

Abstract
Both activating and inactivating mutations in protein tyrosine phosphatase Ptpn11 (encoding Shp2) are associated with tumorigenesis. However, the underlying mechanisms remain unclear. Here we demonstrate that Shp2 plays an important role in mitosis, dysregulation of which results in chromosome instability and cancer predisposition. Depletion of Shp2 compromised the mitotic checkpoint. Shp2-depleted cells exhibited a delay in mitotic entry and an earlier mitotic exit. Moreover, Shp2 deficiency caused defective kinetochore-microtubule attachment, chromosome misalignment, chromosomal congression defects, lagging chromosomes, and chromosome missegregation. Reintroduction of wild-type Shp2, but not a catalytically-deficient mutant, restored the checkpoint function and chromosome alignment at metaphase in Shp2-deficient cells, establishing a requirement for the catalytic activity of Shp2 during mitosis. Further analyses revealed that Shp2 was required for the optimal activation of the mitotic kinases PLK1 and Aurora B and thereby the proper kinetochore localization and phosphorylation of BubR1, a core mitotic checkpoint protein that is also critical for chromosome alignment. Together, our findings demonstrate a previously unrecognized role for Shp2 in the maintenance of chromosome stability and suggest a new mechanism by which dysregulation of Shp2 signaling contributes to malignancy development.
Keywords: Ptpn11 (Shp2), Phosphatase, Mitosis, Chromosome stability
Introduction
Mitosis is the biological process by which a eukaryotic cell precisely separates replicated sister chromatids into two daughter cells during cell division. Deregulated mitotic activities are associated with chromosomal instability and aneuploidy, a key feature of many cancers (1-3). Mitosis is an extraordinarily complex process. Many proteins are involved in the regulation of mitosis. Among these proteins, Polo-like kinases (PLK), especially, PLK1 is involved in a variety of mitotic events including the G2/M transition, centrosome maturation and separation, mitotic spindle formation, chromosome segregation, and cytokinesis (4-6). It exerts its multiple functions by phosphorylating a wide spectrum of substrates. In addition, members of the Aurora kinase family, such as Aurora B, have emerged as key mitotic regulators. They are involved in centrosome separation and maturation, spindle assembly and stability, chromosomal condensation, congression and segregation, and cytokinesis (5, 7, 8). Aurora kinases function also by phosphorylating many substrate proteins.
Multiple fidelity-monitoring checkpoint systems have evolved to ensure correct temporal and spatial coordination of mitosis. The mitotic checkpoint, also known as the spindle assembly checkpoint, is a cellular surveillance mechanism that acts to inhibit entry into anaphase until all chromosomes have successfully attached to spindle microtubules (1, 2, 9). This checkpoint is mediated by a set of highly conserved proteins including Mad1, Mad2, Mad3/BubR1, Bub1, and Bub3 (2). Proper recruitment of these checkpoint proteins to unaligned kinetochores, the protein structures on chromosomes where the spindle microtubules attach, is essential for the mitotic checkpoint to delay anaphase onset. Displacement of these proteins from kinetochores is associated with mitotic checkpoint defects and chromosomal instability. Among them, BubR1 is a central checkpoint protein in term of its essential role for kinetochore localization of other mitotic checkpoint proteins (10). Aurora B kinase is also involved in the regulation of the mitotic checkpoint (11). Inhibition of Aurora B disturbs kinetochore localization of BubR1, Mad2, and Cenp-E, thereby compromising mitotic checkpoint function (12). In addition, a recent study has shown that Aurora B plays a direct role in initiating the mitotic checkpoint by potentiating Mps1 activation (13).
In addition to the mitotic checkpoint, temporal and spatial regulation of kinetochore-microtubule (KT-MT) attachment is essential for error-free chromosome segregation. All chromosomes/kinetochores must be attached to the spindle microtubules and properly aligned to the metaphase plate for accurate chromosome segregation. Depletion of BubR1 not only disturbs the mitotic checkpoint but also leads to severe chromosome misalignment (12), suggesting that BubR1 plays a dual role in both checkpoint signaling and chromosome alignment. Several lines of evidence demonstrate that BubR1 is required for stable chromosome-spindle attachments (14, 15). BubR1 is highly phosphorylated in mitosis, and that phosphorylation of BubR1 is important for maintaining proper KT-MT attachments and chromosome alignment (14, 15). Recent studies have demonstrated that BubR1 is a substrate of PLK1 and that PLK1 regulates chromosome alignment through phosphorylation of BubR1 (14, 16).
Shp2, a ubiquitously expressed Src homology2 domain-containing protein tyrosine phosphatase, has been implicated in diverse cytoplasmic signaling processes activated by growth factors, hormones, cytokines, and extracellular matrix proteins (17, 18). Mutations in Ptpn11 (encoding Shp2) are involved in several human diseases, including cancers (19-21). While activating mutations in Ptpn11 are associated with Noonan syndrome, childhood leukemias, and sporadic solid tumors, Leopard syndrome patients with Ptpn11 inactivating mutations also have increased risks of developing malignancies (22-25). In addition, a recent study has shown that Ptpn11 deficiency enhances diethylnitrosamine-induced hepatocellular carcinoma development (26). However, the molecular mechanisms by which deregulated Shp2 signaling is linked to malignancies are not well understood due to lack of complete understanding of Shp2 function. The cytoplasmic function of Shp2 cannot fully explain how both activating and inactivating mutations in Shp2 induce tumorigenesis. Emerging evidence has indicated that Shp2 may also function in other cell organelles, such as the nucleus (27-31) and the mitochondria (32, 33). Understanding of the novel functions of Shp2 in these organelles may shed light on the molecular mechanisms of Shp2 associated tumorigenesis. In our recent studies investigating effects of activating mutations of Ptpn11 on malignant transformation of hematopoietic cells, we have discovered that gain of function of this phosphatase causes aneuploidy (34), which is known to be associated with aberrant mitosis. This unexpected finding led us to further explore the novel function of Shp2 in mitosis.
