

Background: Sindbis virus RNAs bind the cellular HuR protein and cause its relocalization to the cytoplasm.
Results: HuR relocalization occurs with other alphaviruses but not with several unrelated RNA viruses. It is associated with altered protein phosphorylation.
Conclusion: HuR relocalization is alphavirus-selective and appears to be distinct from other types of HuR shuttling.
Significance: This has potential therapeutic and diagnostic implications for alphavirus infections.
Keywords: Nuclear Transport, RNA Binding Protein, RNA Turnover, RNA Viruses, Virology, Alphavirus, Nuclear-Cytoplasmic Shuttling
Abstract
We have demonstrated previously that the cellular HuR protein binds U-rich elements in the 3′ untranslated region (UTR) of Sindbis virus RNA and relocalizes from the nucleus to the cytoplasm upon Sindbis virus infection in 293T cells. In this study, we show that two alphaviruses, Ross River virus and Chikungunya virus, lack the conserved high-affinity U-rich HuR binding element in their 3′ UTRs but still maintain the ability to interact with HuR with nanomolar affinities through alternative binding elements. The relocalization of HuR protein occurs during Sindbis infection of multiple mammalian cell types as well as during infections with three other alphaviruses. Interestingly, the relocalization of HuR is not a general cellular reaction to viral infection, as HuR protein remained largely nuclear during infections with dengue and measles virus. Relocalization of HuR in a Sindbis infection required viral gene expression, was independent of the presence of a high-affinity U-rich HuR binding site in the 3′ UTR of the virus, and was associated with an alteration in the phosphorylation state of HuR. Sindbis virus-induced HuR relocalization was mechanistically distinct from the movement of HuR observed during a cellular stress response, as there was no accumulation of caspase-mediated HuR cleavage products. Collectively, these data indicate that virus-induced HuR relocalization to the cytoplasm is specific to alphavirus infections and is associated with distinct posttranslational modifications of this RNA-binding protein.
Introduction
Alphaviruses are enveloped arthropod-borne RNA viruses responsible for a range of human and animal diseases. Their genome consists of a capped and polyadenylated positive-sense single-stranded RNA molecule that generates a single subgenomic mRNA during infection that encodes for viral structural proteins (1). Important pathogens of this virus family include Venezuelan, Eastern, and Western equine encephalitis viruses (2) as well as agents (e.g. Chikungunya (3) and Ross River viruses (4)) that cause fever, rash, and epidemic outbreaks of polyarthritis. Understanding the interactions of these viruses with host cells is important to elucidate the mechanistic basis for viral pathogenesis and may also allow the identification of potential targets/strategies for antiviral therapeutics and diagnostics.
Many RNA viruses utilize a variety of cellular RNA-binding proteins for efficient gene expression and replication (5). Maintaining or inducing sufficient quantities of these RNA-binding proteins, as well as ensuring their availability, are therefore important considerations for an optimal RNA virus infection strategy. RNAs from Sindbis virus (SinV)2, a model alphavirus, have been shown to date to interact with four cellular proteins that play a role in the efficiency of viral gene expression/replication. The mosquito La protein interacts with the 3′ end of the negative-sense genomic replication template (6, 7). Enriched levels of hnRNP K protein can be found in membranous fractions of cells containing SinV replication/transcription complexes, and the protein is associated with subgenomic transcripts by coimmunoprecipitation (8). The abundant cellular hnRNP A1 protein binds to the 5′ untranslated region (UTR) of SinV genomic RNA and facilitates translation (9, 10). Finally, we demonstrated that the cellular HuR protein binds to a U-rich element in the 3′ UTR of SinV transcripts and mediates viral RNA stabilization (11, 12). This U-rich element can be found in the 3′ UTR of most, but not all, alphaviruses just upstream of the 3′ terminal conserved sequence element (CSE) that is required for replication (13).
An interesting aspect of these four SinV RNA-protein interactions is that the cellular proteins involved are all predominantly nuclear in normal cells. Thus, the virus presumably induces the movement or relocalization of these proteins from the nucleus to the cytoplasm during infection. This phenomenon of cytoplasmic relocalization has been documented for hnRNP A1 (9) and HuR (12). In this study, we explored aspects of the relocalization of the HuR protein during alphavirus infections.
HuR is a ubiquitously expressed RNA binding protein that has been implicated in regulating cellular gene expression largely through stabilizing mRNAs and influencing translation (14, 15). It consists of three RNA recognition motifs with a flexible hinge region located between RNA recognition motifs 2 and 3 that contains nuclear localization and export signals that direct its shuttling between the nucleus and cytoplasm (16). HuR protein is predominantly nuclear but has been shown to relocalize to the cytoplasm in times of cellular stress and in response to mitogens (17). HuR nuclear import is regulated by its association with Transportin (Trn) 1 and 2 (18, 19). Export of the protein out of the nucleus occurs in conjunction with the nuclear proteins pp32/PHAP1 and April/PHAP2 (20, 21) and appears to involve the Crm1 pathway (21). Furthermore, HuR shuttling can be associated with a variety of protein phosphorylation events, particularly on serine residues in the hinge region (23, 24). The underlying mechanism for HuR relocalization or impaired shuttling in a viral infection, however, has not been elucidated to date. The cytoplasmic accumulation of HuR could be the result of active export from the nucleus or failed/reduced import.
In this study, we have focused on several fundamental aspects of HuR relocalization/impaired shuttling in viral infections. First, we demonstrate increased accumulation of HuR in the cytoplasm in a variety of cell types after infection with several different alphaviruses. Notably, HuR relocalization occurred upon infection with Chikungunya and Ross River virus despite the fact that these viruses lack the conserved U-rich element in their 3′ UTRs that was shown previously to be a high affinity HuR protein binding site. Instead, HuR protein bound to a region upstream of the 3′ CSE in the 3′ UTRs of these viruses with significant affinity. HuR relocalization was not a generic response to any RNA virus infection, as neither measles virus nor dengue virus type 2 infection resulted in significant HuR accumulation in the cytoplasm. Finally, we demonstrate that HuR relocalization/impaired shuttling in a SinV infection is associated with alterations in the posttranslational phosphorylation of the protein. Collectively, these data establish HuR relocalization to the cytoplasm and interaction with the 3′ UTR of viral transcripts as a conserved and fundamental virus-host interaction in both New and Old World alphavirus infections.
