

Abstract
The stepwise formation of bridging (μ-) hydrides of diiron dithiolates is discussed with attention on the pathway for protonation and subsequent isomerizations. Our evidence is consistent with protonations occurring at a single Fe center, followed by isomerization to a series of μ-hydrides. Protonation of Fe2(edt)(CO)4(dppv) (1) gave a single μ-hydride with dppv spanning apical and basal sites, which isomerized at higher temperatures to place the dppv into a dibasal position. Protonation of Fe2(pdt)(CO)4(dppv) (2) followed an isomerization pathway similar to that for [1H]+, except that a pair of isomeric terminal hydrides were observed initially, resulting from protonation at the Fe(CO)3 or Fe(CO)(dppv) site. The first observable product from low temperature protonation of the tris-phosphine Fe2(edt)(CO)3(PMe3)(dppv) (3) was a single μ-hydride wherein PMe3 is apical and the dppv ligand spans apical and basal sites. Upon warming, this isomer converted fully but in a stepwise manner to a mixture of three other isomeric hydrides. Protonation of Fe2(pdt)(CO)3(PMe3)(dppv) (4) proceeded similarly to the edt analogue 3, however a terminal hydride was observed, albeit only briefly and at very low temperatures (−90 °C). Low-temperature protonation of the bis-chelates Fe2(xdt)(CO)2(dppv)2 produced exclusively the terminal hydrides [HFe2(xdt)(μ-CO)(CO)(dppv)2]+ (xdt = edt and pdt), which subsequently isomerized to a pair of μ-hydrides. At room temperature these (dppv)2 derivatives convert to an equilibrium of two isomers, one C2-symmetric and the other Cs-symmetric. The stability of the terminal hydrides correlates with the (C2-isomer)/(Cs-isomer) equilibrium ratio, which reflects the size of the dithiolate. The isomerization was found to be unaffected by the presence of excess acid, by solvent polarity, and the presence of D2O. This isomerization mechanism is proposed to be intramolecular, involving a 120° rotation of the HFeL3 subunit to an unobserved terminal basal hydride as the rate-determining step. The observed stability of the hydrides was supported by DFT calculations, which also highlight the instability of the basal terminal hydrides. Isomerization of the μ-hydride isomers occurs on alternating FeL3 via 120° rotations without generating D2O-exchangeable intermediates.
Introduction
The protonation of di- and polymetallic compounds almost invariably results in products containing μ-hydride ligands.1 The kinetic barriers for protonation are determined by reorganizational energies and are expected to be lower for protonation at main group vs transition metal centers.2 Consistent with this view, low temperature studies on the protonation of anionic metal carbonyls indicate that the carbonyl oxygen is often the kinetic site of protonation followed by a slower transfer of the proton to the metal.3 Polymetallic complexes, in principle, contain multiple possible sites of protonation, each with their own kinetic and thermodynamic preferences.
The interchange of bridging and terminal hydrides in clusters is well established (e.g. for H2Os3(CO)10(PPh3)).3 Some hydrides are known to exist as isomers (Scheme 1).4 As demonstrated in the current report, some diiron dithiolate hydrides protonate at a single metal followed by intramolecular isomerization to a bridging site. The present results suggest that protonation of more complex metal clusters, which invariably affords μ-hydrides, could occur at a single metal followed by rapid transfer of the hydride to a bridging position.
Scheme 1.

Two isomers of HFe(CO)3 (μ-PR2 )Pt(PR′3 )2.
The structure of dimetallic hydrides is relevant to the properties of the two major hydrogenase enzymes, the [FeFe]- and the [NiFe]-hydrogenases.5 The [NiFe]-hydrogenases are proposed to feature μ-hydrides in catalytically significant states, whereas in the [FeFe]-hydrogenases, substrate binding and turnover are assumed to be localized at one apical site of the [2Fe]H cluster (Fig. 1). We have proposed that the regiochemistry of the hydride ligands in these active sites determines the predominant physiological function of the enzyme—the [NiFe] centers are optimal for H2 oxidation and the [FeFe] centers are primarily reducers of protons.6 The (kinetic) reactivity of hydride ligands is well known to be affected by their geometry:7 bridging hydrides are more acidic than terminal hydrides. A parallel pattern has long been noted for the boron hydrides.8
Fig. 1.
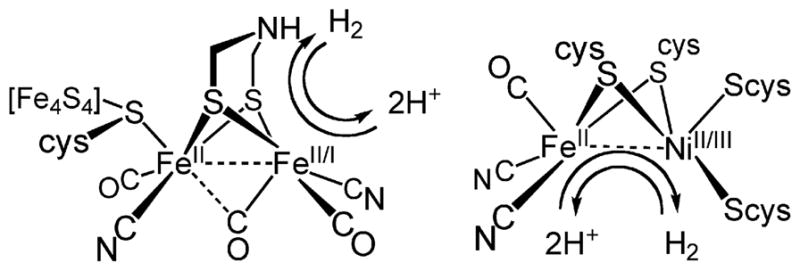
Structures and reactivity of the [FeFe]- and [NiFe]-hydrogenases with proposed oxidation states.
Almost all hydrides derived from protonation of diiron(I) dithiolato complexes are bridging. The only crystallographically characterized terminal hydride [HFe2(edt)(CO)2(PMe3)4]BF4 was found to isomerize unimolecularly to the μ-hydride isomer at room temperature with a half-life of minutes.† The regiochemistry has significant implications for catalysis since the terminal hydrides are more susceptible to protonolysis9 and more readily reduced10 than the μ-hydride isomers. In light of the differing reactivity for the different binding modes, it may be useful to understand the factors leading to the preferential formation of terminal hydride species and the pathway for conversion from terminal- to μ-H species.