Materials and Methods
Cell culture, synchronization, and transfection
HeLa cell line used in this study were originally purchased from ATCC and maintained 10-15 passages in DMEM supplemented with 10% FBS, 100 U/ml penicillin, and 100 μg/ml streptomycin. Mouse embryonic fibroblasts (MEFs) generated from Ptpn11 conditional knock out (Ptpn11fl/fl) mice were provided by Dr. B. G. Neel. They were previously characterized (35) and were maintained as described above. To synchronize cells at the G1 phase, cells were treated with thymidine (2 mM) for 17 hours, washed with phosphate-buffered saline (PBS) 3 times, and then incubated for 9 hours in fresh medium without thymidine. Thymidine (2 mM) was added again for another 16 hours. To arrest the cells in mitosis, cells were treated with nocodazole (200 ng/ml and 400 ng/ml for HeLa cells and MEFs, respectively) for 16 hours. Mitotic cells were then isolated by “shake-off”. For release experiments, isolated mitotic cells were washed 3 times with PBS and plated into fresh medium for the indicated periods of time. To knock down Shp2, HeLa cells were transfected with Shp2 siRNA corresponding to 5′-AAG AAU CCU AUG GUG GAA ACA-3′ using an Amaxa Nucleofector and a transfection kit according to the manufacturer’s protocol (Lonza Group Ltd, Switzerland).
Time-lapse video microscopy
Cells were plated in 35 mm culture dishes. Twenty four hours later, cells were monitored using a Leica DMI600B inverted microscope equipped with an environmental control chamber. Images were taken every 3 or 5 minutes with a Retiga EXI 12 bit camera (Q imaging, Vancouver, B.C.) and analyzed with MetaMorph software (Universal Imaging Corp., Pennsylvania, PA).
Immunostaining and confocal microscopy
For analysis of cold-stable microtubules, cells were fixed for 10 minutes at room temperature with 3.7% formaldehyde in 100 mM Pipes at pH 6.8, 10 mM EGTA, 1 mM MgCl2, and 0.2% Triton X-100. For all other experiments, cells were fixed in 100% methanol at −20°C for 10 minutes. After fixation, cells were permeabilized with 0.25% Triton X-100 in PBS, and then blocked with 2% BSA in PBS for 1 hour. All primary antibodies were incubated at room temperature for 1 hour or at 4°C overnight. Cells were then washed 3 times with PBS and incubated with Alexa 488 or Alexa 568-conjugated secondary antibodies for 1 hour. Nuclei were counterstained with 4′,6-diamidino-2-phenylindole (DAPI). After washing, coverslips were mounted onto glass slides and stained cells were examined with a Nikon fluorescence microscope (Nikon, Japan). All confocal images were acquired using a Zeiss LSM 510 inverted laser-scanning confocal microscope (Zeiss, Germany) and analyzed with Zeiss and MetaMorph software. For staining of chromosome spreads, mitotic cells were obtained by “shake-off” and swelled in a prewarmed 75 mM KCl hypotonic solution at 37°C for 15 minutes. After spun onto slides using Cytospin, chromosomes were fixed and stained with indicated antibodies as above.
For the chromosome alignment assay, control or Shp2 siRNA transfected HeLa cells were synchronized by double thymidine block and released for 10 hours, and then arrested at metaphase by the treatment with MG132 (10 μM) for 2 hours. Cells were fixed and processed for immunostaining. To count unaligned kinetochores, all images were acquired as Z-stack with 0.2 μm spacing using a 100X objective. Misaligned kinetochores were defined as those without localizing to equatorial plate. At least five cells (400 kinetochores) were analyzed for each experiment.
Statistical analysis
Data are presented as mean ± SD. Statistical significance was determined using unpaired two-tailed Student’s t test. * p<0.05; ** p<0.01.
Results
Depletion of Shp2 results in a delay in entering mitosis and causes a failure to sustain the mitotic arrest induced by nocodazole
To test the potential role of Shp2 in mitosis, we transfected HeLa cells with Shp2 specific siRNA and assessed cellular responses to nocodazole, a microtubule depolymerizing agent that activates the mitotic checkpoint and arrests cells at mitosis. Seventy-five percent of control cells were arrested at the mitotic stage defined by 4n DNA content and phospho-histone H3 (Ser10) positive staining. In contrast, knock down of Shp2 substantially decreased nocodazole-induced mitotic arrest in a Shp2 dose-dependent manner (Figure 1A and 1B). Similarly, the percentage of mitotic cells in Shp2-depleted cells treated with taxol, another microtubule poison, was also decreased significantly (Supplementary Figure 1). To minimize potential influence of the cell cycle status on cellular responses to mitotic arrest, asynchronized and synchronized G1 HeLa cells were treated with nocodazole and mitotic cells were assessed at multiple time points. Both asynchronized and synchronized Shp2 knock-down cells showed decreased mitotic arrest (Figure 1C and Supplementary Figure 2). Consistent with the reduction of mitotic cells, phosphorylation of histone H3 in Shp2-depleted cells was much lower than that in control cells and the inhibitory phosphorylation (Tyr15) of Cdc2 kinase (the kinase activity of which is required for mitotic entry) was sustained at higher levels in Shp2 knock-down cells during nocodazole treatment (Figure 1D).
Figure 1. Shp2 depletion compromises mitotic checkpoint function.

(A) HeLa cells were transfected with control or Shp2 siRNA at the indicated concentrations. Forty-eight hours later, transfected cells were treated with nocodazole (200 ng/ml) for 16 hours. Cells were analyzed by FACS after PI and phospho-histone H3 (Ser10) double staining. (B) HeLa cells transfected with control or Shp2 siRNA (400 pmol) were treated with nocodazole for 16 hours. Percentages of mitotic cells were determined as above. (C) HeLa cells transfected with control or Shp2 siRNA were treated with nocodazole for the indicated periods of time. Percentages of mitotic cells were determined. (D) HeLa cells transfected with control or Shp2 siRNA were treated with nocodazole for the indicated periods of time. Whole cell lysates were prepared and analyzed by immunoblotting analyses for phospho-Cdc2 (Tyr15), phospho-histone H3 (Ser10), Shp2, and β-actin. The data shown in this figure are mean±S.D. or representative of three independent experiments.
