

Abstract
The bis(trimethylsilyl)-substituted hydroxycyclopentadienyl ruthenium hydride [2,5-(SiMe3)2-3,4-(CH2OCH2)(η5-C4COH)]Ru(CO)2H (10) is an efficient catalyst for hydrogenation of aldehydes and ketones. Because 10 transfers hydrogen rapidly to aldehydes and ketones and because it does not form an inactive bridging hydride during reaction, hydrogenation of aldehydes and ketones can be performed at room temperature under relatively low hydrogen pressure (3 atm); this is a significant improvement compared with previously developed Shvo type catalysts. Kinetic and 2H NMR spectroscopic studies of the stoichiometric reduction of aldehydes and ketones by 10 established a two-step process for the hydrogen transfer: (1) rapid and reversible hydrogen bond formation between OH of 10 and the oxygen of the aldehyde or ketone, (2) followed by slow transfer of both proton and hydride from 10 to the aldehyde or ketone. The stoichiometric and catalytic activities of complex 10 are compared to those of other Shvo type ruthenium hydrides and related iron hydrides.
Metal-ligand bifunctional catalysts1 for the hydrogenation of polar double bonds provide a “green” alternative to stoichiometric reducing agents such as LiAlH4 or NaBH4.2 Shvo’s hydroxycyclopentadienyl ruthenium hydride 1 was the first reported metal-ligand bifunctional catalyst;3 it is generated in situ by dissociation of the diruthenium bridging hydride 2, which is itself unreactive towards aldehydes and ketones.4 Our group has carried out detailed mechanistic studies of the tolyl analogs of Shvo’s catalytic system. The rates and mechanisms of the steps involved in the catalytic system were separately determined and the kinetics of the overall process was also studied.5 A full kinetic model of the hydrogenation based on rate constants for individual steps in the catalysis was developed that simulates the rate of carbonyl compound hydrogenation and of the amounts of ruthenium species 3 and 4 present during hydrogenation (Scheme 1).6 Although the reduction of benzaldehyde by ruthenium hydride 3 (the active reducing agent) takes place even at −40 °C in toluene, the partitioning between the inactive diruthenium species 4 and the active monoruthenium hydride 3 dictates the efficiency of the catalytic reaction. At 60 °C, the only observable ruthenium species present during catalytic hydrogenation of aldehydes and ketones was the diruthenium complex 4. Consequently, the Shvo and related hydrogenation catalysts typically require relatively high temperatures (> 80 °C) in combination with high hydrogen pressure (35 atm) to be efficient catalysts for hydrogenation of aldehydes and ketones.
Scheme 1.

To develop new more active ruthenium catalysts, systems with structures that interfere with the formation of unreactive M–H–M systems but maintain high reactive of the M–H species are needed. Destabilization of M–H–M formation was achieved by introducing a bulky -NHPh group on the Cp ring in place of -OH, but the low NH acidity resulted in slow stoichiometric reduction of carbonyl groups by 5 (Scheme 2).7 Protonation of 5 to give 6, which possesses a much more acidic -NPhH2+ group, gave a very active catalyst, but acid-catalyzed side reactions led to some ether formation.7 Complex 7, in which PPh3 is substituted for one CO, also prevented formation of unreactive M–H–M complexes.8 While 7 is a very selective catalyst for hydrogenation of aldehydes over ketones and a faster catalyst than 4 for aldehyde hydrogenation below 60 °C, it is a slower catalyst at higher temperatures.
Scheme 2.

Recently, we discovered that the TMS (TMS = trimethylsilyl) substituted hydroxycyclopentadienyl iron hydride 8, first prepared and fully characterized by Knölker,9 is an efficient and chemoselective catalyst for the hydrogenation of ketones (Scheme 3).10 Kinetics studies have suggested that the hydrogen transfer from hydride 8 to ketones is the turnover-limiting step and the hydrogenation rate is independent on hydrogen pressure. Monitoring the stoichiometric reduction by 1H NMR and the catalytic hydrogenation by in situ IR has shown no evidence of a diiron bridging hydride. Distinctly from all other Shvo type systems mentioned above, this iron hydride catalyzes the hydrogenation of both aldehydes and ketones under mild conditions (25 °C, 3 atm of H2). Although the reason for no bridging hydride formation is not fully understood, the remarkable reactivity of 8 has prompted us to investigate the chemistry of ruthenium complexes with a similar ligand set.
Scheme 3.

