

Abstract
Here we report the examination of two convenient strategies, the use of a D-amino acid residue or a glycoside segment, for increasing the proteolytic resistance of supramolecular hydrogelators based on small peptides. Our results show that the introduction of D-amino acid or glycoside to the peptides significantly increases the resistance of the hydrogelators against proteinase K, a powerful endopeptidase. The insertion of D-amino acid in the peptide backbone, however, results relatively low storage moduli of the hydrogels, likely due to the disruption of the superstructures of the molecular assembly. In contrast, the introduction of a glycoside to the C-terminal of peptide enhances the biostability of the hydrogelators without the significant decrease of the storage moduli of the hydrogels. This work suggests that the inclusion of a simple glycogen in hydrogelators is a useful approach to increase their biostability, and the gained understanding from the work may ultimately lead to development of hydrogels of functional peptides for biomedical applications that require long-term biostability.
Introduction
Supramolecular gels are the gels formed by the self-assembly of small molecules via noncovalent molecular interactions in a solvent.1 Made from short L-amino acid sequences to possess inherent and excellent biofunctionality, biocompatibility and biodegradability, small peptide-based supramolecular hydrogels2,3 have received considerable attention and made rapid progress in the past ten years for the development of biomaterials2,4 that serve as scaffolds for tissue engineering,5 matrices for biomineralization,6 dressings for wound healing,7 media for protein chips8 and drug delivery,9 platforms for screening enzyme inhibitors,10 and components for enzyme mimetics.11 Being used in vivo, these small peptide-based hydrogelators may degrade faster than desired because an array of endogenous proteolytic enzymes catalyze the hydrolysis of peptide bonds. Obviously, such an inherent susceptibility towards enzymatic hydrolysis shortens the in vivo lifetime of these small peptide-based hydrogels, thus reducing their efficacy and limiting their scope of applications when long-term bioavailability is required.12
Because of the advantages and limitations of peptides described above, active efforts have focused on designing and synthesizing non-peptide molecules that mimic the structures and functions of peptides or proteins to achieve prolonged or controlled stability and bioavailability in vivo.13 Among the reported works on peptidomimics,14 a large pool of unnatural amino acids, such as D-amino acids15 and β-amino acids,16 have received the most intensive attention for achieving long-term biostability, including the use of D-amino acids and β-amino acids for making hydrogelators.17,18 Though it is possible, it remains a challenge to achieve the functions of native peptides using peptide analogues entirely made of D-amino acids or β-amino acids because the changes of the stereochemistry of the peptide motif, unavoidably, leads to the loss of functions. In nature, biological systems, however, widely use glycosylation as a strategy to enhance the stability of proteins without comprising the functions of the proteins, which implies that the incorporation of glycoside to the C-terminal of amino acid/peptide should be an advantageous approach for developing biostable and functional hydrogels for long-term applications.19,20 Our preliminary works have shown that the supramolecular hydrogelators made from D-amino acid derivatives resist the enzymatic hydrolysis catalyzed by proteinase K and exhibit exceptional biostability comparing to their L-amino acid counterparts.18 This approach provides a hydrogel that can serve as a medium for carrying bioactive molecules or therapeutics agents.18 Encouraged by the works used glycosides to form gels,7,20,21 we found the incorporation of a simple glycoside (e.g., glucosamine) to the C-terminal of short L-peptides confers substantial resistance to proteolytic degradation relative to their unmodified peptidic counterparts.20 These results, clearly, demonstrate that the incorporation of a D-amino acid or a glycoside to the peptide backbone is a useful strategy for enhancing the resistance of the peptide derivatives towards proteolytic digestions. Each of the approaches, however, still has its own limitations. For example, the excessive utilization of D-amino acids or β-amino acids often results in immunogenic response and limits their applications in vivo.22 The employment of glycosylation, though being effective, still poses considerable challenge in synthesis if there is a need of more elaborated glycosides. Thus, it is necessary to compare these two approaches systematically for the development of the hydrogelators that have both the desired functions and biostability.
In this work, we compared the incorporation of D-amino acids and the incorporation of β-amino acids in the same small peptide hydrogelators and evaluated their effects on biostability of the hydrogelators and the rheological behaviors of the corresponding hydrogels. Our results show that a D-amino acid or a glycoside in the peptides significantly increases the resistance of the hydrogelators against proteinase K, a powerful protease, but the D-amino acid in the peptide backbone changes the configuration of the peptides, apparently disrupts the superstructures of the supramolecular self-assemblies, and reduces the elasticity of the hydrogels. On the other hand, the incorporation of glucosamine or chondrosine to the C-terminal of peptide enhances the biostability of hydrogelators without significant decrease of the elasticity of the hydrogels. This work suggests that the incorporation of a simple glycoside to the C-terminal of peptides is a viable way to tailor the stability of hydrogels in complex biological environment and provides useful insights for ultimately expanding the ranges of applications of the hydrogels as biomaterials.