EXPERIMENTAL PROCEDURES
Cell Lines, Virus Propagation, and Plaque Titration
Vero, C2C12, 74B, and 293T cell lines were cultured in DMEM media with 10% FBS. HeLa S3 spinner cells were maintained in JMEM with 10% horse serum. Full-length SinV genomic RNAs were produced by in vitro transcription of either wild-type SinV AR339 or the temperature-sensitive ts6SinV AR339 clone (25, 26), as described previously (11). The ΔURE SinV variant, which contained a 30-base deletion of the URE, was constructed as described (12). Viral titers were determined by plaque titration on Vero cells. Wild-type and temperature-sensitive SinV infections were carried out at an MOI of 5. Because of the reduced infectivity of the SinVΔURE virus, an MOI of 100 was used. RRV, CHIKV, measles virus, and dengue virus type 2 infections were performed at an MOI of 1. Staurosporine was applied to 74B cells at a concentration of 1 μm for 3 h.
Preparation of RNA Substrates
PCR products containing sequences derived from the 3′ UTRs of the T48 strain of Ross River virus (GenBanktm GQ433359.1) and the SL 15649 strain of Chikungunya virus (GenBanktm GU189061.1) were inserted into the EcoRI and PstI sites of pGem4. Primers used for amplification of the 3′ UTR of RRV strain T48 were as follows: forward, 5′-GAA TTC TAA GCT TTA GTT CAA AGG GCC-3′ and reverse, 5′-CTG CAG GTA AAA TAT TAA AAA AAC AAA TTA GAC GCC-3′. The primers used for amplification of the 3′ UTR of CHIKV strain SL15649 were as follows: forward, 5′-GAA TTC TAA CTT GAC AAT TAA GTA TGA AGC-3′ and reverse, 5′-CTG CAG GAA ATA TTA AAA ACA AAA TAA CAT CTC-3′. PCR products were used to generate templates for EMSAs using the following primers for the CHIKV 3′ UTR: forward, 5′-CAT AGC CAT CAT ACG ATT TAG GTG ACA CTA TAG TAA CTT GAC AAT TAA GTA TGA AGG-3′ and reverse, 5′-GAA ATA TTA AAA ACA AAA TAA CAT CTC C-3′. The following primers were used to generate RRV templates for EMSA: forward, 5′-CAT AGC CAT CAT ACG ATT TAG GTG ACA CTA TAG TAA GCT TTA GTT CAA AGG GCC-3′ and reverse, 5′-GTA AAA TAT TAA AAA AAC AAA TTA GAC GCC-3′. For the ΔCSE templates, the same forward primers were used as listed above with the following reverse primers: CHIKV reverse, 5′-AAC ATC TCC TAC GTC CCT GTG G-3′ and RRV reverse, 5′-TAG ACG CCT ACG TCC CCG G-3′. The primers used to clone the CSE into the EcoRI and PstI sites of pGem4 were as follows: forward, 5′-AAT TCA TTT TGT TTT TAA TAT TTC CTG CA-3′ and reverse, 5′-GGA AAT ATT AAA AAC AAA ATG-3′. Templates to generate viral 3′ UTR deletion variants were made using the following oligonucleotides that were inserted into the EcoR1 and BamH1 sites of pGem4: RRV9, 5′-AAT TCT ACT AAT AAA AAT TTA AAA ATC ACT AGA AAT CCA ATC ATT AAA TTA TTA ATT GGC TAG CCG AAC TCT AAG GAG ATG-3′ and 5′-GAT CCA TCT CCT TAG AGT TCG GCT AGC CAA TTA ATA ATT TAA TGA TTG GAT TTC TAG TGA TTT TTA AAT TTT TAT TAG TAG-3′ and CHIKV3, 5′-AAT TCT AGT TTA AAG GGC TAT AAA ACC CCT GAA TAG TAA CAA AAC ATA AAG TTA ATA AAA ATC AAA G-3′ and 5′-GAT CCT TTG ATT TTT ATT AAC TTT ATG TTT TGT TAC TAT TCA GGG GTT TTA TAG CCC TTT AAA CTA G-3′. Internally radiolabeled, capped RNA substrates were generated using SP6 polymerase and purified as described (27).
RNA Electrophoretic Mobility Shift Assays
HuR protein was produced and purified from Escherichia coli using a recombinant human HuR expression plasmid obtained from Dr. N. Curthoys (Colorado State University). Approximately 3 fmoles of internally radiolabeled RNAs containing either an intact alphavirus 3′ UTR or specific fragments thereof were incubated in the presence of recombinant HuR at the indicated concentrations in 15 mm HEPES (pH 7.9), 100 mm KCl, 2.25 mm MgCl2, 5% (v/v) glycerol, 0.15 mm spermidine, and 40 units RNase inhibitor (Fermentas). The complexes were allowed to form for 5 min at 30 °C prior to the addition of 2.6 μg/μl heparin sulfate. The addition of heparin prior to the incubation with recombinant protein gave similar results (data not shown). Following a 5-min incubation on ice, protein-RNA complexes were resolved on a 5% native acrylamide gel and analyzed by phosphorimaging. Values obtained for bound versus free RNA were plotted, and dissociation constants were calculated from the slope of the linear regression line fitted to the data. Standard deviations were calculated from three independent experiments.
Immunofluorescence and Fluorescence in Situ Hybridization Analysis
Cells were grown on glass coverslips, fixed in 4% paraformaldehyde, permeabilized in methanol, and rehydrated in 70% ethanol. Coverslips were blocked in 6% BSA fraction V (Sigma) in PBS for at least 1 h and washed in PBS. Primary antibody (diluted in 0.6% (w/v) BSA in PBS) was added for 1 h at room temperature or overnight at 4 °C and washed in PBS, and secondary antibody (diluted as above) was applied for 1 h. After washing, the coverslips were mounted using ProLong Gold antifade reagent (Invitrogen) with DAPI. Antibodies used were HuR (3A2) (Santa Cruz Biotechnology, Inc.), SinV (ATCC, catalog no. VR-1248AF), CHIKV (ATCC, catalog no. VR-1241AF), RRV (ATCC, catalog no. VR-1246AF), and Cy2 donkey anti-mouse Ig (Jackson ImmunoResearch Laboratories, Inc.).
