

Abstract
There is no confocal microscope optimized for single-molecule imaging in live cells and superresolution fluorescence imaging. By combining the swiftness of the line-scanning method and the high sensitivity of wide-field detection, we have developed a, to our knowledge, novel confocal fluorescence microscope with a good optical-sectioning capability (1.0 μm), fast frame rates (<33 fps), and superior fluorescence detection efficiency. Full compatibility of the microscope with conventional cell-imaging techniques allowed us to do single-molecule imaging with a great ease at arbitrary depths of live cells. With the new microscope, we monitored diffusion motion of fluorescently labeled cAMP receptors of Dictyostelium discoideum at both the basal and apical surfaces and obtained superresolution fluorescence images of microtubules of COS-7 cells at depths in the range 0–85 μm from the surface of a coverglass.
Introduction
A number of fundamental questions of cell biology have been solved by imaging single fluorescent molecules in live cells (1–3). Superresolution fluorescence microscopies such as STORM/dSTORM and PALM/fPALM are also based on single-molecule imaging in cells (4–7). This technique, however, has been used only for a small subset of biological problems, since fluorescence signals from single fluorophores are easily overwhelmed by strong cellular autofluorescence, especially in thick mammalian cells. Even though molecular events on cell membranes can be observed with superior signal/noise ratio at the single-molecule level by using total internal reflection fluorescence (TIRF) microscopy (8), most biological events occurring inside the cell are still out of reach.
To overcome this limit of TIRF microscopy, two methodologies were recently proposed. In highly inclined and laminated optical sheet (HILO) microscopy (9), an illumination sheet several microns thick is generated using an incidence angle slightly smaller than the critical angle. Thus, single-molecule events occurring in the range of a few microns from the glass surface could be monitored. However, aberration of oil-immersion objectives limits the imaging region of this microscopy to up to a few microns away from the glass surface. Its relatively poor optical-sectioning capability has been an additional hindrance to its wide application.
In selective plane illumination microscopy (SPIM), a thin illumination sheet is generated using an independent objective lens positioned perpendicular to an imaging objective lens (10–13). Since the illumination sheet can be vertically scanned synchronously with the imaging objective, single-molecule events in the working range of the imaging objective can be monitored (12,13). However, some disadvantages of the original design of SPIM were that it required special sample preparation processes and was incompatible with conventional imaging techniques. New approaches to overcome the limitations of the original SPIM have been tried, specifically, iSPIM (14) and Bessel beam plane illumination (15), but due to the steric hindrance between the detection and illumination objectives, high-numerical-aperture objectives could not be used for the imaging, and single-molecule sensitivity thus could not be achieved.
Confocal microscopy (16) is a traditional approach to effectively minimize background noise. It is capable of achieving submicron optical sectioning by blocking out-of-focus fluorescence using an optimized pinhole. With high-sensitivity point detectors, it can observe single molecules with good detection efficiency. However, the single-pinhole-based confocal microscopy is not considered as a tool for single-molecule imaging in live cells or superresolution fluorescence imaging, because it is too slow or does not provide enough photons for single-molecule imaging in a high-speed mode. To overcome these problems, spinning-disk and line-scan confocal microscopes are available commercially, but at this time, they are not capable of single-molecule sensitivity (17,18). Considering the advantages of confocal microscopy over HILO microscopy or SPIM for single-molecule imaging—good optical-sectioning capability, compatibility with conventional microscopes, flexibility for various applications—development of a confocal microscope with both high imaging speed and good fluorescence detection efficiency will be a great addition to the available technology. Here, we report the development of a video-rate confocal microscope that is capable of single-molecule imaging in live cells. The compatibility of the microscope with superresolution imaging techniques based on single-molecule localization approaches is also demonstrated.
Materials and Methods
Preparation of a thin fluorescent film for the characterization of optical-sectioning capabilities
We mixed 10 μl of 1 μg/μl Cy3 dissolved in dimethylsulfoxide with 300 μl of poly(methyl methacrylate) solution. A thin film was made on a 24 × 40-mm2 coverslip by spin-coating the mixture at 4000 rpm for 40 s. The thickness of the film was measured as 200 nm using an atomic force microscope (MFP-3D, Asylum Research, Santa Barbara, CA). Measurement of sectioning capability was done by using a 532-nm green laser and the intensity of the laser just before the objective was ∼10 mW. The detection of fluorescence signal was done by using an oil-immersion objective lens, and slit width was 30 μm. The area-averaged fluorescence intensities were recorded during z-scanning using a piezo stage.
Single-molecule FRET measurement
To construct the Holliday junction, we purchased the following DNA strands (written from 5′ to 3′) from IDT (Newark, NJ).
b-strand: Cy5-CCCTAGCAAGCCGCTGCTACGG,
h-strand: Cy3-CCGTAGCAGCGCGAGCGGTGGG,
r-strand: biotin-CCCACCGCTCGGCTCAACTGGG,
x-strand: GGGCGGCGACCTCCCAG TTGAGCGCTTGCTAGGG.