It has long been known that diiron(I) dithiolates protonate to give μ-hydride derivatives. Early studies by Poilblanc led to the phosphine-substituted complexes such as [Fe2(μ-H)(SMe)2(CO)4(PPhMe2)2]+.11 After these compounds were found to resemble the active sites of the [FeFe]-hydrogenases,12 analogous protonations were described for the ethane- and propanedithiolato diiron complexes.12–14 The catalytic properties of the derived μ-hydrido complexes have been examined.12,15
The diiron(I) dithiolates are fluxional species.16,17 For substituted derivatives, the various rotamers could exhibit differing basicities. Recently, the low temperature protonation of several diiron(I) dithiolates has revealed the intermediacy of terminal hydrides, which subsequently form the more stable μ-hydrides. Examination of the protonation of Fe2(pdt)(CO)4(PMe3)2 by low temperature NMR spectroscopy reveals no terminal hydrides but affords three isomeric μ-hydrides, which upon warming to room temperature equilibrate fully to a single isomer.18
In this report, we describe and analyze protonations of a series of substituted diiron(I) dithiolates. Reactions were monitored at low temperature by both 1H and 31P NMR spectroscopies, techniques that usually provide definitive structural assignments, allowing us to deduce reaction sequences. Our results point to a general mechanism for the protonation of electron-rich diiron(I) dithiolates, one that begins with terminal protonation.
Results and discussion
Isomers of [HFe2(edt)(CO)4(dppv)]BF4 and [HFe2(pdt)(CO)4(dppv)]BF4
To provide reference spectra for relatively simple systems, we first examined the protonation of dichloromethane solutions of Fe2(edt)(CO)4(dppv) (1) and Fe2(pdt)(CO)4(dppv) (2) with HBF4·Et2O. The structures of the resulting hydrides can be deduced from the coupling patterns since in diiron dithiolato hydrides trans JP-hydride coupling constants are smaller (J PH ~ 5 Hz) than the corresponding cis coupling constants (J PH > 25 Hz).19 When protonations of 1 and 2 were monitored at lower temperature, we observed intermediates. In the case of 1, the low temperature 31P NMR spectrum featured two signals, corresponding to the 1H NMR signal at δ −16.1 (dd, J PH = 26, 6 Hz). This pattern is consistent with dppv spanning apical and basal sites (Scheme 2). Upon warming to room temperature, this hydride quantitatively converted to a new isomer with a single 31P NMR signal at δ 85 and 1H NMR signal at δ −14.7 (t, J PH = 21 Hz).
Scheme 2.

Course of protonation of 2, Fe2 (pdt)(CO)4 (dppv).
Upon protonation of 2, the resulting hydride isomerized by the same sequence as seen for 1. However, under the same conditions as for the protonation of 1, terminal hydrides were observed upon protonation of 2 at low temperatures. The formation of a pair of isomeric hydrides was indicated by characteristically20–22 low field signals at δ −4.4 (s) and δ −3.3 (t, J PH = 70 Hz). The predominant (~ 92%) product, [2H–Term]+ is characterized by 31P NMR signals at δ 97 and 71, consistent with a terminal hydride on the Fe(CO)3 subunit and an adjacent Fe(CO)(dppv) subunit with an apical–basal dppv. The minor (~ 8%) product, [2H–Term*]+, is an isomeric terminal hydride isomer characterized by a single 31P NMR signal at δ 89, consistent with a terminal-apical hydride on the Fe(CO)(dppv) subunit wherein the dppv is dibasal. Isomerization of the major isomer commences at a convenient rate around −30 °C, and upon warming the sample to room temperature, a single μ-hydride, [2H–A]+, is formed with 31P NMR signals at δ 95 and 92 and 1H NMR signal at δ −14.4 (dd J PH = 25, 5 Hz). The spectrum is consistent with an apical–basal dppv (see Scheme 2, Fig. S3†), identical to the initially observed μ-hydride for the edt example. As in the edt case, the sample further isomerized at room temperature to give exclusively [2H–B]+, a dibasal dppv isomer with a hydride signal at δ −14.8 (t, J PH = 21 Hz). Ezzaher et al. had observed a similar sequence of reactions for the protonation of Fe2(pdt)(CO)4(chel) (chel = dmpe18(1,2-C2H4(PMe2)2 and dppe21 (1,2-C2H4(PPh2)2)—these terminal hydrides convert to the apical–basal then the dibasal isomer of the μ-hydride. The isomerization pathway for the minor isomer of the terminal hydride could not be studied because of inadequate signal/noise ratio.
Protonation of Fe2(edt)(CO)3(PMe3)(dppv) and isomerization of the resulting hydrides
Protonation of room temperature solutions of Fe2(edt)(CO)3-(PMe3)(dppv) (3)23 by HBF4·Et2O was observed to yield a mixture of three isomeric μ-hydrides, as indicated by three multiplets in the high field region of the 1H NMR spectrum.13,24 When this protonation was conducted at −90 °C, only a single μ-hydride was obtained, [3H–A]BF4. In this species, the dppv ligand spans apical and basal sites as indicated by 31P NMR signals at δ 21, 91, 96.23 The 1H NMR spectrum displayed a doublet of triplets at δ −16.9 (J PH1 = 23, J PH2 ~ J PH3 = 3 Hz) (Fig. S6, S7†). This pattern is consistent with the dppv ligand spanning apical (J PH ~ 3 Hz) and basal positions (J PH ~ 23 Hz). Upon warming the sample to 20 °C, [3H–A]+ was found to convert to [3H–B]+. For this isomer, 1H and 31P NMR data indicated that the dppv remains apical–basal, but the PMe3 has shifted to a basal site. Over the course of ~ 20 h at room temperature, two further isomerizations occur giving rise to [3H–C]+ and [3H–D]+. Patterns for 31P and 1H NMR (high-field) spectra also allowed us to uniquely identify the stereochemistry of these species. In [3H–C]+, the PMe3 was found to remain basal, but the dppv shifted to a dibasal orientation. In [3H–D]+, the PMe3 returns to an apical position to give [3H–D]+. The equilibrated (room temperature) solution was found to contain 75% [3H–B]+, 20% [3H–C]+, and 5% [3H–D]+ (Scheme 3).
Scheme 3.

Isomerization pathway for [HFe2 (pdt)(CO)3 (dppv)(PMe3 )]+, [4H]+, and [HFe2 (edt)(CO)3 (dppv)(PMe3 )]+, [3H]+, and their equilibrium isomer distribution.
In a preparative-scale study, 3 was protonated with HCl followed by ion exchange with PF6− to give the thermally and air-stable salt. From an equilibrated solution of [3H]PF6, we grew crystals of exclusively [3H–B]PF6. Crystallographic analysis confirmed the assignment of the dppv orientation as apical–basal and trans-basal PMe3 (Fig. 2). Fresh solutions of these crystals contained only [3H–B]+. Upon allowing its solutions to stand, this species isomerized to the equilibrium mixture of [3H–B]+, [3H–C]+, and [3H–D]+, as discussed above.