Cell Fractionation, Western Blotting, and 2D Gel Electrophoresis
For nuclear-cytoplasmic fractions, SinV was allowed to adsorb into cells for 1 h, at which time prewarmed medium was added to cells and they were incubated at the non-permissive temperature (37 °C) for 12 h. Cells were fractionated, and Western blotting was performed as described previously (12). For 2D gel electrophoresis experiments, 293T cells were infected with SinV for 24 h. Cells were lysed using radioimmune precipitation assay buffer (50 mm Tris-HCl (pH7.4), 150 mm NaCl, 1% Triton X-100, 1% sodium deoxycholate, 0.1% SDS, 1 mm EDTA, protease (Roche), and phosphatase inhibitors (Pierce)), followed by a brief sonication to ensure lysis. Proteins were then purified and precipitated by methanol/chloroform and resuspended in isoelectric focusing sample buffer (8 m urea, 2% IGEPAL, 18 mm DTT, 1% (pH 3–10) immobilized pH gradient buffer (GE Healthcare), bromphenol blue). For λ phosphatase-treated samples, after methanol/chloroform precipitation, samples were resuspended in 1% Triton X-100 and λ phosphatase buffer. Following λ phosphatase treatment for 30 min, proteins were again precipitated by methanol/chloroform and resuspended in isoelectric focusing sample buffer. Isoelectric focusing strips (GE Healthcare) were rehydrated overnight and focused at 2000 voltage hours. Strips were equilibrated, and proteins were resolved in the second dimension by SDS-PAGE electrophoresis followed by blotting on PVDF membranes. Antibodies used for this study were HuR (3A2, Santa Cruz Biotechnology, Inc.), PABPN1 (K-18, Santa Cruz Biotechnology, Inc.), GAPDH (Millipore), and anti-mouse HRP (Santa Cruz Biotechnology, Inc.).
RESULTS
HuR Relocalization to the Cytoplasm Occurs upon Sindbis Virus Infection of Various Mammalian Cell Types
In most vertebrate cell lines, HuR resides in the nucleus. However, under certain cellular stresses, HuR relocalizes to the cytoplasm (17). We have shown previously that after 6–12 h of infection with SinV, HuR protein relocalizes to the cytoplasm in human embryonic kidney 293T cells where it stabilizes viral transcripts (12). Interestingly, in Aedes aegypti (Aag2) and Aedes albopictus (C6/36) mosquito cells, the mosquito HuR homolog is naturally more evenly distributed between the nucleus and cytoplasm, and the portion that resides in the nucleus does not relocalize upon SinV infection (12 and data not shown). Therefore, to distinguish whether the relocalization of HuR from the nucleus to the cytoplasm is a general phenomenon or occurs selectively in 293T cells, we infected a variety of mammalian cell lines (human, mouse, and non-human primate) with SinV and observed the distribution of HuR by immunofluorescence. In each instance, HuR is localized almost exclusively to the nucleus in uninfected cells (Fig. 1A, top panel), but a large percentage accumulates in the cytoplasm after SinV infection (bottom panel). Each cell line exhibited a different percentage of cells showing relocalization of HuR, which roughly corresponds to the number of cells infected as determined by SinV-specific immunofluorescence (supplemental Fig. 1). Therefore, relocalization of HuR is not unique to 293T cells but appears to be a more universal response of mammalian cells to SinV infection.
FIGURE 1.

HuR protein relocalizes to the cytoplasm in alphavirus infections of various types of mammalian cells. A, human (HeLa, 74B), mouse (C2C12), and non-human primate (Vero) cells were either mock-infected (top panels) or infected with SinV. At 24 h post-infection (hpi), cells were fixed and stained with DAPI to identify the nucleus and antibodies to HuR and visualized by fluorescence microscopy. B, 293T cells were infected with WEEV, fixed, stained with DAPI and HuR-specific antibodies at the indicated time points, and analyzed by fluorescence microscopy.
Western Equine Encephalitis Virus also Induces HuR Relocalization
Next we wanted to assess whether the HuR relocalization seen in SinV infections also occurs with other alphaviruses. Western equine encephalitis virus (WEEV), an alphavirus with an identifiable U-rich element (URE) in its 3′ UTR, is closely related to SinV. It arose as a recombinant virus with the nonstructural and capsid genes derived from Eastern equine encephalitis virus and the envelope glycoprotein and downstream regions from SinV (28). We have shown previously that recombinant HuR protein binds the URE of WEEV with a high affinity similar to that for the SinV URE (12). To determine whether WEEV infection induces the accumulation of HuR in the cytoplasm, 293T cells were infected with WEEV, and HuR distribution was assessed by immunofluorescence at 12 and 24 h post-infection. As seen in Fig. 1B, at 12 h post-infection, a large portion of HuR had relocalized to the cytoplasm, similar to the movement observed in SinV infections (Fig. 1A). Therefore, we conclude that HuR relocalization is not specific to SinV but can occur in other alphavirus infections.
Ross River and Chikungunya Viruses Induce HuR Relocalization
Given the close evolutionary relationship between WEEV and SinV and the fact that they both contain high-affinity U-rich HuR binding sites, perhaps it was not surprising that the two viruses would behave similarly with regard to HuR relocalization. Thus, we next sought to determine whether other more distantly related alphaviruses without an obvious URE would also induce HuR relocalization to the cytoplasm. For this study, we chose RRV and CHIKV, both of which lack the common URE in their 3′ UTR. HeLa cells were infected with each virus at a low MOI, and cells were fixed and stained for HuR at various time points. Movement of HuR to the cytoplasm of RRV- and CHIKV-infected cells could be seen as early as 12 h post-infection (data not shown), and by 24 h post-infection HuR had almost completely relocalized to the cytoplasm (Fig. 2). The percentage of cells showing relocalization of HuR corresponds very well to the percentage of cells infected by each virus, as assayed by a viral-specific immunofluorescence (supplemental Fig. 2). Therefore, although these two alphaviruses lack a recognizable high affinity U-rich HuR binding site in their 3′ UTR, they both cause HuR relocalization to the cytoplasm during infection of mammalian cells. Collectively, these data suggest that HuR relocalization is a common property of alphavirus infections.
FIGURE 2.

HuR relocalizes to the cytoplasm during both Ross River and Chikungunya virus infections. HeLa cells were mock-infected or infected with either RRV or CHIKV at an MOI of 1 pfu/cell. At 24 hpi, cells were fixed, stained with DAPI- and HuR-specific antibodies, and analyzed by fluorescence microscopy.
