).
Fig. 8. Proliferation is accelerated in GSK3β inhibitor treated mice compared to similarly injured, but vehicle treated mice.

a) Cyclin D1, c-myc and β-catenin levels by Western blot from renal cortical protein lysates were higher in TDZD-8 treated mice. c) BRDU immunostaining and quantitation of positive nuclei (b & c) are increased in TDZD-8 treated mice on day 4 after HgCl2 treatment. **, P<0.001, x200 original magnification.
5) Regulation of renal GSK3β activity in response to HgCl2 treatment
We next examined whether GSK3β activity was regulated in HgCl2-induced AKI. Protein levels of pGSK3β, the inhibited form of GSK3β phosphorylated at Serine 9, decreased to undetectable levels 6h after HgCl2 injection, gradually increasing by 16h (Fig 9a). By 32h, pGSK3β levels increased above baseline levels and remained high for 5 days. Protein levels of β-catenin, a negatively regulated substrate of GSK3 also increased by 48h, consistent with the GSK3 inhibition. These data indicated that, following HgCl2 treatment, GSK3β (and likely also GSK3α) was initially activated and then, subsequently inhibited.
Fig. 9. Renal GSK3β activity changes in response to HgCl2 treatment.
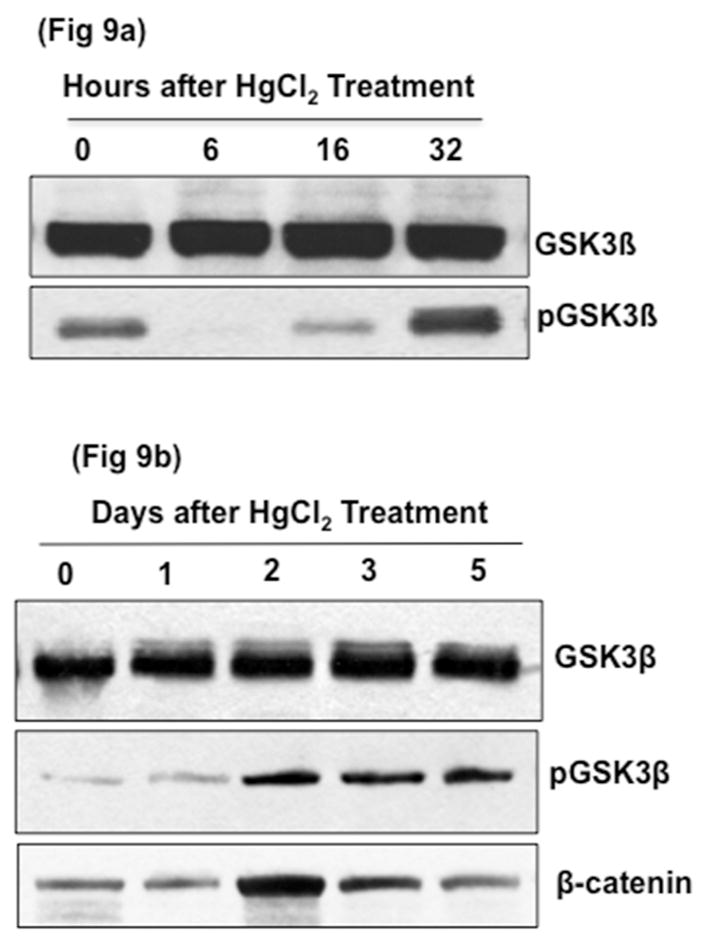
Western blot analysis of tissue lysates from whole renal cortex of WT mice injected once with 8.14mg/kg of HgCl2. a) Inactive phosphorylated GSK3β levels initially decreased within 6 hours after HgCl2 treatment and b) increased to above baseline by 24h. β-Catenin levels also increased on days 2–5.
Discussion
Our studies demonstrate that GSK3β activity in the renal proximal tubules modulates injury and repair in HgCl2-induced AKI. Proximal tubule-specific KO of GSK3β improved survival in HgCl2-treated mice by reducing nephrotoxicity and injury as well as accelerating cell proliferation and repair. These studies show the broader benefits of GSK3β gene deletion or inhibition on reducing injury and more importantly, on repopulating renal proximal tubules after AKI.
