

Abstract
48,XXYY is a rare sex chromosome aneuploidy affecting 1 in 18,000 to 50,000 male births. They present with developmental delay, hypogonadism, gynecomastia, intention tremors, and a spectrum of neurodevelopmental and psychiatric disorders. At one time this condition was considered a variant of Klinefelter syndrome. In clinically suspected cases, 48,XXYY syndrome can be diagnosed by chromosome culture and karyotyping. This patient presented with hypergonadotrophic hypogonadism, attention deficit hyperactive disorder, and renal malformatons. Klinefelter syndrome was clinically suspected. The karyotype confirmed the diagnosis of 48,XXYY syndrome. This is the first reported case of 48,XXYY syndrome from Sri Lanka.
Keywords: 48,XXYY syndrome; hypergonadotrophic hypogonadism; sex chromosome aneuploidy
INTRODUCTION
48,XXYY syndrome is a rare sex chromosomal aneuploidy affecting 1 in 18,000 to 50,000 male births.[1] The first published case was by Sylfest Muldal and Charles H. Ockey in Manchester, England, in 1960.[2] At one time, 48,XXYY syndrome was considered a variant of Klinefelter syndrome due to similar physical and endocrine phenotype including tall stature, hypergonadotrophic hypoganadism and infertility. The presence of an extra X and an extra Y chromosome affects neurodevelopment resulting in developmental delay, learning disability, intellectual disability, and a spectrum of psychological disorders which help to differentiate it from Klinefelter syndrome. Tartaglia et al. described 95 cases of 48,XXYY syndrome in a case series.[3] This is the first reported case of 48,XXYY syndrome from Sri Lanka.
CASE REPORT
A 15-year-old boy was referred for genetic testing due to hypergonadotrophic hypogonadism. He was the third child born to a non-consanguineous couple (mother 34 years, father 45 years).The antenatal period was complicated by gestational hypertension. The baby was delivered by a lower segment caesarean section at 38 weeks. He weighed 2.650 kg at birth. There were no postnatal complications.
His gross motor milestones were age appropriate. The parents had noticed speech delay and hyperactivity at the age of about 2 years. He has been regularly followed up by a child psychiatrist for attention deficit hyperactive disorder and he was on methylphenidate. Except for one episode of febrile convulsions, there was no history of a seizure disorder. He has had intermittent episodes of bronchial asthma and several episodes of food allergies. He has learning disabilities and poor social interactions. He has had recurrent dental caries. A history of peripheral vascular disease and deep vein thrombosis, reported to be associated with the condition, was not elicited.
Examination showed a tall boy with a height of 180 cm and an arm span of 190 cm. He weighed 70 kg. His body mass index was 21.6 kg/m2. There were no facial dysmorphic features. Psychometric assessment found him to have an IQ between 80 and 90 (Test of Nonverbal Intelligence, Third Edition) with a reduction in span of attention. He had reduced facial and body hair, acne, gynaecomastia, small bilateral descended testis (3–4 ml), small penis, and intention tremors. Pubic hair Tanner stage was II.
Investigations showed hypergonadrotropic hypogonadism, testosterone 0.95 ng/ml (range 0.18–1.5 ng/ml), FSH of 54.8 mIU (range 0.7–11.1 mIU/ml), and LH of 22.3 mIU/ ml (range 0.8–7.6 mIU/ml). Thyroid functions were normal [TSH 1.63 μIU/ml (range 0.4–4.0μIU/ml), free T4 0.881 ng/dl (range 0.78–2.19 ng/dl)]. Ultrasound scan of abdomen showed a small right kidney located in the pelvis. DMSA (dimercaptosuccinic acid) scan confirmed the small right kidney located in the pelvis with reduced function (27%) and a small cortical defect in the superior pole. The left kidney was normal with adequate function (73%). Blood urea (26.7 mg/dl) and serum creatinine (0.9 mg/dl) were within the normal range. Fasting plasma glucose level was 109 mg/dl (70–100 mg/dl). No congenital cardiac defects were seen in the echocardiogram.
The karyotype performed on peripheral lymphocytes showed the presence of the 48,XXYY chromosome compliment in all the cells analyzed [Figure 1]. He was commenced on depot testosterone therapy.
Figure 1.