Fig. 2.

Structure of the cation in [Fe2 (edt)(μ-H)(CO)3 (dppv)(PMe3 )]PF6 ([3H–B]+) with thermal ellipsoids drawn at the 50% probability level, phenyl hydrogen atoms omitted for clarity. Key distances (Å): Fe–Fe, 2.5744 (5); Fe(1)–S(1), 2.2581 (7); Fe(1)–S(2), 2.2794 (6); Fe(2)–S(1), 2.2668 (7); Fe(2)–S(2), 2.2760 (6); Fe(1)–P(1), 2.2137 (6); Fe(1)–P(2), 2.2282 (6); Fe(1)–H(1), 1.69 (2); Fe(2)–H(1), 1.64 (2).
Protonation of Fe2(pdt)(CO)3(PMe3)(dppv) and isomerization of the resulting hydrides
As in the protonation of 2, low temperature protonation of Fe2(pdt)(CO)3(PMe3)(dppv) 23 (4) with HBF4·Et2O allowed the observation of kinetic terminal hydrides. At −90 °C, the 1H NMR spectrum displayed signals at both δ −3.2 and −4.0, a chemical shift range that is again diagnostic of terminal hydrides (Fig. S4†).20,21 Both signals were doublets (JPH ~ 70 Hz), consistent with protonation at the Fe(CO)2(PMe3) center, and cis orientation of the terminal-apical hydride and PMe3. The presence of two doublets is attributed to the relative orientation of the propanedithiolate strap, which is known to refold slowly at low temperatures (Scheme 4).25 Alternatively, the data do not exclude the possibility that the two doublets indicate the presence of two stereoisomers arising from either cis or trans basal phosphine ligands (as in [3H–B]+ and [3H–B*]+, see below). 1H NMR spectra indicate that [4H–Term]+ isomerizes quickly at −90 °C (it isomerizes faster than the tetracarbonyl [2H–Term]+). The initial μ-hydride, [4H–A]+, is stereochemically analogous to the edt compound [3H–A]+, having apical–basal dppv and apical PMe3.
Scheme 4.

Possible terminal hydride isomers observed during the low-temperature protonation of 4
1H NMR spectra indicated that [4H–A]+ isomerizes at −30° to afford approximately equal amounts of two isomeric μ-hydrides, labeled [4H–B]+ and [4H–B*]+ (Fig. 3). 1H NMR spectra indicate that both isomers feature two basal and one apical phosphine and that both also feature apical–basal dppv and basal PMe3 ligands (the isomer with dibasal-dppv and apical PMe3 can be eliminated since this isomer corresponds to [4H–D]+, to be described, see Scheme 3). Two species ([4H–B]+ and [4H–B*]+) arise from the same terminal hydride but form via clockwise and anticlockwise turnstile rotations of the HFe(CO)2(PMe3) center, placing the PMe3 either trans-basal ([4H–B]+) or cis-basal ([4H–B*]+). Upon standing at −30 °C, the [4H–B]+/[4H–B*]+ mixture was found to convert entirely to [4H–B]+. We also observed low concentrations of an analogous pair of isomers for the ethanedithiolate (i.e., [3H–B]+ and [3H–B*]+). At room temperature, [4H–B]+ was found to isomerize to [4H–C]+ and [4H–D]+. The equilibrium ratio for [4H]BF4 was 71% [4H–C]+, 18% [4H–B]+ (0% [4H–B*]+), 11% [4H–D]+. The propanedithiolate favors the isomer wherein all phosphines are basal ([4H–C]+), whereas for the edt compound, the major isomer features basal PMe3 but apical–basal dppv ([3H–B]+).
Fig. 3.

1H-NMR spectra (CD2 Cl2, 500 MHz) for protonation of Fe2 (pdt)(CO)3 (dppv)(PMe3 ) (4) with HBF4 ·Et2 O and various stages in the isomerization of the resulting hydrides.
The kinetics of the transformation of [4H–B]+ to [4H–C]+ and [4H–D]+ were measured by 1H NMR spectroscopy. The linear dependence of ln([4H–B]) vs time (Fig. 4) indicated that the isomerization is first-order. The rate constant of isomerization of [4H–B]+ to [4H–C]+ was quantified as 1.1 × 10−5 s−1 at 23 °C (t1/2 ~ 17 h). The rate was unaffected by 1 atm. H2. When the isomerization of [4H–B]+ was conducted under an atmosphere of D2, the hydride ligand did not exchange.
Fig. 4.
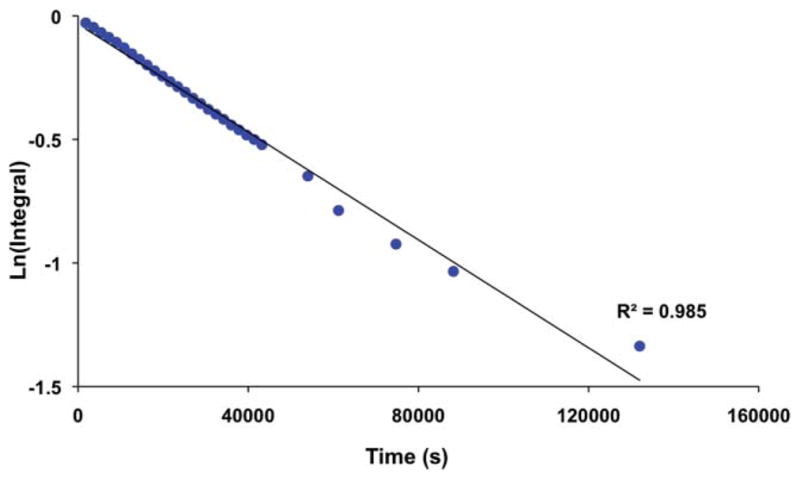
Plot of the isomerization of [4H–B]+ to [4H–C]+, as monitored by 1H NMR spectroscopy.
The equilibration of [4H]+ was examined by high temperature NMR measurements. Between 80 and 120 °C, the signals for two isomers ([4H–C]+ and [4H–D]+) broaden, but coalescence was not observed. Consistent with the proposed dynamics, the two isomers that undergo exchange are related by a single turnstile rotation. After three days in dimethylformamide, samples of [4H]+ were observed to degrade to a mixture of [Fe2(pdt)(μ-H)(CO)2(dppv)2]+ (see below) and unidentified poly-phosphine hydrido complexes.
