

Abstract
Malaria is a deadly disease that causes nearly one million deaths each year. To develop methods to control and eradicate malaria, it is important to understand the genetic basis of Plasmodium falciparum adaptations to antimalarial treatments and the human immune system while taking into account its demographic history. To study the demographic history and identify genes under selection more efficiently, we sequenced the complete genomes of 25 culture-adapted P. falciparum isolates from three sites in Senegal. We show that there is no significant population structure among these Senegal sampling sites. By fitting demographic models to the synonymous allele-frequency spectrum, we also estimated a major 60-fold population expansion of this parasite population ∼20,000–40,000 years ago. Using inferred demographic history as a null model for coalescent simulation, we identified candidate genes under selection, including genes identified before, such as pfcrt and PfAMA1, as well as new candidate genes. Interestingly, we also found selection against G/C to A/T changes that offsets the large mutational bias toward A/T, and two unusual patterns: similar synonymous and nonsynonymous allele-frequency spectra, and 18% of genes having a nonsynonymous-to-synonymous polymorphism ratio >1.
Keywords: P. falciparum, population expansion, base composition, selection
Introduction
Malaria caused by Plasmodium falciparum is one of the major fatal diseases in the world that infects more than 200 million people and causes nearly one million deaths annually (WHO World Malaria Report 2010). The main strategies for controlling the disease have been chemotherapy and mosquito control; however, the emergence of drug-resistant parasites and insecticide-resistant mosquitoes allowed a resurgence of malaria (reviewed in Hartl 2004). Understanding the demographic history and evolutionary forces shaping sequence variation across the genome has practical implications for developing methods of disease control.
Studying the demographic history of malaria parasites is useful for multiple reasons. First, inferences about the timing and magnitude of population size changes can provide clues to the causes of those changes. In addition, demographic history and natural selection are the two major forces affecting genome variation, and therefore attaining a thorough understanding of selection from genome variation requires disentangling its effects from those of demography. Genes involved in evading the natural defenses of the human immune system or offering resistance to antimalarial drugs are under strong selective pressure. To identify selective sweeps and drug-resistant alleles, the influence of demographic history or population substructure on genome variation must be considered. For example, to evaluate evidence for selection, it is important to have a null distribution of a population genetic statistic, such as Tajima’s D (Tajima 1989b), under the proper demographic model.
Previous studies have given evidence for worldwide population structure and identified some regions with signatures of recent selective sweeps in drug-resistant parasites (Anderson et al. 2000; Conway et al. 2000; Joy et al. 2003; Mu et al. 2005; Volkman et al. 2007). However, previous studies of demographic changes of P. falciparum are not completely consistent (Hartl et al. 2002; Su et al. 2003; Hartl 2004), and this question was strongly contested 10 years ago. The original studies could not adequately account for demographic changes because they were based on too few loci, had problems of ascertainment bias, or included too few parasite isolates from each sampled location. Some evidence suggests that the current worldwide population derives from a small number of parasites in the recent past, inferred from the paucity of genetic diversity in synonymous and noncoding regions (Rich et al. 1998; Conway et al. 2000; Volkman et al. 2001); however, regions with high polymorphism suggest that the current worldwide populations are descended from multiple ancient lineages (Verra and Hughes 2000; Hughes and Verra 2001; Polley and Conway 2001; Volkman et al. 2002). Among the clearest studies is that of Joy et al. (2003), who studied the 6 kb mitochondrial genome of 96 worldwide parasites and found evidence supporting a demographic model in which some ancient lineages emerged out of Africa 50,000 to 100,000 years ago and a major population expansion occurred in Africa ∼10,000 years ago followed by extensive migration to other regions. This model helped explain the conflicting patterns of genetic diversity in the previous studies. Because of recombination breaking up linkage, different regions in the genome have different demographic histories. Regions having their most recent common ancestor before migration out of Africa can have high diversity while regions having their most recent common ancestor after migration out of Africa have lower genetic diversity. The estimated times are point estimates, however, and lack of recombination in mitochondrial DNA leaves many details about demographic history in doubt. Moreover, owing again to the lack of recombination, the estimate from mitochondrial sequences could be biased by selection.
To make stronger inferences about demographic history, identify genes under selection, and understand the genome-wide patterns of genetic diversity in P. falciparum, we fully sequenced 25 isolates from a local population in Senegal. Because P. falciparum likely originated in Africa (Conway et al. 2000), studying an African population may yield more information about its population history. Studying a local population in depth has two advantages over worldwide samples. First, polymorphisms within a local population are better at revealing signatures of recent selection than worldwide polymorphisms, and second, the existence of population structure in worldwide samples can result in patterns of linkage disequilibrium (LD) and the allele-frequency spectrum that artifactually resemble those expected from selection.
In this study, we investigated the genome-wide variation patterns of P. falciparum. We used principal component analysis and Bayesian clustering methods to test whether there is any significant population substructure in Senegal; we estimated demographic history using the allele-frequency spectrum; and we identified genes and gene categories under various forces of selection based on deviations from the null allele-frequency spectrum obtained by coalescent simulations with the inferred demographic parameters. Finally, we find evidence that the unusually high A/T composition of the P. falciparum genome (81%; Gardner et al. 2002) is maintained as a dynamic equilibrium between mutation and selection pressures operating on nucleotide composition.
Materials and Methods
Data Set and Data Processing
Twenty-five culture-adapted isolates of P. falciparum from different sites in Senegal were sequenced and investigated in this study. Ten of them are from Pikine (P05.02, P08.04, P09.04, P11.02, P19.04, P26.04, P27.02, P31.01, P51.02, and P60.02), 4 from Velingara (V34.04, V35.04, V42.05, and V92.05), and 11 from Thiès (T074.08, T10.04, T105.07, T113.09, T130.09, T15.04, T230.08, T231.08, T232.08, T26.04, and T28.04). The first letter in the isolate names indicates the site of collection (P for Pikine; V for Velingara; and, T for Thiès), and the last two digits in the isolate names indicate the year of collection (e.g., P05.02 was isolated in 2002).
Sequencing reads (101 bp, paired-end) were generated using Illumina HiSeq machines and were aligned to the P. falciparum 3D7 reference available from PlasmoDB version 7.1 (Gardner et al. 2002) using the Picard pipeline. The Picard pipeline includes BWA aligner (Li and Durbin 2010) and the Samtools data processing tool (Li et al. 2009). Genotypes were called from the reads using the Unified Genotyper (DePristo et al. 2011), which is part of the GATK package, for each isolate separately. Ambiguous calls as well as genotypes with a PHRED-style quality score of less than 30 were discarded. Repeat-rich sequences near the telomeres of each chromosome arm were excluded from the analyses (using the same bounds as in Volkman et al. [2007]). PfEMP1 (var) genes were excluded from the analyses because the reads from these genes are difficult to align to the reference genome correctly due to their extremely high variability and the fact that they undergo ectopic recombination. The average depth (number of reads that map to the same location) is 46 per site. The mean of the number of isolates sequenced per aligned base is 20. More than one-half (51.68%) of sites have nucleotide information of all 25 isolates. Single nucleotide polymorphisms (SNPs) have been submitted to dbSNP (submitter handle BROAD-GENOMEBIO; batch id Pf_0004) and will be released with dbSNP build B136, mid-2012.
Genetic Diversity and LD
We measured genetic diversity using π (the average number of pairwise differences per site among different isolates) and Watterson’s theta θW (number of segregating sites normalized by 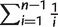 , where n is the number of aligned parasite sequences) (Watterson 1975). Confidence intervals were obtained by bootstrapping 10,000 times over genes or chromosomes. We used the yn00 program in the PAML package (Yang and Nielsen 2000) to calculate the synonymous substitution rate (dS). LD, measured by r2 (Hill and Robertson 1968), was calculated for pairs of SNPs with different physical separation. The r2 measure is the square of the correlation coefficient between two SNPs. Because the distance between SNPs affects the level of LD, sliding LD was calculated only for SNP pairs that were 1–3 kb from each other, and the average distance within each window was also calculated. The background level of LD was estimated by calculating r2 between pairs of SNPs from different chromosomes.
