

Abstract
The follicle-associated epithelium (FAE) overlying the Peyer's patches and the microfold cells (M cells) within it are important sites of antigen transcytosis across the intestinal epithelium. Using a meta-analysis approach, we identified a transcriptional signature that distinguished the FAE from a large collection of mouse cells and tissues. A co-expressed cluster of 21 FAE-specific genes was identified, and the analysis of the transcription factor binding site motifs in their promoter regions indicated that these genes shared an underlying transcriptional programme. This cluster contained known FAE- (Anxa10, Ccl20, Psg18 and Ubd) and M-cell-specific (Gp2) genes, suggesting that the others were novel FAE-specific genes. Some of these novel candidate genes were expressed highly by the FAE and M cells (Calcb, Ces3b, Clca2 and Gjb2), and others only by the FAE (Ascl2, Cftr, Fgf15, Gpr133, Kcna1, Kcnj15, Mycl1, Pgap1 and Rps6kl). We also identified a subset of novel FAE-related genes that were induced in the intestinal epithelium after receptor activator of nuclear factor (NF)-κB ligand stimulation. These included Mfge8 which was specific to FAE enterocytes. This study provides new insight into the FAE transcriptome. Further characterization of the candidate genes identified here will aid the identification of novel regulators of cell function in the FAE.
Keywords: follicle-associated epithelium, M cells, intestine, meta-analysis, clustering
1. Introduction
The luminal surface of the intestine limits the access of pathogenic microorganisms to the underlying host tissues and is protected by a single layer of epithelial cells bound by tight-junctions. Located within the follicle-associated epithelia (FAE) overlying the organized mucosal lymphoid aggregates including Peyer's patches, colonic patches, isolated lymphoid follicles, the nasal-associated lymphoid tissues (NALTs) and occasionally within villous epithelia are microfold cells (M cells). This unique subset of epithelial cells is characterized by an irregular brush border, a reduced glycocalyx and lysosomes and is specialized for the transcytosis of macromolecules and particulate antigens.1,2 Following their uptake and transcytosis by M cells, antigens exit into the intraepithelial pocket on the basolateral membrane where they are subsequently processed by macrophages and classical dendritic cells (DCs). As a consequence, the FAE and the M cells within it enable the host's immune system to efficiently sample the intestinal lumen and mount an appropriate immune response.
Although the FAE as an important site of mucosal antigen sampling, little is known about the genes which influence the development and function of the cells within it. The tumour necrosis factor (TNF) superfamily member receptor activator of nuclear factor (NF)-κB ligand (RANKL) is selectively expressed by subepithelial stromal cells beneath the FAE in Peyer's patches.1 RANKL signals via its receptor RANK which is expressed by epithelial cells throughout the intestine. The production of RANKL by the subepithelial stromal cells beneath the FAE has been shown to be a critical factor that stimulates the differentiation of RANK-expressing enterocytes into M cells.1 Furthermore, M cells are depleted in vivo by RANKL neutralization and are absent in RANKL-deficient mice. Stimulation from other TNF superfamily member ligands via TNF receptor and lymphotoxin β receptor,3 and factors produced by pathogenic bacteria such as cholera toxin (CT)4 have also been shown to influence gene expression in the FAE. Recent data also show that Spi-B is an important transcription factor that acts downstream of RANKL–RANK signalling to control the terminal differentiation of mature M cells.5,6
Our previous meta-analyses of diverse ranges of primary cells and cell lines7 using the novel network graph tool Biolayout Express3D8,9 show that clusters of genes that are correlated in their expression across large data sets give clear insights into the transcriptional networks of specific tissue or cell types (e.g. the haematopoietic and mesenchymal lineages) and specific cellular functions (e.g. phagocytosis, cell cycle, extracellular matrix). In the current study, we reasoned that by examining a diverse collection of publicly available microarray data from distinct mouse cell populations and tissues, we would be able to identify a co-expressed gene signature that was specific to the FAE and thereby gain insights into the genes that regulate the activity of the cells within it. We also compared the effects of in vivo and in vitro RANKL stimulation on gene expression in the intestinal epithelium. As a consequence, a transcriptional signature was identified that distinguished the FAE from all the other cell and tissue data sets included in this analysis. This study also provides new insight into the effects of RANKL stimulation on gene expression in the FAE. Further characterization of the candidate genes identified in the current study will aid the identification of novel regulators of cell function in the FAE.
2. Materials and methods
2.1. Selection of gene expression data sets
Gene expression data sets were selected from the GEO database based on the following three criteria: (i) cell type studied; (ii) chip platform (Affymetrix mouse genome MOE430 2.0 expression arrays) and (iii) availability of raw data (.cel). Raw data (.cel) files were normalized using Robust Multichip Analysis (RMA Express; http://rmaexpress.bmbolstad.com/). Samples were then arranged according to cell-type grouping [intestine, bone marrow (BM) progenitors, myeloid cells, classical DC, lymphocytes, mesenchymal, tissues etc.; Supplementary Table S1]. We also considered data sets from the villous epithelia of mice treated with recombinant RANKL performed on Affymetrix mouse gene 1.0 ST expression arrays5 and in vitro RANKL-stimulated small intestinal organoids performed on Agilent 4 × 44 K whole mouse genome expression arrays (GSE38785).6
2.2. Network analysis
A sample-to-sample correlation matrix was first calculated from the normalized and non-log transformed gene expression data. The matrix was then imported into BioLayout Express3D8,9 and a graph plotted using all sample-to-sample relationships ≥0.84. Each node represents an individual data set (tissue/cell) and the edges which link these data sets represent Pearson's correlation coefficients ≥0.84. Next, a pairwise transcript-to-transcript Pearson correlation matrix was calculated based on each transcript's profile across all samples. A Pearson correlation coefficient cut-off threshold of r ≥ 0.85 was selected and an undirected network graph of these data was generated. In this graph, the nodes represent individual probe sets (genes/transcripts) and the edges between them, Pearson's correlation coefficients ≥0.85. The network was then clustered into groups of probe sets sharing similar profiles using the Markov clustering algorithm using an inflation value (which controls the granularity of clustering)9 of 2.2. Genes in the clusters of interest were assessed for cellular function using literature review and the web-based analysis tools: Ensembl (http://www.ensembl.org/index.html), GSEA MSigDB (http://www.broadinstitute.org/gsea/msigdb/index.jsp) and GOstat (http://gostat.wehi.edu.au).
2.3. Transcription factor binding site motif analysis
RefSeq IDs for each transcript on the Affymetrix MOE430_2 array that were present in the cluster of interest derived from the network graph (i.e. had at least one correlation with another transcript with Pearson's r ≥ 0.85) were obtained from the NetAffx database (https://www.affymetrix.com/analysis/netaffx/index.affx). In order to further improve the accuracy of transcriptional start site (TSS) identification, the FANTOM database of mouse cap analysis of gene expression (CAGE) tags and expression (http://fantom.gsc.riken.jp/4/download/Tables/mouse/CAGE/promoters/tag_clusters/)10 was used to identify true TSS. By sequencing transcripts from the 5′ end and then mapping them to the genome, CAGE provides the state-of-the-art accuracy for the identification of TSS. The most abundantly transcribed CAGE tag in the FANTOM 3 data set within 1000 bp up- or downstream of the annotated RefSeq TSS was taken as the TSS for that gene. Promoter sequences 300 bp upstream and 100 bp downstream of the CAGE-defined TSS were extracted from the mouse genome sequence (version mm9). Transcription factor binding site (TFBS) motifs were identified using the JASPAR CORE 2008 motif set (http://jaspar.cgb.ki.se), and Clover (P ≤ 0.01, score threshold = 6) was used to detect over-represented motifs in the promoters of each gene in cluster 65 compared with a ‘background set’ made up of the 2000-bp upstream sequence from all mouse genes.11
2.4. Mice
Adult (6–12-week-old) C57BL/6 mice and Mfge8−/− mice12 were maintained under specific pathogen-free conditions. All studies using experimental mice and regulatory licences were approved by the University of Edinburgh's Ethical Review Committee and performed under the authority of a UK Home Office Project Licence within the regulations of the UK Home Office ‘Animals (scientific procedures) Act 1986’.