Here we describe the synthesis of ruthenium hydrides with bis(TMS)-substituted cyclopentadienyl ligands. We have examined their reactivity toward the stoichiometric reduction as well as the catalytic hydrogenation of benzaldehyde and acetophenone. These ruthenium hydrides are far more reactive hydrogenation catalysts than other Shvo type complexes and are similar in reactivity to related iron catalysts.
Results
[2,5-(SiMe3)2-3,4-(CH2OCH2)(η5-C4COH)]Ru(CO)2H (10)
Reaction of the known ruthenium tricarbonyl911 with excess NaOH in THF/H2O, followed by acidification with H3PO4, produced the ruthenium hydride 10 in 73 % yield (Scheme 4). These are the same reaction conditions used by Knölker for the synthesis of iron complex 8.9 The structure of 10 was established spectroscopically and confirmed by X-ray crystallography (Figure 1).
Scheme 4.

Figure 1.

X-Ray crystal structure of 10 shown with 50% probability ellipsoids.
In the X-ray crystal structure of 10, one of the two CO ligands lies directly below the hydroxyl group on the Cp ring. A similar conformation was observed in the solid-state structure of iron hydride 8.9 In the solid state, each molecule of 10 is linked to two others by hydrogen bonding between the CpOH and the ether oxygen of the fused ring (Figure 2). However, IR and NMR spectroscopy show that 10 exists primarily as a monomer in toluene at 0.01 M. The IR spectrum shows an O—H band at 3591 cm−1, indicative of a non-hydrogen-bonded hydroxyl. The 1H NMR chemical shift of OH resonance changed from δ 3.56 to δ 3.7212 upon increasing the concentration of 8 from 0.01 M to 0.10 M; these chemical shifts are consistent with little hydrogen bonding in toluene and with little change in aggregation over this concentration range.
Figure 2.

Hydrogen bonding network in the solid-state structure of 10.
Acidity of the Hydroxyl Proton of 10
Since the stoichiometric reactivity of CpOH ruthenium hydrides depends strongly on the acidity of the CpOH group, we sought to determine the acidity of the CpOH proton of 10. The pKa of 3 (the tolyl analogue of the Shvo hydride) was found to be 17.5 in CH3CN using Norton’s IR method.13 The pKa of PPh3-substituted hydride 7 was determined to be 20.7.8a The greater acidity of 3 than 7 was correlated with the greater reactivity of 3 in the stoichiometric reduction of aldehydes and ketones.
Norton’s IR method was used to determine the pKa of 10. When NEt3 (0.040 M, pKa = 18.5 in CH3CN) was added to a 0.040 M CH3CN solution of 10, IR bands due to 10 at 2014 and 1952 cm−1 decreased in intensity and new bands at 1982 and 1915 cm−1 assigned to NEt3H+{[2,5-(SiMe3)2-3,4-(CH2OCH2)(η5-C4CO)]Ru(CO)2H}− (11) grew in. The ratio of 11 : 10 was determined to be 1 : 1.8 by comparing the absorbances with independently determined molar absorptivities. The pKa of 10 was calculated to be 19.0. When 1 equiv of pyridine (pKa = 12.4 in CH3CN) was added to a CH3CN solution of 10, no deprotonation was observed. When 1 equiv of 1,1,3,3-tetramethylguanidine (pKa = 23.3 in CH3CN) was added to a CH3CN solution of 10, complete deprotonation to the guanidinium salt was observed. Ruthenium hydride 10 is 1.5 pKa units less acidic than hydride 3, but 1.7 pKa units more acidic than hydride 7 (Scheme 5).
Scheme 5.

Stoichiometric Reduction of Benzaldehyde by 10
The reaction of 10 with excess benzaldehyde in toluene-d8 at −60 °C was monitored by 1H NMR spectroscopy. Disappearance of the ruthenium hydride resonance of 10 (between δ −10.12 and δ −9.65, vide infra) and concurrent appearance of two sets of benzyl alcohol-like resonances were observed. Complete disappearance of 10 was seen within 30 min at −60 °C. Based on the similarity of these two sets of benzyl alcohol-like resonances to those of the iron alcohol complex produced in the reduction of benzaldehyde by iron hydride 8,14 they were assigned to two isomeric ruthenium alcohol complexes. The two ruthenium alcohol complexes differ only by hydrogen bonding to the two different oxygens of the Cp ring (Scheme 6). Initially one of the alcohol complexes (presumably 12) was the dominant species, but slow isomerization led to a 1:1 equilibrium ratio of 12 : 12’. Above −50 °C, complexes 12 and 12’ decomposed to free benzyl alcohol and some intractable ruthenium species, but no NMR resonances attributable to a diruthenium bridging hydride was observed. The low thermal stability of 12 and 12’ made it impossible to obtain single crystals suitable for X-ray crystallography.
Scheme 6.

When a solution of 10 in toluene-d8 (0.060 M) was treated with excess benzaldehyde in the presence of PPh3 at −60 °C, only the alcohol complexes 12 and 12’ were observed; none of the phosphine-trapping product 13 was seen. When the solution was warmed above −50 °C, ruthenium alcohol complexes 12 and 12’ were cleanly converted to a PPh3-substituted ruthenium complex 13 and the free alcohol (Scheme 7). This establishes that alcohol complexes 12 and 12’ are kinetic products of reduction.
Scheme 7.