Results and Discussion
Scheme 1 shows the molecular structures and modular representations of the hydrogelators based on two known supramolecular hydrogelators (118 and 523). Compound 1 enables many other bioactive molecules to self-assemble in water when it covalently connects to those molecules,24 and 5 represents a new class of hydrogelators that exhibit exceptional biocompatibility.23 The replacement of the L-phenylalanine residue by a D-phenylalanine at the N-terminal or the C-terminal of 1 gives the hydrogelators 2 or 3, respectively. The addition of a disaccharide (e.g., chondrosine) to the C-terminal of 1 affords hydrogelator 4. The addition of D-phenylalanine or glucosamine to the C-terminal of 5 results in hydrogelators 6 or 7, respectively. We used the previous reported procedures7,18,25 to synthesize the new hydrogelators (2, 3, 4, 6, and 7) shown in Scheme 1. Briefly, the synthesis starts with N-hydroxysuccinimide (NHS) assisted coupling of 2-(naphthalen-2-yl) acetic acid (Nap) with L-phenylalanine in two consecutive steps that afford 1. The use of D-phenylalanine in the first coupling step or the second coupling step generates 2 or 3, respectively. After being activated by NHS, 1 couples with chondrosine to form hydrogelator 4. Based on the nucleopeptide (5), we combined solid phase synthesis and simple coupling reactions in liquid phase to attach D-phenylalanine or D-glucosamine to the C-terminal of 5 to obtain hydrogelators 6 or 7, respectively, in a fair total yield (Figure S1).
Scheme 1.
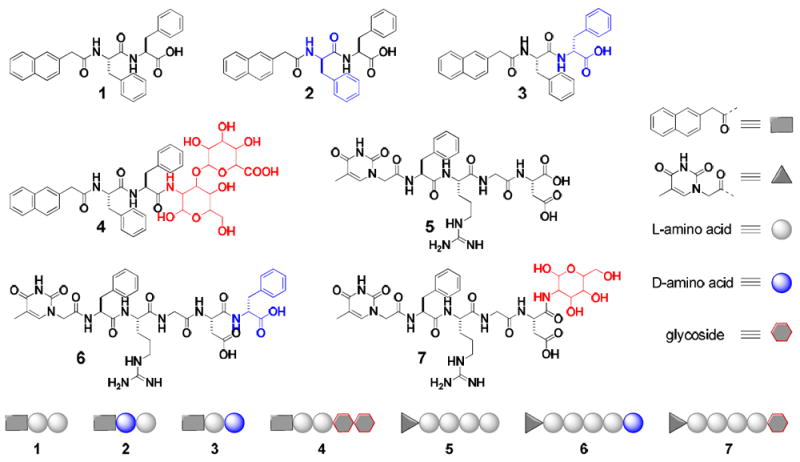
The molecular structures and the modular representations of the hydrogelators.
After the synthesis of the hydrogelators, we examined their ability to form hydrogels. All the analogues of 1 or 5 synthesized in this work behave as hydrogelators to self-assemble in water at proper pHs (Table S1) and concentrations to form the hydrogels. Figure 1 shows the optical images of the hydrogels made of the analogues. As summarized in Table S1, similar to hydrogelator 1, hydrogelators 2 and 3 have good solubility in water at a slightly basic condition and turn into stable hydrogels upon the change of their pH to 7.0. The minimum gelation concentrations of 2 and 3, are 1.0 and 1.5 wt%, respectively, which are higher than that of hydrogelator 1 (0.6 wt%).24 This result suggests that the insertion of D-amino acid in the peptide backbone changes the configuration of the peptides and apparently reduces the intermolecular interactions of the hydrogelators for supramolecular self-assembly. Since the attachment of D-glucosamine to the C-terminal of 1 gives a compound that has the solubility too low to behave as a supramolecular hydrogelator,7 we, in this study, attached chondrosine to 1, to improve the balance between hydrophilicity and hydrophobicity for hydrogelation. Indeed, hydrogelator 4 shows improved solubility in water and is able to form a stable hydrogel at pH 4.0 and the concentration of 2.0 wt%. Containing multiple ionic groups, molecules 5, 6, and 7 all exhibit good solubility in water at pH 7.0 and turn into stable hydrogels at the concentrations of 3.0 wt% upon reducing the solution pH to 4.0.
Figure 1.