, where n is the number of aligned parasite sequences) (Watterson 1975). Confidence intervals were obtained by bootstrapping 10,000 times over genes or chromosomes. We used the yn00 program in the PAML package (Yang and Nielsen 2000) to calculate the synonymous substitution rate (dS). LD, measured by r2 (Hill and Robertson 1968), was calculated for pairs of SNPs with different physical separation. The r2 measure is the square of the correlation coefficient between two SNPs. Because the distance between SNPs affects the level of LD, sliding LD was calculated only for SNP pairs that were 1–3 kb from each other, and the average distance within each window was also calculated. The background level of LD was estimated by calculating r2 between pairs of SNPs from different chromosomes.
Nonsynonymous and synonymous polymorphism (πN and πS) were calculated by dividing the average number of nonsynonymous or synonymous pairwise differences by the number of nonsynonymous or synonymous sites, respectively. The number of nonsynonymous or synonymous sites was calculated using the method described in table 1 of Ina (1995), and the mutation matrix estimated by the divergence of intergenic regions between P. reichenowi and P. falciparum (table 3).
Table 3.
The Observed and Predicted Equilibrium Nucleotide Composition.
| Four-fold degenerate sites |
Intergenic regions |
|||
|---|---|---|---|---|
| Predicted | Observed | Predicted | Observed | |
| A | 0.40 | 0.41 | 0.42 | 0.43 |
| T | 0.38 | 0.41 | 0.41 | 0.42 |
| C | 0.10 | 0.09 | 0.09 | 0.08 |
| G | 0.12 | 0.09 | 0.08 | 0.07 |
| A/T | 0.78 | 0.82 | 0.83 | 0.85 |
Population Substructure
We investigated population structure in Senegal using two kinds of analyses: principal component analysis and a Bayesian model-based clustering method. Principal component analysis (PCA) was conducted using the program SMARTPCA (Patterson et al. 2006) in the software EIGENSOFT 3.0. We applied a local LD correction (nsnpldregress = 2) and calculated the top 10 eigenvectors or principal components from all the SNPs of the Senegal population. The Bayesian model-based clustering method was performed using the program STRUCTURE (version 2.2.3) (Pritchard et al. 2000; Falush et al. 2003). Each run used 20,000 iterations after a burn-in of 10,000 iterations and a model allowing for admixture and correlated allele frequencies. We conducted a series of independent runs with different numbers of clusters. Analyses with or without a prior based on geographical location were both applied. All other parameters were set to the default parameters. To ensure the consistency of results, we performed five independent replicates for each condition.
Demographic Modeling and Inference
We used the deviations in the allele-frequency spectrum from that expected under a neutral, panmictic, constant-size model to assess demographic history. The “folded” allele-frequency spectrum is the frequency spectrum of minor alleles, which is useful when outgroup sequence data is sparse, as is the case with P. reichenowi. Demographic history parameters were inferred using the program ∂a∂i version 1.5.2 (Gutenkunst et al. 2009), which infers demographic parameters by fitting different demographic models to the allele-frequency spectrum. We used the folded allele-frequency spectrum of synonymous SNPs for demographic inference. Two demographic models (a two-epoch model and an exponential growth model) were used. The parameters in these two models are the time in the past at which the change in size began (T) and the ratio of current to ancient population size (NA/N0). Ten independent runs were performed on each model, and the parameter set with the highest log-likelihood was selected as the point estimate of the parameters. For each model, 100 conventional bootstraps were performed to obtain the 95% confidence intervals (CI). Optimized θ from the output of ∂a∂i is equal to 2NAµL (for haploid), where µ is mutation rate and L is the effective sequence length. With L and µ, NA can be calculated and the unit of time (T) can be converted to years. Coalescent simulations under the best-fit demographic models were performed using the ms program of Hudson (2002).
Identifying Genes under Selection
An allele-frequency spectrum-based test, Tajima’s D (Tajima 1989b), a long range haplotype test (Voight et al. 2006), and the ratio of nonsynonymous and synonymous polymorphism (πN/πS) were applied to identify genes or gene categories under selection. The null distribution of Tajima’s D was obtained by coalescent simulations using the ms program and inferred demographic parameters from ∂a∂i. As the null distribution is sensitive to mutation rate per gene, the null distributions of different genes were simulated separately with different mutation rates that are proportional to their gene lengths. We investigated the broader biological basis of evolution by determining whether certain Gene Ontology (GO) terms were overrepresented among the genes found to be significant in Tajima’s D test. The associations between Plasmodb gene IDs and GO terms were downloaded from the FTP site of the Wellcome Trust Sanger Institute (ftp://ftp.sanger.ac.uk/pub/pathogens/Plasmodium/falciparum/3D7/3D7.archive/gene_association_file/gene_association.GeneDB_Pfalciparum.20100519). P values were determined according to a hypergeometric distribution. A Mann-Whitney U test was used to test whether a GO term had significant higher or lower πN/πS than the rest of genes. We considered the problem of multiple testing by estimating q values using the qvalue package (Storey and Tibshirani 2003) in the R-environment.
The significance of πN/πS is determined by the null distribution obtained from sampling 10,000 times two values of πS from the empirical distribution of πS and calculating the ratio of them. This is a very conservative test because the variation in the empirical distribution of πS could be caused by variation in coalescent time between genes due to recombination, whereas πN and πS for the same gene should be based on similar coalescent histories.
The integrated haplotype score (iHS) statistic was computed according to the methods of Voight et al. (2006). Recombination maps were generated with LDhat 2.1 (McVean et al. 2002) using a block penalty of 5.0, 10 million rjMCMC iterations, a missing data cutoff of 20%, minimum minor allele frequency of 8%, and all other parameters set to default values. Because the iHS test does not tolerate missing data, SNPs with data in at least 80% of individuals were imputed with PHASE 2.1.1 (Stephens and Donnelly 2003). As PHASE requires “diploid” data, we dropped the sample with the lowest call rate (SenP60.02) to create an even number of haploid individuals, randomly paired. 17,572 fully imputed SNPs had at least 2 minor alleles among the remaining 24 individuals. The unstandardized iHS was defined as ln (iHHA/iHHD), where iHHA is the integrated EHH in both directions for haplotypes with the major allele at the core SNP, and iHHD is the integrated EHH for haplotypes with the minor allele. These unstandardized iHS scores were then normalized within allele frequency bins. Because the P. reichenowi sequence is sparse and the ancestral allele is not available for most SNPs in our data set, we ignored the sign of the normalized iHS score and instead present the scores as P values based on a two-tailed conversion from a normal distribution.
Nucleotide Transition Matrix and Equilibrium Base Compositions
To obtain empirical nucleotide transition matrices, we used P. reichenowi to infer the ancestral state for observed P. falciparum polymorphisms. The P. reichenowi genomic sequences (Jeffares et al. 2007) were aligned to P. falciparum 3D7 genomic sequence using the NUCmer program from the MUMmer 3.22 package (http://mummer.sourceforge.net) (Kurtz et al. 2004). Reciprocal best hits were selected and trimmed by 10 bases from each end to avoid errors stemming from poor alignment at the ends of segments. Additionally, subtelomeric regions (using the same bounds as in Volkman et al. [2007]) and PfEMP1 genes were also filtered out from the alignment set. Because the P. reichenowi sequence is sparse, only 21.6% of the P. falciparum genome has informative P. reichenowi alleles. We excluded nucleotide positions where there is a lack of a P. reichenowi allele, and for each nucleotide position, we examined all the alleles for the 25 Senegal P. falciparum isolates. If any of these alleles matched the P. reichenowi allele, we used the P. reichenowi allele as the ancestral allele and inferred the nucleotide change, and otherwise we excluded that position. Then, we summed up all the changes to obtain empirical nucleotide transition matrices with the counts, and normalized them by dividing units of each row by the sum of each row (so each row sums up to 1). From the empirical nucleotide transition matrix, we obtained the equilibrium base compositions by solving for the left eigenvector of the empirical nucleotide transition matrix. We did not correct for multiple hits, but the low level of divergence between P. falciparum and P. reichenowi makes this correction negligible.