2.5. Immunohistochemistry
Serial frozen sections (10 µm in thickness) of Peyer's patches were cut on a cryostat and immunostained with the following antibodies: rat anti-mouse GP2 monoclonal antibody (mAb; MBL International, Woburn, MA, USA) or rabbit anti-annexin-A5 (ANXA5) polyclonal antibody (pAb; Abcam, Cambridge, UK) to detect M cells, goat anti-mouse CCL20 pAb (R&D Systems, Abingdon, UK), Alexa Fluor 488-conjugated hamster anti-mouse CD11c mAb (clone N418; eBioscience, Hatfield, UK), biotinylated rat anti-mouse CD68 mAb (clone FA-11; AbD Serotec, Oxford, UK), biotinylated rat anti-E-cadherin (clone DECMA-1; eBioscience), rabbit anti-human KCNJ15 pAb (Protein Tech Group, Chicago, IL, USA), biotinylated rat anti-mouse CD107a (LAMP-1) mAb (clone 1D4B; Biolegend, Cambridge, UK), rat anti-mouse milk fat globule epidermal growth factor (EGF) factor 8 (MFG-E8) mAb (clone FDC-M1; BD Pharmingen, Oxford, UK) and rabbit anti-ubiquitin D pAb (Protein Tech Group). F-actin was detected using Alexa Fluor 647-conjugated phalloidin (Invitrogen, Paisley, UK). Unless indicated otherwise, following the addition of the primary antibody, species-specific secondary antibodies coupled to Alexa Fluor 488 (green) and Alexa Fluor 555 (red) dyes were used (Invitrogen). TUNEL+ cells were detected using the ApopTag Plus Fluorescence In Situ Apoptosis Detection Kit (Merck Millipore, Billerica, MA, USA). Sections were mounted in fluorescent mounting medium (Dako, Ely, UK) and examined using a Zeiss LSM5 confocal microscope (Zeiss, Welwyn Garden City, UK).
2.6. Statistical analysis
Unless indicated otherwise, significant differences between groups were sought by ANOVA. P-values <0.05 were accepted as significant.
3. Results
3.1. Comparison of the cell and tissue gene expression profiles
A total of 130 gene expression data sets generated on a single microarray platform (Affymetrix MOE430-2.0) were collected representing 60 different primary mouse cells, tissues and cell lines (Supplementary Table S1). To examine the relationships between samples, a Pearson correlation matrix was first calculated based on the global expression similarities between these normalized data. The matrix was then imported into Biolayout Express3D8,9 and a graph created of the sample-to-sample correlations using the Pearson correlation relationships r ≥ 0.84 to define edges (Fig. 1). Irrespective of the source of these data sets, the different populations of intestinal, lymphoid, myeloid and mesenchymal cells clustered together and occupied specific regions of the graph. For example, most cells and tissues of intestinal origin clustered together in a distinct region of the graph (clusters 4, 5 and 10; Fig. 1). Similarly, most of the haematopoietic and lymphoid cells (clusters 2 and 13), phagocytes (cluster 3) and cells of mesenchymal origin (cluster 1) clustered together in separate regions of the graph. The CT-stimulated villous epithelium data sets were located within a different cluster from the Peyer's patch M cell and FAE data sets (clusters 5 and 4, respectively) implying distinct expression profiles.
Figure 1.

Clustering of samples based on their global gene expression profile. A Pearson correlation matrix was prepared by comparing data derived from all 130 samples used in this study performed on the Affymetrix mouse genome MOE430 2.0 expression array. A graph was constructed using sample-to-sample relationships greater than r ≥ 0.84. Nodes represent samples (individual chips) and edges are coloured according to the strength of the correlation (red, r = 1.0; blue, r ≥ 0.84). Full descriptions of the sources of each data set are given in Supplementary Table S1.
3.2. Identification of cell- and tissue-specific co-expressed genes signatures
Next, a full probe set-to-probe set Pearson correlation matrix was calculated whereby the similarity in the expression profile of each probe set represented on the array was compared across all 130 samples. The resulting three-dimensional network graph contained 18 213 nodes representing individual probe sets connected by 476 858 edges indicating Pearson's correlations r ≥ 0.85. After clustering, 349 clusters of ≥7 nodes were obtained and their descriptions and contents are provided in Supplementary Tables S2 and S3, respectively. To enable this network graph to be explored in greater detail, these data will be made available as a network graph on the authors' institutional website (http://www.roslin.ed.ac.uk/neil-mabbott).
The network graph's topology is derived from cliques of genes which are co-expressed (Fig. 2). Clusters containing genes with related expression patterns were typically localized within similar neighbourhoods of the network graph (Fig. 2B). For example, clusters 2, 17, 36 and 49 were highly expressed by all the intestine-derived data sets and occupied the same region of the network graph, adjacent to the other intestine-related clusters (Fig. 2). Clusters 3, 25 and 40 were highly expressed by Paneth cells and occupied a distinct region of the graph distant from the other intestine-related clusters, but adjacent to the rest of the Paneth cell-related clusters (Fig. 2). Other clusters, such as those expressed highly by B cells (cluster 22), mononuclear phagocytes (cluster 24) and other lymphocyte, leukocyte and haematopoietic cell lineages occupied a specific region of the graph geographically distant from the intestine-related clusters.
Figure 2.

Network analysis of mouse cell and tissue transcriptomics data. (A) Main component of the network graph derived from 130 samples of mouse cell and tissue populations run on Affymetrix MOE430_2 arrays. Nodes represent transcripts (probe sets) and the edges represent correlations between individual expression profiles above r ≥ 0.85. The boxed area (right) shows three representative clusters derived from the network graph and their mean probe set expression profile across all data sets. BM prog., bone marrow progenitor cells; DC, dendritic cell; pDC, plasmacytoid DC; FDC, follicular dendritic cell; Endo., endothelium; LTi, lymphoid tissue-inducer cells; NH, natural helper. (B) Shadow image of the network graph (edges only indicated by grey lines) with the boundaries of distinct transcriptional networks with related function indicated. Cluster 65 (FAE and Peyer's patch M cell-related) is highlighted.
The average expression profile of the probe sets (and the genes they encode) within the individual clusters can give a strong indication of the biology they encode. Many tissue/cell-specific gene clusters were identified. The mean expression profiles of the genes in a selection of example clusters derived from the network graph are shown in Fig. 3, and the mean expression profiles of every cluster are provided in Supplementary Fig. S1. For example, cluster 13 contained many genes characteristically expressed by mesenchymal cells, especially those encoding components of the extracellular matrix (Fig. 3A). Cluster 22 contained genes characteristically expressed by B cells including those encoding immunoglobulins and the major histocompatibility complex class II complex. Cluster 31 contained genes related to T cell activity, cluster 24 was enriched with genes related to mononuclear phagocyte activity including constituents of the lysosomal proton pump (Atp6v0a1etc.; Fig. 3B). Cluster 20 was enriched with genes expressed by natural helper cells, whereas cluster 78 was enriched with genes expressed by lymphoid tissue-inducer cells (Fig. 3C). Several Paneth cell-related clusters were also identified which contained typical genes related to their activity including (Fig. 3D). Some clusters were expressed highly across all samples and predominantly contained housekeeping genes. For example, clusters 19, 21 and 54 were enriched cell cycle-related genes (Fig. 3E), whereas cluster 16 contained genes involved with RNA processing (Fig. 3F).