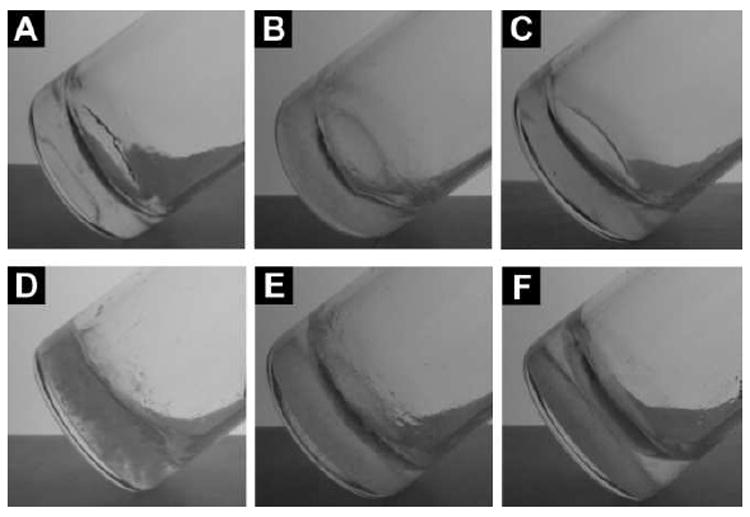
Optical images of the hydrogels of (A) 2 (1.0 wt%, pH 7.0); (B) 3 (1.5 wt%, pH 7.0); (C) 4 (1.5 wt%, pH 7.0); (D) 5 (3.0 wt%, pH 4.0); (E) 6 (3.0 wt%, pH 4.0); and (F) 7 (3.0 wt%, pH 4.0).
Negative staining transmission electronic microscopy (TEM)26 helps reveal the characteristic morphologies of the nanostructures formed by the self-assemblies of the hydrogelators in the hydrogels of 2, 3, 4, 5, 6, or 7. Since the staining occurs after the absorption of the nanofibers on the TEM grid, it is unlikely the staining agent would induce the observed structures. As shown in Figure 2, hydrogelators of 2, 3, and 4 self-assemble into high-aspect-ratio nanofibers with several micrometers in length and 8 nm, 11 nm, and 15 nm in width, respectively, showing resemblance to the morphology of the nanofibers of 1 (8.5±2.5 nm)24 in the corresponding hydrogels. While the change of the C-terminal residue of 1 to D-amino acid leads to little increase of the width of the molecular nanofibers, the attachment of disaccharide at the C-terminal of 1 almost doubles the width of the molecular nanofibers. These results suggest that the use of different motifs at the C-terminal of 1 can be a useful approach for tailoring the nanostructures of the self-assembly of the analogues of 1. TEM images of the hydrogels of 5, 6, and 7 share a common feature of nanoribbons or bundles of nanofibers, such as nanoribbons with 35 nm in width in hydrogel 5, bundles of nanofibers (~26 nm) in hydrogel 6, and untwisted nanoribbons (~33 nm) in hydrogel 7 (Figure 2 and Table S1). The formation of the nanoribbons or bundles indicates the strong interfiber interactions, likely originating from the presence of ionic complementary peptide (i.e., arginine and aspartic acid residues in the RGD segment) on hydrogelators 5, 6 and 7. In addition to nanoribbons or bundles of nanofibers, each of the hydrogels has its own characteristic features. For example, hydrogel 5 mainly contains nanoribbons; hydrogels 6 and 7 have nanofibers of 8 nm and 15 nm in widths, respectively. The TEM of hydrogels 5 and 6 also show large aggregates, which agrees with the opaqueness of the hydrogels 5 and 6. The polymorphism of the nanostructures in hydrogels 5, 6, and 7, reflects the diverse intermolecular interactions and results in diverse nanostructures, which may provide a possible structural foundation for multiple functions from the hydrogelators consisting different classes of building blocks.
Figure 2.

Transmission electron micrograph (TEM) of the negative stained hydrogels formed by (A) 2; (B) 3; (C) 4; (D) 5; (E) 6; and (F) 7 shown in Figure 1 (scale bar = 100 nm).