Results
The genomic sequencing was carried out on a sample of parasites isolated from a relatively small geographical region in Senegal. After verifying that the sample appeared to consist of isolates from a single, genetically homogeneous population, we set out to infer the population’s demographic history, estimate key population genetic parameters, identify the selective forces (balancing, positive, and negative) likely impacting individual genes, and obtain genome-wide estimates of mutation bias.
Polymorphism and LD
Among the 25 isolates from Senegal, we found 78,596 polymorphic sites (SNPs). The average polymorphism (pairwise mismatches, π, and normalized segregating sites, θW) on different chromosomes is shown in supplementary tables S1 and S2, Supplementary Material online. Average pairwise synonymous polymorphism (πS) (0.000601, 95% [CI = 0.000544, 0.000663]) is higher than average pairwise nonsynonymous polymorphism (πN) (0.000317, 95% CI = [0.000298, 0.000337]), indicating that many nonsynonymous changes are harmful and are removed from the population by purifying selection. Moreover, average polymorphism in intergenic regions (πintergenic) (0.000478, 95% CI =[ 0.000415, 0.000544]) and polymorphism in introns (πintron) (0.000420, 95% CI = [0.000378, 0.000464]) are not significantly different, and there are fewer polymorphisms, on average, in both these regions than the average for synonymous polymorphism. The finding of apparent selective constraints in intergenic regions and introns suggests that functionally important nucleotide sites in these regions affecting processes such as gene expression and RNA processing are sufficiently numerous that uniform selective neutrality across these regions should not be assumed. The value of θW for synonymous sites, nonsynonymous sites, introns, and intergenic regions show similar patterns (supplementary table S2, Supplementary Material online).
Furthermore, synonymous polymorphism varies between chromosomes (supplementary fig. S1A, Supplementary Material online). The maximum and minimum average chromosomal πS are 5 × 10−4 and 9 × 10−4, respectively. Three possible explanations are as follows: first, regions of extremely high diversity cause the variation between chromosomes; second, variation in the rate of recombination among chromosomes causes variation in polymorphism reduction due to selective sweeps or background selection at adjacent sites; or, third, mutation rates might vary substantially from one chromosome to the next. To rule out the possibility that this variation is caused by regions of extremely high diversity, such as antigenic genes, we performed a nonparametric test (Mann–Whitney U test) to examine the difference of πS among chromosomes, and the results are consistent.
To understand whether the variation of synonymous polymorphism can be explained by differences in recombination rates, we calculated the correlation between chromosomal recombination rates estimated by Jiang et al. (2011) and synonymous polymorphism. The correlation is significant (Spearman’s rank correlation ρ = 0.54, two-sided P value = 0.048), suggesting that the variation in recombination rates can explain some of the variation in polymorphism among chromosomes. Divergence between species is less affected by recombination rates than polymorphism within species and is a better indicator of mutation rate than polymorphism. Synonymous substitution rates (dS) between P. falciparum and the chimpanzee parasite P. reichenowi also differ among chromosomes (supplementary fig. S1B, Supplementary Material online) and dS is positively correlated with πS (Spearman’s rank correlation ρ = 0.75, two-sided P value = 0.003), suggesting that the variation of πS can at least in part be explained by differences in mutation rate among chromosomes. We emphasize that recombination rates and synonymous substitution rates are not highly correlated (Spearman’s rank correlation test, two-sided P value = 0.10), and therefore we are detecting two different correlations. Furthermore, the difference in πS and dS among chromosomes cannot be explained by the variation of gene density between chromosomes (Spearman’s rank correlation test, two-sided P value = 0.37 and 0.74 for πS and dS, respectively).
We then looked at LD (r2) and polymorphism (π and θW) across all chromosomes using a sliding windows approach (supplementary fig. S2, Supplementary Material online) and determined that LD decays rapidly in the Senegal population (supplementary fig. S3, Supplementary Material online). The average r2 decreases to the baseline level at a distance of only 1 kb, which is consistent with previous studies (Neafsey et al. 2008; Van Tyne et al. 2011); however, the present study is at a finer scale. Low LD suggests high levels of recombination in Senegal. Because most mosquitoes bite only once and meiosis and recombination happen in the mosquito gut, outcrossing (and effective recombination) only happens when patients are infected with multiple strains. Therefore, high levels of recombination in the Senegal population suggest relatively high transmission intensity as compared with regions with higher levels of LD, such as Brazil and Thailand (Neafsey et al. 2008). Moreover, high levels of recombination suggest the potential of fine-scale genetic mapping from genomic sequences. High recombination rates also mean that the signatures of positive selection persist for a shorter period of time. Hence, any significant long-range haplotypes indicate a very recent episode of positive selection.
The abundance of dramatic LD spikes in supplementary figure S2, Supplementary Material online, could be due to both selection and variation in recombination rate. For example, the dramatic LD spikes between 0.75 and 0.85 Mb on chromosome 5 (supplementary fig. S2, Supplementary Material online) are likely to be caused by a selective sweep because the iHS test of selection also captures this region (supplementary table S7, Supplementary Material online). A high variation of recombination rate has also been found in other organisms, such as human and Drosophila (McVean et al. 2004; Kulathinal et al. 2008). Sella et al. (2009) showed that level of synonymous polymorphism is positively correlated with estimated recombination rates.
Evident Absence of Population Substructure
Because population substructure could cause false-positive results when examining the data for signals of selection, we next investigated whether there is any population substructure among the samples from three different cities in Senegal that are less than 250 miles away from each other. Van Tyne et al. (2011) found no population substructure within the Senegal when studying worldwide strains, but that study employed a SNP genotyping array rather than sequencing, and therefore may have been less sensitive than the present approach. We also find no significant principal component for this population using SMARTPCA. The likely cause is the lack of population substructure, however we cannot formally exclude the possibility that unsupervised principal component analysis failed to detect variation among samples from the three locations.
The second analysis for population structure made use of a Bayesian model-based clustering approach implemented in the software STRUCTURE. Similar to PCA, this analysis suggests that there is no obvious population substructure in Senegal. The number of clusters (K) with K = 1 fits the data much better than K = 2 or K = 3, irrespective of whether the sampling location is used as a prior (log-likelihoods of K = 1, K = 2, and K = 3 with the sampling location as a prior are –508380.7, –527733.4, and –529739.8, respectively; log-likelihoods of K = 1, K = 2, and K = 3 without the sampling location as a prior are –508313.1, –518471.7, and –668800.1, respectively). The large differences in log-likelihoods for K = 1, K = 2, and K = 3 mean that, if we assume that the prior probabilities of K = 1, K = 2, and K = 3 are equal, the posterior probability of K = 1 is almost 100% (specifically, 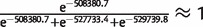 ). In addition, inconsistent individual membership coefficients among five replicate runs when K > 1 support the inference that there is no significant population substructure detected by genetic diversity in Senegal. Thus, if we nevertheless set K = 3, isolates from the three different cities are intermixed among the three clusters, again consistent with the observations that there is a lack of significant substructure among parasites in this study population from Senegal. The estimated ancestry proportion of each isolate when K = 3 is shown in supplementary figure S4A and supplementary table S3, Supplementary Material online. Because isolates that are used in this study are samples from 7 years between 2001 and 2009, we also set K = 7 (supplementary fig. S4B, Supplementary Material online), with the result that isolates from different years have similar patterns of ancestry proportion indicating no significant differences among years.
). In addition, inconsistent individual membership coefficients among five replicate runs when K > 1 support the inference that there is no significant population substructure detected by genetic diversity in Senegal. Thus, if we nevertheless set K = 3, isolates from the three different cities are intermixed among the three clusters, again consistent with the observations that there is a lack of significant substructure among parasites in this study population from Senegal. The estimated ancestry proportion of each isolate when K = 3 is shown in supplementary figure S4A and supplementary table S3, Supplementary Material online. Because isolates that are used in this study are samples from 7 years between 2001 and 2009, we also set K = 7 (supplementary fig. S4B, Supplementary Material online), with the result that isolates from different years have similar patterns of ancestry proportion indicating no significant differences among years.