Renal tubular injury is a distinctive feature of AKI, characterized by apoptosis and necrosis of renal tubules. The extent of injury in our study was similar to previously reported in vivo patterns of HgCl2-induced nephrotoxicity27, 28, insofar as the onset of toxic effects was very rapid and injury was accompanied by apoptosis. Compared to WT mice, the KO mice had better renal function following HgCl2 treatment with lower BUN and plasma creatinine levels. This was linked to less severe injury and accelerated repair observed in KO mice. The reduced injury itself could be at least partially due to a reduced rate of apoptosis in the KO mice.
GSK3β is known to regulate the intrinsic mitochondrial apoptotic pathway39. Bax, a pro-apoptotic protein that is constitutively expressed in the cell, is activated by phosphorylation by GSK3β 40. The activated Bax oligomerizes with the mitochondrial outer membranes and forms pores leading to release of cytochrome c and to other pro-apoptotic factors, which in turn activates caspase dependent or independent pathways 5, 41–43. In our study, activated Bax levels and cleaved caspase 3 levels were lower in the KO mice compared to WT mice. Similarly, Bcl 2, an anti-apoptotic protein, remained unchanged in KO mice while it was reduced in the WT mice following HgCl2 treatment. Bcl 2 is cleaved/degraded by caspase 3 during apoptosis, which in turn leads to mitochondrial pore formation 44. Earlier studies demonstrated that systemic GSK3 inhibition could reduce apoptosis associated with endotoxemia21, 22 and ischemia-reperfusion models of AKI20. While consistent with these studies, our results clearly demonstrated that gene deletion of GSK3β in the proximal tubule per se reduces apoptosis and renal injury and improves survival after AKI.
GSK3β is constitutively active in resting cells and undergoes a rapid and transient inhibition in response to a number of external signals. GSK3β activity increased within 6h of HgCl2 treatment, which coincided with the increased levels of apoptosis and injury seen in WT mice (fig. 8). These results are consistent with similar findings in a rat model of ischemia-reperfusion induced AKI20. Surprisingly, in our study, GSK3β activity was reduced to below pre-HgCl2 treatment levels by 32h and stayed low for 5 days. This period of time (2–5 days post HgCl2 treatment) coincides with a period of active cell proliferation and recovery in WT mice, suggesting that suppression of renal GSK3β activity could be a natural mechanism employed by renal tubules undergoing regeneration following injury. Hence, it could be possible that in the WT mouse kidney, GSK3 is normally inhibited to some extent by the PI3K-AKT, PKA, EGFR and Wnt signaling pathways45–48 that are known to be important for cell proliferation and tissue repair28, 49–52. This response, however, might not be adequate to prevent severe AKI and mortality. Our findings demonstrate that proximal tubule specific gene knockout of GSK3β or the use of a GSK3 inhibitor further significantly protected against injury, and accelerated the repair process.
A rapid proliferative response leading to the restoration of nephron structure and function is very important because episodes of AKI are not always fully reversible and may lead to chronic kidney disease. An interesting observation was that repair occurred at a linear rate in the WT kidney, but at an exponential rate in the KO kidney. Based on injury scores, BUN/creatinine levels and cell proliferation, the phase of renal repair began on day 2 and peaked on day 4 in KO mice, while it started later in WT mice at days 4–6 after initial injury. A paired study using similarly injured WT mice showed that TDZD-8 treatment improved recovery of renal structure and function and the rate of proliferation compared to vehicle treated mice, thus demonstrating that the accelerated rate of repair in KO mice could not be attributed to reduced levels of initial renal injury alone. Cyclin D1, c-myc and β-catenin are pro-proliferative factors and also substrates of GSK3β. Phosphorylation of cyclin D1, c-myc and β-catenin by GSK3β destabilizes them, leading to ubiquitination and degradation 48, 53, 54. We found an accelerated rate of increase in cyclin D1 and c-myc accumulation in the GSK3β knockout mice compared to the WT mice. This was confirmed in studies using TDZD-8. Though β-catenin levels were significantly higher in the TDZD-8 treated mice than in the vehicle treated mice, the KO mice did not show such a change. Past studies have suggested that gene deletion of at least 3 of the 4 alleles of both isoforms of GSK3 α vs. β are required to show an appreciable change in β-catenin levels55. It could also be possible that administration of HgCl2 by itself can increase β-catenin accumulation by activating Wnt-signaling.