Figure 3.
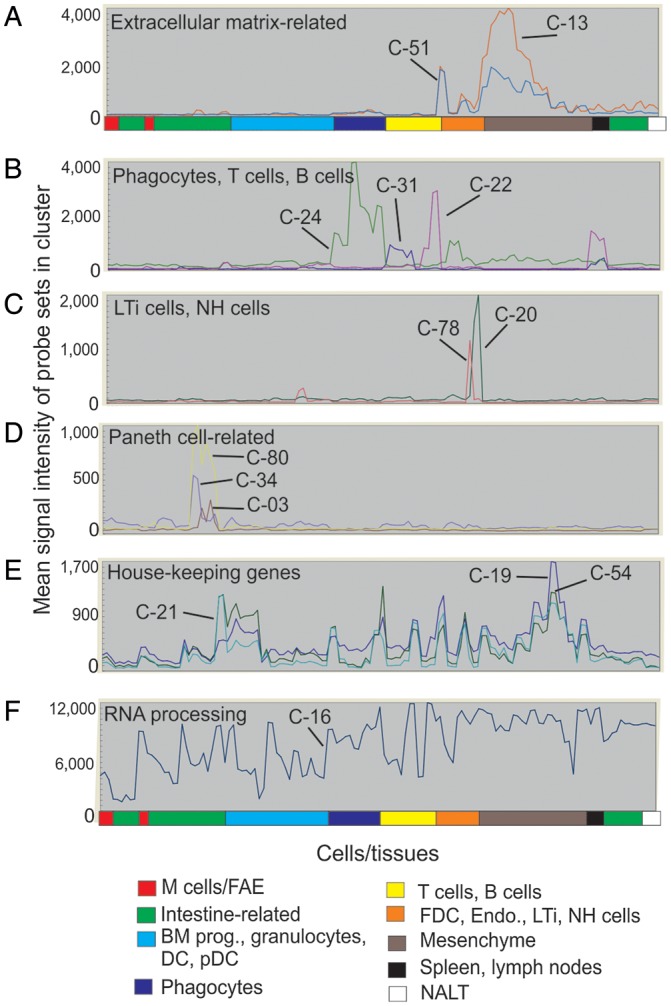
The average expression profiles of the genes in selected clusters over the 130 samples. x-axis shows samples, grouped according to the cell type (in order of presentation in Supplementary Table S1). y-axis shows mean signal expression intensity for the cluster (probe set intensity). Expression profiles of: (A) extracellular matrix-related gene clusters (cluster 13, red; cluster 51, blue); (B) phagocytes (green, cluster 24), B cells (red, cluster 22) and T cells (blue, cluster 31); (C) LTi cells (red, cluster 78) and natural helper cells (green, cluster 20); (D) Paneth cells (red, cluster 3; blue, cluster 34; yellow, cluster 80); (E) house-keeping (e.g. cell cycle) genes (dark blue, cluster 19; light blue, cluster 21; green, cluster 54); (F) RNA processing genes (cluster 16).
3.3. Identification of an FAE-specific co-expressed gene signature
Our main aim was to use this clustering approach to gain further insights into the biology of the FAE. Cluster 65 contained 29 probe sets encoding 21 annotated genes and the mean expression profile of this cluster was restricted to the FAE and M cells (Fig. 4A). The mean expression level of these genes in the FAE samples was significantly higher when compared with the villous epithelium [ileum epithelial cells, small intestine (SI) epithelium and ileum samples; Fig. 4B]. Several of these genes had previously been reported to be expressed highly throughout the FAE (Anxa10, Ccl20, Psg18 and Ubd), whereas Gp2 is specifically expressed by the M cells within it.2,4,13–15 The expression of CCL20, ubiquitin D and GP2 in the FAE is shown in Fig. 4C. By using the principle of ‘guilt-by-association’, these data suggested that the remaining genes in cluster 65 were also related to FAE function. Analysis of the expression profiles of these potentially novel genes across the different data sets indicated that Calcb, Ces3b, Clca2 and Gjb2 were expressed at highly significant levels within the FAE and Peyer's patch M cell samples when compared with the villous epithelium. Others including Ascl2, Cftr, Fgf15, Gpr133, Kcna1, Kcnj15, Mycl1, Pgap1 and Rps6kl were expressed in the FAE, but not by M cells (Fig. 4B).
Figure 4.

Expression profiles of the genes within cluster 65. (A) The mean expression profile of the probe set intensities within cluster 65 over the 130 samples. The red-boxed area indicates the Peyer's patch M cell data sets; the blue-boxed area indicates the FAE data sets. The cluster 65 nodes derived from the network graph are also shown. (B) Heat map showing the mean expression of all probe sets within cluster 65 in samples from enriched Peyer's patch M cells (PP M cells), CT-stimulated villous epithelium (CT-stim. villous epith.), ileum epithelial cells (ileum IEC), FAE, SI epithelium (SI epith.), ileum and NALT. Each column represents the mean (log2) probe set intensity for all samples from each source (n = 2–4). P-values for those genes which were expressed significantly within the FAE at levels >2.0-fold when compared with the villous epithelium (ileum IEC, SI epithelium, ileum) are indicated. (C) IHC confirmation of the expression of proteins encoded by FAE-related genes (CCL20 and ubiquitin D, red) and M cell-related (GP2+ cells, green, arrows) in mouse Peyer's patches. SED, subepithelial dome. Dotted line indicates the luminal surface of the FAE.
CT stimulation is considered to promote the differentiation of villous enterocytes into M cells.4 However, it was noticeable that the CT-stimulated villous epithelium data sets lacked the expression of most of the FAE- and M-cell-related genes in cluster 65 including Gp2 (Fig. 4B) and were situated in a distinct cluster from the FAE and Peyer's patch M cell data sets in the sample-to-sample analysis (Fig. 1). The CT-stimulated villous epithelium data sets were enriched using a mAb which specifically binds sites of α-1,2 fucosylation.4 Although murine M cells express high levels of α-1,2 fucose, the lack of the significant expression of M cell- and FAE-related genes in the CT-stimulated villous epithelium data sets indicates that they most likely contained a mixed population of intestinal epithelial cells that were fucosylated by fucosyltransferase 2 as part of a rapid stress response to oral CT administration.16
3.4. KCNJ15 expression in the SI is restricted to the FAE
Among the genes in cluster 65 that had not previously been reported to be expressed within the FAE was Kcnj15 which encodes for potassium inwardly rectifying channel, subfamily J, member 15 (KCNJ15). The specificity of the Kcnj15 probe sets was confirmed using the RefDIC probe mapping tool (http://refdic.rcai.riken.jp/tools/id_on_map.cgi) which predicts whether or not each probe set recognizes coding regions of target genes17 (Fig. 5A). A large diversity of potassium channels has been described. However, in the intestine, Kcnj15 was the only subfamily J potassium channel-encoding gene represented on the microarray that was significantly and specifically expressed only within the FAE when compared with the villous epithelium samples (Fig. 5B). Immunohistochemistry (IHC) analysis demonstrated that KCNJ15 was distributed within the cytoplasm of most cells throughout the FAE, but appeared to be absent in GP2+ M cells (Fig. 5C). Consistent with the microarray data, the villous epithelium lacked the expression of KCNJ15. These data support the conclusion that the genes within cluster 65 are expressed highly by cells in the FAE.