Kinetics of Hydrogen Transfer from 10 to Benzaldehyde
The reaction of 10 (< 0.010 M) with a large excess of PhCHO (> 10 equiv) in toluene-d8 at −60 °C was monitored by 1H NMR spectroscopy. The peak height of the TMS resonance of 10 relative to that of an internal standard (mesitylene methyl δ 2.15) was followed as a function of time. The disappearance of 10 followed first order kinetics. The observed first order rate constants increased non-linearly as the [PhCHO] increased (Figure 3).
Figure 3.

Plot of kobs for reduction of PhCHO by 10 vs. [PhCHO] in toluene-d8 at −60 °C.
This observation of saturation kinetics supports a mechanism involving rapid pre-equilibrium through hydrogen bonding formation, followed by relatively slow hydrogen transfer (Scheme 8). It was noted that both the RuH and TMS resonances of 10 shifted relative to solvent residual peak (δ 2.09) during reduction. This is consistent with the presence of a changing ratio of free 10 and 14 (a hydrogen bonded adduct of 10 and PhCHO) during reduction (Scheme 8). The OH resonance of 10/14 was not readily seen in the 1H NMR spectrum. However, it was possible to observe shifts in the OD resonance in the 2H NMR spectra of 10-d2/14-d2 during the reaction of 10-d2 with excess PhCHO in toluene. Both the slower reaction of PhCHO with 10-d2 (due to a primary deuterium isotope effect, the reaction was incomplete even after 100 min at −60 °C) and the simplified aromatic range in 2H NMR spectrum provided made these measurements possible. Strong evidence for hydrogen bonding interaction was provided by the shift of the OD resonance from δ 4.7 for pure 10-d2 in toluene to δ 7.8 upon addition of PhCHO.
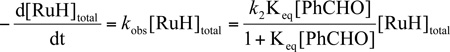 |
(1) |
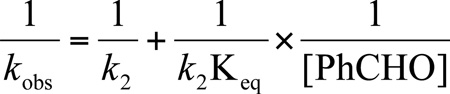 |
(2) |
Scheme 8.

The rate law for the mechanism in Scheme 8 is given by eq 1. Manipulation of eq 1 gives eq 2, which predicts a linear relationship between 1/kobs with 1/[PhCHO]. Such a relationship was observed (Figure 4). Keq and k2 were determined to be 9.3 ± 1.1 M−1 and 3.0 ± 0.1 × 10−3 s−1 from the slope and intercept of Figure 4.
Figure 4.

Plot of 1/kobs as a function of 1/[PhCHO] in toluene-d8 at −60 °C.
The kinetics of the reaction of 10 (< 0.010 M) with a large excess of PhCHO (> 10 equiv) in toluene-d8 was measured over a range of [PHCHO] concentrations between −68 and −54 °C to obtain the equilibrium constants Keq and rate constants k2 as a function of temperature. A van’t Hoff plot gave ΔH° = −8.5 ± 1.7 kcal mol−1 and ΔS° = −35.9 ± 8.2 eu for the hydrogen bonding formation; and an Eyring plot gave ΔH‡ = 13.3 ± 1.3 kcal mol−1 and ΔS‡ = −6.7 ± 6.2 eu for the hydrogen transfer step. The large negative entropy ΔS° is consistent with the hydrogen-bonding formation that brings 10 and PhCHO together prior to hydrogen transfer.
Stoichiometric Reduction of Acetophenone by 10
Reduction of acetophenone by 10 was much slower than reduction of benzaldehyde and took place at 5 °C with or without a trapping agent to give free 1-phenylethanol. In the presence of PPh3, complex 13 was the only observed ruthenium product; in the absence of any external ligand, a complicated mixture of ruthenium species was obtained. In both cases, no bridging hydride was detected.
Kinetics of Hydrogen Transfer from 10 to Acetophenone
Similar to benzaldehyde reduction, the rate of hydrogen transfer from 10 to acetophenone in the presence of PPh3 exhibited a first order in hydride, and kinetic saturation in acetophenone (Figure S6, supporting information). The chemical shift of the ruthenium hydride shifted during the course of reduction, consistent with a changing ratio of free 10 and 15 (a hydrogen bonded adduct of 8 and PhCOMe). The equilibrium constant Keq for hydrogen bond formation and the rate constant k2 for hydrogen transfer step were determined to be 5.7 ± 1.6 M−1 and 3.7 ± 0.5 × 10−3 s−1 respectively at 5 °C.
Using the measured ΔH° = −8.5 ± 1.7 kcal mol−1 and ΔS° = −35.9 ± 8.2 eu for the hydrogen bond formation between 10 and benzaldehyde, Keq was estimated to be 0.068 at 5 °C, which corresponds to only about 1.3% 14 in the presence of 0.2 M PhCHO. This corresponds to an 84 fold smaller Keq for benzaldehyde compared with acetophenone. If this is entirely an enthalphic effect, it is consistent with a 2.4 kcal mol−1 stronger hydrogen bond to the ketone compared with the aldehyde. Ketones (pKa of protonated PhCOMe is 5.2) are known to be significantly stronger bases than aldehydes (pKa of protonated PhCHO is 6.7).15
Comparison of Kinetics of Reduction by Iron and Ruthenium Hydrides
The observation of saturation kinetics for the reduction of acetophenone by ruthenium hydride 10 and the evidence for a hydrogen bonded adduct between 10 and acetophenone stands in strong contrast with the reduction of acetophenone by iron hydride 8, which showed simple first order dependence on both acetophenone and 8.10 In addition, no significant chemical shift change was observed for OD resonance in the 2H NMR spectrum after addition of excess PhCHO or acetophenone to solutions of 8-d2 in toluene if contrast to the substantial shifts for ruthenium hydride 10-d2.
This discrepancy could be due to differences between the metals (Fe or Ru) or differences between the Cp ligands (with or without ether linkage). To separate these two effects, an iron hydride 16 with the same ligand set as ruthenium hydride 10 was synthesized and the kinetics of hydrogen transfer from 16 to acetophenone were examined (Scheme 9).16 As with 10, the reduction of acetophenone by 16 showed a first order dependence on hydride 16, and saturation dependence on acetophenone (supporting material, Figure S7).
Scheme 9.