However, the percentage of phospho-histone H3 negative Shp2-depleted cells with 4n DNA content were increased (Figure 1A). This reflects a delay in G2 or an accumulation of tetraploid G1 cells escaped from mitosis without dividing. To clarify these possibilities, live synchronized cells at the G1 phase were cultured in the presence of nocodazole and monitored by time lapse videomicroscopy. Consistent with the data presented in Figure 1, Shp2 knock down cells showed markedly decreased response to this microtubule poison (Figure 2A and 2B). By carefully monitoring individual cells, we noticed that about 40% of Shp2 knock-down cells failed to enter metaphase, being arrested at the G2 phase. Furthermore, the percentage of Shp2-depleted cells that escaped from mitotic arrest was increased. In addition, Shp2-depleted mitotic cells underwent a higher incidence of death (Figure 2C). We next focused on mitotic cells arrested by nocodazole and quantified the duration of mitotic arrest. In the presence of nocodazole, more than 50% of control cells that were arrested in mitosis stayed in mitosis for up to 16 hours, whereas approximately 80% of Shp2-depleted cells escaped from mitotic arrest without dividing and returned to G1 with tetraploid DNA content within 12 hours (Figure 2D and 2E). These data suggest that Shp2 ablation results in a delay in entering mitosis as well as a premature mitotic exit despite the spindle defects caused by nocodazole.
Figure 2. Shp2 depletion delays entry into mitosis and shortens mitotic arrest in the presence of nocodazole.

(A) HeLa cells transfected with control or Shp2 siRNA were synchronized at G1 by thymidine double block. Synchronized cells were cultured in the presence of nocodazole and monitored by time-lapse videomicroscopy for 24 hours. (B) Cumulative mitotic cell counts were performed by tracking cells in six random fields over 24 hours. The fields were combined and cells entering mitosis (became rounded) were cumulated and reported as a percentage of the total cells in the fields at the start of the experiments. (C) Those cells that changed from the flat morphology to the rounded and stayed rounded at the end of the experiment were classified as “mitotic”, whereas those that changed from the flat morphology to the rounded, but subsequently flattened back were counted as “escaped”, and those that did not flatten back but subsequently shrunk and broke into pieces were identified as “dead”. Those cells that had stayed flat without turning rounded at the end of the experiment were “G2’ cells (these cells contained 4n DNA content as verified by FACS). (D) Asynchronous control or Shp2 siRNA transfected HeLa cells were cultured in the presence of nocodazole and monitored by time-lapse videomicroscopy for 24 hours. Arrows indicate representative cells being evaluated. (E) The length of mitosis was determined morphologically by timing when the cells first became rounded and when they subsequently flattened back. (F) HeLa cells transfected with Shp2 siRNA or scramble RNA were treated with nocodazole for 16 hours. Mitotic cells were collected by “shake-off” and replated in fresh medium without nocodazole. Cells were then harvested at the indicated time points for immunoblotting analyses for PLK1, Cyclin B1, Mad2, phospho-histone H3, and β-actin. The data presented in this figure are representative of at least two independent experiments.
A proper control of anaphase onset/mitotic exit depends on ubiquitin-mediated proteolysis of key regulators at the correct time. We collected nocodazole-arrested mitotic cells and examined levels of Cyclin B1, PLK1, Mad2, and histone H3 after release in regular fresh culture medium. Consistent with the earlier mitotic exit found in the presence of nocodazole, degradation of PLK1 and Cyclin B1 was accelerated in Shp2 knock-down cells in the absence of nocodazole (Figure 2F and Supplementary Figure 3). Phosphorylation of histone H3 was also decreased much faster in Shp2-depleted cells (Figure 2F). In contrast, Mad2 was not significantly degraded in either control or Shp2-depleted cells (Figure 2F).
Catalytic activity is required for Shp2 function in mitosis
Shp2 is a protein tyrosine phosphatase that functions in cell signaling in both catalytically-dependent and - independent manners (36-38). To further elucidate the structural bases for the important role of Shp2 in mitosis, WT Shp2 and mutant Shp2 with C459S mutation that abolishes its catalytic activity were transduced into conditional knockout (Ptpn11fl/fl) mouse embryonic fibroblasts (MEFs). Transduced cells were infected with adenovirus expressing Cre DNA recombinase (to deplete endogenous Shp2) followed by nocodazole treatment. Consistent with the data shown in Figure 1, acute depletion of Shp2 decreased mitotic arrest significantly (Figure 3A). Reintroduction of WT Shp2, but not Shp2 C459S, largely restored responses of Shp2-depleted cells to nocodazole-induced mitotic arrest (Figure 3B and 3C). These rescue experiments, while demonstrating cell autonomous effects of Shp2 deficiency on mitosis, provide evidence that the catalytic activity of Shp2 is required for its function in this process.
Figure 3. Catalytic activity of Shp2 is required for its function in the mitotic checkpoint.
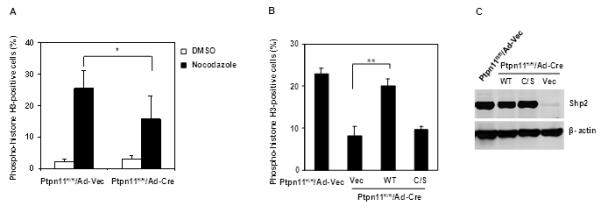
(A) Ptpn11fl/fl MEFs were infected with adenoviruses expressing Cre recombinase (Ad-Cre) or vector (Ad-Vec). Forty-eight hours after adenovirus infection, cells were treated with nocodazole (400 ng/ml) for 16 hours. Percentages of mitotic cells were determined. (B and C) Ptpn11fl/fl MEFs were transfected with plasmids expressing WT Shp2, Shp2 C459S, or control vector. Transfected cells were sorted by FACS according to GFP that is expressed separately by the vector. Sorted cells were infected with adenoviruses, treated with nocodazole, and assessed for mitotic cells as above. Shp2 expression was analyzed by immunoblotting. The data shown in this figure are mean±S.D. of three independent experiments.