Demographic History
A proper null model for identifying genes under selection must incorporate the demographic history of the sampled population. To know whether the Senegal population underwent demographic changes in the past, we compared the folded allele-frequency spectrum of biallelic polymorphic sites in the data with the expected allele-frequency spectrum under the standard Wright–Fisher model with constant population size. In a folded allele-frequency spectrum, the ancestral states of alleles are assumed to be unknown, and hence alleles with frequency x are pooled with those of frequency 25 – x, so the allele frequencies in the folded frequency spectrum range from 1 to 12. In the Senegal data, the folded allele-frequency spectra of all polymorphic sites, genic sites, and intergenic sites are all much more skewed than expected under neutrality with a constant population size (fig. 1). This result suggests that there was a population expansion in the recent past, since the number of low-frequency alleles in the population increases when a population grows in size.
Fig. 1.

Folded allele-frequency spectrum. Allele-frequency spectra of different classes of nucleotide sites all show excess of rare alleles. The neutral allele-frequency spectrum was obtained assuming a constant population size. The excess of rare alleles in the empirical spectrum indicates a population expansion in the past.
The nonsynonymous allele-frequency spectrum is only slightly more skewed than the synonymous allele-frequency spectrum. If nonsynonymous changes are disfavored by selection (as suggested by lower nonsynonymous pairwise polymorphism than nonsynonymous polymorphism), then the nonsynonymous allele-frequency spectrum should be more skewed toward low-frequency variants than the synonymous allele-frequency spectrum. The possible explanations for the lack of a larger difference are discussed later in the context of the ratio of nonsynonymous to synonymous polymorphism.
To further estimate demographic parameters, we fit the synonymous allele-frequency spectrum of observed data to two kinds of demographic models—a two-epoch model and an exponential growth model (fig. 2)—by using likelihood-based software ∂a∂i (Gutenkunst et al. 2009). We used the synonymous allele-frequency spectrum because it is less likely to be affected by selection. Both models include two demographic parameters: the time elapsed since the change in size took place (T) and the ratio of ancient to current population size (NA/N0). The only difference between these two models is that population size changes instantaneously in the two-epoch model whereas population size grows exponentially in the exponential growth model (N0 = NAeαT, where α is the growth rate).
Fig. 2.
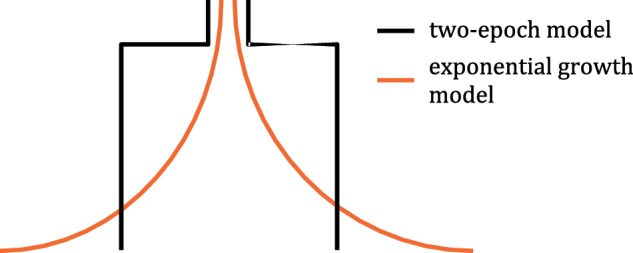
Demographic models. In the exponential growth model, population size starts increasing earlier than in the two-epoch model, and the current population size is larger.
To solve the equations for population size and population expansion time in the two models, an estimate of mutation rate is required. For this estimate, we calculated substitution rates (dS) between P. falciparum and P. reichenowi. As Liu et al. (2010) showed that P. falciparum likely originated from parasites of gorilla origin, we assume that P. falciparum and P. reichenowi diverged at the same time as the ancestors of chimpanzees and gorillas, and used the divergence time between chimpanzees and gorillas, 7.3 million years (Chen and Li 2001), as the credible upper limit of divergence time between P. falciparum and P. reichenowi. The resulting mutation rate was estimated to be 6.82 × 10−9 per site per year. We then applied this estimate to the two models and obtained the estimates of population sizes and expansion time (table 1). The estimated two-epoch model starts with an ancestral population size of 43,000, followed by an increase of 62.03-fold at a time 24,000 years ago. Under the exponential growth model, the ancestral population size is estimated as 40,000 and the population size increases exponentially by 395.93-fold beginning 30,000 years ago. A three-epoch model was also used, but it did not show significantly lower likelihood, and the estimates from replicated runs are different.
Table 1.
Inferred Demographic Parameters Under Two-Epoch Model and Exponential Growth Model.
| Model | Parameter | Estimate | 95% CI |
|---|---|---|---|
| Two-epoch | NA (×105) | 0.43 | 0.34–0.55 |
| N0 (×105) | 26.97 | 22.36–32.16 | |
| T (×104 years) | 2.41 | 2.26–2.55 | |
| N0/NA | 62.03 | 42.84–82.28 | |
| Exponential growth | NA (×105) | 0.40 | 0.20–0.52 |
| N0 (×105) | 107.79 | 1.20–180.39 | |
| T (×104 years) | 3.07 | 2.63–4.02 | |
| N0/NA | 395.93 | 69.77–671.29 |
Note.—CI, confidence interval.
Identification of Genes under Selection
To identify genes under selection, Tajima’s D was calculated for genes that are polymorphic. Positive Tajima’s D is considered as an indicator of balancing selection or partial sweep, and negative Tajima’s D could be caused by either negative selection or selective sweep. Because the null distribution of Tajima’s D under the neutral hypothesis is sensitive to the demographic history and gene length (mutation rate per gene), the significance level was estimated using the null distribution from coalescent simulations that consider both gene length and inferred demographic parameters. Among 4,281 genes examined, 29 genes have extremely high or low Tajima’s D scores and q-values (estimated false discovery rates, Storey and Tibshirani 2003) of less than 0.30 (supplementary table S4, Supplementary Material online). Among these genes, 26 have positive Tajima’s D, including PfAMA1 (PF11_0344), which has been found to be under balancing selection in many previous studies, and genes related to host-parasite interaction, such as acyl-CoA synthetase (PFB0695c) and tryptophan-rich antigen (PF10_0026). Only three of the genes have significant negative Tajima’s D values, and these genes are of unknown function. GO enrichment analysis was used for summarizing the genes with extreme Tajima’s D values to obtain an overall picture of genes that are under selection. GO terms that enriched for significant positive Tajima’s D, such as pathogenesis (GO:0009405), COPI vesicle coat (GO:0030126), and RNA helicase activity (GO:0003724), are listed in supplementary table S5, Supplementary Material online. However, no GO term was found to be enriched for significant negative Tajima’s D values.
The ratio of nonsynonymous and synonymous polymorphism (πN/πS) was also used to find genes that are under potential selection. In contrast to Tajima’s D, πN/πS is less sensitive to demographic history because synonymous sites and nonsynonymous sites are both affected by demographic history. The top 10 highest πN/πS genes are exported protein (PFL0070c), acyl-CoA synthetase PfACS8 (PFB0695c), acyl-CoA synthetase PfACS7 (PFL0035c), sporozoite invasion-associated protein 1 (PFD0425w), cysteine repeat modular protein 1 (PFI0550w), cysteine repeat modular protein 3 (PFL0410w), protein kinase (PFB0520w), oocyst capsule protein (PFC0905c), cysteine-rich surface protein (PF13_0338), and subtilisin-like protease 2 (PF11_0381). Among these, acyl-CoA synthetase was reported to have high genetic diversity and long-range haplotype before (Bethke et al. 2006; Van Tyne et al. 2011). We also tested whether any GO term has significantly higher or lower πN/πS than others. GO categories with significant higher or lower πN/πS (q-values < 0.05) are listed in supplementary table S5, Supplementary Material online.
Because πN/πS is not expected to be higher under positive selection before the favorable allele is fixed (πS is expected to increase with πN in this situation), it is helpful for distinguishing two potential selective forces suggested by positive Tajima’s D, balancing selection and partial sweep. Supplementary figure S6, Supplementary Material online, and figure 3 compare Tajima’s D and πN/πS. Genes with high Tajima’s D and high πN/πS, such as acyl-CoA synthetase (PFL0035c and PFB0695c), are more likely to be under balancing selection. Genes with πN/πS greater than three have significantly higher Tajima’s D than genes with πN/πS lower than three (Mann–Whitney U test, P value = 0.002), suggesting that genes with higher πN/πS tend to be under balancing selection. Genes with high Tajima’s D but not high πN/πS, such as single-strand binding protein (PFE0435c), might indicate partial sweeps caused by positive selection, recent balancing selection (before the selected allele reaches the equilibrium frequency), or balancing selection on noncoding sites like UTRs or introns. Genes having high πN/πS but not high Tajima’s D, such as cysteine repeat modular protein 1 (PFI0550w), might be under balancing selection, and Tajima’s D is not significant due to the occurrence of rare alleles.