The 3′ UTRs of Ross River and Chikungunya Viruses Are Bound by HuR
Because RRV and CHIKV infections cause the relocalization of HuR from the nucleus to the cytoplasm (likely because of impairment of normal HuR shuttling), we wished to determine whether the cellular HuR protein could interact with the viral 3′ UTRs (diagrammed in Fig. 3A) with similar affinity to SinV and other alphaviruses that contain a URE (12). Radiolabeled in vitro transcribed RNAs representing either the 3′UTR of RRV or CHIKV were prepared and analyzed for binding to a recombinant HuR protein by EMSA. As shown in Fig. 3B, recombinant HuR protein interacted with high affinity to the 3′ UTR of RRV (mean dissociation constant 1.3 nm). This interaction is weaker than the interaction of HuR observed with the SinV 3′ UTR (12). However, the dissociation constant is similar to published affinities of HuR for cellular mRNA targets (29). In addition to the URE in SinV, the terminal highly conserved sequence element is fairly U-rich and can also act as a binding element for HuR (supplemental Fig. 3). To assess whether the CSE was entirely responsible for the observed binding of HuR protein to the RRV 3′ UTR, it was deleted from the RRV 3′ UTR RNA and tested this variant transcript for interactions with HuR protein. As seen in Fig. 3B, whereas the binding of HuR to the RRV 3′ UTR lacking the CSE was decreased ∼3-fold, HuR could still interact with the upstream portion of the RRV 3′ UTR with nanomolar affinity. Therefore, although a significant amount of the binding of HuR to the RRV 3′ UTR occurs in the U-rich CSE, additional sequence elements exist upstream of this terminal element that contribute to high affinity binding. As seen in Fig. 3C, the 3′ UTR of CHIKV behaved the same way as the RRV 3′ UTR in these assays. The intact CHIKV 3′ UTR was bound by HuR reasonably well (Kd 3.4 nm), and deletion of the CSE reduced HuR affinity by almost 3-fold. Therefore, we conclude that alphaviruses that naturally lack a URE upstream of their CSE also can effectively interact with the cellular HuR protein. These data suggest that HuR relocalization and binding to the viral 3′ UTR is a common property of most, if not all, alphaviruses.
FIGURE 3.
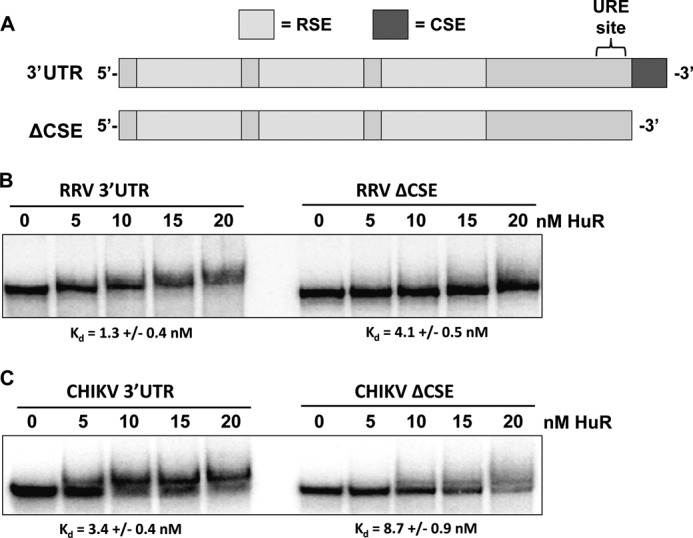
The 3′UTR of Ross River virus and Chikungunya virus binds the HuR protein with high affinity. A, diagram of a “typical” alphavirus 3′ UTR derived from SinV. The CSE, URE, and RSEs are highlighted. B and C, RNAs containing the entire 3′ UTR with (3′UTR) or without the CSE (ΔCSE) of RRV (B) or CHIKV (C) were incubated with the indicated amounts of recombinant HuR protein. RNA-protein complexes were resolved on a 5% non-denaturing gel and visualized by phosphorimaging. The mean Kd for each RNA-protein interaction from three independent experiments (performed using an extended titration of HuR protein over what is depicted in the panel) ± S.D. is indicated below the gel pictures.
To better define the HuR binding regions in RRV and CHIKV, RNAs were prepared from a series of deletion constructs for each of the viral 3′ UTRs. Data from these constructs (nine derived from the RRV 3′ UTR and seven from CHIKV) indicated that the HuR binding site was located in the downstream half of both 3′ UTRs (data not shown). As seen in Fig. 4, A and B, for RRV, a 75-base RNA (RRV9) from a region just downstream of the last repeated sequence element (RSE) was able to effectively interact with HuR with a mean dissociation constant of 79.4 nm. The sequence of the RRV9 fragment is broadly (76%) AU-rich (Fig. 4D), consistent with other previously described HuR binding domains. Interestingly, the HuR binding region in the CHIKV 3′ UTR mapped to the RSE-3 segment of the 3′ UTR (Fig. 4B). The two other highly related RSEs of the viral 3′ UTR failed to interact with HuR protein, as indicated by the lack of a shifted RNA-protein complex despite high amounts of HuR protein for RSE-1 in Fig. 4C. A comparison of the sequences between the HuR-interacting RSE-3 element and the non-interacting RSE-1 element (Fig. 4D) indicates only 12 base differences over the length of the RSE-3 element. Notably, seven of these changes increase AU-richness and, therefore, could influence HuR binding.
FIGURE 4.

Mapping of internal HuR binding sites of the RRV and CHIKV 3′ UTR. A, diagram of the RRV and CHIKV 3′ UTRs along with the small fragments (RRV9 and CHIKV3) that contained HuR binding sites. B, RNAs containing the RRV9 or CHIKV3 3′ UTR segments were incubated with the indicated amounts of recombinant HuR protein. RNA-protein complexes were resolved on a 5% non-denaturing gel and visualized by phosphorimaging. The mean Kd for each RNA-protein interaction from three independent experiments (performed using an extended titration of HuR protein over what is depicted in the panel) ± S.D. is indicated to the right of the gel pictures. C, the RSE-1 segment of CHIKV or RRV, as well as the entire CHIKV 3′ UTR RNA (CHIKV 3′ UTR lanes) were incubated with 600 nm HuR protein. RNA-protein complexes were resolved on a 5% non-denaturing gel and visualized by phosphorimaging. The arrow to the right indicated the shifted HuR-RNA complex formed with the full-length CHIKV 3′ UTR RNA. D, the sequences of the CHIKV RSE-1 and RSE-3 fragments are aligned with differences highlighted with asterisks. The sequence of the RRV9 fragment is shown in the bottom panel.