Our observations on the rapid regenerative response in GSK3β-inactivated renal proximal tubules could be important in light of the current theory that it is the surviving renal tubular epithelial cells that proliferate and repopulate tubules in AKI8. In wild type embryonic stem cells, GSK3 inhibition has been suggested to help in maintaining self-renewal and pluripotency56, 57, and inhibition of GSK3 has been reported to enhance repopulation of the bone marrow by hematopoietic stem cells58. Hence, inhibition of GSK3β could be a key switch that turns on cell proliferation and regeneration of renal proximal tubules in AKI, thus warranting further studies.
In summary, GSK3β mediates injury and repair in HgCl2-induced AKI. While kidneys appear to have an innate ability to suppress GSK3β activity in order to reduce injury and promote regeneration of tubular cells, it is not sufficient to protect mice from a high rate of mortality under conditions of sever injury. Proximal tubule-specific KO of GSK3β not only reduced tubular injury, but also accelerated tubular regeneration. Our study also showed that the mechanism of GSK3β protection is specific to the kidney, as opposed to being a systemic effect that could have indirectly ameliorated organ damage. Thus, GSK3β could be a major regulator of recovery from acute toxic kidney injury. This is clinically relevant since it provides a possible treatment option to accelerate recovery of established AKI. Thus, these findings have potential therapeutic application for a broad range of kidney diseases.
Methods
1) Generating KO mice
GSK3βloxp/loxp mice were bred with transgenic mice in which Cre expression was driven by a promoter fragment of γGT, expressed in the proximal tubule 36. Mice were on a pure C57/BL6J background. Genotype was determined by PCR using tail DNA and primers described earlier 17, 36. Proximal tubules were isolated from WT and KO mice renal cortex by collagenase digestion followed by percoll gradient high-speed centrifugation59, 60.
2) AKI studies
Female WT and KO mouse littermates, 10 weeks old and weighing 24–25g were injected once subcutaneously with HgCl2 at a nephrotoxic dose of 8.16 mg/kg body weight 27. Mice were sacrificed to examine renal morphology and protein expression and blood plasma was collected for BUN and creatinine measurements. For measurement of BrdU incorporation, mice received an injection 2h before sacrifice. In a sub-study (Fig 7), only WT mice were injected with 6.3 mg/kg of body weight of HgCl2. Plasma BUN levels were measured on day 2 and mice with comparable levels of BUN were separated into 2 groups (n=12 each group). One group received two intra-peritoneal injections of an ATP non-competitive GSK3 inhibitor, TDZD-8 dissolved in 75% DMSO (1mg/kg on day 2 and 500Ng/kg on day 3) and the other group received vehicle. All experiments were performed in accordance with the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health (Bethesda, MD) and were approved by the IACUC committee of Vanderbilt University.
3) BUN and creatinine measurements
BUN levels were measured using a QuantiChrom Urea Assay Kit from BioAssay Systems, Hayward, CA). Creatinine levels were measured using HPLC at the Vanderbilt Hormone Assay Core.
4) Western Blot Analysis
Protein (20 μg) was loaded on 12% or 4–20% SDS-PAGE mini-gels and immunoblotting was carried out as described before17. Monoclonal antibody for GSK3β (BD-Transduction Laboratories, San Jose, CA) and polyclonal antibodies for pGSK3β, β-actin, cleaved caspase 3, Bax (Cell Signaling, Danvers, MA) and c-myc (Santa Cruz Biotechnology, CA) were used.
5) Immunohistochemistry and immunofluorescence
Kidneys were collected at sacrifice and a cross section was immediately fixed in 4% paraformaldehyde at 4°C overnight. Samples were dehydrated in a graded alcohol series and embedded in paraffin for histological analysis. To unmask antigens, 5 Nm sections were boiled in Target Retrieval solution (Dako, Carpinteria, CA). Polyclonal antibodies for GSK3β (Santa Cruz Biotechnology) Ki-67 and BrdU (Cell Signaling) were used. LTL (Vector Laboratories), PCNA (Dako), and secondary antibody (Jackson ImmunoResearch Laboratories) conjugated to Cy3 were used. Ki-67, BrdU and PCNA positive nuclei and total nuclei were counted manually at 20X, in 20 high-power fields (HPF) for each kidney sample, and an average percent positivity per HPF was calculated per kidney. Negative controls without primary antibody showed no staining. Images were captured with a Nikon Eclipse E600 microscope and a Nikon DXM1200 digital camera. All assessments were blinded to the treatment protocols.
