

Abstract
Lipid bilayer membranes found in nature are heterogeneous mixtures of lipids and proteins. Model systems, such as supported lipid bilayers (SLBs) are often employed to simplify experimental systems while mimicking the properties of natural lipid bilayers. Here we demonstrate a new method to form SLB arrays by first forming spherical supported lipid bilayers (SSLBs) on sub-micron-diameter SiO2 beads. The SSLBs are then arrayed into microwells using a simple physical assembly method that requires no chemical modification of the substrate, nor modification of the lipid membrane with recognition moieties. The resulting arrays have sub-micron SSLBs with 3 μm periodicity where > 75 % of the microwells are occupied by an individual SSLB. Because the arrays have high density, fluorescence from > 1000 discrete SSLBs can be acquired with a single image capture. We show that 2-component random arrays can be formed and we also use the arrays to determine the equilibrium dissociation constant for cholera toxin binding to ganglioside GM1. SSLB arrays are robust and are stable for at least one week in buffer.
Keywords: Supported lipid bilayer, membrane microarray, membrane curvature, silica beads, cholera toxin, ganglioside GM1
INTRODUCTION
Lipid bilayer membranes are ubiquitous in biology. They define the cytoplasmic boundaries of cells, as well as subcellular organelles. Natural lipid bilayer membranes are mixtures of lipids and proteins with heterogeneous spatial features and diverse chemical and physical properties.1 In order to simplify experimental systems, synthetic model membrane structures such as giant vesicles2 and supported lipid bilayers3 (SLBs) have been used to mimic the chemical and physical properties of natural membranes. SLBs are a convenient model system because they can be prepared by simple methods, such as Langmuir-Blodgett methods4 or spontaneous rupture,5 and their physicochemical properties can be interrogated by a variety of analytical techniques such as fluorescence microscopy,4 scanning probe microscopy,6 Raman spectroscopy7 or mass spectrometry.8 SLBs can also be readily integrated into a variety of optical,9,10 electrical,11,12 and electrochemical13 biosensors.
Arrays of SLBs are of particular interest for applications in sensing14 and for modeling cell-cell interactions.15,16 There are many methods to form SLB patterns and arrays.17,18 Linear arrays of SLBs can be formed in microfluidic channels and be used for immunoassays.19 Alternatively, substrates can be patterned with materials that prevent lipid diffusion, thereby making partitions between adjacent SLB patches.20 Photochemical patterning of SLBs can also be used to define arrays.21,22 SLB patterns can be transferred by microcontact23 or non-contact printing24 and patterned self-assembled monolayers can define patterned hybrid lipid bilayers.25
Here we demonstrate a new method to form high-density SLB arrays by periodic assembly of spherical supported lipid bilayers (SSLBs) in size-tunable microwell array substrates. SSLBs have been previously used by Groves and coworkers for sensing lipid-protein interactions via colloidal assembly.26 However, by immobilizing SSLBs in microwell arrays, this method combines the features of SLB arrays with the advantages of bead array assays,27,28 which include improved signal to noise ratios and the opportunity to generate color-coded random arrays of multiple components, eliminating the need for serial spotting. Serial spotting can be advantageous, however. By using serial deposition in separate arrays on a single chip we show that SSLBs can be coded by their fluorescence emission color (i.e. chemical identity) as well as their position. Akin to bead assays, our method also requires no post-fabrication surface patterning or chemical modification of the substrate, and the SSLBs need no recognition or binding moieties (e.g. streptavidin/biotin). Another advantage of SSLBs is that because they are spherical, there are no exposed lipid bilayer edges which exhibit altered physicochemical properties.29 The motivation for this work stems from a desire to create highly uniform sub-micron lipid bilayer arrays. In earlier work we showed that unilamellar vesicles and natural membrane particles could be arrayed into micro- and nanowells using a simple physical assembly method.30 However, there was no control over the amount of membrane material in each well. By size-matching the SSLB and the microwell we ensure that one and only one SSLB is immobilized in each well. This results in arrays where the amount of lipid membrane is uniform from well-to-well. In fact, since the coefficient of variation of SiO2 bead diameter is smaller than for extruded vesicles,31 the amount of lipid in each well is more uniform than if individual vesicles were immobilized in the wells. With this method we can form arrays with nearly 100,000 discrete SSLBs per mm2.
EXPERIMENTAL SECTION
Formation of phospholipid vesicles and SSLBs
Phospholipid vesicles were prepared by drying and rehydration followed by extrusion. All lipids were obtained from Avanti Polar Lipids, Inc. Lipid mixtures containing the appropriate amounts of egg PC, rhodamine PE (1,2-dimyristoyl-sn-glycero-3-phosphoethanolamine-N-(lissamine rhodamine B sulfonyl) ammonium salt), NBD-PE (1,2-dimyristoyl-sn-glycero-3-phosphoethanolamine-N-(7-nitro-2-1,3-benzoxadiazol-4-yl) ammonium salt) or ganglioside GM1 in chloroform were dried under vacuum for at least 6 hours. All lipid compositions are reported as mole percent. Then the dried lipid films were rehydrated with 0.1 M NaCl to a lipid concentration of 1 mg/mL and allowed to stand overnight. The following day the vesicles were dispersed into solution by vortex mixing and bath sonication. The vesicles were then extruded through 100 nm pore size polycarbonate filters using an Avanti Mini-Extruder. The vesicles were then diluted to 0.1 mg/mL in 0.1 M NaCl. The 700 nm diameter SiO2 beads (Bangs Laboratories) were suspended in 0.1 M NaCl at a concentration of 1.5 × 1010 beads/mL. To form the SSLBs, 25 μL of the SiO2 bead solution was added to 200 μL of the vesicle solution, the mixture was vortex mixed and allowed to rest at room temperature for 1 hour. The SSLBs were separated from unruptured vesicles by three rounds of centrifugation and then resuspended in PBS, pH 7.4. Assuming no loss of SSLBs during the washing steps, the final concentration of SSLBs was 1.7 × 109 SSLBs/mL.