The Holliday junction was annealed by slowly cooling the mixture of b-strand (5 μM), h-strand (2.5 μM), r-strand (1.25 μM), and x-strand (2.5 μM) in 10 mM Tris-HCl (pH 8.0) with 50 mM NaCl from 90°C to 4°C for 2 h. DNA molecules were immobilized on a polyethylene glycol-coated surface via biotin-streptavidin interaction, and imaged in an imaging buffer (10 mM Tris-HCl, pH 8.0, and 100 mM MgCl2) with an oxygen-scavenging system (1 mg/mL glucose oxidase, 0.8% glucose, 0.04 mg/mL catalase, and 1 mM Trolox). To limit the width of the imaging area, an additional slit was vertically attached to the confocal slit, and donor and acceptor signals were separated by using an additional dichroic mirror (Fig. S1 in the Supporting Material). Exposure time of the CCD camera was 100 ms and the width of the confocal slit was 30 μm. For excitation, a 532-nm green laser was used, and the intensity of the laser was ∼40 mW.
Sample preparation for the imaging of cAMP receptors of Dictyostelium discoideum
Dictyostelium discoideum expressing Halo-tagged cAMP receptors was grown in a 50-mm cell culture dish containing HL5 medium, as previously described (19). For the measurements, Dictyostelium amoeba was harvested by mildly pipetting and properly fractioning them. They were moved to a 35-mm cell culture dish 1 day before the measurements. To label cAMP receptors, Dictyostelium was washed with IB buffer (5 mM KH2PO4, 5 mM Na2HPO4, pH 6.4) and incubated with Halo-TMR (50 nM; G8252, Promega, Fitchburg, WI) dissolved in IB buffer for 30 min with mild shaking. After the incubation, Dictyostelium was washed with IB buffer three times. The interval between washing steps was 10 min. The cells were harvested by mildly pipetting, moved to a chambered coverglass (Lab Tek II, Nunc, Penfield, NY), and incubated for 10 min for the attachment of the cells to the surface. The chambered coverglass was cleaned just before starting the experiments by sonicating it sequentially in deionized water, 1 M KOH, and ethanol, and finally dried by using N2 gas. For imaging of the cAMP receptor, a 532-nm green laser was used with an intensity of ∼20 mW. The exposure time of the CCD camera was 50 ms, the width of the confocal slit was 40 μm, and an oil immersion objective was used.
Diffusion-coefficient analysis
To track single cAMP receptors, imageJ plug-in program was used (Mosaic, ETH Zurich). Diffusion coefficients of the receptors were calculated as described previously (19). Briefly, a data set of displacement with various lag times was obtained from the trajectories of the molecule. Then, a cumulative histogram of displacement was constructed and the mean-squared displacement was obtained by fitting the histogram with the equation . We repeated the process above for various lag times and obtained a diffusion coefficient by linear fitting of the mean-squared displacement plot. In our analysis, only the particles emitting photons >15 frames before photobleaching were used to calculate the diffusion coefficient. Trajectories from a single amoeba cell generated a single diffusion-coefficient value. Diffusion coefficients for the basal and apical surfaces were obtained from different cells.
Sample preparation for superresolution imaging
The COS-7 cells (Korean Cell Line Bank) of 20–30 passages were grown in a cell culture flask containing Dulbecco's modified Eagle's medium (DMEM, 11995065, Life Technologies, Carlsbad, CA) supplemented with 10% fetal bovine serum and 1% penicillin-streptomycin at 37°C. During the cell culture, CO2 level was maintained at 5%. For experiments, the cells were harvested and moved to a precleaned 18-mm square coverslip and grown for a day. The cells were then fixed using cold methanol at −20°C and washed with PBS buffer. To block nonspecific binding of antibodies, the fixed cells were incubated with 1% bovine serum albumin (BSA) for 1 h. After BSA blocking, the cells were incubated with a primary α-tubulin antibody (T6199, Sigma Aldrich,, St. Louis, MO) for 1 h, and washed three times with 1% BSA solution. Primary antibody was 1/250 times diluted in 1% BSA solution just before the experiments. A secondary antibody (A10538, Life Technologies) was labeled by mixing it with Cy5 NHS-ester (PA15101, GE Healthcare, Waukesha, WI) in NaHCO3 buffer (50 μl secondary antibody, 6 μl of 1 M NaHCO3, and 0.3 μl of 0.018 mg/μl Cy5 in dimethylsulfoxide). Cy5-labeled secondary antibody diluted 1/150 times was incubated for 15 min and washed three times with 1% BSA solution. A sample chamber was made by attaching a coverslip on a slide glass using double-sided tape and sealed using epoxy. For superresolution imaging at deep imaging depth, the cells were grown on a 76.2 × 25.4 × 1 mm quartz slide instead of a coverslip. The quartz slide was used instead of a glass slide to avoid high autofluorescence background from thick slide glass. After the same fixation process, a sample chamber was made by attaching the coverslip to a quartz slide using double-sided tape, and the chamber was sealed with epoxy. The distance from the surface of the coverslip to the cells on the quartz slide was ∼85 μm.