Depletion of Shp2 causes chromosome misalignment and defective kinetochore-microtubule attachment
To further determine the role of Shp2 in mitosis, unperturbed mitotic progression was directly visualized in a greater detail. HeLa cells stably expressing a histone H2B-GFP fusion protein that enables monitoring chromosomes in live cells with high resolution were transfected with control or Shp2 siRNA, and then monitored under a videomicroscope. The percentages of mitotic cells with misaligned and lagging chromosomes were markedly increased after Shp2 depletion (Figure 4A, 4B, and 4C). Moreover, some Shp2-depleted cells entered anaphase without chromosome aligning to the metaphase plate, indicative of chromosome congression defects (Figure 4B and 4C), and in some cases, metaphase chromosomes underwent decondensation before the onset of anaphase (Figure 4B). As a result, the unperturbed mitosis of Shp2-depleted cells was significantly prolonged due to these mitotic defects (Figure 4D), similar to that of PLK1 deficient cells (39). In agreement with these chromosome analyses results, Shp2 knock down primary MEFs developed increased aneuploidy compared to control cells (Figure 4E).
Figure 4. Depletion of Shp2 causes chromosome misalignment, congression defects, and chromosome missegregation.

(A) HeLa-H2B-GFP cells were transfected with control or Shp2 siRNA. Forty-eight hours following transfection mitotic cells were monitored using real-time videomicroscopy. Arrows point to misaligned chromosomes in a metaphase cell and lagging chromosomes in a telophase cell. (B) HeLa-H2B-GFP cells transfected with control or Shp2 siRNA were monitored using time-lapse videomicroscopy. Images were taken every 3 minutes with a 20X objective. Entry into mitosis (set as time 0:00) was determined based on the loss of nuclear integrity and the condensation of chromosomes. (C) Mitotic cells with misaligned chromosomes, lagging chromosomes, and chromosome congression defects were quantified. (D) Duration of mitosis of individual mitotic cells was recorded and percentages of mitotic cells with the indicated times in mitosis were summarized. (E) Primary MEFs at passage 3 were transfected with control or Shp2 siRNA and karyotyped 48 hours later. Cells with either more or fewer than 40 chromosomes were scored as aneuploid cells. The data shown in this figure are mean±S.D. or representative of three independent experiments.
To rule out the possibility that chromosome misalignment in Shp2-depleted cells was due to premature mitotic exit, we blocked the anaphase onset using the proteasome inhibitor MG132. Under these conditions, Shp2-depleted metaphase cells with chromosome misalignment (Figure 5A) and telophase cells with lagging chromosomes (Supplementary Figure 4) were still significantly increased. Further rescue experiments showed that add-back of WT Shp2, but not catalytically deficient Shp2 C459S, restored chromosomal alignment at metaphase (Figure 5B), reaffirming the catalytic-dependent role of Shp2 in this process. Consistent with chromosome misalignment, more detailed visualization of kinetochores illustrated that Shp2-depleted metaphase cells had increased unaligned kinetochores (Figure 5C). The chromosome misalignment phenotype suggests that Shp2 might be required for attachment of kinetochores to the spindle. Indeed, unattached kinetochores were frequently observed in Shp2-depleted cells (Figure 5C, insets). To address the underlying cause of chromosome misalignment, we examined the stability of kinetochore-microtubule (KT-MT) attachment by exposing MG132-arrested mitotic cells to low temperature (0 °C), which depolymerized unstable microtubules. In control cells, the spindle remained relatively intact, with most CREST-stained kinetochores clearly attached to microtubule fibers. In contrast, Shp2-depleted cells showed only few cold-stable KT-MT attachments, and most kinetochores had no microtubules attached to them (Figure 5D).
Figure 5. Depletion of Shp2 results in chromosome misalignment and loss of kinetochore-microtubule attachment.

(A) HeLa cells were transfected with control or Shp2 siRNA. Forty-eight hours following transfection cells were synchronized by double thymidine block and then released for 10 hours. These cells were treated with MG132 (10 μM) for 2 hours to enrich metaphase cells. Cells were then fixed and stained with CREST and α-tubulin antibodies, and counterstained with DAPI. Metaphase cells with misaligned chromosomes were quantified. At least 100 metaphase cells were examined in each experiment. (B) Ptpn11fl/fl MEFs were transfected and cultured as described in Figure 3B. Cells were then treated with MG132 and stained with CREST and α-tubulin antibodies and DAPI. Metaphase cells with misaligned chromosomes were quantified. At least 50 metaphase cells were examined in each experiment. (C) HeLa cells transfected with control or Shp2 siRNA were synchronized, treated with MG132, and then stained with CREST and anti-α-tubulin antibodies. Insets (1-4) show individual kinetochores with (1, 2) or without (3, 4) microtubule attachments. To quantify unaligned kinetochores, all images were acquired as Z-stack with 0.2 μM spacing using a 100X objective. Unaligned kinetochores defined as those without localizing to equatorial plates were counted. At least 400 kinetochores (5 cells) were analyzed in each experiment. (D) MG132 arrested cells were incubated on ice for 20 minutes before fixation. Cells were stained with CREST and anti-α-tubulin antibodies. The data shown in this figure are mean±S.D. or representative of three independent experiments.