The biostability test of the hydrogelators shows that hydrogelators 2, 3, 4, 6, and 7 exhibit increased resistance towards the proteolytic digestion catalysed by proteinase K,27 a powerful endopeptidase that degrades a wide range of peptidic substrates and dictates various regulated cascades in biological processes. The concentration of each hydrogelator in its solution is at 0.02 wt%, a concentration that is significantly lower than the critical gelation concentration of the hydrogelators, so that there is little resistance contributed from the hydrogelation. As shown in Figure 3, hydrogelator 2, in which the D-amino acid is at C-terminal, exhibits the highest biostability to resist the enzymatic digestion; more than 92 % of 2 remain after 24 hrs of incubation with proteinase K. When the D-amino acid is at the middle of the hydrogelator (3), only 42% of 3 remain after the incubation with proteinase K for 24 hrs. The presence of the chondrosine at the C-terminal of the hydrogelator 4 results in a considerable increase of the resistance of 4 towards proteinase K (e.g., 57% of 4 remain intact after 24 hrs of incubation with proteinase K). Comparing to the fact of complete degradation of hydrogelator 1 within 4 hrs of proteinase K treatment, the use of D-amino acid at the C-terminal of dipeptide-based hydrogelator could be an exceptional effective approach to increase the resistance towards proteolysis. Unlike the case of 2, only 15% of 6 remain after 24 hrs of incubation with proteinase K, suggesting that the D-amino acid at the C-terminal of 5 confers the limited proteolytic resistance to the hydrogelator. This result is hardly surprising because proteinase K, as an endopeptidase, will likely have high probability to catalyze the bond cleavages of longer peptides. Interestingly, almost half of hydrogelator 7 remain after 24 hrs of incubation with proteinase K, which clearly proves that the attachment of glycoside at the C-terminal of peptides is still an effective approach to increase the biostability of the hydrogelators. Because hydrogelators 1, 2, and 3 have similar hydrophobicity and morphology of their nanofibers, the difference of their proteolytic resistance indicates that the hydrophobicity of the hydrogelators and the morphology of the nanostructures, though may affect the rate of proteolysis, unlikely are the critical factor for proteolytic resistance in the case of supramolecular hydrogels in which self-assembly and disassembly are in equilibrium. In addition, since the biostability of the hydrogelators at the physiological pH determines the biostability (not the physical stability) of the supramolecular hydrogels in vivo, it is more relevant to evaluate the stability of the hydrogelators at the physiological pH than at the pH of the hydrogelation.
Figure 3.
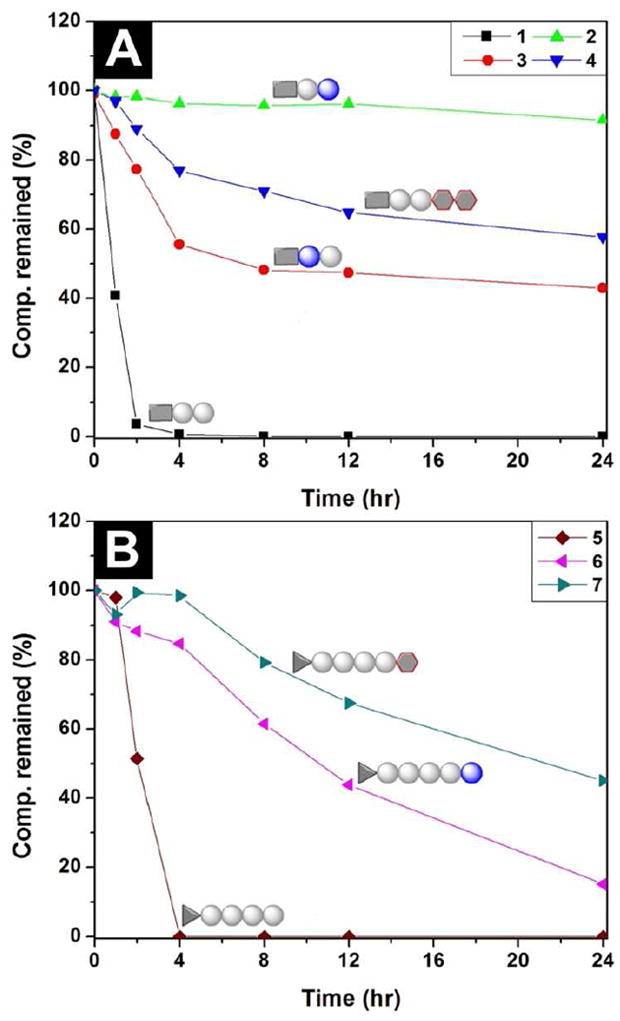
The digestions of hydrogelators (A) 1, 2, 3 and 4; and (B) 5, 6 and 7 over the course of the incubation with proteinase K for 24 hours. All the hydrogelators are at the concentrations of 0.2 wt%.