Measles and Dengue Viruses Do Not Cause Relocalization of HuR
The next question we addressed was whether HuR relocalization from the nucleus to the cytoplasm was specific for alphaviruses or a general reaction of the cell to a virus infection. Previous work has shown, for example, that HuR protein can relocalize to the cytoplasm in response to a variety of cellular stresses (17). To assess the specificity of HuR relocalization in virus infections, we chose another positive-sense cytoplasmic virus, dengue virus, and a negative-sense cytoplasmic virus, measles virus. To date, neither dengue virus nor measles virus transcripts have been shown to interact with the cellular HuR protein. 293T and Vero cells were infected with dengue and measles viruses, respectively, and HuR-specific immunofluorescence was performed as described above for alphavirus infections. As seen in Fig. 5, neither dengue nor measles virus infection induced a redistribution of HuR from the nucleus to the cytoplasm at 24 hpi (or any other time point assayed (data not shown)). The syncytia formed in the measles virus samples clearly indicate that virus-induced cytopathology is occurring in infected cells in the absence of HuR redistribution. Collectively, these data indicate that HuR relocalization is not a general response to viral infection and may be specific for alphavirus infections.
FIGURE 5.

HuR relocalization to the cytoplasm does not occur in measles or Dengue virus infections. Human 293T cells were infected with dengue virus, and Vero cells were infected with measles virus at an MOI of 1. At 24 hpi, cells were fixed and stained with DAPI- and HuR-specific antibodies and analyzed by fluorescence microscopy. The HuR panel documents staining with HuR antibody, whereas the overlay panel includes both the DAPI and HuR staining.
Alphaviral Gene Expression Is Required for HuR Relocalization
Because HuR relocalization appears to be somewhat specific for an alphavirus infection, it may occur through a novel mechanism. Hence, we wished to obtain some clues to the mechanism of induction of HuR redistribution. To determine whether viral gene expression is required for HuR relocalization, we used a temperature-sensitive SinV mutation that renders the viral polymerase non-functional at the non-permissive temperature of 37 °C, effectively halting the vast majority of viral gene expression and replication (11). Cells were infected with the temperature-sensitive mutant SinV, and the virus was allowed to adsorb for 1 h. Post-adsorption, cells were incubated at the non-permissive temperature for the remainder of the experiment and then fractionated into nuclear and cytoplasmic portions. As seen in Fig. 6A, the majority of the HuR resides in the nucleus, as expected in uninfected cells. When the temperature-sensitive virus is used to infect cells, the large majority of HuR still remains nuclear at a time point at which we previously showed that much of the HuR relocalizes to the cytoplasm (Fig. 1, Ref. 12). Therefore, SinV gene expression and/or replication are required for HuR relocalization.
FIGURE 6.

HuR relocalization during a SinV infection requires viral gene expression and appears to be distinct from a typical cellular stress response. A, 293T cells were either mock-infected (Uninfected) or infected by an MOI of 3 with a SinV variant that contains a temperature-sensitive mutation in the viral polymerase and switched to the non-permissive temperature post-absorption. Cells were biochemically separated into nuclear (N) and cytoplasmic (C) fractions, and proteins from the fractions were separated on by SDS-PAGE and analyzed by Western blotting using the antibodies indicated in the right panel. B, 293T cells were mock-infected, infected with SinV, or infected with a SinV variant virus that contained a deletion of the URE element in its 3′ UTR (SinVΔURE). At 24 hpi, cells were fixed and stained with DAPI- and HuR-specific antibodies and analyzed by fluorescence microscopy. The HuR panel documents staining with HuR antibody, whereas the overlay includes both the DAPI and HuR staining. C, 293T cells were infected with SinV or 74B cells were treated with staurosporine (STS). At the indicated times post-infection, cells were lysed, and proteins were separated by SDS-PAGE and analyzed by Western blotting with an HuR-specific antiserum. The positions of the intact HuR protein (HuR) and the CP1 cleavage product (HuR-CP1) are indicated on the right.
The SinV 3′UTR URE High-affinity Binding Site Is Not Required for HuR Relocalization
We next determined whether the high-affinity URE binding site for HuR (Kd = 0.16 nm) in the SinV 3′ UTR was necessary for HuR relocalization (12). Note that we could not delete the CSE element because it is absolutely required for viral replication (13). When the URE is removed from the UTR, SinV replicates poorly in cells, with a 10-fold decrease in the amount of viral progeny produced during a one-step growth curve (12). Cells were infected with a SinV variant containing the URE deletion in its 3′ UTR (SinVΔURE), and HuR protein relocalization was assessed by immunofluorescence. As seen in Fig. 6B and supplemental Fig. 4, SinVΔURE virus still caused movement of HuR to the cytoplasm. Thus, we conclude that the presence of a URE high-affinity HuR binding site in the 3′UTR of SinV is not required for HuR relocalization.
HuR Relocalization in SinV Infection Is Not Mediated by HuR Cleavage
In cellular stress responses, HuR relocalization has been linked with caspase-mediated cleavage of the protein (30). To examine whether HuR is cleaved upon SinV infection, cells were infected with SinV for 24 h, total protein was collected, and a HuR-specific Western blot analysis was performed. As a control, cells were treated with staurosporine to induce a stress response and HuR cleavage. As seen in Fig. 6C, staurosporine treatment induced the cleavage of HuR into a smaller band (HuR-CP). However, even after 24 h of SinV infection, no cleavage of HuR was observed, despite the fact that the majority of the protein was now cytoplasmic (Fig. 1). Thus we conclude that, unlike the cellular stress responses where the relocalization of HuR is associated with caspase-mediated cleavage of the protein, no cleavage products accumulate when HuR is relocalized in a SinV infection. This strongly suggests that aspects of the mechanism of HuR relocalization are different in SinV infection and stress situations.
HuR Is Differentially Phosphorylated upon SinV Infection
Finally, several studies have shown that changes in the phosphorylation or methylation state of HuR can be associated with the relocalization of the protein to the cytoplasm (23, 24, 31–37). To examine whether HuR is differentially posttranslationally modified upon SinV infection, cells were infected for 24 h, and 2D gel electrophoresis was performed, followed by HuR-specific Western blotting. As seen in Fig. 7 (upper panels), although the majority of HuR migrates close to the pI predicted for unmodified protein (∼9) in mock-infected cells, several more acidic spots are detected that would be consistent with phosphorylation or other acidic posttranslational modification. In SinV-infected extracts, there is a distinct shift of the modified HuR to a more basic position. To examine whether this basic shift in HuR protein upon SinV infection is due to phosphorylation or some other posttranslational modification, SinV-infected or mock-infected extracts were treated with phosphatase and then resolved by 2D gel electrophoresis. As seen in Fig. 7 (lower panels), in both infected and uninfected cells, phosphatase treatment resulted in complete loss of the acidic shifted protein. Therefore, we conclude that in uninfected cells HuR is phosphorylated but that upon infection this modification is partially removed. To the best of our knowledge, Sindbis virus infection is the first example of a cellular stress causing dephosphorylation of HuR. These data indicate that upon Sindbis infection, there are changes in phosphorylation of HuR protein that may be correlated with redistribution of the protein from the nucleus to the cytoplasm.