Protonation of Fe2(xdt)(CO)2(dppv)2 and isomerization of the resulting hydrides (where xdt = edt, pdt)
Low-temperature protonation of Fe2(edt)(CO)2(dppv)2 produced the terminal hydride [HFe2(edt)(μ-CO)(CO)(dppv)2]+, [5H–Term]+. For protonations of the (dppv)2 derivatives, we selected [H(Et2O)2]BArF4 to ensure precise stoichiometry, however, replicate experiments with HBF4·Et2O provided qualitatively similar results. At −20 °C, this terminal hydride was found to rapidly isomerize to a μ-hydride, [5H–A]BArF4, wherein both dppv ligands chelate in the apical–basal orientation. The situation was similar for [HFe2(pdt)(CO)2(dppv)2]BArF4 ([6H–Term]BAr F4) although the conversion of [6H–Term]BArF4 to [6H–A]BArF4 was much slower. Between −10 and 20 °C, this isomerization was found to be first-order with respect to [6H–Term]+ with a half-life of ~ 12 min at 23 °C. Qualitatively the isomerization was found to be unaffected by the presence of excess acid. The rate of isomerization was indistinguishable for CD3CN and CD2Cl2. Additionally, no H/D exchange was observed when a solution of [5H–Term]+ was allowed to isomerize in the presence of D2O at 10 °C. Upon warming solutions of [5H–A]+ and [6H–A]+ to room temperature we observed the appearance of the unsymmetrical isomers, [5H–B]+ and [6H–B]+, respectively. 1H NMR spectra of these isomers display hydride signals—doublet-of-doublet-of-doublet-of-doublets and four signals in the 31P NMR spectrum—consistent with the one dppv ligand in the apical–basal orientation and the other in the dibasal orientation.
DFT investigation of the hydrides [HFe2(edt)(CO)2(PMe3)(dppv)]+ [HFe2(edt)(CO)3(PH3)3]+ and [HFe2(pdt)(CO)2(dppv)2]+
Protonation of Fe2(edt)(CO)3(PMe3)(dppv) (3) can in principle occur at apical sites, at basal terminal sites, or at the Fe–Fe bond (Scheme 5). Protonation at sulfur has also been analyzed computationally.26 As expected, and in line with DFT results 27 the obtained for other organometallic [2Fe]H model complexes, μ-H isomers are predicted to be significantly more favored than the terminal hydride isomers (by ca. 9.0 kcal/mol). DFT optimization for the hydride complex [3H]+ indicates that the four bridging hydrides observed experimentally are close in energy, differing only by a few kcal/mol. The calculated stabilities for the isomeric μ-hydrides agreed quite well with the observed equilibrium values, with [3H–B]+ being lowest in energy and [3H–C]+, [3H–D]+, and [3H–A]+ being higher in energy by 1.2, 0.5, and 2.8 kcal/mol, respectively (Scheme 5).
Scheme 5.

Computed relative thermodynamic stability of relevant [(dppv)(CO)Fe(edt)Fe(CO)2(PMe3 )(H)]+ isomers, energies in kcal/mol.
Within the manifold of terminal hydrides, the apical hydrides are predicted to be more stable than the isomeric basal hydrides, with the most stable apical hydride featuring a basal PMe3 and apical–basal dppv. This observation conforms to the trend wherein the strongly σ-donating hydride ligand stabilizes the trans π-acceptor CO ligand. In basal isomers, the hydride is trans to the S center of the chelating edt ligand. In agreement with experimental findings, protonation at the Fe(CO)2(PMe3) subunit is slightly favored (1.6 kcal/mol) relative to the Fe(CO)(dppv) side.
In order to elucidate how the steric bulk of the phosphines affects the relative stability of isomers of [3H]+, we conducted calculations on simplified analogues in which dppv and PMe3 ligands were replaced by the sterically less demanding PH3 ligands, i.e., [HFe2(edt)(CO)3(PH3)3]+. In general, the energetic ranking of the isomers remains similar—μ-hydrides are more stable and basal hydrides are least stable. In these uncrowded complexes, the most stable μ-H isomer features two trans CO ligands, whereas in the organophosphine derivatives steric effects slightly favor only one trans CO (Scheme 6). Among the terminal hydrides, isomers with FeH(CO)2(PH3) and FeH(CO)(PH3)2 subunits are almost equienergetic.
Scheme 6.

Computed relative thermodynamic stability of relevant [(PH3 )2 (CO)Fe(edt)Fe(CO)2(PH3 )(H)]+ isomers, energies in kcal/mol.
The influence of ligand bulk on the protonation regiochemistry is even more evident for [(dppv)(CO)Fe(pdt)Fe(dppv)(CO)] (Scheme 7). Again, we observe the usual energetic trend of μ-hydride < apical–hydride < basal-hydride, but the gap between the basal and apical hydrides is twice that seen for the (PMe3)(dppv) system.
Scheme 7.

Computed relative thermodynamic stability of relevant [(dppv)(CO)Fe(edt)Fe(CO)(dppv)(H)]+ isomers, energies in kcal/mol.
Discussion
The first suggestion of isomeric hydrides in the diiron dithiolates came from the characterization of [Fe2(edt)(μ-H)(CO)4(CN)2−.13] The 1H NMR spectrum shows two hydride signals that are assigned to isomers of [(CO)2(CN)Fe(μ-edt)(μ-H)Fe(CO)2(CN)]−.28 Related derivatives of the type [Fe2(edt)(μ-H)(CO)3L(CN)2]− also exist as isomers (L = PPh3, CN−).28 Additionally, Scholl-hammer and Hogarth et al. have demonstrated isomerism in [HFe2(pdt)(CO)4(chel)]+ (chel = dmpe,18 dppe,21 phen22).29
Terminal protonation
Diiron complexes with terminal hydrides are rarely observed.30 Poilblanc proposed that protonation of the diiron dithiolates occurs directly at the Fe–Fe bond.31 However, when these protonations are monitored at low temperatures by NMR spectroscopy, terminal hydrides are often observed, especially for complexes containing chelating ligands.10,18,21,22 The chelating phosphine ligands elevate the barrier for the conversion of the terminal hydrides to the bridging hydrides. Even though terminal hydrides are not observed consistently, results suggest that terminal hydrides are almost always the kinetic product of protonation. In cases where the terminal hydrides have not been observed spectroscopically,— 3, and Fe2(xdt)(CO)4(PMe3)2 (xdt = adt, edt, pdt)—the initially observed μ-hydrides are explicable as arising via intramolecular rearrangement from the analogous terminal hydrides.