Fig. 3.
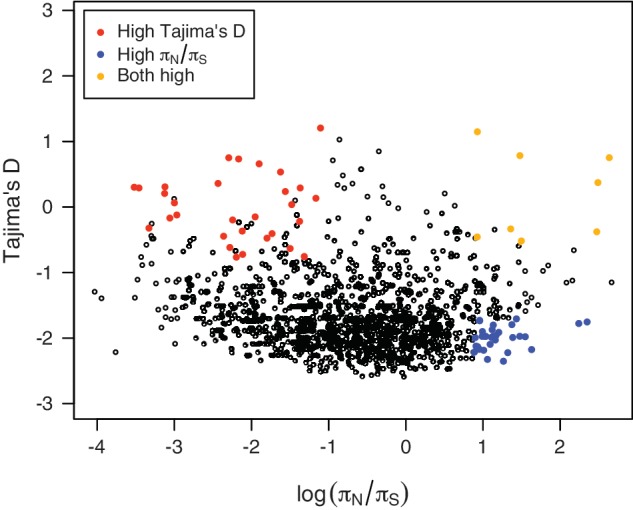
Tajima’s D versus log(πN/πS). Tajima’s D and πN/πS do not always have the same pattern. By considering both together, genes under selection can more readily be identified. Orange dots show genes with both high Tajima’s D (P value < 0.05) and πN/πS (top 5%), red dots show genes with only high Tajima’s D, and blue dots represent genes with only high πN/πS.
Interestingly, 18% of genes with synonymous polymorphism greater than 0 have πN/πS greater than 1 (supplementary fig. S7 and supplementary table S6, Supplementary Material online). Such a high value is very unusual and to our knowledge has not been reported in any other organism. This finding, together with the observed similar synonymous and nonsynonymous spectra, may be due to one or more of the following. First, one must consider sequencing artifacts or faulty annotation. Sequencing error could increase the number of singletons, and if the number of false singletons is much larger than the number of true ones, the difference between synonymous and nonsynonymous spectra would be diminished. Sequencing error could also increase πN/πS. To test this possibility, we increased the quality score threshold from 30 to 50, and found that the nonsynonymous and synonymous frequency spectra are still very similar (supplementary fig. S5, Supplementary Material online), with 17% of genes having πN/πS greater than 1. In regard to annotation, incorrect annotation could cause high πN/πS and similar synonymous and nonsynonymous spectra. However, this is unlikely to be a major factor because 456 out of 556 genes (82% of genes) with πN/πS greater than 1 were found to have mass spectrometry-based evidence of expression (PlasmoDB, http://plasmodb.org/plasmo/).
A second possibility is relaxed selection. Relaxed constraint or inefficient selection could contribute to the patterns. The possible relaxed constraint or inefficient selection could be caused by population expansion, recent intervention to reduce transmission, and clonal interference within the human host. Deleterious mutations have more chance to stay in population under population expansion. Recent intervention by drug treatments or bed net distribution may have reduced the effective population size very recently and hence reduced the efficacy of selection. This effect would not be captured by the allele frequency spectrum because it is so recent, and also because the effect is smaller than that of population expansion. Clonal interference within the human host could also increase the probability of deleterious mutations remaining in the population if they are linked to beneficial mutations. The finding of significantly lower polymorphism in nonsynonymous sites compared with synonymous sites indicates that some genes at least are under effective purifying selection.
Another factor might be purifying selection. Purifying selection on synonymous sites could increase πN/πS and reduce the difference between synonymous and nonsynonymous spectra. Finally, the abundance of genes under long-term balancing selection or diversifying selection due to strong selective pressure from the host immune system could explain the pattern. It is possible that the effect on the nonsynonymous frequency spectrum of genes that are under balancing or diversifying selection offsets the effect of purifying selection, and genes under long-term multi-allelic balancing selection or diversifying selection tend to have higher πN/πS ratio. We observed 26 genes with significantly positive Tajima’s D, and 17 genes have significant πN/πS ratio. However, it requires very high number of genes under multi-allelic balancing selection or diversifying selection to explain the whole pattern, and this high amount has never been seen in other organisms.
LD-based tests were also used for finding regions affected by putative selective sweeps. A quantile–quantile plot of iHS is shown in supplementary figure S8, Supplementary Material online. We performed an iHS test for positive selection on 17,572 imputed SNPs (fig. 4). Contiguous regions of positive selection were identified by taking each genome-wide significant core SNP from the iHS test, and extending out in each direction until the extended haplotype of the major allele at a site (EHHA) and the extended haplotype of the minor allele at a site (EHHD) both decayed below 0.05. Each core SNP thereby defined a window of putative positive selection, and overlapping windows were merged to define the regions described in supplementary table S7, Supplementary Material online. The linkage-based tests suggest several regions of recent positive selection, including areas near the known drug resistance loci pfcrt (MAL7P1.27) and pfmdr1 (PFE1150w). Additional regions include areas on chromosomes 2, 4, 5, 6, 7, 8, and 12. Because partial selective sweep also causes positive Tajima’s D, Tajima’s D values are also listed in supplementary table S7, Supplementary Material online, for comparison.
Fig. 4.

Manhattan plot of iHS P-values on a negative log10 scale. The dashed line indicates a Bonferroni threshold for significance. Dots are colored by chromosome, and their sizes are scaled according to P value. Several regions of recent positive selection are suggested by the iHS statistic, including areas near the known drug resistance loci pfcrt and pfmdr1.
Selection on Base Composition
The genomes of P. falciparum and P. reichenowi have an A/T base composition of 81% (Pollack et al. 1982; Gardner et al. 2002; Jeffares et al. 2007). This is much higher than what is observed in the genomes of other primate malarial parasites like P. vivax and P. knowlesi, which have an A/T content of ∼60% (Williamson et al. 1985; Carlton 2003; Pain et al. 2008). To study the dynamics of changes in base composition, we used P. reichenowi to infer the ancestral state for observed P. falciparum polymorphisms. We first generated empirical nucleotide transition matrices and calculated the equilibrium states (tables 2 and 3). We found that the observed mean A/T composition at 4-fold degenerate sites in coding regions as well as in intergenic regions are close to the predicted equilibrium A/T composition based on the empirical nucleotide transition matrices. This suggests that the nucleotide composition in P. falciparum may be close to equilibrium.
Table 2.
Empirical Nucleotide Transition Matrices.
| A | T | C | G | |
|---|---|---|---|---|
| Four-fold degenerate sites | ||||
| A | 0.9905 | 0.0033 | 0.0020 | 0.0043 |
| T | 0.0033 | 0.9903 | 0.0043 | 0.0021 |
| C | 0.0078 | 0.0154 | 0.9717 | 0.0051 |
| G | 0.0141 | 0.0062 | 0.0043 | 0.9754 |
| Intergenic regions | ||||
| A | 0.9947 | 0.0028 | 0.0007 | 0.0019 |
| T | 0.0028 | 0.9945 | 0.0020 | 0.0007 |
| C | 0.0042 | 0.0084 | 0.9853 | 0.0020 |
| G | 0.0085 | 0.0042 | 0.0021 | 0.9852 |
Within-species polymorphism provides suitable material to study to what extent the high A/T percentage in the genome is due to mutation pressure offset by selection. We generated separate derived allele-frequency spectra for mutations that would serve to decrease the A/T composition (“A/T to G/C”) and mutations that would increase the A/T composition “G/C to A/T”) in silent coding and intergenic regions (fig. 5). The G/C to A/T spectrum has more rare derived alleles and fewer high frequency derived alleles than the A/T to G/C spectrum in both intergenic and silent coding regions, suggesting purifying selection against A/T nucleotides and/or positive selection favoring C/G nucleotides. That is, on average, mutations that would result in a higher A/T composition of the genome are acted against by selection, and/or mutations that would result in lower G/C composition are selected for, and therefore the high equilibrium AT composition in P. falciparum likely reflects a dynamic equilibrium balanced between A/T biased mutation pressure and selection to augment the relative fixation probability of G/C mutations. Although an incorrect ascertainment of ancestral state due to ancestral polymorphism could be responsible for a small fraction of the high frequency derived alleles observed, we do not expect a difference in the accuracy of ancestral state inferences between the two classes of mutations; therefore, the observation of a relative difference in the frequencies of mutations that would further increase versus decrease A/T composition should be robust.