Figure 5.

KCNJ15 is expressed throughout the FAE, but not by M cells or the villous epithelium. (A) The Affymetrix probe sets registered for Kcnj15 which were co-expressed within cluster 65 were surveyed with the RefDIC probe-mapping tool to confirm that they recognized exons within this gene (red bars). NM_019664 and NM_001039056 accession numbers of reference sequences. (B) Heat map showing the expression of profile throughout the intestine of probe sets encoding KCNJ potassium channels (a representative probe set is shown when multiple probe sets for a gene were present on the array). Each column represents the mean probe set intensity (log2) for all samples from each source (n = 2–4). Kcnj15 was the only gene which was expressed significantly within the FAE at levels >2.0-fold when compared with the villous epithelium. The red-boxed area indicates the mAb NKM 16-2-4-enriched Peyer's patch M cell data sets; the blue-boxed area indicates the FAE data sets. (C) IHC analysis of mouse Peyer's patches shows the cytoplasmic expression of KCNJ15 (red) throughout cells within the FAE, but not in M cells (GP2+ cells, green, arrows) or the villous epithelium. SED, subepithelial dome. Dotted line indicates the border of the FAE.
3.5. TFBS motif analysis of the promoters of genes within cluster 65
We next determined whether the genes within cluster 65 shared transcription factor-responsive elements in their promoters. For each transcript identified in cluster 65, the region −300 to +100 bp of the CAGE-defined TSS was extracted from the mouse genome. Of the probe sets within cluster 65, one (1419775_at) did not have an associated RefSeq ID. For the remaining probe sets, 28 unique RefSeq IDs were identified in the RefSeq mm9 database; however, for three probe sets (1449426_a_at, 1422639_at and 1421658_x_at), no CAGE TSS was found.
Significantly over-represented TFBS enrichment in the set of gene promoters within cluster 65 was then assessed using Clover analysis (P-value threshold ≤0.01).11 The Clover score estimates the degree of over-representation of a given sequence by estimating the thermodynamic binding energy of a transcription factor to the promoter sequences of genes in a given cluster and comparing this to the binding energy of the transcription factor to a control sequence (in this study, the 2000 bp region upstream of all mouse gene promoters).18 The transcription factors that had the highest positive correlations are shown in Supplementary Table S4. The genes within cluster 65 were significantly enriched with 11 candidate TFBS motifs supporting the view that the cluster was distinct and derived from an underlying transcriptional programme. For example, among these, significantly enriched motifs were those for NF-κB and two of its subunits Rel and RelA. The implications of these data are discussed below.
3.6. Effect of RANKL stimulation on the expression of FAE-related genes in the villous epithelium
We next examined gene expression data from the villous epithelium of mice treated with recombinant RANKL performed on Affymetrix mouse gene 1.0 ST expression arrays.5 These data were normalized, annotated and the expression levels of the genes present within cluster 65 compared. Unfortunately, as these samples were analysed on a different microarray platform, it was not possible to combine these data sets with those used in the cluster analysis above. Expression data for 18 orthologs of the 21 annotated genes within cluster 65 were available. Among those genes, nine demonstrated a significant >2.0-fold increase in the villous epithelium after RANKL-treatment including Ccl20, Clca2, Gjb2, Gpr133, Mycl1, Ubd and the M cell-specific marker Gp2 (Supplementary Fig. S2). However, the expression of Anxa10, Ascl2, Kcna1, Kcnj15 and Rps6kl1 was not significantly influenced. These data show that although stromal cells beneath the FAE express high levels of RANKL, not all FAE-related genes are directly dependent on RANKL stimulation for their expression.
We next determined the effect of RANKL stimulation on the expression of other FAE- and M cell-related genes. To do so, we identified the genes which were significantly induced >2.0-fold by RANKL stimulation in the villous epithelium and also expressed highly (significantly >2.0-fold) in the enriched Peyer's patch M cells and FAE samples, when compared with the other intestine-related samples. This analysis identified 369 individual genes which were expressed highly by the Peyer's patch M cells and FAE samples, and of these, orthologs for 48 were significantly induced by RANKL stimulation in the villous epithelium (Supplementary Table S5). Among those genes were several, in addition to those in cluster 65, which were known to be expressed highly in the FAE and by M cells including Anxa5, Ccl9, Clu, Ctsh, Icam1, Krt7, Marcksl1, Prnp, Scg5, Spib and Tnfaip2.2,4,5,13,19–22 However, the expression of 32 genes had not implying that they were novel RANKL-dependent FAE-specific genes.
In order to further exclude the possibility that the expression of some genes may be due to the presence of mononuclear phagocytes in the original samples, we also analysed expression data from RANKL-stimulated in vitro-cultivated small intestinal organoids. Although important additional stimuli such as those provided by lymphocytes22,23 may be absent in the in vitro organoids, the expression of 30 of the RANKL-dependent M cell- and/or FAE-related genes identified above were also induced in the in vitro organoids data sets (Supplementary Table S5).
3.7. Milk fat globule EGF factor 8 is expressed highly by FAE enterocytes
Among the novel genes that were significantly up-regulated by RANKL treatment in the villous epithelium in vivo, and in small intestinal organoids in vitro, was Mfge8 which encodes MFG-E8 (Supplementary Table S5). IHC was used to confirm the expression of MFG-E8 in the FAE. As anticipated, high levels of MFG-E8 were detected in association with mononuclear phagocytes in the B cell follicles and subepithelial domes (SEDs) of Peyer's patches (Fig. 6A, arrowheads). However, strong MFG-E8 immunolabelling was also detected within epithelial cells within the FAE (Fig. 6A, arrow). No MFG-E8 expression was detected in Mfge8−/− mice (Fig. 6A). Since both the anti-GP2 mAb and anti-MFG-E8 mAb were raised in rats, ANXA5 was used as an alternative marker to detect M cells.20 Our analysis showed that MFG-E8 was strongly expressed within FAE enterocytes but not by M cells, since ANXA5+ cells (Fig. 6B, arrows) lacked MFG-E8. Enterocytes in the FAE, but not the villous epithelium, contain large LAMP1+ endosomes towards their apical surfaces.24 In FAE enterocytes, the MFG-E8 was preferentially co-localized to these large LAMP1+ endosomes (Fig. 6C).
Figure 6.

IHC analysis of MFG-E8 expression in the SI epithelium. (A) Strong MEG-E8 immunolabelling was detected in the FAE of WT mice with increasing intensity towards the apex (green, left-hand panels, arrow). No MFG-E8 expression was detected throughout the villous epithelium of WT mice. Arrowheads indicate high levels of MFG-E8 in association with mononuclear phagocytes. No MFG-E8 expression was detected in the intestines of Mfge8−/− mice (right-hand panels). SED, subepithelial dome; FO, B cell follicles. (B) Inclusions of MEG-E8 (red) were detected towards the apical surfaces, FAE enterocytes, but not ANXA5+ M cells (green, arrows). (C) MEG-E8 (red) preferentially co-localized to the large LAMP1+ endosomes (green) of FAE enterocytes (arrows). (D) Although occasional CD11c+ cells were detected within the FAE (green; arrow-heads), and some appeared to contain cytoplasmic inclusions of MFG-E8 (arrow), the majority of the MFG-E8 (red) was detected on the apical surfaces of epithelial cells. (E) Many large MFG-E8-containing CD68+ macrophages were detected in the SED (arrowheads), but were rarely detected in the FAE. Dotted lines indicate the border of the FAE.