FIGURE 7.
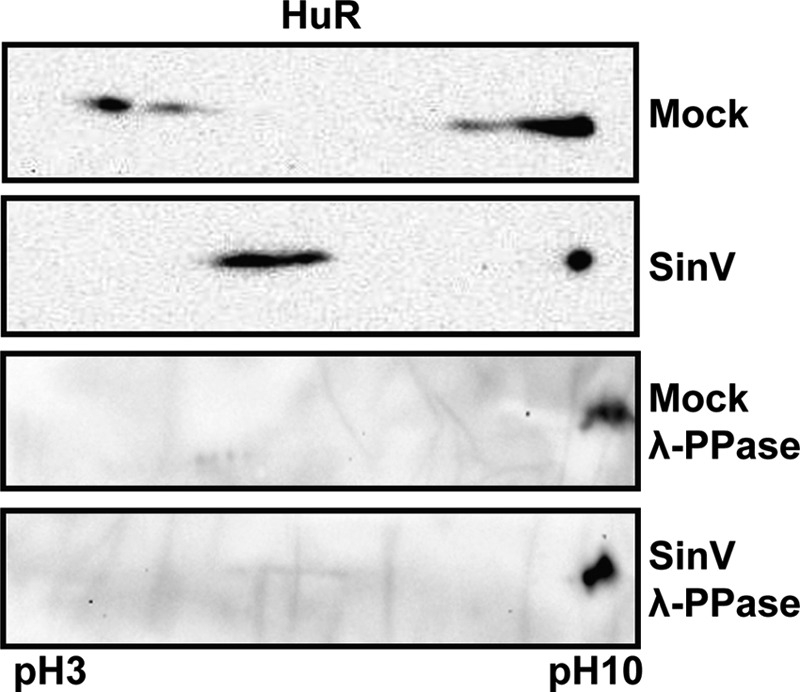
HuR protein undergoes dephosphorylation upon SinV infection. 293T cells were either mock-infected or infected with SinV. At 24 hpi post-infection, cells were lysed, and proteins were separated by 2D gel electrophoresis either before (Mock and SinV) or after treatment with a protein phosphatase (λ-PPase). The second dimension was analyzed by Western blotting with HuR-specific antisera. The pH limits of the isoelectric focusing step are indicated in the bottom panel.
DISCUSSION
This study makes several observations that provide insights into alphavirus-host cell interactions. First, we demonstrate that four independent alphaviruses cause the relocalization/impaired shuttling of the cellular HuR protein from the nucleus to the cytoplasm during infection. This strongly suggests that HuR relocalization/impaired shuttling may be a conserved phenomenon for most, if not all, alphavirus infections. Second, results shown here and in our previous publication (Figs. 3 and 4, Ref. 12) demonstrate that the HuR protein can interact with significant affinity (Kd in the nanomolar range) with the 3′ UTR of all seven of the alphaviruses tested to date. This reinforces the model that HuR plays a conserved role in maintaining alphavirus mRNA stability during infection. Next, we localized a binding element with affinity for HuR to an unexpected region of the 3′ UTR of RRV and CHIKV. These data may help expand our appreciation of HuR target sequences in other viral and cellular mRNAs. Finally, we detected a novel dephosphorylation event on the HuR protein that may be directly related to its relocalization during alphavirus infection. Collectively, these data provide biological and mechanistic insight into the conserved role of HuR protein in alphavirus infections.
The observations made in this study regarding HuR relocalization/impaired shuttling in alphavirus infections have two potential translational applications. First, because many alphaviruses appear to induce the relocalization of HuR from the nucleus to the cytoplasm during infection (and this does not appear to be a common occurrence in other RNA virus infections), one might be able to monitor the subcellular localization of HuR protein as an indication whether or not a tissue/cell line is infected with an alphavirus. Such a rapid, broad-spectrum alphavirus diagnostic aid could help narrow down the identity of an emerging virus. Second, because HuR appears to be relocalizing to the cytoplasm by a novel mechanism, this may represent a valid target for the development of broad spectrum anti-alphavirus small molecule therapeutics. Exploiting the observations in this study along either of these lines awaits future experimentation.
The data presented in Figs. 3 and 4 demonstrate that sequences upstream of the 3′ terminal CSE region of both RRV and CHIKV can interact with HuR with nanomolar affinity. Previous work on HuR protein-RNA interactions have shown a clear preference for U-rich and AU-rich binding sites (38), perhaps influenced by local RNA structure (39). Surprisingly, the internal HuR binding site in the 3′ UTR of CHIKV lies within the third RSE, in which small sequence variations from the other two RSEs have created AU-rich segments that likely serve as the HuR binding platform. On the basis of our prior analysis of RSE segments from SinV (11, 12), the CHIKV RSE-3 is the only RSE identified to date that possesses the ability to interact with HuR. It will be interesting to see if this adaptation of a single RSE to interact with a cellular RNA binding protein plays any role in CHIKV infections in human or mosquito cells.
Upon certain cellular signaling events, HuR has been shown previously to relocalize from the nucleus to the cytoplasm. For instance, a variety of cellular stresses have been shown to alter HuR phosphorylation at several residues (Ser-88, Ser-100, Thr-118, Ser-158, Ser-202, Ser-221, Ser-242, Ser-318), causing a shift of the protein out of the nucleus and a change in the interaction of the protein with target mRNAs (23, 24, 31–37). Several of these phosphorylation events occur in the hinge region of the protein that contains the important nuclear localization and export signals that play a role in HuR shuttling. A portion of the translocated HuR protein is also cleaved in a caspase 1-dependent fashion under stress conditions (30). The modifications that occur to the HuR protein in the context of an alphavirus infection, however, appear to be distinct from the changes that occur in the context of a cellular stress response. Following SinV infection, HuR protein is dephosphorylated rather than gaining additional phosphate residues (Fig. 7). We could find no evidence for HuR cleavage products (Fig. 6C), and HuR protein does not appear to have diminished RNA binding capacity, as it interacts very avidly with viral RNAs during infection (12). We are currently investigating the phosphatase involved in dephosphorylation of HuR or, alternatively, which kinase is inactivated in SinV infection, to gain additional mechanistic insights into the fate of HuR during infection. Interestingly, phosphorylation of the Ser-242 residue of the hinge region near the HuR nucleocytoplasmic shuttling sequence (residues 205–237) has been shown to hinder HuR cytoplasmic localization (36), making this perhaps an attractive target for alphavirus-induced dephosphorylation. We are also using a variety of SinV mutants and transfected viral gene products to identify the virus-specific factors involved in usurping the HuR protein.