6) TUNEL assay
TUNEL assay was performed to evaluate apoptosis using an In Situ Cell Death Detection Kit (Roche Applied Science, Indianapolis, IN). In brief, 4 μm renal sections were exposed to a TUNEL reaction mixture containing terminal deoxynucleotidyl transferase and nucleotides, including tetramethylrhodamine–labeled (TMR-labeled) dUTP. Total and TUNEL positive nuclei were counted as described for PCNA.
7) Tubule injury and fibrosis measurements
Tubular injury score was determined by assessing hematoxylin and eosin stained paraffin sections. Loss of brush border, tubular dilation, cast formation, cell lysis, vacuolization and sloughing were scored on a scale of 0–4, where 0 represents no injury, 1= 5%–25%; 2= 25%–50%; 3= 50%–75%; 4= >75% tubules having necrosis, dilatation or cell swelling. The observer was unaware of the protocol assignments of the mice. Fibrosis was measured by morphometric analysis of collagen fibril by sirius red staining. Deparaffinized sections were stained with 0.1% picrosirius red 61.
Statistical Analysis
Comparisons between WT and KO mice or TDZD-8 treated and vehicle treated mice were analyzed by the unpaired Student’s t-test. Comparisons of multiple points were made using ANOVA with the Bonferroni correction. P<0.05 was taken as significant. Data are expressed as means ± SEM.
Supplementary Material
Acknowledgments
We thank Dr Alan Yu for critically reading the manuscript. These studies were supported by a Center of Excellence in Pediatric Nephrology Pilot and Feasibility grant- 3P50DK044757-18S, an American Society of Nephrology Carl W. Gottschalk Research Scholar Grant and R01 DK-083525 grants to RR, a 3P50DK044757 to ABF and P30 DK079341, 5R01 DK62794, DK51265 and VA Merit Review grants to RCH.
This work was partly done at Vanderbilt University by RR and CH. CH is currently at Tennessee State University, Nashville, TN
Footnotes
DISCLOSURES
None
References
- 1.Bonventre JV, Yang L. Cellular pathophysiology of ischemic acute kidney injury. J Clin Invest. 2011;121:4210–4221. doi: 10.1172/JCI45161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lieberthal W, Nigam SK. Acute renal failure. I. Relative importance of proximal vs. Distal tubular injury. Am J Physiol. 1998;275:F623–631. doi: 10.1152/ajprenal.1998.275.5.F623. [DOI] [PubMed] [Google Scholar]
- 3.Sharfuddin AA, Molitoris BA. Pathophysiology of ischemic acute kidney injury. Nat Rev Nephrol. 2011;7:189–200. doi: 10.1038/nrneph.2011.16. [DOI] [PubMed] [Google Scholar]
- 4.Schrier RW. Early intervention in acute kidney injury. Nat Rev Nephrol. 2010;6:56–59. doi: 10.1038/nrneph.2009.170. [DOI] [PubMed] [Google Scholar]
- 5.Havasi A, Borkan SC. Apoptosis and acute kidney injury. Kidney Int. 2011;80:29–40. doi: 10.1038/ki.2011.120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Baum M, Quigley R. Proximal tubule water transport-lessons from aquaporin knockout mice. Am J Physiol Renal Physiol. 2005;289:F1193–1194. doi: 10.1152/ajprenal.00283.2005. [DOI] [PubMed] [Google Scholar]
- 7.Pfaller W, Gstraunthaler G, Willinger CC. Morphology of renal tubular damage from nephrotoxins. Toxicol Lett. 1990;53:39–43. doi: 10.1016/0378-4274(90)90092-z. [DOI] [PubMed] [Google Scholar]
- 8.Humphreys BD, Valerius MT, Kobayashi A, Mugford JW, Soeung S, Duffield JS, McMahon AP, Bonventre JV. Intrinsic epithelial cells repair the kidney after injury. Cell stem cell. 2008;2:284–291. doi: 10.1016/j.stem.2008.01.014. [DOI] [PubMed] [Google Scholar]