Potential exceptions to the universality of terminal protonation come from studies on the protonation of diiron dithiolates that are constrained by special chelating ligands. The complex Fe2(pdt)(Cy2PCH2PCy2)(CO)4, where the diphosphine links the two Fe centers, protonates rapidly to give the μ-hydride.32 The two isomers of Fe2(SCH2CH2PPh2)2(CO)4, protonate with retention of geometry (eqn (1)).33 These protonations are slow at room temperature, however, possibly reflecting the high barrier to protonation of the Fe–Fe bond. A recent report suggests that protonation at a Fe–Fe bond can be accelerated by ligands that can serve as proton relays.34 In such cases however, it is difficult to exclude relay to a single Fe center followed by rapid rearrangement to the μ-hydride isomer. In contrast, the protonation of the complexes described in this paper occur within the time of mixing at room temperature.‡
Mechanism of terminal to μ-hydride isomerization
In those cases where they can be observed, terminal hydrides do not readily deprotonate. For example, separate NMR signals for hydrides and their conjugate bases can be resolved readily, consistent with a relatively large barrier (> 12 kcal/mol) to intermolecular proton transfer. Indicative of their significant kinetic robustness is the high barrier for deprotonation. Thus, the terminal hydride [HFe2(pdt)(CO)2(dppv)2]+ resists deprotonation by a large excess of NEt3, which is far more basic than the diiron center (ΔpK a ~ 6).35 Thus, the isomerization of the terminal hydrides to the μ-hydrides does not occur via a deprotonation–reprotonation pathway. Consistent with this view, conversion of [4H–Term]+ to [4H–A]+ occurs without H–D exchange with D2O.
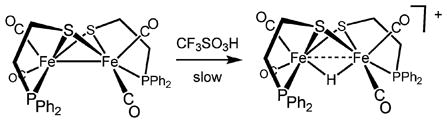 |
(1) |
The terminal hydrides are however unstable with respect to conversion to the μ-hydrides. The rate of this isomerization is strongly affected by the dithiolate, the pdt derivatives being slower to isomerize than the analogous edt compounds.36 Basicity effects alone do not explain the stability trends for the terminal hydrides as indicated by the finding that [HFe2(pdt)(CO)3(PMe3)(dppv)]BF4is significantly less stable than [HFe2(pdt)(CO)4(dppv)]BF4 ([2H–Term]BF4). The terminal hydride [4H–Term]+ is observed only fleetingly at −90 °C, whereas [2H–Term]BF4 is stable until ca. −40 °C.§ We suggest the first step in the isomerization is a rate-determining 120° “turnstile” rotation that shifts the terminal apical hydride to a terminal basal site. DFT calculations predict that the basal hydrides are significantly less stable than the apical hydrides, consistent with the fact that basal hydrides species have so far never been observed (Scheme 8).29 The higher barrier for the isomerization of [2H–Term]+, in comparison to [4H–Term]+, is attributed to the instability of [2H–Basal]+ where CO is trans to μ-CO. The second step in the isomerization involves the interchange of the basal hydride ligand with the μ-CO ligand. Precedent for the conversion of the terminal to the μ-hydride comes from the pairwise exchange of terminal and μ-hydrides in species such as HOs3(μ-H)(CO)10L.37
Scheme 8.

Schematic reaction coordinate for the isomerization of the apical-terminal hydrides. Basal-terminal hydrides are not observed, indicating their rapid conversion to the μ-hydrides.
Interconversions within the μ-hydride manifold
Two or more isomers of the μ-hydrides are observed in all cases reported here. In general, these isomers exist in dynamic equilibrium, and the overall isomer distribution is dictated by the energetic advantage associated with CO ligands trans to the hydride balanced against the relief of steric congestion.28 The μ-hydrides are proposed to interconvert via 120° turnstile rotations of FeL3 subunits. The isomerization occurs at alternating Fe sites. Particularly telling mechanistically is the observation of pairs of μ-H isomers in the case of the pdt-(PMe3)(dppv) system ([4H–B]+/[4H–B*]+). These rotamers contain basal PMe3 ligands that are mutually cis and trans with respect to the apical–basal dppv ligand. These apparent rotamers arise from the clockwise and anticlockwise rotations of the Fe(CO)2(PMe3) center. A high temperature 1H NMR spectrum of [4H]+ showed that a pair of isomers related by a single 120° rotation of the Fe(CO)2(PMe3) subunit ([4H–C]+ and [4H–D]+) approached coalescence. The scission of individual Fe–μ-H bonds has been proposed to occur upon photolysis of [Fe2(pdt)(μ-H)(CO)4(PMe3)2]+.14,38
Concluding remarks
The protonation of the substituted diiron dithiolato carbonyls provides new insights into protonation of polynuclear complexes, a broadly important theme.39 The main pathway entails protonation at a single metal followed by isomerization to a series of μ-hydrides. As we have described recently,6 the terminal hydrides are mechanistically related to the active site of the [FeFe]-hydrogenases, whereas the μ-hydrides are more closely related to the active sites of the [NiFe]-hydrogenases.
Experimental
Manipulations were conducted using standard Schlenk techniques. Solvents were filtered through activated alumina and subsequently degassed. 1H and 31P NMR spectra were acquired on a Unity Varian 500 or a Unity Varian 600 spectrometer. IR spectra were collected on a Mattson Infinity Gold FTIR spectrometer. Fe2(edt)(CO)4(dppv), Fe2(pdt)(CO)4(dppv), Fe2(edt)(CO)3-(PMe3)(dppv), Fe2(pdt)(CO)3(PMe3)(dppv), Fe2(xdt)(CO)2-(dppv)2,10,23[H(Et2O)2]BArF440 were prepared according to literature procedures. HBF4·Et2O and dppv (cis-1,2-bis(diphenylphosphino)ethylene) were purchased from Aldrich and used as received.