The mechanical properties of peptide-based hydrogels are essential because the mechanical strength or viscoelastic behaviors of the hydrogels directly affect their applications (e.g., modulating the differentiation of stem cells28). Thus we used oscillatory rheology to assess the elasticity of the hydrogels made of the hydrogelators. As shown in Figure 4 and Table S1, the hydrogel formed by 2 or 3 shows liquid-like rheological behavior with the magnitude of the storage moduli (G’) close to the value of the loss moduli (G”) within the oscillating strain limit investigated during strain sweep, which suggests the weak mechanical properties of hydrogels of 2 and 3. The absence of a stable value for hydrogel 2 or 3 during frequency sweep (Figure 4C) is consistent with the features of a weak hydrogel. Though the rheological measurement indicates the hydrogels of 2 and 3 are weak gels when the concentration of the hydrogelators at 1.0 wt%, it is feasible to enhance the strength of the gels by the increase of the amount of hydrogelators 2 and 3. Containing the D-amino acid in its peptide backbone, hydrogelator 6 also exhibits a considerable lower storage modulus (G’=0.4 kPa) than that of its parent hydrogel (hydrogel of 5; G’ = 40 kPa). The frequency sweep (Figure 4D) indicates that the storage modulus of the hydrogel of 6 is independent to the frequency, suggesting that the hydrogel of 6 is solid-like. These results, collectively, suggest that the incorporation of D-amino acid to the peptidic backbones likely causes the distortion of their molecular packing, thus weakening the intermolecular interactions that are critical for the physical crosslinking of the nanofibers of the hydrogelators. On the other hand, the attachment of the glycoside at the C-terminal of the peptides 1 or 5, results in little drop of the storage moduli of the hydrogels 4 and 7. For example, hydrogel 7 exhibits essentially same storage modulus as that of hydrogel 5. Although it is difficult to link the rheology to the sequence, the only difference between 1 and 4 (or 5 and 7) is the addition of glycoside. Therefore, it is reasonable to evaluate the effect of the addition of the glycoside to the rheological properties since the peptide sequences remain the same between 1 and 4 (or 5 and 7). As shown in Figure 4C and 4D, the G’ and G” values measured in the frequency sweep experiment are independent to the oscillatory frequency of the sweep, indicating the elastic feature of the hydrogels 4 and 7. In addition, the storage modulus (G’) of hydrogel 4 is higher than its loss moduli (G”), suggesting a solid-like rheological behavior. These results indicate that the attachment of glycoside at the C-terminal of peptides will be an effective approach to construct supramolecular hydrogel with little compromise of the elasticity of the resulting hydrogels.
Figure 4.
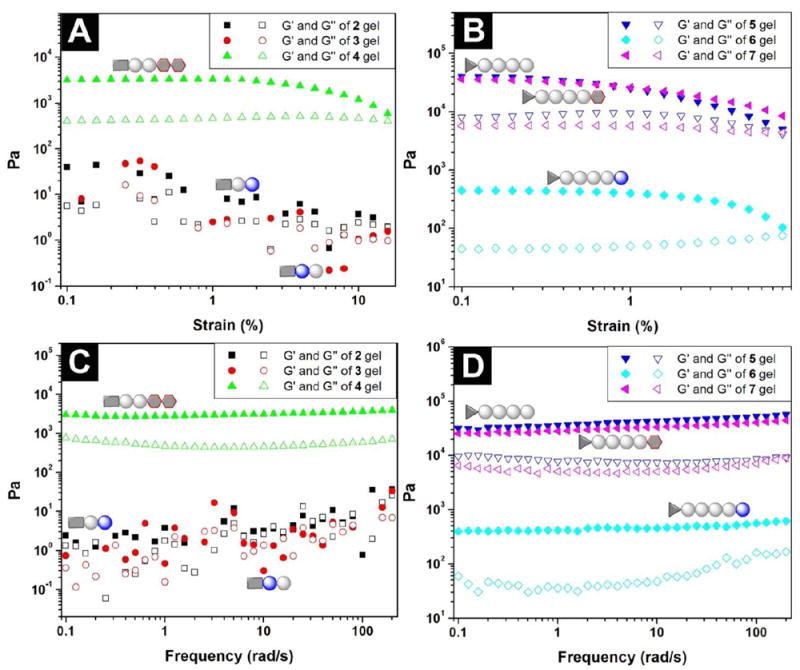
(A) Strain dependence of the dynamic storage moduli (G’) and the loss moduli (G”) of the hydrogels of 2, 3 and 4 shown in Figure 1; (B) strain dependence of the dynamic storage moduli (G’) and the loss moduli (G”) of the hydrogels of 5, 6 and 7; (C) frequency dependence of the dynamic storage moduli (G’) and the loss moduli (G”) of the hydrogels of 2, 3 and 4 shown in Figure 1; (D) frequency dependence of the dynamic storage moduli (G’) and the loss moduli (G”) of the hydrogels of 5, 6 and 7 shown in Figure 1.