- 9.Doble BW, Woodgett JR. Gsk-3: Tricks of the trade for a multi-tasking kinase. J Cell Sci. 2003;116:1175–1186. doi: 10.1242/jcs.00384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Woodgett JR, Force T. Unique and overlapping functions of gsk-3 isoforms in cellular differentiation, proliferation, and cardiovascular development. J Biol Chem. 2008 doi: 10.1074/jbc.R800077200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Woodgett JR. Molecular cloning and expression of glycogen synthase kinase-3/factor a. EMBO J. 1990;9:2431–2438. doi: 10.1002/j.1460-2075.1990.tb07419.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bain J, Plater L, Elliott M, Shpiro N, Hastie CJ, McLauchlan H, Klevernic I, Arthur JS, Alessi DR, Cohen P. The selectivity of protein kinase inhibitors: A further update. Biochem J. 2007;408:297–315. doi: 10.1042/BJ20070797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kaidanovich-Beilin O, Woodgett JR. Gsk-3: Functional insights from cell biology and animal models. Frontiers in molecular neuroscience. 2011;4:40. doi: 10.3389/fnmol.2011.00040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ge Y, Si J, Tian L, Zhuang S, Dworkin LD, Gong R. Conditional ablation of glycogen synthase kinase 3beta in postnatal mouse kidney. Lab Invest. 2011;91:85–96. doi: 10.1038/labinvest.2010.142. [DOI] [PubMed] [Google Scholar]
- 15.Rao R, Hao CM, Breyer MD. Hypertonic stress activates glycogen synthase kinase 3beta-mediated apoptosis of renal medullary interstitial cells, suppressing an nfkappab-driven cyclooxygenase-2-dependent survival pathway. J Biol Chem. 2004;279:3949–3955. doi: 10.1074/jbc.M309325200. [DOI] [PubMed] [Google Scholar]
- 16.Rao R, Hao CM, Redha R, Wasserman DH, McGuinness OP, Breyer MD. Glycogen synthase kinase 3 inhibition improves insulin-stimulated glucose metabolism but not hypertension in high-fat-fed c57bl/6j mice. Diabetologia. 2007;50:452–460. doi: 10.1007/s00125-006-0552-5. [DOI] [PubMed] [Google Scholar]
- 17.Rao R, Patel S, Hao C, Woodgett J, Harris R. Gsk3beta mediates renal response to vasopressin by modulating adenylate cyclase activity. J Am Soc Nephrol. 2010;21:428–437. doi: 10.1681/ASN.2009060672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rao R, Zhang MZ, Zhao M, Cai H, Harris RC, Breyer MD, Hao CM. Lithium treatment inhibits renal gsk-3 activity and promotes cyclooxygenase 2-dependent polyuria. Am J Physiol Renal Physiol. 2005;288:F642–649. doi: 10.1152/ajprenal.00287.2004. [DOI] [PubMed] [Google Scholar]
- 19.Yao HB, Shaw PC, Wong CC, Wan DC. Expression of glycogen synthase kinase-3 isoforms in mouse tissues and their transcription in the brain. J Chem Neuroanat. 2002;23:291–297. doi: 10.1016/s0891-0618(02)00014-5. [DOI] [PubMed] [Google Scholar]
- 20.Wang Z, Havasi A, Gall J, Bonegio R, Li Z, Mao H, Schwartz JH, Borkan SC. Gsk3{beta} promotes apoptosis after renal ischemic injury. J Am Soc Nephrol. 2010;21:284–294. doi: 10.1681/ASN.2009080828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dugo L, Collin M, Allen DA, Patel NS, Bauer I, Mervaala EM, Louhelainen M, Foster SJ, Yaqoob MM, Thiemermann C. Gsk-3beta inhibitors attenuate the organ injury/dysfunction caused by endotoxemia in the rat. Crit Care Med. 2005;33:1903–1912. doi: 10.1097/01.ccm.0000178350.21839.44. [DOI] [PubMed] [Google Scholar]