Superresolution imaging of microtubule
Measurements were taken in the presence of an oxygen-scavenging system (10% w/v glucose, 50 mM Tris-HCl, pH 8.0, 1 mg/mL glucose oxidase, and 0.04 mg/mL catalase) supplemented with 142 mM β-mercaptoethanol (125470010, Acros Organics, Geel, Belgium). Cy5 molecules were excited by a 640-nm laser (Cube640-100C, Coherent, Santa Clara, CA), and the typical intensity of this laser was ∼80 mW. During imaging, 532-nm or 473-nm lasers (Blues TM50, Cobalt, Seattle, WA) were turned on at all times to maintain a proper density of activated molecule in each frame. To obtain single superresolution images, typically, 10,000 frames were recorded with a 10-Hz filming rate. Imaging at different z-positions was done by translating samples with a three-axis piezo stage (P-561.3DD, Physik Instrumente, Auburn, MA). When an oil immersion objective was used, imaging depth was corrected by multiplying by a factor of 0.79. The width of the confocal slit was typically 40 μm.
Analysis for superresolution images
Only pixels with photon counts greater than a threshold value were selected for image fitting. 7x7 pixels around the local maximum were fitted to a 2-D elliptical Gaussian function. Images that were too dim, too elliptic, too small, or too large were discarded. Bright spots observed in sequential frames within one pixel were regarded as the same molecule and fitted after summing all the frames for better localization precision. For rendering of a high-resolution image and a conventional fluorescence image, each localization result was represented as a Gaussian function with 15-nm width for the high-resolution image or 150-nm width for the low-resolution image. The unit pixel size was 5 nm for high-resolution images and 50 nm for low-resolution images. Lateral drift was corrected using an image cross-correlation method (20) for every 50 frames. To get cross-correlation, the 50 frames were averaged. All analysis steps were done using a home-built program written in MATLAB (MathWorks, Natick, MA).
Results
Scheme of the line-scan confocal microscope
The optical setup of the microscope is described in Fig. 1 a. To obtain an optimum line illumination, the width and thickness of the excitation beam were independently controlled by using three cylindrical lenses (CL1–CL3) and a spherical lens (L1). Specifically, the CL1/CL3 pair expanded the excitation beam in the horizontal direction (x-direction) by 6.25 times, resulting in 48-μm illumination width for an oil immersion objective (UPLSAPO100XO, Olympus, Center Valley, PA) or 80-μm illumination width for a water immersion objective (UPLSAPO60XW, Olympus). On the other hand, the CL2/L1 pair generated a diffraction-limited line illumination on the sample plane by expanding the excitation beam in the vertical direction (y-direction) by 6.25 times to slightly overfill the back aperture of the objective in a vertical direction. (The beam size in the vertical direction at the back aperture of the objective was 9 mm, whereas the back-aperture sizes of the oil- and water-immersion objectives were 7 mm and 8.5 mm, respectively.) To scan the illumination line in a vertical direction, a galvanometric scanning mirror (GM1) was used. The telescopic lens pair (L2/L3) maintained the illumination beam at the center of the objective back aperture regardless of the illumination beam angle controlled by GM1.
Figure 1.