Kinetochore localization of BubR1 and Aurora B kinase activity are reduced in Shp2-depleted cells
Proper localization to kinetochores is critical for mitotic proteins to execute their functions (40), and displacement of these proteins from kinetochores is associated with mitotic checkpoint defects. To elucidate the mechanisms of the defective mitotic checkpoint caused by Shp2 depletion, we examined the kinetochore localization of BubR1 and Mad2. In control mitotic cells, BubR1 was nicely localized to kinetochores. However, kinetochore signals of BubR1 were diminished in Shp2-depleted cells (Figure 6A). To confirm this result, we performed centromeric staining of chromosome spreads. As illustrated in Figure 6B, the percentage of the metaphase spreads with strong BubR1 signals at kinetochores also was decreased significantly in Shp2-depleted cells. In contrast, the kinetochore localization of Mad2 was unaffected (data not shown). Previous studies have shown that Aurora B activity is important for kinetochore localization of BubR1 (12, 14, 41). We tested whether mis-localization of BubR1 might be associated with abnormal Aurora B activation and found that Aurora B kinase activity determined by phosphorylation of Thr232 (Figure 6C) and kinase assays (Figure 6D) was significantly decreased in Shp2-depleted cells. These results suggest that decreased Aurora B activity may be responsible for the diminished kinetochore localization of BubR1 in Shp2 knock-down cells.
Figure 6. Kinetochore localization of BubR1 and Aurora B kinase activity are decreased in Shp2-depleted cells.

(A) HeLa cells were transfected with control or Shp2 siRNA. Forty-eight hours following transfection, cells were immunostained with anti-BubR1 and CREST antibodies. Colocalization of BubR1 with CREST (kinetochores) was determined under a confocal microscope. Percentages of the cells with strong, medium, and weak BubR1 colocalization with CREST were determined. (B) HeLa cells transfected with control or Shp2 siRNA were treated with nocodazole (200 ng/ml) for 16 hours. Mitotic cells were collected. Metaphase chromosome spreads were prepared and immunostained with anti-BubR1 and CREST antibodies. Percentages of the cells with strong, medium, and weak BubR1 colocalization with CREST (centromeres) were determined as above. (C) HeLa cells transfected with control or Shp2 siRNA were treated with nocodazole as above. Mitotic cells, left-over attached cells, and untreated cells were analyzed for activation of Aurora B kinase by immunoblotting with anti-phospho-Aurora B (Thr232) antibody. Blots were stripped and reprobed with anti-Aurora B, anti-Shp2, and anti-β-actin antibodies. (D) Mitotic cells were analyzed for Aurora B kinase activity by the Aurora B kinase assay using histone H3 as the substrate. Phosphorylation of histone H3 was visualized by autoradiography. Levels of Aurora B in the immunoprecipitates used in the kinase assay were examined by immunoblotting with anti-Aurora B antibody. The data shown in this figure are mean±S.D. or representative of three independent experiments.
Phosphorylation of BubR1 and PLK1 activity are decreased in Shp2-depleted cells
The kinetochore localization of BubR1 has been shown to be important for its phosphorylation which is essential for KT-MT attachment and proper chromosome alignment (14, 16). We determined phosphorylation levels of BubR1 in mitotic cells. In response to nocodazole and taxol, phosphorylation of BubR1 was substantially decreased in Shp2-depleted cells compared with those in control cells (Figure 7A). To exclude the possibility that the decreased phosphorylation of BubR1 was due to the reduction of mitosis in Shp2-depleted cells, phosphorylation levels of BubR1 were also determined in synchronized “shake-off” mitotic cells. Similar results were obtained from Shp2-depleted mitotic cells (Figure 7B). Recent studies have demonstrated that BubR1 is a substrate of PLK1 (14, 16). We assessed PLK1 kinase activity in Shp2-depleted mitotic cells. As seen in Figure 7C and 7D, PLK1 activity determined by phosphorylation of Thr210 and kinase assays was decreased significantly in Shp2 knock down cells. These results suggest that decreased PLK1 activity, followed by the consequent reduction of BubR1 phosphorylation, leads to chromosome misalignment in Shp2-depleted cells.
Figure 7. Phosphorylation of BubR1 and PLK1 activity are decreased in Shp2-depleted cells.

(A) HeLa cells were transfected with control or Shp2 siRNA. Forty-eight hours following transfection, cells were treated with nocodazole (200 ng/ml) or taxol (10 μM) for the indicated periods of time. Whole cell lysates were prepared and analyzed by immunoblotting with anti-BubR1 antibody. The blot was stripped and reprobed with anti-β-actin antibody. (B) Asynchronized (Asy) and mitotic “shake-off” (shake-off) cells were collected, and whole cell lysates were examined by immunoblotting with anti-BubR1 antibody. (C) HeLa cells transfected with control or Shp2 siRNA were treated with nocodazole for 16 hours as above. Mitotic cells, left-over attached cells, and untreated cells were analyzed for PLK1 kinase activity by immunoblotting with anti-phospho-PLK1 (Thr210) antibody. Blots were stripped and reprobed with anti-PLK1 and anti-β-actin antibodies. (D) Mitotic cells and untreated cells were analyzed for PLK1 kinase activity by the PLK1 kinase assay using α-Casein as the substrate. Phosphorylation of α-Casein was visualized by autoradiography. Levels of PLK1 in the immunoprecipitates used in the kinase assay were examined by immunoblotting with anti-PLK1 antibody. (E) Proposed model of the role of Shp2 in the maintenance of chromosomal stability. The data shown in this figure are mean±S.D. or representative of three independent experiments.
Discussion
Shp2, a ubiquitously expressed protein tyrosine phosphatase, has been well recognized for its important role in the signal transduction of growth factors, cytokines, and extracellular matrix proteins (17, 18). It is implicated in multiple signaling pathways by interacting with numerous signaling proteins. However, the information gathered from previous studies represents the role of Shp2 in interphase, likely G0/G1 cells, since the cells used in those studies were usually growth factor-starved. By focusing on mitotic cells we now demonstrate that Shp2 also plays an important role in maintenance of chromosome stability. Both activating mutations and inactivating mutations in Ptpn11 (Shp2) are associated with tumorigenesis. Leopard syndrome patients with Ptpn11 inactivating mutations also have increased risks of developing malignancies (23-25). As inactivated Leopard syndrome mutant Shp2 functions in a dominant negative manner due to the increased binding of mutant Shp2 to signaling partners (22), it is possible that the mutant Shp2 contributes to malignancy development by decreasing Shp2 catalytic activity and thereby causing chromosomal instability although it may reduce cell growth.