Conclusions
By integration of a D-amino acid residue or a simple glycoside segment into the backbones of small peptides, we developed two new types of biostable supramolecular hydrogels. Our results suggest that the attachment of the glycoside to the C-terminal of peptides that have more than two residues appears to be a more viable approach to enhance the biostability and preserve the mechanical strength of the hydrogels. Unlike the use of oligomeric or polymeric ethylene glycol (PEG) for increasing the biostability of molecules, the much smaller size of a simple glycoside (comparing to PEG) causes little decrease of effective collisions in the binding process. The results suggest that the use of a single glycoside for extending the biostability of biofunctional hydrogels is a more attractive approach than that of the use of PEG. Moreover, since there are various bioactive peptides or molecular recognition motifs in nature, it is highly desirable to incorporate them in the hydrogelators to form hydrogels for long-term applications. In fact, the RGD (Arg-Gly-Asp) motif in hydrogelator 5, 6, and 7 is a well-established ligand to bind with cells through αvβ3 and αvβ5 integrin receptors,29 and the incorporation of glycoside to the C-terminal not only improves the biostability of the hydrogels, but also offers a new way to use hydrogelators like 7 to mimic the functions of glycoproteins, which is an area under our active investigation.20,25
Supplementary Material
Acknowledgments
This work was partially supported by a NIH grant (R01CA142746), a HFSP grant (RGP0056/2008), and start-up fund from Brandeis University. We thank the assistance of Brandeis EM facility.
Footnotes
Supporting Information. Synthesis of hydrogelators 1, 2, 3, 4, 5, 6, and 7, TEM, rheological measurements, and biostability tests. This material is available free of charge via the Internet at http://pubs.acs.org.”
References
- 1.Terech P, Weiss RG. Chem Rev. 1997;97:3133–3159. doi: 10.1021/cr9700282. [DOI] [PubMed] [Google Scholar]; Estroff LA, Hamilton AD. Chem Rev. 2004;104:1201–1217. doi: 10.1021/cr0302049. [DOI] [PubMed] [Google Scholar]; Hirst AR, Coates IA, Boucheteau TR, Miravet JF, Escuder B, Castelletto V, Hamley IW, Smith DK. J Am Chem Soc. 2008;130:9113–9121. doi: 10.1021/ja801804c. [DOI] [PubMed] [Google Scholar]; Sahoo P, Adarsh NN, Chacko GE, Raghavan SR, Puranik VG, Dastidar P. Langmuir. 2009;25:8742–8750. doi: 10.1021/la9001362. [DOI] [PubMed] [Google Scholar]; Suzuki M, Hanabusa K. Chem Soc Rev. 2009;38:967–975. doi: 10.1039/b816192e. [DOI] [PubMed] [Google Scholar]; Chen L, Morris K, Laybourn A, Elias D, Hicks MR, Rodger A, Serpell L, Adams DJ. Langmuir. 2010;26:5232–5242. doi: 10.1021/la903694a. [DOI] [PubMed] [Google Scholar]; Yan C, Pochan DJ. Chem Soc Rev. 2010;39:3528–3540. doi: 10.1039/b919449p. [DOI] [PMC free article] [PubMed] [Google Scholar]; Dawn A, Shiraki T, Haraguchi S, Tamaru S, Shinkai S. Chem Asian J. 2011;6:266–282. doi: 10.1002/asia.201000217. [DOI] [PubMed] [Google Scholar]; Steed JW. Chem Commun. 2011;47:1379–1383. doi: 10.1039/c0cc03293j. [DOI] [PubMed] [Google Scholar]; Das D, Kar T, Das PK. Soft Matter. 2012;8:2348–2365. [Google Scholar]; Yan C, Mackay ME, Czymmek K, Nagarkar RP, Schneider JP, Pochan DJ. Langmuir. 2012;28:6076–6087. doi: 10.1021/la2041746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zhang SG. Nat Biotechnol. 2003;21:1171–1178. doi: 10.1038/nbt874. [DOI] [PubMed] [Google Scholar]