- 22.Wang Y, Huang WC, Wang CY, Tsai CC, Chen CL, Chang YT, Kai JI, Lin CF. Inhibiting glycogen synthase kinase-3 reduces endotoxaemic acute renal failure by down-regulating inflammation and renal cell apoptosis. Br J Pharmacol. 2009;157:1004–1013. doi: 10.1111/j.1476-5381.2009.00284.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kerkela R, Kockeritz L, Macaulay K, Zhou J, Doble BW, Beahm C, Greytak S, Woulfe K, Trivedi CM, Woodgett JR, Epstein JA, Force T, Huggins GS. Deletion of gsk-3beta in mice leads to hypertrophic cardiomyopathy secondary to cardiomyoblast hyperproliferation. J Clin Invest. 2008;118:3609–3618. doi: 10.1172/JCI36245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Peng J, Ramesh G, Sun L, Dong Z. Impaired wound healing in hypoxic renal tubular cells: Roles of hypoxia-inducible factor-1 and glycogen synthase kinase 3beta/beta-catenin signaling. J Pharmacol Exp Ther. 2012;340:176–184. doi: 10.1124/jpet.111.187427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Torres AM, Dnyanmote AV, Bush KT, Wu W, Nigam SK. Deletion of multispecific organic anion transporter oat1/slc22a6 protects against mercury-induced kidney injury. J Biol Chem. 2011;286:26391–26395. doi: 10.1074/jbc.M111.249292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yang H, Xu Z, Liu W, Deng Y, Xu B. The protective role of procyanidins and lycopene against mercuric chloride renal damage in rats. Biomed Environ Sci. 2011;24:550–559. doi: 10.3967/0895-3988.2011.05.015. [DOI] [PubMed] [Google Scholar]
- 27.Langworthy M, Zhou B, de Caestecker M, Moeckel G, Baldwin HS. Nfatc1 identifies a population of proximal tubule cell progenitors. J Am Soc Nephrol. 2009;20:311–321. doi: 10.1681/ASN.2008010094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wang Z, Chen JK, Wang SW, Moeckel G, Harris RC. Importance of functional egf receptors in recovery from acute nephrotoxic injury. J Am Soc Nephrol. 2003;14:3147–3154. doi: 10.1097/01.asn.0000098681.56240.1a. [DOI] [PubMed] [Google Scholar]
- 29.Bridges CC, Zalups RK. Molecular and ionic mimicry and the transport of toxic metals. Toxicol Appl Pharmacol. 2005;204:274–308. doi: 10.1016/j.taap.2004.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bridges CC, Zalups RK. Transport of inorganic mercury and methylmercury in target tissues and organs. J Toxicol Environ Health B Crit Rev. 2010;13:385–410. doi: 10.1080/10937401003673750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Diamond GL, Zalups RK. Understanding renal toxicity of heavy metals. Toxicol Pathol. 1998;26:92–103. doi: 10.1177/019262339802600111. [DOI] [PubMed] [Google Scholar]
- 32.Tanaka-Kagawa T, Suzuki M, Naganuma A, Yamanaka N, Imura N. Strain difference in sensitivity of mice to renal toxicity of inorganic mercury. J Pharmacol Exp Ther. 1998;285:335–341. [PubMed] [Google Scholar]
- 33.Yanagisawa H. hgcl2-induced acute renal failure and its pathophysiology. Nihon Eiseigaku Zasshi. 1998;52:618–623. doi: 10.1265/jjh.52.618. [DOI] [PubMed] [Google Scholar]
- 34.Zalups RK. Molecular interactions with mercury in the kidney. Pharmacol Rev. 2000;52:113–143. [PubMed] [Google Scholar]
- 35.Hoeflich KP, Luo J, Rubie EA, Tsao MS, Jin O, Woodgett JR. Requirement for glycogen synthase kinase-3beta in cell survival and nf-kappab activation. Nature. 2000;406:86–90. doi: 10.1038/35017574. [DOI] [PubMed] [Google Scholar]