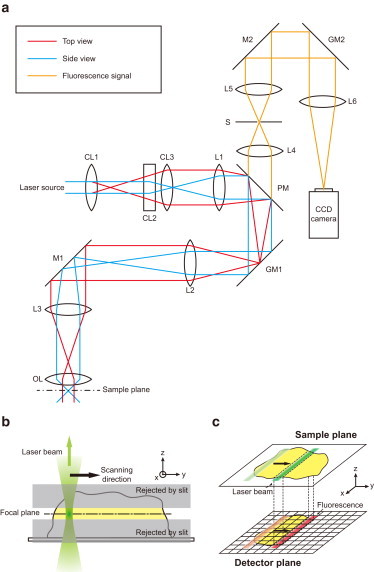
Scheme of the line-scan confocal microscope. (a) An optical design of the line-scan confocal microscope. The new microscope was built by modifying a commercial inverted microscope (IX71, Olympus). For versatility of the microscope, three lasers—473-nm (Blues TM50, Cobalt), 532-nm (Compass215M, Coherent), and 640-nm (Cube640-100C, Coherent)—were used as light sources. CL1, a cylindrical lens (f = 40 mm, LJ1125L1-A, Thorlabs, Newton, NJ); CL2, a cylindrical lens (f = 50 mm, LJ1695L2-A, Thorlabs); CL3, a cylindrical lens (f = 250 mm, LJ1267L1-A, Thorlabs); L1, a spherical lens (f = 250 mm, LA1461-A, Thorlabs); L2, a spherical achromatic lens (f = 250 mm, LAO-250.0-50.0/075, Melles-Griot); L3, a spherical achromatic lens (f = 300 mm, LAO-300.0-50.0/075, Melles-Griot); L4 and L5, spherical achromatic lenses (f = 100 mm, LAO-100.0-50.0/075, Melles-Griot, Albuquerque, NM); L6, a spherical achromatic lens (LAO-350.0-40.0/Specialty HEBBAR coating for 415nm to 700nm, Melles Griot); PM, a polychroic mirror (z473/532/633rpc-xt, Chroma, Muenster, Germany); GM1 and GM2, galvanometric mirrors (VM1000+, General Scanning); OL, an objective lens (UPLSAPO100XO, or UPLSAPO60XW, Olympus); S, a slit (S30R or S40R, Thorlabs). (b) Side view of the sample plane. The illumination beam (green) is focused in a line shape on the sample plane. Only the fluorescence signal near the focal plane (yellow) can filter through a confocal slit and thus be detected. (c) Scheme of 2-D image generation. The illumination line on the sample plane (green) and the fluorescence image on a CCD camera (red) are simultaneously scanned once per filming cycle of the camera.
The fluorescence signal from the sample was collected through the same objective that was used for excitation, guided in a direction opposite that of the excitation beam until it diverged from the excitation-beam path after a polychroic mirror (PM), and then focused on a confocal slit (S), which rejected out-of-focus background noise (Fig. 1 b). To maximally reject out-of-focus background without significantly hampering the signal-detection efficiency, the slit sizes were selected to be slightly larger than 1 Airy unit of the illumination beam: 37 μm for an oil-immersion objective, and 26 μm for a water-immersion objective. After the slit, the fluorescence signal was guided and focused on a charge-coupled-device (CCD) camera. The magnification of the image was controlled by using the L5/L6 lens pair so that the full pixels of the CCD camera could be utilized for imaging. The resulting imaging area of the microscope was 35 μm for the oil-immersion objective and 57 μm for the water-immersion objective.
Compared to the commercialized line-scan confocal microscope based on an insensitive line-CCD camera (LSM 7 LIVE, Zeiss, Oberkochen, Germany), we used a highly sensitive 2-D array electron-multiplying CCD (EM-CCD) camera (ProEM, Princeton Instruments, Trenton, NJ) as a detector. To generate 2-D images on the camera, we developed a double-scanning method in which the fluorescence image of the slit was scanned on the camera synchronously with the illumination beam on the sample plane (Fig. 1 c) by using the second galvanometric scanning mirror (Fig. 1 a, GM2). The scanning of the illumination beam and fluorescence signal was synchronized with the data acquisition of the CCD camera by connecting the transistor-transistor-logic output signal of the camera to the controller (SC2000, General Scanning, Cambridge, MA) of the drivers of the galvanometric scanning mirrors (MiniSAX II, General Scanning). The beam scanning was controlled by using a LabVIEW program provided by the manufacturer, and image acquisition was done using a home-built program written in C++. We used a sawtooth waveform for scanning so that scanning mirrors rotated with a constant velocity from the original position to the final position during the data acquisition period of the CCD camera (the slow phase of the sawtooth waveform). During the time interval between two adjacent filming events (∼300 μs), the mirrors returned quickly to the original position (the fast phase of the sawtooth waveform), resulting in one scan per frame. At the full frame rate of the camera (33 Hz), therefore, it is estimated that single fluorophores are excited for 0.23 ms/frame for the oil-immersion objective (the exposure time was multiplied by the diffraction-limited beam width (∼266 nm, or half of 532 nm) divided by the scanning distance (35 μm)).To obtain the best image quality, laser power was controlled in the range 30–50 mW. Despite the complexity of the optical setup of the new microscope, we measured the fluorescence-collection efficiency of the new microscope as 90% of that of a single-molecule TIRF microscope. To obtain images at varying heights, the z-position of the sample stage was controlled using a piezo stage (P-561.3DD, Physik Instrumente) in the case of line-scan confocal microscopy, or the z-position of the objective lens was controlled using a piezo actuator (P-721.CLQ, Physik Instrumente) in the case of both epifluorescence and HILO microscopies.