- 3.Rajagopal K, Schneider JP. Curr Opin Struct Biol. 2004;14:480–486. doi: 10.1016/j.sbi.2004.06.006. [DOI] [PubMed] [Google Scholar]; Ulijn RV, Smith AM. Chem Soc Rev. 2008;37:664–675. doi: 10.1039/b609047h. [DOI] [PubMed] [Google Scholar]; Yang Z, Liang G, Xu B. Acc Chem Res. 2008;41:315–326. doi: 10.1021/ar7001914. [DOI] [PubMed] [Google Scholar]; Cui H, Webber MJ, Stupp SI. Biopolymers. 2010;94:1–18. doi: 10.1002/bip.21328. [DOI] [PMC free article] [PubMed] [Google Scholar]; Matson JB, Stupp SI. Chem Commun. 2012;48:26–33. doi: 10.1039/c1cc15551b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Yang Z, Xu B. J Mater Chem. 2007;17:2385–2393. [Google Scholar]
- 5.Silva GA, Czeisler C, Niece KL, Beniash E, Harrington DA, Kessler JA, Stupp SI. Science. 2004;303:1352–1355. doi: 10.1126/science.1093783. [DOI] [PubMed] [Google Scholar]; Jayawarna V, Ali M, Jowitt TA, Miller AE, Saiani A, Gough JE, Ulijn RV. Adv Mater. 2006;18:611–+. [Google Scholar]; Shekaran A, Garcia AJ. BBA-Gen Subjects. 2011;1810:350–360. doi: 10.1016/j.bbagen.2010.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Schnepp ZAC, Gonzalez-McQuire R, Mann S. Adv Mater. 2006;18:1869–+. [Google Scholar]; Ndao M, Keene E, Amos FF, Rewari G, Ponce CB, Estroff L, Evans JS. Biomacromolecules. 2010;11:2539–2544. doi: 10.1021/bm100738r. [DOI] [PubMed] [Google Scholar]
- 7.Yang Z, Liang G, Ma M, Abbah AS, Lu WW, Xu B. Chem Commun. 2007:843–845. doi: 10.1039/b616563j. [DOI] [PubMed] [Google Scholar]
- 8.Kiyonaka S, Sada K, Yoshimura I, Shinkai S, Kato N, Hamachi I. Nat Mater. 2004;3:58–64. doi: 10.1038/nmat1034. [DOI] [PubMed] [Google Scholar]
- 9.Li XM, Li JY, Gao YA, Kuang Y, Shi JF, Xu B. J Am Chem Soc. 2010;132:17707–17709. doi: 10.1021/ja109269v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yang ZM, Xu B. Chem Commun. 2004:2424–2425. doi: 10.1039/b408897b. [DOI] [PubMed] [Google Scholar]
- 11.Wang Q, Yang Z, Zhang X, Xiao X, Chang CK, Xu B. Angew Chem Int Ed. 2007;46:4285–4289. doi: 10.1002/anie.200700404. [DOI] [PubMed] [Google Scholar]
- 12.Jun HW, Yuwono V, Paramonov SE, Hartgerink JD. Adv Mater. 2005;17:2612–+. [Google Scholar]
- 13.Wender PA, Mitchell DJ, Pattabiraman K, Pelkey ET, Steinman L, Rothbard JB. Proc Natl Acad Sci U S A. 2000;97:13003–13008. doi: 10.1073/pnas.97.24.13003. [DOI] [PMC free article] [PubMed] [Google Scholar]; Umezawa N, Gelman MA, Haigis MC, Raines RT, Gellman SH. J Am Chem Soc. 2002;124:368–369. doi: 10.1021/ja017283v. [DOI] [PubMed] [Google Scholar]; Wender PA, Rothbard JB, Jessop TC, Kreider EL, Wylie BL. J Am Chem Soc. 2002;124:13382–13383. doi: 10.1021/ja0275109. [DOI] [PubMed] [Google Scholar]; Farrera-Sinfreu J, Giralt E, Castel S, Albericio F, Royo M. J Am Chem Soc. 2005;127:9459–9468. doi: 10.1021/ja051648k. [DOI] [PubMed] [Google Scholar]; Chongsiriwatana NP, Patch JA, Czyzewski AM, Dohm MT, Ivankin A, Gidalevitz D, Zuckermann RN, Barron AE. Proc Natl Acad Sci U S A. 2008;105:2794–2799. doi: 10.1073/pnas.0708254105. [DOI] [PMC free article] [PubMed] [Google Scholar]; Czyzewski AM, Barron AE. Aiche J. 2008;54:2–8. [Google Scholar]
- 14.Chen F, Zhu NY, Yang D. J Am Chem Soc. 2004;126:15980–15981. doi: 10.1021/ja044493+. [DOI] [PubMed] [Google Scholar]; Li X, Yang D. Chem Commun. 2006:3367–3379. doi: 10.1039/b602230h. [DOI] [PubMed] [Google Scholar]; Ma B, Lin G, Yang D. Drug Metab Rev. 2010;42:294–294. [Google Scholar]