Fig. 5.

Derived site-frequency spectrum. The unfolded site-frequency spectrum was generated by using P. reichenowi as an outgroup. The G/C to A/T spectrum is more skewed toward low frequencies than the A/T to G/C spectrum, suggesting positive selection favoring C/G nucleotides or purifying selection against A/T nucleotides.
Discussion
In this study, we carried out whole-genome sequencing of 25 isolates of P. falciparum from Senegal and used population genetic methods to investigate the evolutionary forces shaping genetic diversity in this local African population. Fully sequenced population genomic data provide an important resource for understanding genome-wide patterns of diversity as well as the evolution of genes of particular interest.
Previous studies of the evolutionary history of P. falciparum are not completely consistent (Hartl et al. 2002; Su et al. 2003; Hartl 2004), which may in part reflect variation in the evolutionary history and genetic diversity across different regions or genes in the genome due to both selection and recombination. With the complete genome sequences of a deep sample from a single African population, we have been able to use genome-wide synonymous changes that are known to be minimally affected by selective forces (especially when LD is low) to better estimate demographic parameters than in previous studies which relied on only a few loci. Moreover, the absence of any obvious population substructure, which can also affect the allele-frequency spectrum (Tajima 1989a), suggests that our estimates for Senegal are not biased by substructure. Because the effect on the synonymous frequency spectrum of selection on AT content (fig. 5) may bias the estimates of demographic parameters, we also fitted the same model to the synonymous frequency spectrum with only A/T to T/A and C/G to G/C changes, and the estimates of demographic parameters were consistent. However, it should be noted that selection on linked sites could affect synonymous sites and bias our estimates of demographic parameters, even if average LD over the genome is low.
We estimate a major 60-fold population expansion approximately 20,000–40,000 years ago by fitting demographic models to the synonymous allele-frequency spectrum. The effective population size prior to the population expansion was about 20,000–55,000. Although the effective population size after the expansion is sensitive to the demographic model (varying from 2.2 million to 18 million), it is consistently very large. Our estimate of population expansion time in Africa is close to the 95% confidence interval (2,000–14,500) of the estimate in Joy et al. (2003) based on mitochondrial DNA of pan-African strains and different methods of analysis. Moreover, it was recently suggested that P. falciparum first infected human ancestors 365,000 years ago (Baron et al. 2011). This indicates that this event was well before the population expansion we identified and our estimates are unlikely biased by it. However, it should be noted that our estimates of population sizes and population expansion time are highly dependent on the estimate of mutation rate, and therefore on the choice of divergence time between P. falciparum and P. reichenowi. Although we have curated our data as carefully as possible, any remaining sequencing error, reference-sequence bias, and alignment error caused by nearby insertions or deletions, would all potentially bias the allele-frequency spectrum and therefore our estimate of demographic parameters. Sequencing errors and reference-sequence bias both can skew the site-frequency spectrum to rare alleles, resulting in an overestimate of the changes in population sizes or the population expansion time.
Our estimate of population expansion time of 20,000 to 40,000 years overlaps with the Upper Paleolithic era (10,000 to 40,000 years ago) and the Mousterian Pluvial (30,000 to 50,000 years ago) and is immediately after human migration out of Africa (40,000 to 130,000 years ago). During the Mousterian Pluvial, northern Africa was a land of lakes, swamps, and rivers, and this may have increased the spread of malaria parasites by mosquitoes and facilitated epidemic transmission. As such, we believe that our estimate of the date of expansion offers a good fit to archaeological and climatological explanations.
The recent availability of large-scale genomic data sets such as this one affords an opportunity to refine our understanding of the general evolutionary patterns in P. falciparum. First, we found that both synonymous polymorphism and synonymous substitution rates differ among chromosomes, suggesting that the mutation rates are not the same for all chromosomes. Since population genetic statistics, such as Tajima’s D per gene, are sensitive to mutation rate, it is desirable to correct for the variation in mutation rate when conducting this test, provided that reliable estimates of mutation rate for each chromosome are available. Second, lower intergenic and intronic polymorphism as compared with synonymous polymorphism suggests weak selection constraints might be common in intergenic regions and introns. Divergence between P. falciparum and P. reichenowi is also lower in intergenic regions and introns (Neafsey et al. 2005). Third, low LD indicates the potential of fine-scale mapping of selection or association signals and fewer problems when applying analytic tools that assume SNPs are unassociated with each other, such as ∂a∂i and STRUCTURE. Fourth, we reported two unusual findings, the similar synonymous and nonsynonymous allele-frequency spectra, and the large number of genes with high nonsynonymous-to-synonymous polymorphism, and discussed some possible explanations.
Fifth, we found that the nucleotide composition of P. falciparum, which has the highest A/T content of any eukaryotic genomes described so far, is at or near equilibrium. Moreover, we showed that the unfolded site-frequency spectra of G/C to A/T polymorphic sites in both synonymous sites and intergenic regions have more rare alleles than that of A/T to C/G polymorphic sites, which suggests that selection acts against A/T at least at certain sites in the P. falciparum genome. In contrast, the empirical nucleotide transition matrices (table 3) support the trend toward high A/T composition, because A, T, C, and G all tend to change to A or T, and A and T also change less often. Because the empirical nucleotide transition matrix is influenced by both mutation and selection and we know that selection acts, on average, against C/G to A/T changes, it can be inferred that mutations are toward A/T. This result differs from that of Escalante et al. (1998), who found a trend toward higher A/T but no clear evidence of A/T mutational bias in studies of 10 highly polymorphic genes including six encoding surface antigens; signals of strong selection on five of the genes may have obscured a signal of A/T biased mutation. Mutational bias toward A/T has been found in other organisms (e.g., Hershberg and Petrov 2010), and selective advantages of biased mutation rates have been discussed (Rocha and Danchin 2002; Dalpke et al. 2006). It was suggested that G and C were less favored by natural selection in obligatory pathogens and symbionts because GTP and CTP nucleotides cost more energy and there is less availability of GTP and CTP in the cell (Rocha and Danchin 2002). Also, as toll-like receptor 9 specifically recognizes nonmethylated CpG dinucleotides (Dalpke et al. 2006), and there is lack of evidence of DNA methylation in P. falciparum (Choi et al. 2006), reducing G and C in the genome would be one mechanism to reduce the innate immune response and therefore be favored by selection. However, when mutations are strongly biased to A/T, it may affect amino acid composition. Singer and Hickey (2000) found that A/T codon bias in P. falciparum was so severe that it was affecting amino acid composition. Although mutational bias can change amino acid composition in some genes, genes under strong selective constraint are less influenced by mutational bias. It was shown in P. falciparum that amino acids encoded by GC-rich codons are significantly more frequent in highly expressed genes (Chanda et al. 2005), perhaps because highly expressed genes are more conserved, and the ancestral state presumably exhibited less biased AT content. Our findings that suggest a mutation-selection balance in the A/T content of the genome (with mutation favoring higher A/T and selection against A/T) add a new layer of understanding to this otherwise puzzling aspect of P. falciparum’s genome composition.