Since the strength of a hydrogen bond correlates with the acidity of the hydrogen donor, we determined the acidity of the OH proton of 16 using the IR method. The pKa of iron complex 16 was found to be 19.0, the same as that of analogous ruthenium complex 10, establishing that the metal had little influence on the acidity. In contrast, replacing the oxygen-containing five-membered ring in iron complex 16 with an all-carbon six-member ring decreased the OH proton acidity; the pKa value of OH proton in 8 was found to be 20.6. Apparently the electron withdrawing ether oxygen increases the acidity of the CpOH group of 16 relative to 8. In accordance with the pKa values, the hydride complexes 10 and 16 with the more acidic CpOH protons are prone to form hydrogen-bonded intermediates before transfer of hydrogens. In contrast, the less acidic CpOH proton of 8 does not lead to an observable hydrogen bonded intermediate and proton transfer and hydride transfer take place simultaneously.
Synthesis of the Ruthenium Analog 18 of Iron Complex 8
The ruthenium analog [2,5-(SiMe3)2-3,4-[(CH2)4](η5-C4COH)]Ru(CO)2H (18) related to iron complex 8 was synthesized for comparison (Scheme 10). For unknown reasons, ruthenium hydride 18 was always contaminated with small amounts (< 10 mol %) of a bridging hydride complex. The reaction of 18 with PhCHO in toluene-d8 at −60 °C generated a single alcohol complex 19 (there is only one oxygen available for intramolecular hydrogen bonding, but two oxygens are available in 12 and 12’).
Scheme 10.