Shp2 appears to facilitate the mitotic checkpoint and chromosome alignment by promoting PLK1 and Aurora B activation. PLK1 and Aurora B are multifunctional kinases that are critical for all stages of mitosis. Overexpression of these kinases has been observed in tumors, suggesting that these kinases may act as oncogenes (42-45). However, missense mutations in PLK1 have also been identified in several cancer cell lines (46). Moreover, inhibition of PLK1 leads to delayed mitotic entry and cells are arrested in mitosis with a monopolar or disorganized spindle (39, 47). Inhibition of Aurora B in cells causes misaligned chromosomes, perturbed cytokinesis, and resistance to taxol-induced mitotic arrest (12). As both PLK1 and Aurora B kinase activities were decreased in Shp-2 depleted cells (Figure 6C, 6D, 7C, and 7D), the phenotypes displayed by these cells may reflect combined effects of PLK1 and Aurora B deficiencies. Delayed entry into mitosis and prolonged duration of unperturbed mitosis are likely the consequences of decreased PLK1 activity while failure to sustain microtubule poisons-induced mitotic arrest, chromosomal misalignment, chromosomal congression defects, and chromosomal missegregation might result from decreased Aurora B activity. Aurora B has been shown to be required for the kinetochore localization of BubR1 (12, 14, 48), a core mitotic checkpoint protein. This accounts for the diminished kinetochore localization of BubR1 in Shp2-depleted cells (Figure 6A and 6B). Since recruitment of BubR1 to unattached kinetochores is necessary to maintain the mitotic checkpoint, it is likely that loss of Shp2 compromises this checkpoint by interrupting kinetochore localization of BubR1. PLK1 and Aurora B have been shown to be required for the phosphorylation of BubR1 (12, 14, 16), which is important for chromosome alignment. BubR1 phosphorylation is significantly decreased in Shp2 knock-down cells (Figure 7A and 7B). Thus, it appears that Shp2 depletion causes chromosome misalignment also by reducing PLK1 and Aurora B activation. A proposed model of Shp2 function in the maintenance of genomic stability is shown in Figure 7E.
How Shp2 promotes activation of PLK1 and Aurora B kinases remains to be further determined. Shp2 plays an overall positive role in the activation of receptor and cytosolic kinases despite its direct function in protein dephosphorylation (17, 18). The underlying mechanism is completely unclear. As such, it is not understood why PLK1 activity is decreased in Shp2-depleted mitotic cells. Aurora B kinase activity is also decreased by depletion of Shp2 (Figure 6C and 6D). This is likely the result of decreased PLK1 activity due to the well recognized extensive interplay between PLK1 and Aurora B kinases (5). Further studies are needed to define the detailed molecular mechanisms by which Shp2 promotes PLK1 activity. Shp2 appears to function in this context in a catalytic-dependent manner as only WT Shp2, but not catalytically-deficient Shp2 C459S, restores nocodazole-induced mitotic arrest and chromosome alignment in Shp2-depleted cells (Figure 3 and 5B). It seems that a general mechanism might be involved in the functional interactions between Shp2 and its interacting kinases. Elucidation of rudimental biochemical activities of this phosphatase may shed new light on its acting mechanisms in these signaling pathways.
Supplementary Material
Acknowledgments
We are extremely grateful to Dr. Hongtao Yu of the University of Texas Southwestern Medical Center and Howard Hughes Medical Institute for providing reagents, advice on experimental designs, and critical reading of the manuscript.
Grant Support This work was supported by The National Institutes of Health grants HL068212 and HD070716 (to C.K.Q.).
Footnotes
Author contributions X.L. and H.Z. conducted the research and summarized the data. C.K.Q. designed the experiments and provided technical training to the first two authors. X.L. and C.K.Q. wrote the manuscript.
Competing financial interests: The authors declare no competing financial interests.
References
- 1.Holland AJ, Cleveland DW. Boveri revisited: chromosomal instability, aneuploidy and tumorigenesis. Nat Rev Mol Cell Biol. 2009;10:478–87. doi: 10.1038/nrm2718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kops GJ, Weaver BA, Cleveland DW. On the road to cancer: aneuploidy and the mitotic checkpoint. Nat Rev Cancer. 2005;5:773–85. doi: 10.1038/nrc1714. [DOI] [PubMed] [Google Scholar]
- 3.Thompson SL, Bakhoum SF, Compton DA. Mechanisms of chromosomal instability. Curr Biol. 2010;20:R285–95. doi: 10.1016/j.cub.2010.01.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Archambault V, Glover DM. Polo-like kinases: conservation and divergence in their functions and regulation. Nat Rev Mol Cell Biol. 2009;10:265–75. doi: 10.1038/nrm2653. [DOI] [PubMed] [Google Scholar]
- 5.Lens SM, Voest EE, Medema RH. Shared and separate functions of polo-like kinases and aurora kinases in cancer. Nat Rev Cancer. 2010;10:825–41. doi: 10.1038/nrc2964. [DOI] [PubMed] [Google Scholar]
- 6.Lane HA, Nigg EA. Antibody microinjection reveals an essential role for human polo-like kinase 1 (Plk1) in the functional maturation of mitotic centrosomes. J Cell Biol. 1996;135:1701–13. doi: 10.1083/jcb.135.6.1701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Shannon KB, Salmon ED. Chromosome dynamics: new light on Aurora B kinase function. Curr Biol. 2002;12:R458–60. doi: 10.1016/s0960-9822(02)00945-4. [DOI] [PubMed] [Google Scholar]