Characterization of the microscope
To characterize the optical-sectioning capability of the new microscope, we prepared a thin (200-nm) fluorescent film on a coverglass (Materials and Methods) and measured the area-averaged fluorescence intensities as a function of sample z-position by using the oil-immersion objective. We note that there was focal shift due to refractive index mismatch when the film was positioned below the focal plane. When imaging depth was not large (a few micrometers from the surface), focal shift was approximately proportional to the imaging depth. Therefore, focal shift was corrected by multiplying the movement of the objective by a factor of 0.79 (21,22). The optical-sectioning capability of the new microscope (1.0 μm) thus obtained was better than that of either HILO microscopy (6.7 μm) or SPIM (13) (Fig. 2 a). The fact that the measured optical-sectioning thickness of our HILO microscope was similar to the previously reported value (9) indicates that our HILO microscope was well optimized.
Figure 2.
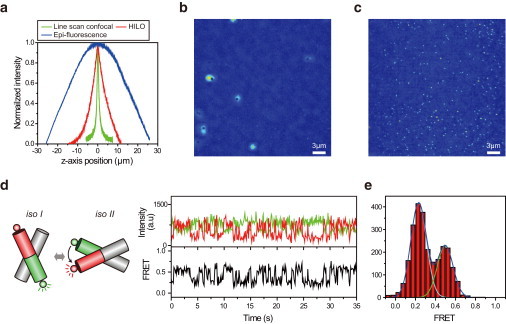
Characterization of the line-scan confocal microscope. (a) Comparison of the optical-sectioning capabilities of epifluorescence microscopy (31.7 μm), HILO (6.7 μm), and the line-scan confocal microscopy (1.0 μm). (b) A line-scan confocal image of Cy3-labeled single-stranded DNA on a coverslip in the presence of 10 nM free Cy3. To make the images, 10 frames with 100-ms exposure time were averaged and background fluorescence was subtracted. Scale bar, 3μm. (c) Same as in (b), except that the image was acquired in a HILO microscope. (d) Observation of Holliday junction dynamics via line-scan confocal microscopy. (left) A model of two-state conformational dynamics of the Holliday junction. For FRET measurements, donor (green circle) and acceptor (red circle) dye labels were attached to the ends of the Holliday junction. (right) Representative fluorescence intensity (green for donor and red for acceptor) and corresponding FRET-efficiency (black) time traces. Exposure time, 100 ms. (e) FRET histogram generated from 44 molecules. The oil-immersion objective was used for all experiments.
To demonstrate the improved optical-sectioning capability and single-molecule detection sensitivity of the new microscope compared to the HILO microscope, we imaged Cy3-labeled DNA molecules in the presence of 10 nM free Cy3. For the imaging, single-stranded DNA labeled with biotin and Cy3 was immobilized on a quartz surface via streptavidin-biotin interaction. Although single molecules were hardly recognized using the HILO microscope, due to overwhelming background fluorescence (Fig. 2 b), they were clearly visualized using our line-scan confocal microscope (Fig. 2 c). The large spots in Fig. 2 b are not single-molecule images but blurs due to nonuniform illumination of the HILO microscope. In the absence of free dye, the quality of single-molecule images obtained using our HILO microscope was similar to that obtained using the line-scan confocal microscope (Fig. S1), indicating that both microscopes were properly optimized.
We also demonstrated that the line-scan confocal microscope is compatible with single-molecule fluorescence resonance energy transfer (FRET) measurements. To do FRET experiments, the optical setup in Fig. 1 a was slightly modified (Fig. S2). We could successfully monitor the two-state dynamics of the Holliday junction by monitoring fluorescence intensities of donors and acceptors labeled at the ends of the Holliday junction (Fig. 2 d, Materials and Methods). The existence of two dominant populations was clear in the FRET histogram (Fig. 2 e).
Single-molecule imaging in live cells
As a test of the new microscope for single-molecule live-cell imaging, we prepared Dictyostelium discoideum cells with TMR-labeled cAMP receptors (Materials and Methods). Single cAMP receptors could be clearly visualized on both the basal and apical surfaces of the cell (Fig. 3, a and b, Movie S1, and Movie S2). In HILO microscopy, clear single-molecule images could not be obtained either on the basal surface or on the apical surface (Fig. S3). In TIRF microscopy, only molecules on the basal surface could be visualized (19,23).
Figure 3.
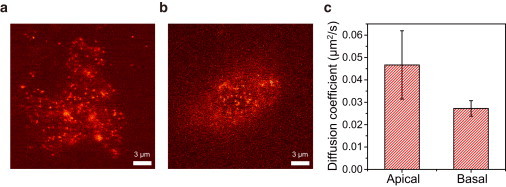
Single-molecule imaging in live cells. TMR-labeled cAMP receptors of Dictyostelium discoideum were imaged on both the basal (a) and apical surfaces (b). Scale bars, 3 μm. (c) Diffusion coefficients of cAMP receptors at the apical and basal surfaces of the cell. Molecules surviving for >15 frames (54 molecules/cell for the basal surface and 30 molecules/cell for the apical surface on average) were used for the analysis. Molecules from the same amoeba cell were used to get a single diffusion-coefficient value for either the basal surface or the apical surface. The data presented are the averages of 13 cells for the basal surface, and 11 cells for the apical surface. The oil-immersion objective was used for the experiments, and the exposure time was 50 ms.