Ketone and Aldehyde Hydrogenation Catalyzed by Ruthenium Hydrides 10 and 18
Having established that the stoichiometric reduction of carbonyls by 10 occurred at low temperature and that no inactive bridging hydride was generated during these reactions, we anticipated that 10 might serve as an aldehyde and ketone hydrogenation catalyst under mild conditions. Indeed, the hydrogenation of acetophenone (0.3 M in toluene) was catalyzed by 10 (2 mol %) at 25 °C under 35 atm of H2. The exponential disappearance of acetophenone (1690 cm−1) was monitored using a ReactIR apparatus; the only observed ruthenium species was 10 (2020 and 1960 cm−1).17 The half-life of the hydrogenation was determined to be 1.9 h. A similar hydrogenation was also carried out under much lower hydrogenation pressure (3 atm) using a Fisher-Porter apparatus, and reaction progress was periodically monitored by taking 1H NMR spectrum of the aliquots withdrawn from the reaction mixture. A half-life of 2.0 h was determined, suggesting that the rate of hydrogenation was independent on H2 pressure. The hydrogenation product 1-phenylethanol was isolated in 80 % yield after 20 h of hydrogenation (Table 1).
Table 1.
Hydrogenation of Carbonyls Catalyzed by Ruthenium Hydrides 10 and 18 and by Iron Hydride 8.a
| substrate | catalyst | Time (h) | conversion (%) | yield (%)b |
|---|---|---|---|---|
| PhCHO | 10 | 1 | 100 | 91 |
| PhCHO | 18 | 1 | 100 | 92 |
| PhCHO | 8 | 1 | 100 | 90 |
| PhCOCH3 | 10 | 20 | 99 | 80 |
| PhCOCH3 | 18 | 24 | 97 | 83 |
| PhCOCH3 | 8 | 20 | 99 | 83 |
Conditions: [substrate] = 0.30 M, [catalyst] = 0.0060 M, p(H2) = 3 atm, in toluene at 25 °C.
isolated yield.
The hydrogenation of benzaldehyde catalyzed by 10 was more rapid. With the same concentration of substrate and catalyst loading under 3 atm of H2 pressure, the reaction was complete within one hour. The hydrogenation product benzyl alcohol was isolated in 91 % yield.
Although a small amount of bridging hydride was always seen during the synthesis and reactions of hydride 18, the hydrogenation of carbonyls catalyzed by 18 was reasonably efficient. The catalytic activity of ruthenium complex 18 was comparable to that of iron complex 810 (Table 1).
Discussion
In an effort to develop a more active ruthenium hydrogenation catalyst related to the Shvo system, we synthesized the bis(trimethylsilyl) substituted Cp complex 10 in a successful effort to avoid formation of unreactive bridging hydrides analogous to 2 and 4. Since we have not seen detectable amounts of bridging hydrides related to either bis(trimethylsilyl)-substituted Cp iron complex 8 or ruthenium complex 10, we suggest that the sterically large TMS groups prevent formation of bridging hydrides. The reduction of carbonyls catalyzed by 10 is fast at low temperature; under H2 atmosphere, we suggest that a coordinatively unsaturated species is generated and rapidly trapped by hydrogen to regenerate the hydride catalyst (Scheme 11). With sufficient H2 present, the hydrogenation is independent on the H2 pressure and the turnover-limiting step is the hydrogen transfer step.
Scheme 11.

Experimental Evidence for a Hydrogen Bonded Intermediate Prior to Hydride Transfer from 10
The stoichiometric reduction of PhCHO by 10 displayed saturation kinetics that provided the first direct evidence for the hydrogen bonded adduct 14 prior to hydride transfer. Substantial shifts of NMR resonances of 10 upon addition of PhCHO also supported the formation of the hydrogen-bonded complex 14. The Keq values determined from the saturation kinetics analysis indicate that at −60 °C about 60% of the ruthenium species present at 0.2 M PhCHO is the hydrogen bonded adduct 14. This is the first system in which a hydrogen-bonded CpOH--O=CHR interaction has been directly seen. Why wasn’t it seen before for other Ru and Fe complexes? We suggest that this is related to the higher acidity of the CpOH group of 10 compared with iron complex 8 and other Ru complexes such as 7. The greater acidity of 10 results in a stronger hydrogen bond to the carbonyl group. The greater acidity also increases the reactivity of 10; the faster rate of reaction of 10 allows kinetic measurements at lower temperatures where the hydrogen-bonded adduct 14 is more thermodynamically stable (the large negative ΔS° = −36 eu diminishes Keq as the temperature is increased).
Overall, a wide range of subtle differences in kinetic behavior has been seen for the transfer of hydride and proton from species related to the Shvo catalyst. In the case of 10, there is rapid and reversible formation of substantial amounts of hydrogen-bonded intermediate 14 prior to hydride and proton transfer. In the case of 2, computations suggest the reversible formation of substantial amounts of hydrogen-bonded intermediate, but not enough of it is formed to obtain direct experimental evidence for its presence.18 In the case of reduction of p-MeO-C6H4CMe=N-Ph by 1, the absence of a kOH/kOD isotope effect was interpreted as resulting from proton transfer to nitrogen occurring prior to rate limiting hydride transfer.19 In the case of reduction of TolCH=N-CHMe2 by 3, both substrate isomerization and inverse isotope effects provided information that both proton and hydride transfer were rapid and reversible and amine coordination to Ru was rate limiting.20
Comparison of Stoichiometric Reduction Rates of 10 and other Hydrides
The rate constants and activation parameters determined previously5,6,10 allow comparison of the reactivity of hydride 10 and related hydrides. For benzaldehyde at −60 °C, hydride 10 is about 20–40 times more reactive than the tolyl analog of Shvo’s hydride 3 and is significantly more reactive than PPh3-subsituted hydride 7 (Scheme 12). Interestingly, for acetophenone reduction at 5 °C, hydride 10 is 4–8 times less reactive than 3; however, it is comparable to 8 and slightly more reactive than the iron analog 16 (Scheme 13). There is almost no reaction between 7 and acetophenone at 5 °C. The close similarity of the rates for analogous Fe and Ru complexes 8 and 10 is very significant. It suggests that the more economical iron catalysts are attractive alternatives to ruthenium catalysts.
Scheme 12.