- 8.Carmena M, Ruchaud S, Earnshaw WC. Making the Auroras glow: regulation of Aurora A and B kinase function by interacting proteins. Curr Opin Cell Biol. 2009;21:796–805. doi: 10.1016/j.ceb.2009.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Musacchio A, Salmon ED. The spindle-assembly checkpoint in space and time. Nat Rev Mol Cell Biol. 2007;8:379–93. doi: 10.1038/nrm2163. [DOI] [PubMed] [Google Scholar]
- 10.Chen RH. BubR1 is essential for kinetochore localization of other spindle checkpoint proteins and its phosphorylation requires Mad1. J Cell Biol. 2002;158:487–96. doi: 10.1083/jcb.200204048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Santaguida S, Vernieri C, Villa F, Ciliberto A, Musacchio A. Evidence that Aurora B is implicated in spindle checkpoint signalling independently of error correction. EMBO J. 2011;30:1508–19. doi: 10.1038/emboj.2011.70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ditchfield C, Johnson VL, Tighe A, Ellston R, Haworth C, Johnson T, et al. Aurora B couples chromosome alignment with anaphase by targeting BubR1, Mad2, and Cenp-E to kinetochores. J Cell Biol. 2003;161:267–80. doi: 10.1083/jcb.200208091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Saurin AT, van der Waal MS, Medema RH, Lens SM, Kops GJ. Aurora B potentiates Mps1 activation to ensure rapid checkpoint establishment at the onset of mitosis. Nat Commun. 2011;2:316. doi: 10.1038/ncomms1319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Elowe S, Hummer S, Uldschmid A, Li X, Nigg EA. Tension-sensitive Plk1 phosphorylation on BubR1 regulates the stability of kinetochore microtubule interactions. Genes Dev. 2007;21:2205–19. doi: 10.1101/gad.436007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lampson MA, Kapoor TM. The human mitotic checkpoint protein BubR1 regulates chromosome-spindle attachments. Nat Cell Biol. 2005;7:93–8. doi: 10.1038/ncb1208. [DOI] [PubMed] [Google Scholar]
- 16.Matsumura S, Toyoshima F, Nishida E. Polo-like kinase 1 facilitates chromosome alignment during prometaphase through BubR1. J Biol Chem. 2007;282:15217–27. doi: 10.1074/jbc.M611053200. [DOI] [PubMed] [Google Scholar]
- 17.Liu X, Qu CK. Protein Tyrosine Phosphatase SHP-2 (PTPN11) in Hematopoiesis and Leukemogenesis. J Signal Transduct. 2011;2011:195239. doi: 10.1155/2011/195239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chan G, Kalaitzidis D, Neel BG. The tyrosine phosphatase Shp2 (PTPN11) in cancer. Cancer Metastasis Rev. 2008;27:179–92. doi: 10.1007/s10555-008-9126-y. [DOI] [PubMed] [Google Scholar]
- 19.Tartaglia M, Mehler EL, Goldberg R, Zampino G, Brunner HG, Kremer H, et al. Mutations in PTPN11, encoding the protein tyrosine phosphatase SHP-2, cause Noonan syndrome. Nat Genet. 2001;29:465–8. doi: 10.1038/ng772. [DOI] [PubMed] [Google Scholar]
- 20.Tartaglia M, Niemeyer CM, Fragale A, Song X, Buechner J, Jung A, et al. Somatic mutations in PTPN11 in juvenile myelomonocytic leukemia, myelodysplastic syndromes and acute myeloid leukemia. Nat Genet. 2003;34:148–50. doi: 10.1038/ng1156. [DOI] [PubMed] [Google Scholar]
- 21.Bentires-Alj M, Paez JG, David FS, Keilhack H, Halmos B, Naoki K, et al. Activating mutations of the noonan syndrome-associated SHP2/PTPN11 gene in human solid tumors and adult acute myelogenous leukemia. Cancer Res. 2004;64:8816–20. doi: 10.1158/0008-5472.CAN-04-1923. [DOI] [PubMed] [Google Scholar]
- 22.Kontaridis MI, Swanson KD, David FS, Barford D, Neel BG. PTPN11 (Shp2) mutations in LEOPARD syndrome have dominant negative, not activating, effects. J Biol Chem. 2006;281:6785–92. doi: 10.1074/jbc.M513068200. [DOI] [PubMed] [Google Scholar]
- 23.Kratz CP, Rapisuwon S, Reed H, Hasle H, Rosenberg PS. Cancer in Noonan, Costello, cardiofaciocutaneous and LEOPARD syndromes. Am J Med Genet C Semin Med Genet. 2011;157:83–9. doi: 10.1002/ajmg.c.30300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Keren B, Hadchouel A, Saba S, Sznajer Y, Bonneau D, Leheup B, et al. PTPN11 mutations in patients with LEOPARD syndrome: a French multicentric experience. J Med Genet. 2004;41:e117. doi: 10.1136/jmg.2004.021451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Merks JH, Caron HN, Hennekam RC. High incidence of malformation syndromes in a series of 1,073 children with cancer. Am J Med Genet A. 2005;134A:132–43. doi: 10.1002/ajmg.a.30603. [DOI] [PubMed] [Google Scholar]
- 26.Bard-Chapeau EA, Li S, Ding J, Zhang SS, Zhu HH, Princen F, et al. Ptpn11/Shp2 acts as a tumor suppressor in hepatocellular carcinogenesis. Cancer Cell. 2011;19:629–39. doi: 10.1016/j.ccr.2011.03.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wu TR, Hong YK, Wang XD, Ling MY, Dragoi AM, Chung AS, et al. SHP-2 is a dual-specificity phosphatase involved in Stat1 dephosphorylation at both tyrosine and serine residues in nuclei. J Biol Chem. 2002;277:47572–80. doi: 10.1074/jbc.M207536200. [DOI] [PubMed] [Google Scholar]
- 28.Chughtai N, Schimchowitsch S, Lebrun JJ, Ali S. Prolactin induces SHP-2 association with Stat5, nuclear translocation, and binding to the beta-casein gene promoter in mammary cells. J Biol Chem. 2002;277:31107–14. doi: 10.1074/jbc.M200156200. [DOI] [PubMed] [Google Scholar]