Finally, the movement of HuR into the cytoplasm, as well as the potential for preferential binding to viral rather than cellular transcripts, could have significant effects on cellular gene expression. HuR is a well studied RNA stability factor involved in regulating many cellular mRNAs through binding AU- or U-rich sequences in the 5′ or 3′ UTR. It also has been shown to influence the translation of several mRNAs (40). Several of the targets of HuR include mRNAs that are important in cellular survival (e.g. Bcl-2, Mcl-1, SIRT1, p21, and Mdm-2) as well as cytokines and factors involved in innate immunity. The dysregulation of HuR subcellular localization/shuttling and competition for binding with viral RNAs could significantly alter the relative stability and translation of these key cellular transcripts. In addition, HuR has also been associated with the nuclear processes of splicing and polyadenylation (22, 38, 41). Thus, the relocalization of the protein to the cytoplasm/disruption of the normal shuttling of HuR back to the nucleus could affect other aspects of the posttranscriptional control of cellular gene expression. The work described in this study lays the foundation for future insights into this alphavirus-host interaction that may have significant impact on host cell processes and pathogenesis.
Acknowledgments
We thank Dr. Jerome Lee for preparing recombinant HuR, Dr. Norm Curthoys for providing the human HuR expression plasmid, Dr. Alan Schenkel for microscopy assistance, Stephanie Moon for dengue infections, Aaron Phillips for WEEV infections, and members of the Wilusz laboratory for helpful suggestions.
These studies were supported, in whole or in part, by National Institutes of Health Grant AI063434 and NIAID, National Institutes of Health award U54 AI-065357 through the Rocky Mountain Regional Center of Excellence (to J. W.).

This article contains supplemental Figs. 1–4.
- SinV
- Sindbis virus
- UTR
- untranslated region
- CSE
- conserved sequence element
- MOI
- multiplicity of infection
- 2D
- two-dimensional
- WEEV
- Western equine encephalitis virus
- URE
- U-rich element
- RRV
- Ross River virus
- CHIKV
- Chikungunya virus
- RSE
- repeated sequence element.
REFERENCES
- 1. Jose J., Snyder J. E., Kuhn R. J. (2009) A structural and functional perspective of alphavirus replication and assembly. Future Microbiol. 4, 837–856 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Zacks M. A., Paessler S. (2010) Encephalitic alphaviruses. Vet. Microbiol. 140, 281–286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Schwartz O., Albert M. L. (2010) Biology and pathogenesis of Chikungunya virus. Nat. Rev. Microbiol. 8, 491–500 [DOI] [PubMed] [Google Scholar]
- 4. Jupille H. J., Oko L., Stoermer K. A., Heise M. T., Mahalingam S., Gunn B. M., Morrison T. E. (2011) Mutations in nsP1 and PE2 are critical determinants of Ross River virus-induced musculoskeletal inflammatory disease in a mouse model. Virology 178, 32–40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Li Z., Nagy P. D. (2011) Diverse roles of host RNA binding proteins in RNA virus replication. RNA Biol. 8, 305–315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Pardigon N., Lenches E., Strauss J. H. (1993) Multiple binding sites for cellular proteins in the 3′ end of Sindbis alphavirus minus-sense RNA. J. Virol. 67, 5003–5011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Pardigon N., Strauss J. H. (1996) Mosquito homolog of the La autoantigen binds to Sindbis virus RNA. J. Virol. 70, 1173–1181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Burnham A. J., Gong L., Hardy R. W. (2007) Heterogeneous nuclear ribonuclear protein K interacts with Sindbis virus nonstructural proteins and viral subgenomic mRNA. Virology 367, 212–221 [DOI] [PubMed] [Google Scholar]
- 9. Lin J. Y., Shih S. R., Pan M., Li C., Lue C. F., Stollar V., Li M. L. (2009) hnRNP A1 interacts with the 5′ untranslated regions of enterovirus 71 and Sindbis virus RNA and is required for viral replication. J. Virol. 83, 6106–6114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Gui H., Lu C. W., Adams S., Stollar V., Li M. L. (2010) hnRNP A1 interacts with the genomic and subgenomic RNA promoters of Sindbis virus and is required for the synthesis of G and SG RNA. J. Biomed. Sci. 17, 59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Garneau N. L., Sokoloski K. J., Opyrchal M., Neff C. P., Wilusz C. J., Wilusz J. (2008) The 3′ untranslated region of Sindbis virus represses deadenylation of viral transcripts in mosquito and mammalian cells. J. Virol. 82, 880–892 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Sokoloski K. J., Dickson A. M., Chaskey E. L., Garneau N. L., Wilusz C. J., Wilusz J. (2010) Sindbis virus usurps the cellular HuR protein to stabilize its transcripts and promote productive infections in mammalian and mosquito cells. Cell Host Microbe 8, 196–207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Hardy R. W., Rice C. M. (2005) Requirements at the 3′ end of the Sindbis virus genome for efficient synthesis of minus-strand RNA. J. Virol. 79, 4630–4639 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. von Roretz C., Di Marco S., Mazroui R., Gallouzi I. E. (2011) Turnover of AU-rich-containing mRNAs during stress. A matter of survival. Wiley Interdiscip. Rev. RNA 2, 336–347 [DOI] [PubMed] [Google Scholar]
- 15. Rivas-Aravena A., Ramdohr P., Vallejos M., Valiente-Echeverría F., Dormoy-Raclet V., Rodríguez F., Pino K., Holzmann C., Huidobro-Toro J. P., Gallouzi I. E., López-Lastra M. (2009) The Elav-like protein HuR exerts translational control of viral internal ribosome entry sites. Virology 392, 178–185 [DOI] [PubMed] [Google Scholar]
- 16. Hinman M. N., Lou H. (2008) Diverse molecular functions of Hu proteins. Cell Mol. Life Sci. 65, 3168–3181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Doller A., Pfeilschifter J., Eberhardt W. (2008) Signalling pathways regulating nucleo-cytoplasmic shuttling of the mRNA-binding protein HuR. Cell Signal 20, 2165–2173 [DOI] [PubMed] [Google Scholar]