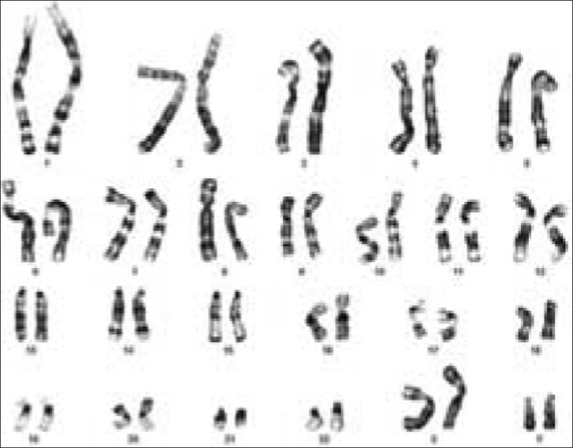
Karyogram showing the 48,XXYY karyotype
DISCUSSION
Although 48,XXYY syndrome was initially considered a variant of Klinefelter syndrome, it is now considered as a distinct clinical and genetic entity with neurodevelopmental and psychological involvement. 48,XXYY syndrome results from non-disjunction during gametogenesis. The extra set of chromosomes almost always comes from the sperm due to non-disjunction during meiosis 1 and meiosis 2.[4] This results in a sperm with three sex chromosomes (one X chromosome and two Y chromosomes). When that sperm fertilizes a normal egg with one X chromosome, an offspring with the 48,XXYY chromosome complement will be produced. In a small percentage of patients, mitotic non-disjunction occurring during the early stages of a 46, XY zygote can result in 48,XXYY syndrome.
48,XXYY affects male sexual development due to the presence of a small testis and low testosterone levels. At puberty, adolescents do not develop normal secondary sexual characteristics resulting in having reduced facial and body hair, poor muscle development, and gynecomastia.[4] Most of them (71%) usually develop intention tremors in adolescence which worsen with age.[3,5] Neurodevelopmental and psychological disorders are common but variable in severity.[4] Other associated features include dental defects, peripheral vascular disease, deep vein thrombosis, dysmorphic features, elbow abnormalities, as well as allergy and asthma (50%).[1] The risk of developing type 2 diabetes is high.[6]
This patient was referred for karyotyping due to hypergonadotropic hyogonadism, tall stature, and behavioral problems. He was clinically suspected to have Klinefelter syndrome. Despite the fact that the patient has an average IQ, he had learning difficulties, especially reading problems. The wide range of psychological and medical problems has been attributed to skewed X chromosome inactivation.[7] He had a small ectopic right kidney showing reduced function with a small cortical defect in the superior pole. One case of 48,XXYY syndrome with unilateral renal aplasia has been previously reported.[8] He has had impaired glucose tolerance.
Since most children with 48,XXYY syndrome have learning disabilities, a complete neurodevelopment assessment is needed at the time of diagnosis. A multidisciplinary team consisting of an endocrinologist, pediatrician, psychiatrist, speech therapist, behavioral therapist, and an occupational therapist is required to care for these patients.[1]
Footnotes
Source of Support: Nil
Conflict of Interest: None declared.
REFERENCES
- 1.Visootsak J, Graham JM., Jr Klinefelter syndrome and other sex chromosomal aneuploidies. Orphanet J Rare Dis. 2006;1:42. doi: 10.1186/1750-1172-1-42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Muldal S, Ockey CH, Thompson M, White LL. ‘Double male’-a new chromosome constitution in the Klinefelter syndrome. Acta Endocrinol (Copenh) 1962;39:183–203. doi: 10.1530/acta.0.0390183. [DOI] [PubMed] [Google Scholar]
- 3.Tartaglia N, Davis S, Hench A, Nimishakavi S, Beauregard R, Reynolds A, et al. A new look at XXYY syndrome: Medical and psychological features. Am J Med Genet A. 2008;146A:1509–22. doi: 10.1002/ajmg.a.32366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Genetic Home Reference. XXYY Syndrome. 2010. Jan, [cited 2011 Jun 16].
- 5.Tartaglia N, Borodyanskaya M, Hall DA. Tremor in 48,XXYY syndrome. Mov Disord. 2009;24:2001–7. doi: 10.1002/mds.22700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dubois S, Illouz F, Pinson L, Bonneau D, Rohmer V, Guichet A. Endocrine function in a 48,XXYY adult with type 2 diabetes: Case report with a review of the literature. Ann Endocrinol (Paris) 2007;68:384–8. doi: 10.1016/j.ando.2007.07.002. [DOI] [PubMed] [Google Scholar]
- 7.Iitsuka Y, Bock A, Nguyen DD, Samango-Sprouse CA, Simpson JL, Bischoff FZ. Evidence of skewed X-chromosome inactivation in 47, XXY and 48,XXYY Klinefelter patients. Am J Med Genet. 2001;98:25–31. [PubMed] [Google Scholar]
- 8.Zantour B, Sfar MH, Younes S, Alaya W, Kamoun M, Mkaouar E, et al. 48XXYY Syndrome in an adult with type 2 diabetes mellitus, unilateral renal aplasia, and pigmentary retinitis. Case Report Med. 2010;2010:612315. doi: 10.1155/2010/612315. [DOI] [PMC free article] [PubMed] [Google Scholar]