- 36.Iwano M, Plieth D, Danoff TM, Xue C, Okada H, Neilson EG. Evidence that fibroblasts derive from epithelium during tissue fibrosis. J Clin Invest. 2002;110:341–350. doi: 10.1172/JCI15518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lipina TV, Kaidanovich-Beilin O, Patel S, Wang M, Clapcote SJ, Liu F, Woodgett JR, Roder JC. Genetic and pharmacological evidence for schizophrenia-related disc1 interaction with gsk-3. Synapse. 2011;65:234–248. doi: 10.1002/syn.20839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Martinez A, Alonso M, Castro A, Perez C, Moreno FJ. First non-atp competitive glycogen synthase kinase 3 beta (gsk-3beta) inhibitors: Thiadiazolidinones (tdzd) as potential drugs for the treatment of alzheimer’s disease. Journal of medicinal chemistry. 2002;45:1292–1299. doi: 10.1021/jm011020u. [DOI] [PubMed] [Google Scholar]
- 39.Beurel E, Jope RS. The paradoxical pro- and anti-apoptotic actions of gsk3 in the intrinsic and extrinsic apoptosis signaling pathways. Prog Neurobiol. 2006;79:173–189. doi: 10.1016/j.pneurobio.2006.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Linseman DA, Butts BD, Precht TA, Phelps RA, Le SS, Laessig TA, Bouchard RJ, Florez-McClure ML, Heidenreich KA. Glycogen synthase kinase-3beta phosphorylates bax and promotes its mitochondrial localization during neuronal apoptosis. J Neurosci. 2004;24:9993–10002. doi: 10.1523/JNEUROSCI.2057-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Juhaszova M, Zorov DB, Kim SH, Pepe S, Fu Q, Fishbein KW, Ziman BD, Wang S, Ytrehus K, Antos CL, Olson EN, Sollott SJ. Glycogen synthase kinase-3beta mediates convergence of protection signaling to inhibit the mitochondrial permeability transition pore. J Clin Invest. 2004;113:1535–1549. doi: 10.1172/JCI19906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Somervaille TC, Linch DC, Khwaja A. Growth factor withdrawal from primary human erythroid progenitors induces apoptosis through a pathway involving glycogen synthase kinase-3 and bax. Blood. 2001;98:1374–1381. doi: 10.1182/blood.v98.5.1374. [DOI] [PubMed] [Google Scholar]
- 43.Tan J, Zhuang L, Leong HS, Iyer NG, Liu ET, Yu Q. Pharmacologic modulation of glycogen synthase kinase-3beta promotes p53-dependent apoptosis through a direct bax-mediated mitochondrial pathway in colorectal cancer cells. Cancer Res. 2005;65:9012–9020. doi: 10.1158/0008-5472.CAN-05-1226. [DOI] [PubMed] [Google Scholar]
- 44.Kirsch DG, Doseff A, Chau BN, Lim DS, de Souza-Pinto NC, Hansford R, Kastan MB, Lazebnik YA, Hardwick JM. Caspase-3-dependent cleavage of bcl-2 promotes release of cytochrome c. J Biol Chem. 1999;274:21155–21161. doi: 10.1074/jbc.274.30.21155. [DOI] [PubMed] [Google Scholar]
- 45.Cross DA, Alessi DR, Cohen P, Andjelkovich M, Hemmings BA. Inhibition of glycogen synthase kinase-3 by insulin mediated by protein kinase b. Nature. 1995;378:785–789. doi: 10.1038/378785a0. [DOI] [PubMed] [Google Scholar]
- 46.Fang X, Yu SX, Lu Y, Bast RC, Jr, Woodgett JR, Mills GB. Phosphorylation and inactivation of glycogen synthase kinase 3 by protein kinase a. Proc Natl Acad Sci U S A. 2000;97:11960–11965. doi: 10.1073/pnas.220413597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ojeda L, Gao J, Hooten KG, Wang E, Thonhoff JR, Dunn TJ, Gao T, Wu P. Critical role of pi3k/akt/gsk3beta in motoneuron specification from human neural stem cells in response to fgf2 and egf. PLoS One. 2011;6:e23414. doi: 10.1371/journal.pone.0023414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Yost C, Torres M, Miller JR, Huang E, Kimelman D, Moon RT. The axis-inducing activity, stability, and subcellular distribution of beta-catenin is regulated in xenopus embryos by glycogen synthase kinase 3. Genes Dev. 1996;10:1443–1454. doi: 10.1101/gad.10.12.1443. [DOI] [PubMed] [Google Scholar]