[Fe2(edt)(μ-H)(CO)4(dppv)]PF6 ([1H]PF6)
A solution of 0.110 g (0.155 mmol) of Fe2(edt)(CO)4(dppv) in 10 mL of CH2Cl2 was treated with 0.20 mL (0.40 mmol) of a 2.0 M solution HCl in Et2O, the reaction flask was immediately stoppered. After the reaction solution stirred for 3 h, solvent was removed in vacuum. The residue, a red powder, was then redissolved in 5 mL of MeOH. The red solution was then treated with 0.025 g of NH4PF6 (0.160 mmol) in 5 mL of MeOH, precipitating the product. The red solid was collected via filter cannula and washed with 2 ×10 mL Et2O. Anal. Calcd for C32H27Fe2O4F6P3S2 (found): C, 44.78 (44.70); H, 3.17 (3.02). 1H NMR (CD2Cl2): δ 7.2–8.3 (m, 22H, P(C6H 5)2; C2H 2(PPh2)2); 0.7–2.9 (m, 4H, S2C2H 4); δ −15 (t, 1H, Fe–H). 31P NMR (CD2Cl2): δ 85. FT-IR (CH2Cl2): 2098 (s), 2052 (m), 2038 (m), 1980 (m) cm−1.
[Fe2(pdt)(μ-H)(CO)4(dppv)]PF6 ([2H]PF6)
A solution of 0.066 g (0.091 mmol) of Fe2(pdt)(CO)4(dppv) in 10 mL of CH2Cl2 was treated with 0.2 mL (0.40 mmol) of a 2.0 M solution HCl in Et2O, the reaction flask was then stoppered. After the reaction solution stirred for 3 h, solvent was removed in vacuum. The residue, a red powder, was then re-dissolved in 5 mL of MeOH. The red solution was then treated with 0.015 g of NH4PF6 (0.092 mmol) in 5 mL of MeOH, precipitating the product. The red solid was collected via filter cannula and washed with 2 × 10 mL Et2O. 1H NMR (CD2Cl2): δ 7.2–8.5 (m, 22H, P(C6H 5)2; C2H 2(PPh2)2); 2.3– 3.6 (m, 6H, S2C3H 6 ); δ −14.7 (t, 1H, Fe–H, J PH = 23 Hz). 31P NMR (CD2Cl2): δ 85.6. FT-IR (CH2Cl2): 2098 (s), 2052 (m), 2038 (m), 1980 (m) cm−1.
[Fe2(edt)(μ-H)(CO)3(PMe3)(dppv)]PF6 ([3H]PF6)
A solution of 0.125 g (0.164 mmol) of 3 in 25 mL of MeCN was treated with 8.2 mL (0.820 mmol) of a 0.10 M solution of 12 M HCl in MeCN. After the reaction solution stirred for 3 h, solvent was removed in vacuum. The residue, an orange powder, was rinsed with 30 mL of Et2O and then re-dissolved in 5 mL of MeOH. This extract was treated with a solution of 0.026 g of NH4PF6 (0.164 mmol) in 5 mL of MeOH, and the product was precipitated by the addition of 30 mL of H2O. The orange solid was collected and rinsed with 15 mL each of H2O and Et2O. Crystals were grown via slow diffusion of hexanes into a CH2Cl2 solution. Anal. Calcd for C34H36Fe2O3F6P4S2 (found): C, 45.06 (44.89); H, 4.00 (3.93). 1H NMR (CD3CN): δ 7.2–8.5 (m, 22H, P(C6H 5)2; C2H 2(PPh2)2); 0.4–2.8 (m, 4H, S2C2H 4); 1.6 (d, 9H, PMe3); δ −17 (td, 1H, Fe-H). 31P NMR (CD3CN): δ 92.3, 90.7 (d, t, dppv); δ22.8 (d, PMe3). FT-IR (MeCN): 2031, 1978 cm−1.
[Fe2(pdt)(μ-H)(CO)3(PMe3)(dppv)]PF6 ([4H]PF6)
The sample was prepared following the procedure for [3H]PF6.Anal. Calcd for C35H38Fe2O3F6P4S2 (found): C, 45.67 (46.20); H, 4.16 (4.22). 1H NMR (CD3CN): δ 7.2–8.5 (m, 22H, P(C6H 5)2; C2H 2(PPh2)2); 0.4–2.8 (m, 6H, S2C3H 6 ); 1.6 (d, 9H, PMe3); δ −15 (td, 1H, Fe-H). 31P NMR (CD3CN): δ 91.6, 90.6 (t, d, dppv); δ21.6 (d, PMe3). FT-IR (MeCN): 2028, 1973 cm−1.
Procedure for protonation of 1, 2, 3, 4 at low temperature
Into a J-Young NMR tube dried CD2Cl2 was distilled onto 5 mg diiron dithiolate. To the frozen solution ~ 5 μL HBF4·Et2O (6.91 M) (0.035 mmol, 1 to 10 equiv.) was added was freeze-pump-thawed to ensure a vacuum atmosphere. The frozen J-Young tube was then placed directly into a dry ice/Acetone bath (−78 °C), thawed slowly, and characterized by low temperature NMR studies with a pre-cooled NMR spectrometer probe.
Isomerization of [HFe2(pdt)(CO)3(PMe3)(dppv)]PF6, [4H]PF6
In a J-Young NMR tube dried CD2Cl2 was distilled onto a solid mixture of ~ 5 mg (0.005 mmol) of [HFe2(pdt)(CO)3(PMe3)-(dppv)]PF6 ([4H]PF6) and ~ 1 mg (0.006 mmol) hexamethylbenzene (as internal standard). The frozen J-Young tube was thawed carefully and immediately inserted into the NMR spectrometer. The signals of interest (the PCH 3 signals and the hydride signals) were integrated relative to the internal standard over the course of several weeks.
H/D exchange from D2 + [HFe2(pdt)(CO)3(PMe3)(dppv)]PF6
In a J-Young tube and following the conditions for the isomerization of [4H]PF6, D2 (~1.0 atmosphere) was added to the freshly prepared and freshly frozen solution of [4H]PF6 in that solvent. The sample was then thawed carefully, shaken (to ensure complete and immediate saturation of solution), and immediately inserted into the NMR spectrometer for kinetic characterization. Experiments were conducted in both ambient light and dark, and no measurable differences were observed.