With the unique capability of the new microscope to monitor single cAMP receptors on both the basal and apical surfaces, we compared the diffusion coefficients of the receptors on the two surfaces. Single cAMP receptors were tracked using the imageJ plug-in program (Mosaic, ETH Zürich, Zürich, Switzerland), and diffusion coefficients of the receptors were calculated as described previously (19). The diffusion coefficient of cAMP receptors on the basal surface was similar to the previously reported value obtained using TIRF microscopy (19), whereas the diffusion coefficient on the apical surface was 30% larger than that on the basal surface (Fig. 3 c), probably indicating differences in the local environments on the two surfaces. The exact reasons for the difference between the diffusion constants on the two surfaces are out of the scope of this work, but we notice that our observation is consistent with that of the previous report, namely, that membrane protein motion on the basal surface can be perturbed by direct interactions between a cell and a glass (24).
Superresolution fluorescence imaging using the new microscope
Finally, we obtained superresolution fluorescence images using the new microscope. Microtubules of COS-7 cell were coated with Cy5-labeled antibodies (Materials and Methods), and the bottom of the cell was imaged using dSTORM (Fig. 4 a). Microtubules not distinguishable using conventional fluorescence microscopy could be clearly distinguished with this superresolution imaging (Fig. 4 a, inset). The full width at half-maximum (FWHM) of microtubules of the dSTORM image was ∼59 nm (Fig. 4 b), which is similar to the value previously reported (25). Characterization of localization errors of the new microscope indicates that the precision of single-molecule localization is affected little by the instability of the scanning mirrors; rather, it is mainly determined by the photon numbers collected from single molecules (Fig. S4). The precision of the final image estimated by photon number, pixel size, background level, and scanning error was ∼9.5 nm, and the corresponding resolution was ∼22 nm.
Figure 4.
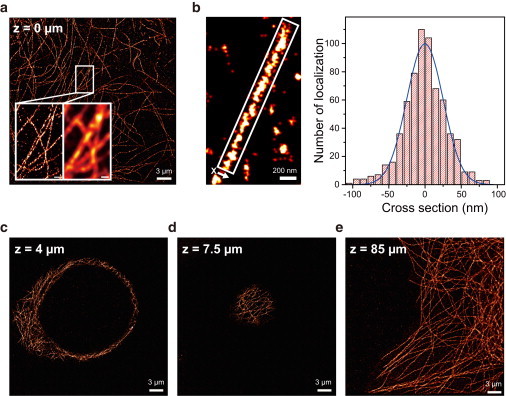
Superresolution fluorescence imaging. (a) Microtubules at the bottom of COS-7 were imaged via dSTORM. (Inset) Comparison between dSTORM (left) and conventional fluorescence microscopy (right). (b) Microtubule width in the dSTORM image. The cross-section profile of the microtubule (right) was generated by collecting a number of localization spots along the line perpendicular to the length of the microtubule in the white box in the dSTORM image. The histogram was well fitted to a Gaussian function with FWHM of 59 nm (right, blue lines). (c–e) Superresolution images at depths of 4 μm (c), 7.5 μm (d), and 85 μm (e), respectively. The image in e was obtained from a cell fixed on the glass slide side of the channel using the water-immersion objective. For all other experiments, the oil-immersion lens was used. Exposure time, 100 ms.
Since single-molecule imaging could be done deep inside the cell, we could also obtain superresolution images in the middle and at the top of the cell (Fig. 4, c and d). The localization precision at different depths could be well explained by the aberration of an oil-immersion objective (21) (Fig. S5). When an oil-immersion objective was replaced with a water-immersion objective, resolution of dSTORM was slightly impaired, but superresolution imaging could be done in much deeper regions with negligible aberration (Fig. 3 e and Fig. S5). This result suggests a potential of the new microscope for single-molecule imaging at the tissue level. However, it is true that our experimental conditions are different from those in tissues, and single-molecule imaging at the tissue level has yet to be demonstrated.
Discussion
It is well recognized that for cellular imaging, confocal microscopy has a number of advantages over HILO and SPIM. However, due to the poor sensitivity of currently available video-rate confocal microscopes, this imaging technique is not used for single-molecule studies in live cells or for superresolution fluorescence imaging. Is this a fundamental limit of confocal microscopy? It is known that the rapid scanning mode of single-pinhole-based confocal microscopes does not provide enough photons to distinguish single molecules from background noise. Spinning-disk or line-scan type confocal microscopes do not have the same problem. We asked whether these confocal microscopes could be optimized to provide single-molecule sensitivity. In the case of spinning-disk confocal microscopes, single-molecule images can barely be obtained using a highly sensitive camera as a detector (26), and it is generally agreed that single-molecule images of satisfactory quality and photostability cannot be obtained using commercial spinning-disk confocal microscopes, probably due to significant signal loss in the detection path (2,18).