- 49.Gu J, Sun P, Zhao H, Watts HR, Sanders RD, Terrando N, Xia P, Maze M, Ma D. Dexmedetomidine provides renoprotection against ischemia-reperfusion injury in mice. Crit Care. 2011;15:R153. doi: 10.1186/cc10283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Okusa MD, Linden J, Huang L, Rosin DL, Smith DF, Sullivan G. Enhanced protection from renal ischemia-reperfusion [correction of ischemia:Reperfusion] injury with a(2a)-adenosine receptor activation and pde 4 inhibition. Kidney Int. 2001;59:2114–2125. doi: 10.1046/j.1523-1755.2001.00726.x. [DOI] [PubMed] [Google Scholar]
- 51.Wang Z, Havasi A, Gall JM, Mao H, Schwartz JH, Borkan SC. Beta-catenin promotes survival of renal epithelial cells by inhibiting bax. J Am Soc Nephrol. 2009;20:1919–1928. doi: 10.1681/ASN.2009030253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lin SL, Li B, Rao S, Yeo EJ, Hudson TE, Nowlin BT, Pei H, Chen L, Zheng JJ, Carroll TJ, Pollard JW, McMahon AP, Lang RA, Duffield JS. Macrophage wnt7b is critical for kidney repair and regeneration. Proc Natl Acad Sci U S A. 2010;107:4194–4199. doi: 10.1073/pnas.0912228107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Diehl JA, Cheng M, Roussel MF, Sherr CJ. Glycogen synthase kinase-3beta regulates cyclin d1 proteolysis and subcellular localization. Genes Dev. 1998;12:3499–3511. doi: 10.1101/gad.12.22.3499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Yada M, Hatakeyama S, Kamura T, Nishiyama M, Tsunematsu R, Imaki H, Ishida N, Okumura F, Nakayama K, Nakayama KI. Phosphorylation-dependent degradation of c-myc is mediated by the f-box protein fbw7. EMBO J. 2004;23:2116–2125. doi: 10.1038/sj.emboj.7600217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Doble BW, Patel S, Wood GA, Kockeritz LK, Woodgett JR. Functional redundancy of gsk-3alpha and gsk-3beta in wnt/beta-catenin signaling shown by using an allelic series of embryonic stem cell lines. Developmental cell. 2007;12:957–971. doi: 10.1016/j.devcel.2007.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Sato N, Meijer L, Skaltsounis L, Greengard P, Brivanlou AH. Maintenance of pluripotency in human and mouse embryonic stem cells through activation of wnt signaling by a pharmacological gsk-3-specific inhibitor. Nat Med. 2004;10:55–63. doi: 10.1038/nm979. [DOI] [PubMed] [Google Scholar]
- 57.Storm MP, Bone HK, Beck CG, Bourillot PY, Schreiber V, Damiano T, Nelson A, Savatier P, Welham MJ. Regulation of nanog expression by phosphoinositide 3-kinase-dependent signaling in murine embryonic stem cells. J Biol Chem. 2007;282:6265–6273. doi: 10.1074/jbc.M610906200. [DOI] [PubMed] [Google Scholar]
- 58.Trowbridge JJ, Xenocostas A, Moon RT, Bhatia M. Glycogen synthase kinase-3 is an in vivo regulator of hematopoietic stem cell repopulation. Nat Med. 2006;12:89–98. doi: 10.1038/nm1339. [DOI] [PubMed] [Google Scholar]
- 59.Vinay P, Gougoux A, Lemieux G. Isolation of a pure suspension of rat proximal tubules. Am J Physiol. 1981;241:F403–411. doi: 10.1152/ajprenal.1981.241.4.F403. [DOI] [PubMed] [Google Scholar]
- 60.Zhong Q, Terlecky SR, Lash LH. Diabetes increases susceptibility of primary cultures of rat proximal tubular cells to chemically induced injury. Toxicol Appl Pharmacol. 2009;241:1–13. doi: 10.1016/j.taap.2009.08.007. [DOI] [PubMed] [Google Scholar]
- 61.Castano AP, Lin SL, Surowy T, Nowlin BT, Turlapati SA, Patel T, Singh A, Li S, Lupher ML, Jr, Duffield JS. Serum amyloid p inhibits fibrosis through fc gamma r-dependent monocyte-macrophage regulation in vivo. Science translational medicine. 2009;1:5ra13. doi: 10.1126/scitranslmed.3000111. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.