The absence of population substructure in Senegal means that signatures of selection can be identified with greater confidence. Here, we used the Tajima’s D test, the ratio of nonsynonymous to synonymous polymorphism, and the iHS test, and successfully detected signatures of selection in some genes identified previously, such as pfcrt and pfmdr1, as well as some new candidate genes, such as two acyl-CoA synthetases (PFL0035c and PFB0695c). Additionally, we identified the gene categories that are more likely to be under negative selection (e.g., ribosome and translation) and balancing/diversifying selection (e.g., membrane and attachment of GPI anchor to protein). The identification of these GO categories not only provides a broader view about types of selection in various biological processes but also helps with functional characterization of genes whose function is unknown.
Our study provides the one of first population genomic analyses of a deeply sampled local population of P. falciparum. The estimation of demographic parameters by genome-wide SNPs offers, for the first time, a proper null distribution for identifying genes under various selective forces. Genes identified here could be validated by follow-up functional assays, and the results have practical implications for finding functional variants of medical relevance and developing methods of disease control. If whole genome sequences of closely related species, such as P. reichenowi and other species from chimpanzee and gorilla, are available in the future, our data set can be used for investigating evolutionary questions with more confidence, such as controlling for variation in mutation rates and detecting selection using the ratio of divergence to polymorphism.
Supplementary Material
Supplementary tables S1–S7 and figures S1–S8 are available at Molecular Biology and Evolution online (http://www.mbe.oxfordjournals.org/).
Acknowledgments
The authors thank John Wakeley, Pleuni Pennings, Yu-Ping Poh, Grace Yuh Chwen Lee, and Timothy Sackton for their helpful discussion and suggestions. This study is supported by the Bill and Melinda Gates Foundation, National Institutes of Health (Grants 1R01AI075080-01A1, 1R56AI082589, and 5R01GM061351), ExxonMobil Foundation, National Institutes of Health Fogarty Grant (D43TW001503), and Broad SPARC. D.J.P. is supported by a National Science Foundation Graduate Research Fellowship. P.C.S. is supported by fellowships from the Burroughs Wellcome and Packard Foundations.
References
- Anderson TJ, Haubold B, Williams JT, et al. (16 co-authors) Microsatellite markers reveal a spectrum of population structures in the malaria parasite Plasmodium falciparum. Mol Biol Evol. 2000;17(10):1467–1482. doi: 10.1093/oxfordjournals.molbev.a026247. [DOI] [PubMed] [Google Scholar]
- Baron JM, Higgins JM, Dzik WH. A revised timeline for the origin of Plasmodium falciparum as a human pathogen. J Mol Evol. 2011;73(5–6):297–304. doi: 10.1007/s00239-011-9476-x. [DOI] [PubMed] [Google Scholar]
- Bethke LL, Zilversmit M, Nielsen K, Daily J, Volkman SK, Ndiaye D, Lozovsky ER, Hartl DL, Wirth DF. Duplication, gene conversion, and genetic diversity in the species-specific acyl-CoA synthetase gene family of Plasmodium falciparum. Mol Biochem Parasitol. 2006;150(1):10–24. doi: 10.1016/j.molbiopara.2006.06.004. [DOI] [PubMed] [Google Scholar]
- Carlton J. The Plasmodium vivax genome sequencing project. Trends Parasitol. 2003;19(5):227–231. doi: 10.1016/s1471-4922(03)00066-7. [DOI] [PubMed] [Google Scholar]
- Chanda I, Pan A, Dutta C. Proteome composition in Plasmodium falciparum: higher usage of GC-rich nonsynonymous codons in highly expressed genes. J Mol Evol. 2005;61(4):513–523. doi: 10.1007/s00239-005-0023-5. [DOI] [PubMed] [Google Scholar]
- Chen FC, Li WH. Genomic divergences between humans and other hominoids and the effective population size of the common ancestor of humans and chimpanzees. Am J Hum Genet. 2001;68(2):444–456. doi: 10.1086/318206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi SW, Keyes MK, Horrocks P. LC/ESI-MS demonstrates the absence of 5-methyl-2′-deoxycytosine in Plasmodium falciparum genomic DNA. Mol Biochem Parasitol. 2006;150(2):350–352. doi: 10.1016/j.molbiopara.2006.07.003. [DOI] [PubMed] [Google Scholar]
- Conway DJ, Fanello C, Lloyd JM, et al. (12 co-authors) Origin of Plasmodium falciparum malaria is traced by mitochondrial DNA. Mol Biochem Parasitol. 2000;111(1):163–171. doi: 10.1016/s0166-6851(00)00313-3. [DOI] [PubMed] [Google Scholar]
- Dalpke A, Frank J, Peter M, Heeg K. Activation of toll-like receptor 9 by DNA from different bacterial species. Infect Immun. 2006;74(2):940–946. doi: 10.1128/IAI.74.2.940-946.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DePristo MA, Banks E, Poplin R, et al. (18 co-authors) A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat Genet. 2011;43(5):491–498. doi: 10.1038/ng.806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Escalante AA, Lal AA, Ayala FJ. Genetic polymorphism and natural selection in the malaria parasite Plasmodium falciparum. Genetics. 1998;149(1):189–202. doi: 10.1093/genetics/149.1.189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falush D, Stephens M, Pritchard JK. Inference of population structure using multilocus genotype data: linked loci and correlated allele frequencies. Genetics. 2003;164(4):1567–1587. doi: 10.1093/genetics/164.4.1567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gardner MJ, Hall N, Fung E, et al. (45 co-authors) Genome sequence of the human malaria parasite Plasmodium falciparum. Nature. 2002;419(6906):498–511. doi: 10.1038/nature01097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gutenkunst RN, Hernandez RD, Williamson SH, Bustamante CD. Inferring the joint demographic history of multiple populations from multidimensional SNP frequency data. PLoS Genet. 2009;5(10):e1000695. doi: 10.1371/journal.pgen.1000695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartl DL. The origin of malaria: mixed messages from genetic diversity. Nat Rev Microbiol. 2004;2(1):15–22. doi: 10.1038/nrmicro795. [DOI] [PubMed] [Google Scholar]
- Hartl DL, Volkman SK, Nielsen KM, Barry AE, Day KP, Wirth DF, Winzeler EA. The paradoxical population genetics of Plasmodium falciparum. Trends Parasitol. 2002;18(6):266–272. doi: 10.1016/s1471-4922(02)02268-7. [DOI] [PubMed] [Google Scholar]
- Hershberg R, Petrov DA. Evidence that mutation is universally biased towards AT in bacteria. PLoS Genet. 2010;6(9):e1001115. doi: 10.1371/journal.pgen.1001115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill WG, Robertson A. Linkage disequilibrium in finite populations. Theor Appl Genet. 1968;38:226–231. doi: 10.1007/BF01245622. [DOI] [PubMed] [Google Scholar]
- Hudson RR. Generating samples under a Wright-Fisher neutral model of genetic variation. Bioinformatics. 2002;18(2):337–338. doi: 10.1093/bioinformatics/18.2.337. [DOI] [PubMed] [Google Scholar]
- Hughes AL, Verra F. Very large long-term effective population size in the virulent human malaria parasite Plasmodium falciparum. Proc Biol Sci. 2001;268(1478):1855–1860. doi: 10.1098/rspb.2001.1759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ina Y. New methods for estimating the numbers of synonymous and nonsynonymous substitutions. J Mol Evol. 1995;40(2):190–226. doi: 10.1007/BF00167113. [DOI] [PubMed] [Google Scholar]
- Jeffares DC, Pain A, Berry A, et al. (15 co-authors) Genome variation and evolution of the malaria parasite Plasmodium falciparum. Nat Genet. 2007;39(1):120–125. doi: 10.1038/ng1931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang H, Li N, Gopalan V, et al. (16 co-authors) High recombination rates and hotspots in a Plasmodium falciparum genetic cross. Genome Biol. 2011;12(4):R33. doi: 10.1186/gb-2011-12-4-r33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joy DA, Feng X, Mu J, et al. (12 co-authors) Early origin and recent expansion of Plasmodium falciparum. Science. 2003;300(5617):318–321. doi: 10.1126/science.1081449. [DOI] [PubMed] [Google Scholar]