To address the question, we adopted the line-scanning method for the new microscope. Different versions of line-scan confocal microscopes have been developed over recent years (27–30), and some of these have been commercialized (Meridian, InSIGHT PLUS; Bio-Rad, DVC 250; Zeiss, LSM 7 LIVE). However, none of these models provide single-molecule detection capability. We developed a line-scan confocal microscope with superior single-molecule detection sensitivity. The microscope is based on our unique double-scanning method; the illumination line on the sample plane and the fluorescence image on the CCD camera were synchronously scanned using independent galvanometric scanners. Compared to HILO microscopy, the new technique has the advantage that single-molecule imaging can be done in much deeper regions and with several times better signal/noise ratio. Compared to SPIM in the original design, which requires special optical design and sample preparation processes, the new microscopy is fully compatible with conventional cell-imaging techniques, and therefore, single-molecule imaging can be much more easily done at depths up to several hundred microns from the glass surface. Although iSPIM and Bessel-beam plane illumination cannot yet provide single-molecule images, clear single-molecule images can be easily obtained using the new microscope.
Conclusion
Single-molecule fluorescence imaging in cells has been pivotal to our understanding of several fundamental questions of cell biology, but its application deep inside the cell has been difficult to achieve due to technical limits of the currently available microscopes. We have developed video-rate confocal microscopy with excellent single-molecule detection efficiency and optical-sectioning capability, and we have demonstrated that this technology makes it easy to undertake single-molecule studies in conditions under which they would previously have been very difficult. We expect that the new microscope will be actively used for both single-molecule studies in live cells and superresolution fluorescence imaging in thick samples.
Acknowledgments
We thank Juhun Park for his help with the fluorescent film preparation.
This work was supported by Creative Research Initiatives (Physical Genetics Laboratory, 2009-0081562) of the National Research Foundation of Korea, and by a World Class University program to S.H. J.L. was financially supported by Hi Seoul Science (Humanities) Fellowship from the Seoul Scholarship Foundation.
Supporting Material
References
- 1.Li G.W., Xie X.S. Central dogma at the single-molecule level in living cells. Nature. 2011;475:308–315. doi: 10.1038/nature10315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lord S.J., Lee H.L., Moerner W.E. Single-molecule spectroscopy and imaging of biomolecules in living cells. Anal. Chem. 2010;82:2192–2203. doi: 10.1021/ac9024889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Huang B., Babcock H., Zhuang X. Breaking the diffraction barrier: super-resolution imaging of cells. Cell. 2010;143:1047–1058. doi: 10.1016/j.cell.2010.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rust M.J., Bates M., Zhuang X. Sub-diffraction-limit imaging by stochastic optical reconstruction microscopy (STORM) Nat. Methods. 2006;3:793–795. doi: 10.1038/nmeth929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Heilemann M., van de Linde S., Sauer M. Subdiffraction-resolution fluorescence imaging with conventional fluorescent probes. Angew. Chem. Int. Ed. Engl. 2008;47:6172–6176. doi: 10.1002/anie.200802376. [DOI] [PubMed] [Google Scholar]
- 6.Betzig E., Patterson G.H., Hess H.F. Imaging intracellular fluorescent proteins at nanometer resolution. Science. 2006;313:1642–1645. doi: 10.1126/science.1127344. [DOI] [PubMed] [Google Scholar]
- 7.Hess S.T., Girirajan T.P., Mason M.D. Ultra-high resolution imaging by fluorescence photoactivation localization microscopy. Biophys. J. 2006;91:4258–4272. doi: 10.1529/biophysj.106.091116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sako Y., Minoghchi S., Yanagida T. Single-molecule imaging of EGFR signalling on the surface of living cells. Nat. Cell Biol. 2000;2:168–172. doi: 10.1038/35004044. [DOI] [PubMed] [Google Scholar]
- 9.Tokunaga M., Imamoto N., Sakata-Sogawa K. Highly inclined thin illumination enables clear single-molecule imaging in cells. Nat. Methods. 2008;5:159–161. doi: 10.1038/nmeth1171. [DOI] [PubMed] [Google Scholar]
- 10.Fuchs E., Jaffe J., Azam F. Thin laser light sheet microscope for microbial oceanography. Opt. Express. 2002;10:145–154. doi: 10.1364/oe.10.000145. [DOI] [PubMed] [Google Scholar]