In the SED, numerous CD11c+ cells (indicative of classical DC) were observed with cytoplasmic inclusions of MFG-E8 (Fig. 6D, arrow). Although occasional CD11c+ cells were detected in the FAE (Fig. 6D, arrowheads), and some contained cytoplasmic inclusions of MFG-E8 (Fig. 6D, arrow), the majority of the MFG-E8 was epithelial cell-associated. MFG-E8-containing CD68+ macrophages were also detected in the SED (Fig. 6E, arrowheads), but were rare in the FAE. These data clearly show that in the FAE, MFG-E8 expression was restricted to the large LAMP1+ endosomes of the enterocytes and was not simply due to the presence of mononuclear phagocytes. This conclusion is consistent with the induction of Mfge8 expression in the in vitro small intestinal organoids which contained only epithelial cells (Supplementary Table S5).6
3.8. Mfge8 deficiency does not impair apoptotic cell engulfment in the SED
MFG-E8 aids the phagocytic clearance of apoptotic cells by mononuclear phagocytes.12,25 The mononuclear phagocytes in Peyer's patches comprise a heterogeneous population of classical DC and macrophages, and our data show that the majority of those in the FAE and B cell follicles (including tingible body macrophages) express CD11c.26 In the SED of Peyer's patches, cytoplasmic inclusions of MFG-E8 and the epithelial cell marker E-cadherin were detected in mononuclear phagocytes implying the uptake of apoptotic epithelial cells (Fig. 7A and B, arrows). We therefore determined whether the expression of MFG-E8 by the FAE was important for the engulfment of apoptotic cells by mononuclear phagocytes in the underlying SED by comparing the number of phagocyte-associated apoptotic cells in the Peyer's patches of WT and Mfge8−/− mice. In the SED, the mean number of phagocyte-bound apoptotic cells was similar in Peyer's patches from each group of mice indicating that the engulfment of apoptic cells was unaffected in the absence of MGF-E8 (Fig. 7C). In contrast, the apoptotic cell load of phagocytes in the B cell follicles was significantly higher in Peyer's patches from Mfge8−/− mice (Fig. 7D) consistent with previous data.25 These data show that MFG-E8 expression in the FAE does not regulate the engulfment of apoptotic cells by mononuclear phagocytes within the SED.
Figure 7.

Mfge8 deficiency does not impair apoptotic cell engulfment in the SED. (A) Heavy cytoplasmic inclusions of MEG-E8 (green) and E-cadherin (red) were detected within MNP within the SED (arrows). (B) In the SED of WT and Mfge8−/− mice, strong cytoplasmic inclusions of E-cadherin (red) were detected within CD11c+ MNP (green) indicative of the uptake of apoptotic epithelial cells (arrows). (C and D) Apoptotic cells (TUNEL+, green) and CD11c+ MNP (red) were detected by IHC in the SED (C) and B cell follicles (D) of Peyer's patches from WT and Mfge8−/− mice. Bar charts show the mean number of MNP-bound apoptotic cells in the SED (C) and B cell follicles (D) of Peyer's patches of WT (open bars) and Mfge8−/− mice (closed bars). FO, follicle. Each bar represents data from the analysis of 100–300 MNP from each mouse strain.
4. Discussion
Here, we used a meta-analysis approach to identify a co-expressed transcriptional signature that distinguished the FAE from a large collection of mouse primary cells, tissues and cell lines. A co-expressed gene cluster (cluster 65) was identified which was highly expressed only in the FAE and by M cells and was enriched with known M-cell- (Gp2) or FAE-specific (Anxa10, Ccl20, Psg18 and Ubd)2,4,13–15 genes. Such clusters of co-expressed genes provide an opportunity to identify novel lineage markers and regulators of cell function, as well as functions for uncharacterized genes. For example, recent studies show that the broad complex, tramtrack, bric-a-brac, and zinc finger transcription factor Zbt46 is specifically expressed by all classical DC lineages.27,28 Retrospective analysis of data from our previous comparison of the transcriptomes of different mouse lymphocyte and leukocyte populations shows that Zbtb46 was co-expressed in a cluster of 12 genes whose expression was restricted to classical DC (cluster 79 in Mabbott et al.7). By applying the principle of ‘guilt-by-association’, this suggested that the other genes co-expressed within cluster 65 in the current study were also FAE-related. These novel FAE-related genes could be separated into two groups: those expressed highly within the FAE and by M cells (Calcb, Ces3b, Clca2 and Gjb2) and those expressed in the FAE, but not by M cells (Ascl2, Cftr, Fgf15, Gpr133, Kcna1, Kcnj15, Mycl1, Pgap1 and Rps6kl). These genes could also be further subdivided depending on their requirement for RANKL-stimulation for their expression. The potential functions of the FAE-related genes identified in this study are summarized in Fig. 8 and examples discussed below.
Figure 8.

Cartoon illustrating the putative functions of the FAE- and M-cell-related genes identified in this study. Genes were classified into groupings of related cellular function based on published data from literature searches. Red font, RANKL-dependent genes; plain font, RANKL-independent genes.
Cells in the FAE express high levels of the glycosylphosphatidylinisotol (GPI)-anchored proteins GP2 and cellular prion protein (PrPC, encoded by Prnp).2,4,15 GPI inositol deacylation by post-GPI attachment to proteins 1 (PGAP1, encoded by Pgap1) is important for the efficient transport of GPI-anchored proteins from the endoplasmic reticulum to the Golgi.29 Pgap1 was co-expressed with Gp2 within cluster 65 implying an important role in the expression of GPI-anchored proteins in the FAE. GP2 and PrPC may each act as a transcytotic receptors for mucosal antigens on M cells.15,30 Prnp did not cluster with other genes across this data set, most likely reflecting its ubiquitous cellular expression.
Potassium channels control the apical recycling of potassium ions. Our analysis of the novel genes within cluster 65 revealed that Kcnj15 was specific to the FAE, but was not induced in the villous epithelium after RANKL stimulation. The ion channels Cftr, Clca2 and Kcna1 were also co-expressed within cluster 65, and others (Clca4 and Kctd12) were significantly induced by RANKL stimulation. The role of KCNJ15 and the other ion channels in the FAE is uncertain. Enterocytes within the FAE, unlike those in the villous epithelium, contain large LAMP1+ endosomes.24 These genes may help maintain ion concentrations in the large endosomes of FAE enterocytes.
Although RANKL stimulation is important for the maturation and maintenance of M cells, Tnfrsf11a (which encodes RANK) did not cluster with any other gene across this data set. This is most likely due to the additional roles for RANK–RANKL signalling in the function of osteoclasts31 and lymphoid tissue-inducer cells32 that were included in the analysis. Since RANKL may also promote the survival of BM-derived DC and stimulate their expression of pro-inflammatory cytokines,33 our subtraction analysis identified only those RANKL-induced genes which were highly expressed in the FAE and also by Peyer's patch M cells when compared with the villous epithelium. This approach identified 48 genes, many of which were known to be expressed in the FAE and by M cells including Anxa5, Ccl9, Ccl20, Clu, Ctsh, Gp2, Icam1, Krt7, Marcksl1, Prnp, Scg5, Spib, Tnfaip2 and Ubd or were also identified in the current study in cluster 65 (Clca2, Gjb2 and Mycl1).2,4,5,13,20,21,34,35 This implies that the remaining 32 genes represent novel RANKL-dependent FAE-related genes.