Relative activity for stoichiometric PhCHO reduction in toluene-d8 at −60 °C.
Scheme 13.

Relative activity for stoichiometric PhCOCH3 reduction in toluene-d8 at 5 °C.
Comparison of Catalytic Reactivity of 10 and Other Hydrides
The activity of catalysts related to the Shvo system depend not only on their stoichiometric reduction rates, but also on whether the dominant species is an active monoruthenium hydride or an unreactive bridging hydride. For example, PPh3-subsituted hydride 7 catalyzes the hydrogenation of PhCHO at room temperature under 2.5 atm of H2 pressure, while hydride 3 is completely unreactive under similar conditions. While 3 is a faster stoichiometric hydrogen donor than 7, it is a slow catalyst because it is rapidly converted to the inactive diruthenium hydride 4. The ruthenium hydrides 10 and 18 reported here are highly active catalysts both because of rapid stoichiometric rates and because they are present as active monoruthenium species. The hydrogenation of PhCHO catalyzed by 10 or 18 is complete within 1 h at 25 °C under 3 atm of H2 pressure as opposed to 20 h for 7 (Scheme 14). More remarkably, the hydrogenation of acetophenone catalyzed by 10 or 18 takes place at room temperature and low H2 pressure (3 atm); previously developed Shvo type ruthenium catalysts are ineffective under these mild conditions.
Scheme 14.