- 15.Tugyi R, Uray K, Ivan D, Fellinger E, Perkins A, Hudecz F. Proc Natl Acad Sci U S A. 2005;102:413–418. doi: 10.1073/pnas.0407677102. [DOI] [PMC free article] [PubMed] [Google Scholar]; Welch BD, VanDemark AP, Heroux A, Hill CP, Kay MS. Proc Natl Acad Sci U S A. 2007;104:16828–16833. doi: 10.1073/pnas.0708109104. [DOI] [PMC free article] [PubMed] [Google Scholar]; Kolodkin-Gal I, Romero D, Cao S, Clardy J, Kolter R, Losick R. Science. 2010;328:627–629. doi: 10.1126/science.1188628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Seebach D, Abele S, Schreiber JV, Martinoni B, Nussbaum AK, Schild H, Schulz H, Hennecke H, Woessner R, Bitsch F. Chimia. 1998;52:734–739. [Google Scholar]; Sadowsky JD, Murray JK, Tomita Y, Gellman SH. ChemBioChem. 2007;8:903–916. doi: 10.1002/cbic.200600546. [DOI] [PubMed] [Google Scholar]; Aguilar MI, Purcell AW, Devi R, Lew R, Rossjohn J, Smitha AI, Perlmutter P. Org Biomol Chem. 2007;5:2884–2890. doi: 10.1039/b708507a. [DOI] [PubMed] [Google Scholar]
- 17.Yang Z, Liang G, Ma M, Gao Y, Xu B. Small. 2007;3:558–562. doi: 10.1002/smll.200700015. [DOI] [PubMed] [Google Scholar]
- 18.Liang GL, Yang ZM, Zhang RJ, Li LH, Fan YJ, Kuang Y, Gao Y, Wang T, Lu WW, Xu B. Langmuir. 2009;25:8419–8422. doi: 10.1021/la804271d. [DOI] [PubMed] [Google Scholar]
- 19.Sola RJ, Griebenow K. J Pharm Sci. 2009;98:1223–1245. doi: 10.1002/jps.21504. [DOI] [PMC free article] [PubMed] [Google Scholar]; Culyba EK, Price JL, Hanson SR, Dhar A, Wong CH, Gruebele M, Powers ET, Kelly JW. Science. 2011;331:571–575. doi: 10.1126/science.1198461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Li X, Kuang Y, Shi J, Gao Y, Lin H-C, Xu B. J Am Chem Soc. 2011;133:17513–17518. doi: 10.1021/ja208456k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Vassilev VP, Simanek EE, Wood MR, Wong CH. Chem Commun. 1998:1865–1866. [Google Scholar]; Luboradzki R, Gronwald O, Ikeda M, Shinkai S, Reinhoudt DN. Tetrahedron. 2000;56:9595–9599. [Google Scholar]; Wang G, Sharma V, Cheuk S, Williams K, Dakessian L, Thorton Z. Carbohydr Res. 2006;341:705–716. doi: 10.1016/j.carres.2006.01.023. [DOI] [PubMed] [Google Scholar]; Griffiths PC, Knight DW, Morgan IR, Ford A, Brown J, Davies B, Heenan RK, King SM, Dalgliesh RM, Tomkinson J, Prescott S, Schweins R, Paul A. Beilstein J Org Chem. 2010;6:1079–1088. doi: 10.3762/bjoc.6.123. [DOI] [PMC free article] [PubMed] [Google Scholar]; Zhao F, Weitzel CS, Gao Y, Browdy HM, Shi J, Lin H-C, Lovett ST, Xu B. Nanoscale. 2011;3:2859–2861. doi: 10.1039/c1nr10333d. [DOI] [PMC free article] [PubMed] [Google Scholar]; Li X, Kuang Y, Xu B. Soft Matter. 2012;8:2801–2806. doi: 10.1039/C2SM06920B. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Benevenga NJ, Steele RD. Annu Rev Nutr. 1984;4:157–181. doi: 10.1146/annurev.nu.04.070184.001105. [DOI] [PubMed] [Google Scholar]; Fuchs SA, Berger R, Klomp LWJ, de Koning TJ. Mol Genet Metab. 2005;85:168–180. doi: 10.1016/j.ymgme.2005.03.003. [DOI] [PubMed] [Google Scholar]
- 23.Li X, Kuang Y, Lin H-C, Gao Y, Shi J, Xu B. Angew Chem Int Ed. 2011;50:9365–9369. doi: 10.1002/anie.201103641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhang Y, Kuang Y, Gao Y, Xu B. Langmuir. 2011;27:529–537. doi: 10.1021/la1020324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Li X, Du X, Gao Y, Shi J, Kuang Y, Xu B. Soft Matter. 2012 doi: 10.1039/C1032SM25725D. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hayat MA. Principles and Techniques of Electron Microscopy: Biological Applications. 4. Cambridge University Press; 2000. [Google Scholar]
- 27.Bromme D, Peters K, Fink S, Fittkau S. Arch Biochem Biophys. 1986;244:439–446. doi: 10.1016/0003-9861(86)90611-9. [DOI] [PubMed] [Google Scholar]
- 28.Rehfeldt F, Engler AJ, Eckhardt A, Ahmed F, Discher DE. Adv Drug Deliv Rev. 2007;59:1329–1339. doi: 10.1016/j.addr.2007.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ruoslahti E. Annu Rev Cell Dev Biol. 1996;12:697–715. doi: 10.1146/annurev.cellbio.12.1.697. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.