H/D exchange from D2O + [HFe2(pdt)(CO)2(dppv)2]BArF4
In a J-Young NMR tube dried CD2Cl2 was distilled onto a solid mixture of 10 mg (0.009 mmol) Fe2(pdt)(CO)2(dppv)2 and ~ 10 mg (0.010 mmol) [H(Et2O)2]BArF4. The frozen J-Young tube was then thawed carefully at −40 °C (CH3CN: dry ice) and then after complete thawing (~ 1 min) was frozen in liquid N2 and 1 drop D2O was added. The sample was then evacuated on the high-vac line and carefully thawed at −40 °C and immediately inserted into the NMR spectrometer for characterization.
X-ray crystallography
Structure was phased by direct methods. Systematic conditions suggested the ambiguous space group. The space group choice was confirmed by successful convergence of the full-matrix least-squares refinement on F2. The highest peaks in the final difference Fourier map were in the vicinity of atoms F1, C9, and F4; the final map had no other significant features. A final analysis of variance between observed and calculated structure factors showed no dependence on amplitude or resolution.
DFT calculations
DFT calculations were carried out using the B-P86 functional and a valence triple-ζ basis set with polarization on all atoms (TZVP),41 a level of theory which has been found to give reliable results for analogous organometallic compounds.42 Stationary points of the energy hypersurface have been located by means of energy gradient techniques and full vibrational analysis has been carried out to further characterize each stationary point. Relevant IR bands, computed for all intermediate species, which are involved in the mechanism of interconversion, are available as ESI.†
All energy differences have been computed by correcting gas phase data with the inclusion of an implicit treatment of solvent effect (COSMO).43 The dielectric constant value has been set to 37.5 (corresponding to acetonitrile).
Supplementary Material
Acknowledgments
This research was supported by the U.S. National Institutes of Health and the Petroleum Research Fund. We thank Terésa Prussak-Wieckowska for assistance with the crystallographic analyses.
Footnotes
Electronic supplementary information (ESI) available: Experimental details. CCDC reference number 733498. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/b910147k
Although fast at room temperature, the protonation of Fe2 (edt)(CO)4 -(dppv) is noticeably slower than the protonation of Fe2 (pdt)(CO)4 -(dppv) at −80 °C.
For similar reactions and identical rates at 250 vs 200 K, the difference in the activation energies is ca. 20%.
Contributor Information
Luca De Gioia, Email: luca.degioia@unimib.it.
Thomas B. Rauchfuss, Email: Rauchfuz@uiuc.edu.
References
- 1.Dyson PJ, McIndoe JS. Transition Metal Carbonyl Cluster Chemistry. Taylor & Francis; London, UK: 2000. [Google Scholar]
- 2.Kramarz KW, Norton JR. Prog Inorg Chem. 1994;42:1. [Google Scholar]
- 3.Rosenberg E. Polyhedron. 1989;8:383. [Google Scholar]
- 4.Powell J, Gregg MR, Sawyer JF. J Chem Soc, Chem Commun. 1987:1029. [Google Scholar]
- 5.Fontecilla-Camps JC, Volbeda A, Cavazza C, Nicolet Y. Chem Rev. 2007;107:4273. doi: 10.1021/cr050195z. [DOI] [PubMed] [Google Scholar]
- 6.Mealli C, Rauchfuss TB. Angew Chem, Int Ed. 2007;46:8942. doi: 10.1002/anie.200703413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Justice AK, Nilges M, Rauchfuss TB, Wilson SR, De Gioia L, Zampella G. J Am Chem Soc. 2008;130:5293. doi: 10.1021/ja7113008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cavanaugh MA, Fehlner TP, Stramel R, O’Neill ME, Wade K. Polyhedron. 1985;4:687. [Google Scholar]
- 9.Justice AK, Linck RC, Rauchfuss TB, Wilson SR. J Am Chem Soc. 2004;126:13214. doi: 10.1021/ja0455594. [DOI] [PubMed] [Google Scholar]
- 10.Barton BE, Rauchfuss TB. Inorg Chem. 2008;47:2261. doi: 10.1021/ic800030y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fauvel K, Mathieu R, Poilblanc R. Inorg Chem. 1976;15:976. [Google Scholar]
- 12.Gloaguen F, Lawrence JD, Rauchfuss TB. J Am Chem Soc. 2001;123:9476. doi: 10.1021/ja016516f. [DOI] [PubMed] [Google Scholar]
- 13.Zhao X, Georgakaki IP, Miller ML, Yarbrough JC, Darensbourg MY. J Am Chem Soc. 2001;123:9710. doi: 10.1021/ja0167046. [DOI] [PubMed] [Google Scholar]
- 14.Zhao X, Georgakaki IP, Miller ML, Mejia-Rodriguez R, Chiang CY, Darensbourg MY. Inorg Chem. 2002;41:3917. doi: 10.1021/ic020237r. [DOI] [PubMed] [Google Scholar]
- 15.Borg SJ, Tye JW, Hall MB, Best SP. Inorg Chem. 2007;46:384. doi: 10.1021/ic061211t. [DOI] [PubMed] [Google Scholar]; Felton GAN, Vannucci AK, Chen J, Lockett LT, Okumura N, Petro BJ, Zakai UI, Evans DH, Glass RS, Lichtenberger DL. J Am Chem Soc. 2007;129:12521. doi: 10.1021/ja073886g. [DOI] [PubMed] [Google Scholar]; Wang F, Wang M, Liu X, Jin K, Dong W, Sun L. Dalton Trans. 2007:3812. doi: 10.1039/b706178a. [DOI] [PubMed] [Google Scholar]