Relative catalytic activity for the hydrogenation of PhCHO in toluene at 25 °C under 3 atm of H2 pressure.
Experimental Section
General Procedures
All air-sensitive compounds were prepared and handled under a nitrogen atmosphere using standard Schlenk and inert-atmosphere glove box techniques. Toluene was deoxygenated and dried in a solvent purification system by passing through an activated alumina column and an oxygen-scavenging column under nitrogen.21 Toluene-d8 and C6D6 were distilled from Na and benzophenone under a nitrogen atmosphere. CH3CN and Et3N were dried over CaH2 and distilled under a nitrogen atmosphere. Methanol, glyme, and benzene were degassed by bubbling N2 through the solvents for 15 min before use. {2,5-(SiMe3)2-3,4-[(CH2)4](η5-C4COH)}Fe(CO)2H (8),9 [2,5-(SiMe3)2-3,4-(CH2OCH2)(η4-C4CO)]Ru(CO)3 (9),11 2,5-(SiMe3)2-3,4-[(CH2)4](C4CO), 22Me3SiC≡C(CH2)4C≡CSiMe3,23 and [2,5-(SiMe3)2-3,4-(CH2OCH2)(η4-C4CO)]Fe(CO)324 were prepared as described in the literature.
[2,5-(SiMe3)2-3,4-(CH2OCH2)(η5-C4COH)]Ru(CO)2H (10)
Under a nitrogen atmosphere, a degassed solution of NaOH (0.80 g, 20 mmol) in 25 mL of H2O was added to a solution of 9 (1.13 g, 2.5 mmol) in 50 mL THF. The resulting biphasic mixture was vigorously stirred at room temperature for 2.5 h before 85 wt. % of H3PO4 in H2O (about 0.8 mL) was added to neutralize the reaction mixture. The organic layer was transferred via cannula into a Schlenk flask under nitrogen and the aqueous layer was extracted with Et2O several times. The combined organic layers were concentrated under vacuum, dissolved in degassed benzene, dried over Na2SO4, and filtered into another Schlenk flask. The resulting solution was pumped to dryness to afford 10 as a yellow powder (0.78 g, 73% yield). X-ray quality crystals of 10 were grown via slow diffusion of hexanes into a saturated solution of 10 in CH2Cl2 at −30 °C. 1H NMR (toluene-d8, 300 MHz) δ −10.12 (s, RuH, 1H), 0.17 (s, Si(CH3)3, 18H), 3.58 (br s, OH, 1H), 4.28–4.58 (m, CH2, 4H). 13C{1H} NMR (toluene-d8, 126 MHz) δ 0.35 (Si(CH3)3), 68.31 (CH2), 70.99, 111.07, 151.31 (COH), 201.43 (Ru(CO)2). IR (toluene, cm−1) 2018, 1959. HRMS (ESI) calcd (found) for [C15H24O4Si2Ru+Na]+• 449.0154 (449.0174).
pKa Determination of CpOH Proton of 10
Triethylamine (2.8 µL, 20 µmol) was mixed with a solution of 10 (8.5 mg, 20 µmol) in CH3CN (500 µL). An aliquot of the solution was syringed into a 0.1 mm CaF2 IR cell. Two resonances at 2014 and 1952 cm−1 were assigned to 10 and two at 1982 and 1915 cm−1 were assigned to NEt3H+[2,5-(SiMe3)2-3,4-(CH2OCH2)(η5-C4CO)]Ru(CO)2H}− (11). The absolute concentrations of the two species were determined by obtaining the molar absorptivities of 10 and 11. For 10: 2014 (1640 M−1 cm−1) and 1952 cm−1 (1800 M−1 cm−1). For 11: 1982 (1480 M−1 cm−1) and 1915 cm−1 (1670 M−1 cm−1).25 From the molar absorptivities, the ratio of 10 : 11 was determined to be 1.8 : 1. By using the known pKa of NEt3 (pKa = 18.5) and the observed equilibrium constant (Keq = 0.30), a pKa of 19.0 was determined for the CpOH proton of 10.
Stoichiometric Reduction of Benzaldehyde by 10
A Teflon capped NMR tube containing a solution of 10 (12.6 mg, 30 µmol) in toluene-d8 (400 µL) was chilled in a dry ice/acetone bath, and a solution of PhCHO (3.6 µL, 36 µmol) in toluene-d8 (100 µL) was added via a gas tight syringe through the Teflon cap. The two solutions were carefully mixed while cold, and the NMR tube was immediately placed into an NMR probe at −60 °C (calibrated). The progress of reduction was monitored by 1H NMR spectroscopy. After 30 min, almost all 10 had disappeared and two sets of peaks corresponding to ruthenium alcohol complexes 12 and 12’ grew in. The major complex slowly isomerized to the minor complex, and finally reached an equilibrium ratio of 1 : 1. Major alcohol complex 12: 1H NMR (toluene-d8, 360 MHz) δ 0.33 (s, Si(CH3)3, 18H), 3.82 and 4.14 (AB, JAB = 10.8 Hz, CH2, 4H), 4.03 (s, PhCH2OH, 2H), 6.67–7.43 (m, Ar, 5H), and OH resonance was not located. Minor alcohol complex 12’: 1H NMR (toluene-d8, 360 MHz) δ 0.25 (s, Si(CH3)3, 18H), 3.97 and 4.37 (AB, JAB = 10.8 Hz, CH2, 4H), 4.41 (s, PhCH2OH, 2H), 6.67–7.43 (m, Ar, 5H), and OH resonance was not located.
NMR Measurements of Rate Constants for the Reduction of Benzaldehyde by 10
Under a nitrogen atmosphere a solution of PhCHO (1.0 M) in toluene-d8 was mixed with a solution of 10 and mesitylene (internal standard) in a resealable NMR tube at −78 °C. The first 1H NMR spectrum was recorded within 5 min of mixing, and the reaction was monitored for three to five half-lives. Zero filling was used to ensure adequate digital resolution. The height of the Si(CH3)3 resonance of 10 (which steadily shifted from δ 0.34 to δ 0.38) was compared to that of mesitylene (δ 2.15). The NMR probe temperature was calibrated using a 100 % methanol standard.26 The results are summarized in Tables S1–S5 and S1–S5 in the supporting information.