- 11.Huisken J., Swoger J., Stelzer E.H. Optical sectioning deep inside live embryos by selective plane illumination microscopy. Science. 2004;305:1007–1009. doi: 10.1126/science.1100035. [DOI] [PubMed] [Google Scholar]
- 12.Ritter J.G., Veith R., Kubitscheck U. Light sheet microscopy for single molecule tracking in living tissue. PLoS ONE. 2010;5:e11639. doi: 10.1371/journal.pone.0011639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cella Zanacchi F., Lavagnino Z., Diaspro A. Live-cell 3D super-resolution imaging in thick biological samples. Nat. Methods. 2011;8:1047–1049. doi: 10.1038/nmeth.1744. [DOI] [PubMed] [Google Scholar]
- 14.Wu Y., Ghitani A., Shroff H. Inverted selective plane illumination microscopy (iSPIM) enables coupled cell identity lineaging and neurodevelopmental imaging in Caenorhabditis elegans. Proc. Natl. Acad. Sci. USA. 2011;108:17708–17713. doi: 10.1073/pnas.1108494108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Planchon T.A., Gao L., Betzig E. Rapid three-dimensional isotropic imaging of living cells using Bessel beam plane illumination. Nat. Methods. 2011;8:417–423. doi: 10.1038/nmeth.1586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Webb R.H. Confocal optical microscopy. Rep. Prog. Phys. 1996;59:427. [Google Scholar]
- 17.Xie X.S., Choi P.J., Lia G. Single-molecule approach to molecular biology in living bacterial cells. Annu. Rev. Biophys. 2008;37:417–444. doi: 10.1146/annurev.biophys.37.092607.174640. [DOI] [PubMed] [Google Scholar]
- 18.Yildiz A. Single-molecule fluorescent particle tracking. In: Hinterdorfer P., Oijen A., editors. Handbook of Single-Molecule Biophysics. Springer; New York: 2009. pp. 1–18. [Google Scholar]
- 19.Miyanaga Y., Matsuoka S., Ueda M. Single-molecule imaging techniques to visualize chemotactic signaling events on the membrane of living Dictyostelium cells. Methods Mol. Biol. 2009;571:417–435. doi: 10.1007/978-1-60761-198-1_28. [DOI] [PubMed] [Google Scholar]
- 20.Guizar-Sicairos M., Thurman S.T., Fienup J.R. Efficient subpixel image registration algorithms. Opt. Lett. 2008;33:156–158. doi: 10.1364/ol.33.000156. [DOI] [PubMed] [Google Scholar]
- 21.Egner A., Hell S.W. Aberrations in confocal and multi-photon fluorescence microscopy induced by refractive index mismatch. In: Pawley J.B., editor. Handbook of Biological Confocal Microscopy. Springer; New York: 2006. pp. 404–413. [Google Scholar]
- 22.Huang B., Wang W., Zhuang X. Three-dimensional super-resolution imaging by stochastic optical reconstruction microscopy. Science. 2008;319:810–813. doi: 10.1126/science.1153529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ueda M., Sako Y., Yanagida T. Single-molecule analysis of chemotactic signaling in Dictyostelium cells. Science. 2001;294:864–867. doi: 10.1126/science.1063951. [DOI] [PubMed] [Google Scholar]
- 24.Lee H.L., Dubikovskaya E.A., Wender P.A. Single-molecule motions of oligoarginine transporter conjugates on the plasma membrane of Chinese hamster ovary cells. J. Am. Chem. Soc. 2008;130:9364–9370. doi: 10.1021/ja710798b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bates M., Huang B., Zhuang X. Multicolor super-resolution imaging with photo-switchable fluorescent probes. Science. 2007;317:1749–1753. doi: 10.1126/science.1146598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tadakuma H., Yamaguchi J., Funatsu T. Imaging of single fluorescent molecules using video-rate confocal microscopy. Biochem. Biophys. Res. Commun. 2001;287:323–327. doi: 10.1006/bbrc.2001.5574. [DOI] [PubMed] [Google Scholar]
- 27.Burns D.H., Hatangadi R.B., Spelman F.A. Scanning slit aperture confocal microscopy for three-dimensional imaging. Scanning. 1990;12:156–160. [Google Scholar]
- 28.Brakenhoff G.J., Visscher K. Confocal imaging with bilateral scanning and array detectors. J. Microsc. 1992;165:139–146. [Google Scholar]
- 29.Wolleschensky R., Zimmermann B., Kempe M. High-speed confocal fluorescence imaging with a novel line scanning microscope. J. Biomed. Opt. 2006;11:064011. doi: 10.1117/1.2402110. [DOI] [PubMed] [Google Scholar]
- 30.Poher V., Kennedy G.T., Neil M.A. Improved sectioning in a slit scanning confocal microscope. Opt. Lett. 2008;33:1813–1815. doi: 10.1364/ol.33.001813. [DOI] [PubMed] [Google Scholar]
- 31.Thompson R.E., Larson D.R., Webb W.W. Precise nanometer localization analysis for individual fluorescent probes. Biophys. J. 2002;82:2775–2783. doi: 10.1016/S0006-3495(02)75618-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.