The genes induced by RANKL stimulation encode proteins with a range of FAE- and M-cell-related activities and are summarized in Fig. 8. Many were associated with cytoskeletal regulation (Epb4.4l2, Fyb, Marksl1, Mreg, Myo1b, Reep1, S100a11 and Tnfaip2) and may play a role in the adaptation of the cell towards one specialized for transcytosis.4,21,36–39 The expression of Anxa5, Clu, Gp2, Mfge8 and Prnp may encode proteins that bind to and regulate the host's response to antigen and pathogens in the intestinal lumen.2,15,25,40,41 Similarly, the expression of Ctsh, Ncf4, Serpina1a/1b, Rnase1 and Ubd suggests a role in antigen processing or pathogen degradation. Chemokines and chemokine receptors mediate the attraction of lymphocytes and leukocytes to lymphoid tissues and control their positioning within them. The chemokine genes Ccl9, Ccl20 and Cxcl11 were up-regulated by RANKL stimulation indicating a role in the attraction of specific cell populations towards the FAE. For example, in Peyer's patches, the CCL20-mediated recruitment of a specific B cell subset towards the FAE is important for maintaining M differentiation.22,42 Consistent with this role, Ccl20 was co-expressed with the other FAE-related genes in cluster 65.
Expression of MFG-E8 by the FAE had not previously been reported. The expression of MFG-E8 was significantly up-regulated by RANKL treatment and was preferentially co-localized to the large LAMP1+ endosomes of FAE enterocytes. In the B cell follicles, MFG-E8 produced by stromal follicular DCs aids the phagocytosis of apoptotic cells by acting as a bridging molecule between phosphatidylserine residues on apoptotic cells and integrins on mononuclear phagocytes.12,25 However, the engulfment of apoptotic cells by mononuclear phagocytes within the SED beneath the FAE was not impaired in MFG-E8-deficient mice implying an alternative role for FAE-derived MFG-E8. Data show that MFG-E8 can also act as an anti-inflammatory modulator by inhibiting the LPS-mediated release of pro-inflammatory cytokines.43 The phosphatidylserine-binding protein ANXA5 is also expressed highly in the FAE by M cells.20 In addition to its role in regulating the phagocytosis of apoptotic cells, ANXA5 can bind to Gram-negative bacteria with high affinity via the lipid A domain of LPS, blocking the endotoxin activity.40 Thus, in the FAE, the expression of MFG-E8 by enterocytes and ANXA5 by M cells may each play important anti-inflammatory roles to protect against LPS-mediated endotoxemia.
The promoter analysis of the cluster 65 genes identified over-representation of 11 candidate TFBS motifs supporting the conclusion that the genes in this cluster shared an underlying transcriptional programme. Many of these motifs bind transcription factors that control epithelial cell and enterocyte differentiation and homeostasis. The nuclear transcription factor Y subunit α can regulate the differentiation of a human epithelial colorectal adenocarcinoma cell line.44 Similarly, hepatocyte nuclear factor 1α maintains enterocyte terminal differentiation and cell fate commitment in the intestinal epithelium.45 Two high mobility group box transcription factors were also identified. Sox9 is expressed in intestinal epithelium stem/progenitor cells and in Paneth cells and can regulate homeostasis in the intestinal epithelium.46 Sox17 can promote the growth and differentiation of respiratory epithelial cells.47,48 Motifs for basic helix-loop-helix (bHLH) transcription factors were also identified by the promoter analysis. Furthermore, genes encoding two bHLH transcription factors, L-myc (encoded by Mycl1) and achaete scute-like 2 (encoded by Ascl2), were co-expressed in cluster 65. These transcription factors are important regulators of cell fate in the intestine49 implying a similar role in the FAE.
NF-κB functions as a dimeric transcription factor and plays a central role in regulating gene expression during intestinal inflammation. The identification of enriched binding motifs for NF-κB transcription factor family members is consistent with the role for RANK–RANKL signalling in controlling M cell differentiation.1 RelA has been shown to be required for the homeostatic regulation of cell death and division in the intestinal epithelium under steady-state conditions and for protection during acute inflammation.50 Similarly, in the large intestine, Rel regulates the innate inflammatory response to the gut microflora.51
The chronological comparison of the effects of RANKL stimulation on the expression of the cluster 65 genes revealed that although stromal cells beneath the FAE express high levels of RANKL, not all of the FAE-related genes identified in this study are directly induced by RANKL stimulation. Furthermore, although Ccl20, Clca2, Gpr133 and Ubd were rapidly up-regulated following RANKL stimulation, Gp2 and Psg18 were induced at later times. The promoter analysis indicated that Ccl20, Clca2, Gpr133 and Ubd all contained NF-κB family TFBS motifs, whereas they were absent in Gp2 and Psg18. These data indicate that although RANKL stimulation is important for the induction of M cell differentiation from enterocytes, it does not directly regulate the expression of genes such as Gp2 which are associated with terminally differentiated M cells. High levels of the E26 transformation-specific family transcription factor transcription factor Spi-B (encoded by Spib) were induced in the villous epithelium after RANKL stimulation. Recent data show that Spi-B is an important transcription factor which acts downstream of RANKL–RANK signalling to control the subsequent terminal differentiation of functionally mature M cells.5,6
Data in this study show that gene expression signatures specifically associated with the FAE and the M cells within it can be readily identified from the meta-analysis of a large collection of publicly available gene expression data. Little is known of the genes that specifically regulate the differentiation and function of cells within the FAE. Further characterization of the candidate genes identified in the current study will aid the identification of novel regulators of FAE and M cell function and homeostasis.
Supplementary Data
Supplementary Data are available at www.dnaresearch.oxfordjournals.org.
Funding
This work was supported by project and Institute Strategic Programme Grant funding from the Biotechnology and Biological Sciences Research Council (to N.A.M). A.K. is supported by a Japan Society for the Promotion of Science Fellowship for Research Abroad and natural sciences grant funding from the Mitsubishi Foundation. J.K.B. is supported by a Wellcome Trust Clinical Fellowship. I.R.W. is supported by a grant from the National Institutes of Health (DK-064730).
Supplementary Material
Acknowledgements
We thank Simon Cumming, Bob Fleming, Fraser Laing, Mick Watson and the Pathology Services Group (University of Edinburgh, UK) for excellent technical support.