Supplementary Material
Acknowledgement
Financial support from the Department of Energy, Office of Basic Energy Sciences is gratefully acknowledged. Grants from NSF (CHE-9208463 and CHE-9709065) and NIH (1 S10 RR0 8389-01) for the purchase of NMR spectrometers are acknowledged. We thank Dr. Ilia Guzei for assistance with X-ray crystallography and Dr. Charles Fry for assistance with NMR spectroscopy.
Footnotes
Supporting Information Available: Preparation of 10-d2, 16, 17, and 18; kinetics of reduction of PhCHO by 10; kinetics of reduction of PhCOMe by ruthenium and iron hydrides; catalytic hydrogenation of carbonyls by 10 and 18; and X-Ray Crystal Structure of 10. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Noyori introduced this nomenclature for catalysts that transfer a hydride from a metal center and a proton from a ligand. Yamakawa M, Ito H, Noyori R. J. Am. Chem. Soc. 2000;122:1466.
- 2.Fehring V, Selke R. Angew. Chem. Int. Ed. 1998;37:1827. [Google Scholar]
- 3.(a) Shvo Y, Czarkie D, Rahamim Y, Chodosh DF. J. Am. Chem. Soc. 1986;108:7400. [Google Scholar]; (b) Menashe N, Shvo Y. Organometallics. 1991;10:3885. [Google Scholar]; (c) Menashe N, Salant E, Shvo Y. J. Organomet. Chem. 1996;514:97. [Google Scholar]
- 4.For recent reviews of Shvo’s Catalyst: Warner MC, Casey CP, Bäckvall J-E. Top. Organomet. Chem. 2011;37:85. Conley BL, Pennington-Boggio MK, Boz E, Williams TJ. Chem. Rev. 2010;110:2294. doi: 10.1021/cr9003133.
- 5.(a) Casey CP, Singer SW, Powell DR, Hayashi RK, Kavana M. J. Am. Chem. Soc. 2001;123:1090. doi: 10.1021/ja002177z. [DOI] [PubMed] [Google Scholar]; (b) Casey CP, Johnson JB, Singer SW, Cui Q. J. Am. Chem. Soc. 2005;127:3100. doi: 10.1021/ja043460r. [DOI] [PubMed] [Google Scholar]; (c) Casey CP, Johnson JB. Can. J. Chem. 2005;83:1339. [Google Scholar]
- 6.Casey CP, Beetner SE, Johnson JB. J. Am. Chem. Soc. 2008;130:2285. doi: 10.1021/ja077525c. [DOI] [PubMed] [Google Scholar]
- 7.Casey CP, Vos TE, Singer SW, Guzei IA. Organometallics. 2002;21:5038. [Google Scholar]
- 8.(a) Casey CP, Strotman NA, Beetner SE, Johnson JB, Priebe DC, Vos TE, Khodavandi B, Guzei IA. Organometallics. 2006;25:1230. [Google Scholar]; (b) Casey CP, Strotman NA, Beetner SE, Johnson JB, Priebe DC, Guzei IA. Organometallics. 2006;25:1236. [Google Scholar]
- 9.Knölker HJ, Baum E, Goesmann H, Klauss R. Angew. Chem. Int. Ed. 1999;38:2064. doi: 10.1002/(SICI)1521-3773(19990712)38:13/14<2064::AID-ANIE2064>3.0.CO;2-W. [DOI] [PubMed] [Google Scholar]
- 10.Casey CP, Guan H. J. Am. Chem. Soc. 2007;129:5816. doi: 10.1021/ja071159f. [DOI] [PubMed] [Google Scholar]
- 11.Yamamoto Y, Miyabe Y, Itoh K. Eur. J. Inorg. Chem. 2004:3651. [Google Scholar]
- 12.This chemical shift change amounts to 48 Hz using a 300 MHz NMR spectrometer.
- 13.(a) Jordan RF, Norton JR. J. Am. Chem. Soc. 1982;104:1255. [Google Scholar]; (b) Moore EJ, Sullivan JM, Norton JR. J. Am. Chem. Soc. 1986;108:2257. doi: 10.1021/ja00269a022. [DOI] [PubMed] [Google Scholar]
- 14.Casey CP, Guan H. J. Am. Chem. Soc. 2009;131:2499. doi: 10.1021/ja808683z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Arnett EM, Quirk RP, Larsen JW. J. Am. Chem. Soc. 1970;92:3977. [Google Scholar]
- 16.The reduction of PhCHO by iron hydrides 8 and 16 were fast even at −72 °C. The rates were not measured with accuracy.
- 17.Based on Figure S4, Keq for acetophenone at 25 °C is expected to be small, and the reduction could be first order dependent on both hydride 10 and the ketone.
- 18.(a) Casey CP, Bikzhanova GA, Cui Q, Guzei IA. J. Am. Chem. Soc. 2005;127:14062. doi: 10.1021/ja053956o. [DOI] [PubMed] [Google Scholar]; (b) Comas-Vives A, Ujaque G, Lledós A. Organometallics. 2007;26:4135. [Google Scholar]
- 19.(a) Samec JSM, Éll AH, Åberg JB, Privalov T, Eriksson L, Bäckvall J-E. J. Am. Chem. Soc. 2006;128:14293. doi: 10.1021/ja061494o. [DOI] [PubMed] [Google Scholar]; (b) Éll AH, Johnson JB, Bäckvall JE. Chem. Commun. 2003:1652. [Google Scholar]
- 20.Casey CP, Johnson JB. J. Am. Chem. Soc. 2005;127:1883. doi: 10.1021/ja044450t. [DOI] [PubMed] [Google Scholar]
- 21.Pangborn AB, Giardello MA, Grubbs RH, Rosen RK, Timmers FJ. Organometallics. 1996;15:1518. [Google Scholar]
- 22.Takahashi T, Tsai FY, Li Y, Nakajima K. Organometallics. 2001;20:4122. [Google Scholar]
- 23.Bushnell LPM, Evitt ER, Bergman RG. J. Organomet. Chem. 1978;157:445. [Google Scholar]
- 24.(a) Knölker HJ, Heber J, Mahler CH. Synlett. 1992:1002. [Google Scholar]; (b) Knölker HJ, Heber J. Synlett. 1993:924. [Google Scholar]
- 25.The molar absorptivities of the 1,1,3,3-tetramethylguanidinium salt (1976 and 1908 cm−1) were used to determine the concentration of 11.
- 26.(a) van Geet AL. Anal. Chem. 1970;42:679. [Google Scholar]; (b) Raidford DS, Fisk CL, Becker ED. Anal. Chem. 1979;51:2050. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.