- 18. Güttinger S., Mühlhäusser P., Koller-Eichhorn R., Brennecke J., Kutay U. (2004) Transportin2 functions as importin and mediates nuclear import of HuR. Proc. Natl. Acad. Sci. U.S.A. 101, 2918–2923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Rebane A., Aab A., Steitz J. A. (2004) Transportins 1 and 2 are redundant nuclear import factors for hnRNP A1 and HuR. RNA 10, 590–599 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Brennan C. M., Gallouzi I. E., Steitz J. A. (2000) Protein ligands to HuR modulate its interaction with target mRNAs in vivo. J. Cell Biol. 151, 1–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Gallouzi I. E., Brennan C. M., Steitz J. A. (2001) Protein ligands mediate the CRM1-dependent export of HuR in response to heat shock. RNA 7, 1348–1361; Partial Retraction (2003) RNA9, 1410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Zhou H. L., Hinman M. N., Barron V. A., Geng C., Zhou G., Luo G., Siegel R. E., Lou H. (2011) Hu proteins regulate alternative splicing by inducing localized histone hyperacetylation in an RNA-dependent manner. Proc. Natl. Acad. Sci. U.S.A. 108, E627–635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Kim H. H., Gorospe M. (2008) Phosphorylated HuR shuttles in cycles. Cell Cycle 7, 3124–3126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Doller A., Schlepckow K., Schwalbe H., Pfeilschifter J., Eberhardt W. (2010) Tandem phosphorylation of serines 221 and 318 by protein kinase Cdelta coordinates mRNA binding and nucleocytoplasmic shuttling of HuR. Mol. Cell Biol. 30, 1397–1410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Barton D. J., Sawicki S. G., Sawicki D. L. (1988) Demonstration in vitro of temperature-sensitive elongation of RNA in Sindbis virus mutant ts6. J. Virol. 62, 3597–3602 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Bick M. J., Carroll J. W., Gao G., Goff S. P., Rice C. M., MacDonald M. R. (2003) Expression of the zinc-finger antiviral protein inhibits alphavirus replication. J. Virol. 77, 11555–11562 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Wilusz J., Shenk T. (1988) A 64-kd nuclear protein binds to RNA segments that include the AAUAAA polyadenylation motif. Cell 52, 221–228 [DOI] [PubMed] [Google Scholar]
- 28. Weaver S. C., Kang W., Shirako Y., Rumenapf T., Strauss E. G., Strauss J. H. (1997) Recombinational history and molecular evolution of Western equine encephalomyelitis complex alphaviruses. J. Virol. 71, 613–623 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Epis M. R., Barker A., Giles K. M., Beveridge D. J., Leedman P. J. (2011) The RNA-binding protein HuR opposes the repression of ERBB-2 gene expression by microRNA miR-331–3p in prostate cancer cells. J. Biol. Chem. 286, 41442–41454 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Mazroui R., Di Marco S., Clair E., von Roretz C., Tenenbaum S. A., Keene J. D., Saleh M., Gallouzi I. E. (2008) Caspase-mediated cleavage of HuR in the cytoplasm contributes to pp32/PHAP-I regulation of apoptosis. J. Cell Biol. 180, 113–127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Yu T. X., Wang P. Y., Rao J. N., Zou T., Liu L., Xiao L., Gorospe M., Wang J. Y. (2011) Chk2-dependent HuR phosphorylation regulates occludin mRNA translation and epithelial barrier function. Nucleic Acids Res. 39, 8472–8487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Doller A., Winkler C., Azrilian I., Schulz S., Hartmann S., Pfeilschifter J., Eberhardt W. (2011) High-constitutive HuR phosphorylation at Ser-318 by PKC{delta} propagates tumor relevant functions in colon carcinoma cells. Carcinogenesis 32, 676–685 [DOI] [PubMed] [Google Scholar]
- 33. Kim H. H., Abdelmohsen K., Gorospe M. (2010) Regulation of HuR by DNA damage response kinases. J. Nucleic Acids doi: 10.4061/2010/98148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. von Roretz C., Gallouzi I. E. (2010) Protein kinase RNA/FADD/caspase-8 pathway mediates the proapoptotic activity of the RNA-binding protein human antigen R (HuR). J. Biol. Chem. 285, 16806–16813 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Liu L., Rao J. N., Zou T., Xiao L., Wang P. Y., Turner D. J., Gorospe M., Wang J. Y. (2009) Polyamines regulate c-Myc translation through Chk2-dependent HuR phosphorylation. Mol. Biol. Cell 20, 4885–4898 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Kim H. H., Yang X., Kuwano Y., Gorospe M. (2008) Modification at HuR(S242) alters HuR localization and proliferative influence. Cell Cycle 7, 3371–3377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Kim H. H., Abdelmohsen K., Lal A., Pullmann R., Jr., Yang X., Galban S., Srikantan S., Martindale J. L., Blethrow J., Shokat K. M., Gorospe M. (2008) Nuclear HuR accumulation through phosphorylation by Cdk1. Genes Dev. 22, 1804–1815 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Mukherjee N., Corcoran D. L., Nusbaum J. D., Reid D. W., Georgiev S., Hafner M., Ascano M., Jr., Tuschl T., Ohler U., Keene J. D. (2011) Integrative regulatory mapping indicates that the RNA-binding protein HuR couples pre-mRNA processing and mRNA stability. Mol. Cell 43, 327–339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Kim H. S., Wilce M. C., Yoga Y. M., Pendini N. R., Gunzburg M. J., Cowieson N. P., Wilson G. M., Williams B. R., Gorospe M., Wilce J. A. (2011) Different modes of interaction by TIAR and HuR with target RNA and DNA. Nucleic Acids Res. 39, 1117–1130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Abdelmohsen K., Gorospe M. (2010) Posttranscriptional regulation of cancer traits by HuR. Wiley Interdiscip. Rev. RNA 1, 214–229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Zhu H., Zhou H. L., Hasman R. A., Lou H. (2007) Hu proteins regulate polyadenylation by blocking sites containing U-rich sequences. J. Biol. Chem. 282, 2203–2210 [DOI] [PubMed] [Google Scholar]