Footnotes
Edited by Osamu Ohara
References
- 1.Knoop K.A., Kumar N., Butler B.R., et al. RANKL is necessary and sufficient to initiate development of antigen-sampling M cells in the intestinal epithelium. J. Immunol. 2009;183:5738–47. doi: 10.4049/jimmunol.0901563. doi:10.4049/jimmunol.0901563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Nakato G., Fukuda S., Hase K., Goitsuka R., Cooper M.D., Ohno H. New approach for M-cell-specific molecules by screening comprehensive transcriptome analysis. DNA Res. 2009;16:227–35. doi: 10.1093/dnares/dsp013. doi:10.1093/dnares/dsp013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wang J., Lopez-Fraga M., Rynko A., Lo D.D. TNFR and LTβR agonists induce follicle-associated epithelium and M cell specific genes in rat and human intestinal epithelial cells. Cytokine. 2009;47:69–76. doi: 10.1016/j.cyto.2009.05.001. doi:10.1016/j.cyto.2009.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Terahara K., Yoshida M., Igarashi O., et al. Comprehensive gene expression profiling of Peyer's patch M cells, villous M-like cells, and intestinal epithelial cells. J. Immunol. 2008;180:7840–6. doi: 10.4049/jimmunol.180.12.7840. [DOI] [PubMed] [Google Scholar]
- 5.Kanaya T., Hase K., Takahashi D., et al. The Ets transcription factor Spi-B is essential for the differentiation of intestinal microfold cells. Nat. Immunol. 2012;13:729–36. doi: 10.1038/ni.2352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Peyer's patch M cells derived from Lgr5+ stem cells require SpiB and are induced by RankL in cultured “miniguts”. Mol. Cell. Biol. 2012;32:3639–47. doi: 10.1128/MCB.00434-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mabbott N.A., Baillie J.C., Hume D.A., Freeman T.C. Meta-analysis of co-expressed gene signatures in mouse leukocyte populations. Immunobiology. 2010;215:724–36. doi: 10.1016/j.imbio.2010.05.012. doi:10.1016/j.imbio.2010.05.012. [DOI] [PubMed] [Google Scholar]
- 8.Freeman T.C., Goldovsky L., Brosch M., et al. Construction, visualisation, and clustering of transcriptional networks from microarray expression data. PLoS Comp. Biol. 2007;3:2032–42. doi: 10.1371/journal.pcbi.0030206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Theocharidis A., van Dongen S., Enright A.J., Freeman T.C. Network visualisation and analysis of gene expression data using Biolayout Express(3D) Nat. Protocols. 2009;4:1535–50. doi: 10.1038/nprot.2009.177. doi:10.1038/nprot.2009.177. [DOI] [PubMed] [Google Scholar]
- 10.Carninci P., Kasukawa T., Katayama S., et al. The transcriptional landscape of the mammalian genome. Science. 2005;309:1559–63. doi: 10.1126/science.1112014. [DOI] [PubMed] [Google Scholar]
- 11.Frith M.C., Fu Y., Yu L., Chen J.F., Hansen U., Weng Z. Detection of functional DNA motifs via statistical over-representation. Nucleic Acids Res. 2004;32:1372–81. doi: 10.1093/nar/gkh299. doi:10.1093/nar/gkh299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hanamaya R., Tanaka M., Miyasaka K., et al. Autoimmune disease and impaired uptake of apoptotic cells in MFG-E8-deficient mice. Science. 2004;304:1147–50. doi: 10.1126/science.1094359. doi:10.1126/science.1094359. [DOI] [PubMed] [Google Scholar]
- 13.Hase K., Ohshima D., Kawano K., et al. Distinct gene expression profiles characterize cellular phenotypes of follicle-associated epithelium and M cells. DNA Res. 2005;12:127–37. doi: 10.1093/dnares/12.2.127. doi:10.1093/dnares/12.2.127. [DOI] [PubMed] [Google Scholar]
- 14.Kawano K., Ebisawa M., Hase K., et al. Psg18 is specifically expressed in follicle-associated epithelium. Cell Struct. Func. 2007;32:115–26. doi: 10.1247/csf.07014. doi:10.1247/csf.07014. [DOI] [PubMed] [Google Scholar]
- 15.Hase K., Kawano K., Nochi T., et al. Uptake through glycoprotein 2 of FimH+ bacteria by M cells initiates mucosal immune responses. Nature. 2009;462:226–31. doi: 10.1038/nature08529. doi:10.1038/nature08529. [DOI] [PubMed] [Google Scholar]
- 16.Terahara K., Nochi T., Yoshida M., et al. Distinct fucosylation of M cells and epithelial cells by Fut1 and Fut2, respectively, in response to intestinal environmental stress. Biochem. Biophys. Res. Commun. 2011;404:822–8. doi: 10.1016/j.bbrc.2010.12.067. doi:10.1016/j.bbrc.2010.12.067. [DOI] [PubMed] [Google Scholar]
- 17.Hijikata A., Kitamura H., Kimura Y., et al. Construction of an open-access database that integrates cross-reference information from the transcriptome and proteome of immune cells. Bioinformatics. 2007;23:2934–41. doi: 10.1093/bioinformatics/btm430. doi:10.1093/bioinformatics/btm430. [DOI] [PubMed] [Google Scholar]
- 18.Frith M.C., Li M.C., Weng Z. Cluster-binding: finding dense clusters of motifs in DNA sequences. Nucleic Acids Res. 2003;31:3666–8. doi: 10.1093/nar/gkg540. doi:10.1093/nar/gkg540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hopkins A.M., Baird A.W., Nusrat A. ICAM-1: targeted docking for exogenous as well as endogenous ligands. Adv. Drug Delivery Rev. 2004;56:763–78. doi: 10.1016/j.addr.2003.10.043. doi:10.1016/j.addr.2003.10.043. [DOI] [PubMed] [Google Scholar]
- 20.Verbrugghe P., Waelput W., Dieriks B., Waeytens A., Vandesompele J., Cuvelier C.A. Murine M cells express annexin V specifically. J. Pathol. 2006;209:240–9. doi: 10.1002/path.1970. doi:10.1002/path.1970. [DOI] [PubMed] [Google Scholar]
- 21.Hase K., Kimura S., Takatsu H., et al. M-Sec promotes membrane nanotube formation by interacting with Ral and the exocyst complex. Nat. Cell Biol. 2009;11:1427–32. doi: 10.1038/ncb1990. doi:10.1038/ncb1990. [DOI] [PubMed] [Google Scholar]
- 22.Ebisawa M., Hase K., Takahashi D., et al. CCR6hiCD11cint B cells promote M-cell differentiation in Peyer's patch. Int. Immunol. 2011;23:261–9. doi: 10.1093/intimm/dxq478. doi:10.1093/intimm/dxq478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hsieh E.N., Fernandez X., Wang J., et al. CD137 is required for M cell functional maturation but not lineage commitment. Am. J. Pathol. 2010;177:666–76. doi: 10.2353/ajpath.2010.090811. doi:10.2353/ajpath.2010.090811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kujala P., Raymond C., Romeijn M., et al. Prion uptake in the gut: identification of the first uptake and replication sites. PLoS Pathog. 2011;7:e1002449. doi: 10.1371/journal.ppat.1002449. doi:10.1371/journal.ppat.1002449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kranich J., Krautler N.J., Heinen E., et al. Follicular dendritic cells control engulfment of apoptotic bodies by secreting Mfge8. J. Exp. Med. 2008;205:1293–302. doi: 10.1084/jem.20071019. doi:10.1084/jem.20071019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bradford B.M., Sester D., Hume D.A., Mabbott N.A. Defining the anatomical localisation of subsets of the murine mononuclear phagocyte system using integrin alpha X (ITGAX) and colony stimulating factor 1 receptor (CSF1-R) expression fails to discriminate dendritic cells from macrophages. Immunobiology. 2011;216:1228–37. doi: 10.1016/j.imbio.2011.08.006. doi:10.1016/j.imbio.2011.08.006. [DOI] [PubMed] [Google Scholar]