- Kulathinal RJ, Bennett SM, Fitzpatrick CL, Noor MA. Fine-scale mapping of recombination rate in Drosophila refines its correlation to diversity and divergence. Proc Natl Acad Sci U S A. 2008;105(29):10051–10056. doi: 10.1073/pnas.0801848105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurtz S, Phillippy A, Delcher AL, Smoot M, Shumway M, Antonescu C, Salzberg SL. Versatile and open software for comparing large genomes. Genome Biol. 2004;5(2):R12. doi: 10.1186/gb-2004-5-2-r12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Durbin R. Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics. 2010;26(5):589–595. doi: 10.1093/bioinformatics/btp698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R. The sequence alignment/map format and SAMtools. Bioinformatics. 2009;25(16):2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu W, Li Y, Learn GH, et al. (22 co-authors) Origin of the human malaria parasite Plasmodium falciparum in gorillas. Nature. 2010;467(7314):420–425. doi: 10.1038/nature09442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McVean G, Awadalla P, Fearnhead P. A coalescent-based method for detecting and estimating recombination from gene sequences. Genetics. 2002;160(3):1231–1241. doi: 10.1093/genetics/160.3.1231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McVean GA, Myers SR, Hunt S, Deloukas P, Bentley DR, Donnelly P. The fine-scale structure of recombination rate variation in the human genome. Science. 2004;304(5670):581–584. doi: 10.1126/science.1092500. [DOI] [PubMed] [Google Scholar]
- Mu J, Awadalla P, Duan J, McGee KM, Joy DA, McVean GA, Su XZ. Recombination hotspots and population structure in Plasmodium falciparum. PLoS Biol. 2005;3(10):e335. doi: 10.1371/journal.pbio.0030335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neafsey DE, Hartl DL, Berriman M. Evolution of noncoding and silent coding sites in the Plasmodium falciparum and Plasmodium reichenowi genomes. Mol Biol Evol. 2005;22(7):1621–1626. doi: 10.1093/molbev/msi154. [DOI] [PubMed] [Google Scholar]
- Neafsey DE, Schaffner SF, Volkman SK, et al. (28 co-authors) Genome-wide SNP genotyping highlights the role of natural selection in Plasmodium falciparum population divergence. Genome Biol. 2008;9(12):R171. doi: 10.1186/gb-2008-9-12-r171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pain A, Bohme U, Berry AE, et al. (54 co-authors) The genome of the simian and human malaria parasite Plasmodium knowlesi. Nature. 2008;455(7214):799–803. doi: 10.1038/nature07306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patterson N, Price AL, Reich D. Population structure and eigenanalysis. PLoS Genet. 2006;2(12):e190. doi: 10.1371/journal.pgen.0020190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pollack Y, Katzen AL, Spira DT, Golenser J. The genome of Plasmodium falciparum. I: DNA base composition. Nucleic Acids Res. 1982;10(2):539–546. doi: 10.1093/nar/10.2.539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polley SD, Conway DJ. Strong diversifying selection on domains of the Plasmodium falciparum apical membrane antigen 1 gene. Genetics. 2001;158(4):1505–1512. doi: 10.1093/genetics/158.4.1505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pritchard JK, Stephens M, Donnelly P. Inference of population structure using multilocus genotype data. Genetics. 2000;155(2):945–959. doi: 10.1093/genetics/155.2.945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rich SM, Licht MC, Hudson RR, Ayala FJ. Malaria’s eve: evidence of a recent population bottleneck throughout the world populations of Plasmodium falciparum. Proc Natl Acad Sci U S A. 1998;95(8):4425–4430. doi: 10.1073/pnas.95.8.4425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rocha EP, Danchin A. Base composition bias might result from competition for metabolic resources. Trends Genet. 2002;18(6):291–294. doi: 10.1016/S0168-9525(02)02690-2. [DOI] [PubMed] [Google Scholar]
- Sella G, Petrov DA, Przeworski M, Andolfatto P. Pervasive natural selection in the Drosophila genome? PLoS Genet. 2009;5(6):e1000495. doi: 10.1371/journal.pgen.1000495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singer GA, Hickey DA. Nucleotide bias causes a genomewide bias in the amino acid composition of proteins. Mol Biol Evol. 2000;17(11):1581–1588. doi: 10.1093/oxfordjournals.molbev.a026257. [DOI] [PubMed] [Google Scholar]
- Stephens M, Donnelly P. A comparison of bayesian methods for haplotype reconstruction from population genotype data. Am J Hum Genet. 2003;73(5):1162–1169. doi: 10.1086/379378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Storey JD, Tibshirani R. Statistical significance for genomewide studies. Proc Natl Acad Sci U S A. 2003;100(16):9440–9445. doi: 10.1073/pnas.1530509100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su XZ, Mu J, Joy DA. The "Malaria’s Eve" hypothesis and the debate concerning the origin of the human malaria parasite Plasmodium falciparum. Microbes Infect. 2003;5(10):891–896. doi: 10.1016/s1286-4579(03)00173-4. [DOI] [PubMed] [Google Scholar]
- Tajima F. DNA polymorphism in a subdivided population: the expected number of segregating sites in the two-subpopulation model. Genetics. 1989a;123(1):229–240. doi: 10.1093/genetics/123.1.229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tajima F. Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics. 1989b;123(3):585–595. doi: 10.1093/genetics/123.3.585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Tyne D, Park DJ, Schaffner SF, et al. (31 co-authors) Identification and functional validation of the novel antimalarial resistance locus PF10_0355 in Plasmodium falciparum. PLoS Genet. 2011;7(4):e1001383. doi: 10.1371/journal.pgen.1001383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verra F, Hughes AL. Evidence for ancient balanced polymorphism at the Apical Membrane Antigen-1 (AMA-1) locus of Plasmodium falciparum. Mol Biochem Parasitol. 2000;105(1):149–153. doi: 10.1016/s0166-6851(99)00162-0. [DOI] [PubMed] [Google Scholar]
- Voight BF, Kudaravalli S, Wen X, Pritchard JK. A map of recent positive selection in the human genome. PLoS Biol. 2006;4(3):e72. doi: 10.1371/journal.pbio.0040072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volkman SK, Barry AE, Lyons EJ, Nielsen KM, Thomas SM, Choi M, Thakore SS, Day KP, Wirth DF, Hartl DL. Recent origin of Plasmodium falciparum from a single progenitor. Science. 2001;293(5529):482–484. doi: 10.1126/science.1059878. [DOI] [PubMed] [Google Scholar]
- Volkman SK, Hartl DL, Wirth DF, et al. (11 co-authors) Excess polymorphisms in genes for membrane proteins in Plasmodium falciparum. Science. 2002;298(5591):216–218. doi: 10.1126/science.1075642. [DOI] [PubMed] [Google Scholar]
- Volkman SK, Sabeti PC, DeCaprio D, et al. (28 co-authors) A genome-wide map of diversity in Plasmodium falciparum. Nat Genet. 2007;39(1):113–119. doi: 10.1038/ng1930. [DOI] [PubMed] [Google Scholar]
- Watterson GA. On the number of segregating sites in genetical models without recombination. Theor Popul Biol. 1975;7(2):256–276. doi: 10.1016/0040-5809(75)90020-9. [DOI] [PubMed] [Google Scholar]
- WHO World Malaria Report. World Malaria Report. Geneva (Switzerland): World Health Organization; 2010. Available from: http://www.who.int/malaria/world_malaria_report_2010. [Google Scholar]
- Williamson DH, Wilson RJ, Bates PA, McCready S, Perler F, Qiang BU. Nuclear and mitochondrial DNA of the primate malarial parasite Plasmodium knowlesi. Mol Biochem Parasitol. 1985;14(2):199–209. doi: 10.1016/0166-6851(85)90038-6. [DOI] [PubMed] [Google Scholar]
- Yang Z, Nielsen R. Estimating synonymous and nonsynonymous substitution rates under realistic evolutionary models. Mol Biol Evol. 2000;17(1):32–43. doi: 10.1093/oxfordjournals.molbev.a026236. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.