- 29.Yuan L, Yu WM, Qu CK. DNA damage-induced G2/M checkpoint in SV40 large T antigen-immortalized embryonic fibroblast cells requires SHP-2 tyrosine phosphatase. J Biol Chem. 2003;278:42812–20. doi: 10.1074/jbc.M305075200. [DOI] [PubMed] [Google Scholar]
- 30.Yuan L, Yu WM, Xu M, Qu CK. SHP-2 phosphatase regulates DNA damage-induced apoptosis and G2/M arrest in catalytically dependent and independent manners, respectively. J Biol Chem. 2005;280:42701–6. doi: 10.1074/jbc.M506768200. [DOI] [PubMed] [Google Scholar]
- 31.Jakob S, Schroeder P, Lukosz M, Buchner N, Spyridopoulos I, Altschmied J, et al. Nuclear protein tyrosine phosphatase Shp-2 is one important negative regulator of nuclear export of telomerase reverse transcriptase. J Biol Chem. 2008;283:33155–61. doi: 10.1074/jbc.M805138200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Salvi M, Stringaro A, Brunati AM, Agostinelli E, Arancia G, Clari G, et al. Tyrosine phosphatase activity in mitochondria: presence of Shp-2 phosphatase in mitochondria. Cell Mol Life Sci. 2004;61:2393–404. doi: 10.1007/s00018-004-4211-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lee I, Pecinova A, Pecina P, Neel BG, Araki T, Kucherlapati R, et al. A suggested role for mitochondria in Noonan syndrome. Biochim Biophys Acta. 2010;1802:275–83. doi: 10.1016/j.bbadis.2009.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Xu D, Liu X, Yu WM, Meyerson HJ, Guo C, Gerson SL, et al. Non-lineage/stage-restricted effects of a gain-of-function mutation in tyrosine phosphatase Ptpn11 (Shp2) on malignant transformation of hematopoietic cells. J Exp Med. 2011;208:1977–88. doi: 10.1084/jem.20110450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yang W, Klaman LD, Chen B, Araki T, Harada H, Thomas SM, et al. An Shp2/SFK/Ras/Erk signaling pathway controls trophoblast stem cell survival. Dev Cell. 2006;10:317–27. doi: 10.1016/j.devcel.2006.01.002. [DOI] [PubMed] [Google Scholar]
- 36.Li W, Nishimura R, Kashishian A, Batzer AG, Kim WJ, Cooper JA, et al. A new function for a phosphotyrosine phosphatase: linking GRB2-Sos to a receptor tyrosine kinase. Mol Cell Biol. 1994;14:509–17. doi: 10.1128/mcb.14.1.509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yu WM, Hawley TS, Hawley RG, Qu CK. Catalytic-dependent and -independent roles of SHP-2 tyrosine phosphatase in interleukin-3 signaling. Oncogene. 2003;22:5995–6004. doi: 10.1038/sj.onc.1206846. [DOI] [PubMed] [Google Scholar]
- 38.Bennett AM, Tang TL, Sugimoto S, Walsh CT, Neel BG. Protein-tyrosine-phosphatase SHPTP2 couples platelet-derived growth factor receptor beta to Ras. Proc Natl Acad Sci U S A. 1994;91:7335–9. doi: 10.1073/pnas.91.15.7335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Liu X, Lei M, Erikson RL. Normal cells, but not cancer cells, survive severe Plk1 depletion. Mol Cell Biol. 2006;26:2093–108. doi: 10.1128/MCB.26.6.2093-2108.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Cleveland DW, Mao Y, Sullivan KF. Centromeres and kinetochores: from epigenetics to mitotic checkpoint signaling. Cell. 2003;112:407–21. doi: 10.1016/s0092-8674(03)00115-6. [DOI] [PubMed] [Google Scholar]
- 41.Hauf S, Cole RW, LaTerra S, Zimmer C, Schnapp G, Walter R, et al. The small molecule Hesperadin reveals a role for Aurora B in correcting kinetochore-microtubule attachment and in maintaining the spindle assembly checkpoint. J Cell Biol. 2003;161:281–94. doi: 10.1083/jcb.200208092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yamamoto Y, Matsuyama H, Kawauchi S, Matsumoto H, Nagao K, Ohmi C, et al. Overexpression of polo-like kinase 1 (PLK1) and chromosomal instability in bladder cancer. Oncology. 2006;70:231–7. doi: 10.1159/000094416. [DOI] [PubMed] [Google Scholar]
- 43.Takahashi T, Sano B, Nagata T, Kato H, Sugiyama Y, Kunieda K, et al. Polo-like kinase 1 (PLK1) is overexpressed in primary colorectal cancers. Cancer Sci. 2003;94:148–52. doi: 10.1111/j.1349-7006.2003.tb01411.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Smith SL, Bowers NL, Betticher DC, Gautschi O, Ratschiller D, Hoban PR, et al. Overexpression of aurora B kinase (AURKB) in primary non-small cell lung carcinoma is frequent, generally driven from one allele, and correlates with the level of genetic instability. Br J Cancer. 2005;93:719–29. doi: 10.1038/sj.bjc.6602779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sorrentino R, Libertini S, Pallante PL, Troncone G, Palombini L, Bavetsias V, et al. Aurora B overexpression associates with the thyroid carcinoma undifferentiated phenotype and is required for thyroid carcinoma cell proliferation. J Clin Endocrinol Metab. 2005;90:928–35. doi: 10.1210/jc.2004-1518. [DOI] [PubMed] [Google Scholar]
- 46.Simizu S, Osada H. Mutations in the Plk gene lead to instability of Plk protein in human tumour cell lines. Nat Cell Biol. 2000;2:852–4. doi: 10.1038/35041102. [DOI] [PubMed] [Google Scholar]
- 47.van Vugt MA, van de Weerdt BC, Vader G, Janssen H, Calafat J, Klompmaker R, et al. Polo-like kinase-1 is required for bipolar spindle formation but is dispensable for anaphase promoting complex/Cdc20 activation and initiation of cytokinesis. J Biol Chem. 2004;279:36841–54. doi: 10.1074/jbc.M313681200. [DOI] [PubMed] [Google Scholar]
- 48.Zachos G, Black EJ, Walker M, Scott MT, Vagnarelli P, Earnshaw WC, et al. Chk1 is required for spindle checkpoint function. Dev Cell. 2007;12:247–60. doi: 10.1016/j.devcel.2007.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.