- 27.Satpathy A.T., Wumesh K.C., Albring J.C., et al. Zbt46 expression distinguishes classical dendritic cells and their committed progenitors from other immune lineages. J. Exp. Med. 2012;209:1135–52. doi: 10.1084/jem.20120030. doi:10.1084/jem.20120030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Meredith M.M., Liu K., Darrasse-Jeze G., et al. Expression of the zinc finger transcription factor zDC (Zbtb46, Btd4) defines the classical dendritic cell lineage. J. Exp. Med. 2012;209:1153–65. doi: 10.1084/jem.20112675. doi:10.1084/jem.20112675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tanaka S., Maeda Y., Tashima Y., Kinoshita T. Inositol deacylation of glycosylphosphatidylinositol-anchored proteins is mediated by mammalian PGAP1 and yeast Bst1p. J. Biol. Chem. 2004;279:14256–63. doi: 10.1074/jbc.M313755200. doi:10.1074/jbc.M313755200. [DOI] [PubMed] [Google Scholar]
- 30.Nakato G., Hase K., Suzuki M., et al. Cutting edge: Brucella abortus exploits a cellular prion protein on intestinal M cells as an invasive receptor. J. Immunol. 2012;189:1540–4. doi: 10.4049/jimmunol.1103332. [DOI] [PubMed] [Google Scholar]
- 31.Kong Y.Y., Yoshida H., Sarosi I., et al. OPGL is a key regulator of osteoclastogenesis, lymphocyte development and lymph node organogenesis. Nature. 1999;397:315–23. doi: 10.1038/16852. doi:10.1038/16852. [DOI] [PubMed] [Google Scholar]
- 32.Kim D., Mebius R.E., MacMicking J.D., et al. Regulation of peripheral lymph node genesis by the tumour necrosis factor family member TRANCE. J. Exp. Med. 2000;192:1467–78. doi: 10.1084/jem.192.10.1467. doi:10.1084/jem.192.10.1467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chino T., Draves K.E., Clark E.A. Regulation of dendritic cell survival and cytokine production by osteoprotegerin. J. Leukoc. Biol. 2009;86:933–40. doi: 10.1189/jlb.0708419. doi:10.1189/jlb.0708419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ueki T., Mizuno M., Uesu T., Kiso T., Tsuji S. Expression of ICAM-1 on M cells covering isolated lymphoid follicles of the human colon. Acta Med. Okayama. 1995;49:145–51. doi: 10.18926/AMO/30412. [DOI] [PubMed] [Google Scholar]
- 35.Wong N.A.C.S., Herriot M., Rae F. An immunohistochemical study and review of potential markers of human intestinal M cells. Eur. J. Histochem. 2003;47:143–50. doi: 10.4081/820. [DOI] [PubMed] [Google Scholar]
- 36.Pauker M.H., Riecher B., Fried S., Perl O., Barda-Saad M. Functional cooperation between the proteins Nck and ADAP is fundamental for actin reorganization. Mol. Cell Biol. 2011;31:2653–66. doi: 10.1128/MCB.01358-10. doi:10.1128/MCB.01358-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Montenegro G., Rebelo A.P., Connell J., et al. Mutations in the ER-shaping protein reticulon 2 cause the axon-degenerative disorder hereditary spastic paraplegia type 12. J. Clin. Invest. 2012;122:538–44. doi: 10.1172/JCI60560. doi:10.1172/JCI60560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.He H., Li J., Weng S., Li M., Yu Y. S100A11: diverse function and pathology corresponding to different target proteins. Cell Biochem. Biophys. 2009;55:117–26. doi: 10.1007/s12013-009-9061-8. doi:10.1007/s12013-009-9061-8. [DOI] [PubMed] [Google Scholar]
- 39.Ohbayashi N., Maruta Y., Ishida M., Fukuda M. Melanoregulin regulates retrograde melanosome transport through interaction with the RILP-p150Glued complex in melanocytes. J. Cell Sci. 2012;125:1508–18. doi: 10.1242/jcs.094185. doi:10.1242/jcs.094185. [DOI] [PubMed] [Google Scholar]
- 40.Rand J.H., Wu X.-X., Lin E.Y., Griffel A., Gialanella P., McKitrick J.C. Annexin A5 binds to lipopolysaccharide and reduces its endotoxin activity. mBio. 2012;3:e00292–311. doi: 10.1128/mBio.00292-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Watarai M., Kim S., Erdenebaatar J., et al. Cellular prion protein promotes Brucella infection into macrophages. J. Exp. Med. 2003;198:5–17. doi: 10.1084/jem.20021980. doi:10.1084/jem.20021980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Iwasaki A., Kelsalla B.A. Localization of distinct Peyer's patch dendritic cell subsets and their recruitment by chemokines macrophage inflammatory protein (MIP)-3, MIP-3β, and secondary lymphoid organ chemokine. J. Exp. Med. 2000;191:1381–94. doi: 10.1084/jem.191.8.1381. doi:10.1084/jem.191.8.1381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Aziz M.M., Ishihara S., Mishima Y., et al. MFG-E8 attenuates intestinal inflammation in murine experimental colitis by modulating osteopontin-dependent αvβ3 integrin signaling. J. Immunol. 2009;182:7222–32. doi: 10.4049/jimmunol.0803711. doi:10.4049/jimmunol.0803711. [DOI] [PubMed] [Google Scholar]
- 44.Bevilaccqua M.A., Faniello M.C., Iovine B., Russo T., Cimino F., Costanzo F. Transcription factor NF-Y regulates differentiation of CaCo-2 cells. Arch. Biochem. Biophys. 2002;407:39–44. doi: 10.1016/s0003-9861(02)00436-8. doi:10.1016/S0003-9861(02)00436-8. [DOI] [PubMed] [Google Scholar]
- 45.D'Angelo A., Bluteau O., Garcia-Gonzalez M.A., et al. Hepatocyte nuclear factor 1 alpha and beta control terminal differentiation and cell fate in the gut epithelium. Development. 2010;137:1573–82. doi: 10.1242/dev.044420. doi:10.1242/dev.044420. [DOI] [PubMed] [Google Scholar]
- 46.Bastide P., Darido C., Pannequin J., et al. Sox9 regulates cell proliferation and is required for Paneth cell differentiation in the intestinal epithelium. J. Cell Biol. 2007;178:635–48. doi: 10.1083/jcb.200704152. doi:10.1083/jcb.200704152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Parks K.S., Wells J.M., Zorn A.M., Wert S.E., Whitsett J.A. Sox17 influences the differentiation of respiratory epithelial cells. Dev. Biol. 2006;294:192–202. doi: 10.1016/j.ydbio.2006.02.038. doi:10.1016/j.ydbio.2006.02.038. [DOI] [PubMed] [Google Scholar]
- 48.Lange A.W., Keiser A.R., Wells J.M., Whitsett J.A. Sox17 promotes cell cycle progression and inhibits TGF-beta/Smad3 signaling to initiate progenitor cell behavior in the respiratory epithelium. PLoS One. 2009;4:e5711. doi: 10.1371/journal.pone.0005711. doi:10.1371/journal.pone.0005711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.van der Flier L.G., van Gijn M.E., Hatzis P., et al. Transcription factor Aschaete scute-like 2 controls intestinal stem cell fate. Cell. 2009;136:903–12. doi: 10.1016/j.cell.2009.01.031. doi:10.1016/j.cell.2009.01.031. [DOI] [PubMed] [Google Scholar]
- 50.Steinbrecher K.A., Harmel-Laws E., Sitcheran R., Baldwin A.S. Loss of epithelial RelA results in deregulated intestinal proliferative/apoptotic homeostasis and susceptibility to inflammation. J. Immunol. 2008;180:2588–99. doi: 10.4049/jimmunol.180.4.2588. [DOI] [PubMed] [Google Scholar]
- 51.Wang Y., Rickman B.H., Poutahidis T., et al. c-Rel is essential for the development of innate and T cell-induced colitis. J. Immunol. 2008;180:8118–25. doi: 10.4049/jimmunol.180.12.8118. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.