- 16.Adams RD, Cotton FA, Cullen WR, Hunter DL, Mihichuk L. Inorg Chem. 1975;14:1395. [Google Scholar]
- 17.Tye JW, Darensbourg MY, Hall MB. Inorg Chem. 2006;45:1552. doi: 10.1021/ic051231f. [DOI] [PubMed] [Google Scholar]; Olsen MT, Bruschi M, De Gioia L, Rauchfuss TB, Wilson SR. J Am Chem Soc. 2008;130:12021. doi: 10.1021/ja802268p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ezzaher S, Capon JF, Gloaguen F, Kervarec N, Pétillon FY, Pichon R, Schollhammer P, Talarmin J. C R Chim. 2008;11:906. doi: 10.1021/ic0703124. [DOI] [PubMed] [Google Scholar]
- 19.Meakin P, Muetterties EL, Jesson JP. J Am Chem Soc. 1973;95:75. [Google Scholar]; Bellachioma G, Cardaci G, Macchioni A, Zuccaccia C. J Organomet Chem. 2001;628:255. doi: 10.1021/ja015959g. [DOI] [PubMed] [Google Scholar]
- 20.Van Der Vlugt JI, Rauchfuss TB, Whaley CM, Wilson SR. J Am Chem Soc. 2005;127:16012. doi: 10.1021/ja055475a. [DOI] [PubMed] [Google Scholar]
- 21.Ezzaher S, Capon JF, Gloaguen F, Pétillon FY, Schollhammer P, Talarmin J, Pichon R, Kervarec N. Inorg Chem. 2007;46:3426. doi: 10.1021/ic0703124. [DOI] [PubMed] [Google Scholar]
- 22.Orain PY, Capon JF, Kervarec N, Gloaguen F, Pétillon F, Pichon R, Schollhammer P, Talarmin J. Dalton Trans. 2007:3754. doi: 10.1039/b709287c. [DOI] [PubMed] [Google Scholar]
- 23.Justice AK, Zampella G, De Gioia L, Rauchfuss TB, Van Der Vlugt JI, Wilson SR. Inorg Chem. 2007;46:1655. doi: 10.1021/ic0618706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Schwartz L, Eilers G, Eriksson L, Gogoll A, Lomoth R, Ott S. Chem Commun. 2006:520. doi: 10.1039/b514280f. [DOI] [PubMed] [Google Scholar]
- 25.Lyon EJ, Georgakaki IP, Reibenspies JH, Darensbourg MY. J Am Chem Soc. 2001;123:3268. doi: 10.1021/ja003147z. [DOI] [PubMed] [Google Scholar]
- 26.Tye JW, Darensbourg MY, Hall MB. Theochem. 2006;771:123. [Google Scholar]
- 27.Bruschi M, Greco C, Fantucci P, De Gioia L. Inorg Chem. 2008;47:6056. doi: 10.1021/ic8006298. [DOI] [PubMed] [Google Scholar]; Bruschi M, Greco C, Kaukonen M, Fantucci P, Ryde U, De Gioia L. Angew Chem, Int Ed. 2009;48:3503. doi: 10.1002/anie.200900494. [DOI] [PubMed] [Google Scholar]
- 28.Boyke CA. PhD Thesis. University of Illinois; Urbana-Champaign: 2005. [Google Scholar]
- 29.Adam FI, Hogarth G, Kabir SE, Richards I. C R Chim. 2008;11:890. [Google Scholar]
- 30.Ohki Y, Suzuki H. Angew Chem, Int Ed. 2000;39:3120. [PubMed] [Google Scholar]; Cheah MH, Borg SJ, Bondin MI, Best SP. Inorg Chem. 2004;43:5635. doi: 10.1021/ic049746e. [DOI] [PubMed] [Google Scholar]
- 31.Arabi MS, Mathieu R, Poilblanc R. J Organomet Chem. 1979;177:199. [Google Scholar]
- 32.Adam FI, Hogarth G, Richards I. J Organomet Chem. 2007;692:3957. [Google Scholar]
- 33.Zhao X, Hsiao YM, Lai CH, Reibenspies JH, Darensbourg MY. Inorg Chem. 2002:699. doi: 10.1021/ic010741g. [DOI] [PubMed] [Google Scholar]
- 34.Ezzaher S, Capon JF, Gloaguen F, Petillon FY, Schollhammer P, Talarmin J, Kervarec N. Inorg Chem. 2009;48:2. doi: 10.1021/ic801369u. [DOI] [PubMed] [Google Scholar]
- 35.Izutsu K. Acid–Base Dissociation Constants in Dipolar Aprotic Solvents. Blackwell Scientific Publications; 1990. [Google Scholar]; Eilers G, Schwartz L, Stein M, Zampella G, de Gioia L, Ott S, Lomoth R. Chem–Eur J. 2007;13:7075. doi: 10.1002/chem.200700019. [DOI] [PubMed] [Google Scholar]; Stanley JL, Heiden ZM, Rauchfuss TB, Wilson SR, De Gioia L, Zampella G. Organometallics. 2007;27:119. doi: 10.1021/om7009599. [DOI] [PMC free article] [PubMed] [Google Scholar]; Barton BE, Olsen MT, Rauchfuss TB. J Am Chem Soc. 2008;130:16834. doi: 10.1021/ja8057666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Justice AK, De Gioia L, Nilges MJ, Rauchfuss TB, Wilson SR, Zampella G. Inorg Chem. 2008;47:7405. doi: 10.1021/ic8007552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rosenberg E, Anslyn EV, Barner-Thorsen C, Aime S, Osella D, Gobetto R, Milone L. Organometallics. 1984;3:1790. [Google Scholar]
- 38.Zhao X, Chiang CY, Miller ML, Rampersad MV, Darensbourg MY. J Am Chem Soc. 2003;125:518. doi: 10.1021/ja0280168. [DOI] [PubMed] [Google Scholar]
- 39.Peruzzini M, Poli R. Recent Advances in Hydride Chemistry. New York: 2001. [Google Scholar]
- 40.Brookhart M, Grant B, Volpe AF. Organometallics. 1992;11:3920. [Google Scholar]
- 41.Schaefer A, Huber C, Ahlrichs R. J Chem Phys. 1994;100:5829. [Google Scholar]
- 42.Niu S, Hall MB. Chem Rev. 2000;100:353. doi: 10.1021/cr980404y. [DOI] [PubMed] [Google Scholar]; Bruschi M, Zampella G, Fantucci P, De Gioia L. Coord Chem Rev. 2005;249:1620. doi: 10.1021/ja0508424. [DOI] [PubMed] [Google Scholar]; Siegbahn PEM, Tye JW, Hall MB. Chem Rev. 2007;107:4414. doi: 10.1021/cr050185y. [DOI] [PubMed] [Google Scholar]
- 43.Klamt A, Schüürmann G. J Chem Soc, Perkin Trans. 1993;2:799. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.