Fabrication of microwell array substrates
The microwell arrays were prepared using photolithography.30 MEGAPOSIT SPR-955 photoresist (Rohm and Haas) was spin-coated on silicon wafers with a 100 nm-thick thermal oxide and exposed with an i-line stepper (Canon 2500 i3) using a mask with 5 μm holes in a hexagonal array with periodicity 15 μm over a 10 mm × 10 mm area. This was reduced 5× resulting in 1 μm holes with 3 μm center-to-center spacing over a 2 mm × 2 mm area and stepped across an entire wafer with 5 mm steps, leaving 3 mm spacing between the microwell arrays. The wafer was developed for 90 seconds in MF CD 26 (Rohm and Haas) using a CEE 200X (Brewer Science) spray developer. Using the resist as an etch mask, the thermal oxide layer was etched using RIE (STS model 320) with CF4 and Ar gases at 100 W for 6 minutes. The silicon was etched to a depth of approximately 500 nm in a deep trench silicon etcher (Plasma Therm SLR-770) using the oxide and resist as a mask. The microwells were soaked in acetone for 5 minutes to remove the resist, cleaned in piranha solution, a 1:1 mixture of sulfuric acid and hydrogen peroxide, for 5 minutes, and then the oxide mask was removed in a BOE bath. Piranha solution is a strong oxidizing agent and extreme caution should be exercised in its use and disposal. Then the microwell surface was covered with a conformal 100 nm-thick layer of Al2O3 by ALD. ALD was carried out at 250 °C, and the deposition rate was about 1.1 Å per cycle.
Formation of SSLB arrays and GM1/cholera toxin binding
The SSLB arrays were prepared in a manner similar to previous work.30 Briefly, 5 μL drops of SSLB solution (1.7 × 109 SSLBs/mL) were placed upon 2 mm × 2 mm microwell arrays and the SSLBs were allowed to settle for 30 minutes. Then array was gently washed with PBS to remove loosely adherent SSLBs and then placed in shallow dish containing PBS. A PDMS block was then gently pulled by hand across the surface at an angle 5 times to remove SSLBs that were not immobilized in the recessed wells.32 The array was then rinsed with PBS. For CTX binding experiments the SSLB arrays were first blocked by incubating in 2 mg/mL BSA for 1 hour. Then the arrays were exposed to various concentrations of CTX-Alexa 488 (Invitrogen) for 1 hour. After CTX exposure the SSLB arrays were rinsed with copious amounts of PBS and imaged with either a Nikon Eclipse LV100 upright microscope with a 50× objective or an Olympus FV1000 upright confocal microscope with a 60× water immersion objective with N.A. = 0.9. Images were analyzed with ImageJ, version 1.44j.
RESULTS AND DISCUSSION
To form the SSLB arrays, SiO2 beads (700 nm diameter) were first coated with lipid bilayer membranes by vesicle rupture.33 Phospholipid vesicles containing phosphatidylcholine (egg PC) and fluorescent lipids (rhodamine or NBD-tagged phosphatidylethanolamine; Rho-PE or NBD-PE) and/or a lipid receptor (ganglioside GM1) were prepared by drying and rehydration followed by extrusion. Then the vesicle solution was mixed with a solution containing SiO2 beads and allowed to rest for 1 hour. The resulting SSLBs were separated from residual vesicles by centrifugation and resuspended in phosphate buffered saline (PBS, pH = 7.4). Figure 1a shows a schematic illustration of a SSLB on SiO2 bead.
Figure 1.

Spherical supported lipid bilayers (SSLBs) and periodic assembly. (a) Illustration of a SSLB with egg PC (black), fluorescent rhodamine-tagged PE (red) and ganglioside GM1 (blue) lipids. (b–d) Illustration showing the assembly process starting with SSLBs dispersed on a microwell substrate (b), removal of SSLBs not immobilized in microwells with the use of a PDMS block “squeegee” (c) and the resulting periodic assembly of SSLBs (d).
The microwell array substrates were fabricated by photolithography of silicon wafers, and the microwell diameter was precisely tuned by atomic layer deposition (ALD) of Al2O3. The as-fabricated microwells had 1 μm diameter and ~500 nm depth and had 3 μm center-to-center spacing in a hexagonal array. Each array was 2 mm × 2 mm. Conformal ALD of 100 nm of Al2O3 reduced the diameter to approximately 800 nm while the depth remained constant. SSLB arrays were prepared by placing 5 μL of the SSLB solution (1.7 × 109 SSLBs/mL) on each microwell array, and the SSLBs were allowed to settle for 30 minutes. (Figure 1b) After 30 minutes the substrates were gently washed with PBS and submerged in a PBS bath. Then a PDMS block was used as a squeegee to remove SSLBs that were not immobilized in the recessed microwells. (Figure 1c) This approach extends a previously reported method by Wittenberg et al., where a PDMS squeegee was used to form natural membrane microarrays, to assemble ordered arrays of single SSLBs on sub-micron beads.30 After removing excess SSLBs the substrate was completely devoid of SSLBs on the top surface, resulting in a high-density periodic SSLB array. (Figure 1d)
Figure 2a shows a scanning electron micrograph of SSLBs immobilized in a microwell array. In this image it is clear that the microwells are either empty or occupied by an individual SSLB. This is due to precise tuning of microwell dimensions via ALD. Figure 2b shows a single bead occupying an individual microwell. The lighter color layer coating the microwell is the 100 nm thick Al2O3 which facilitates the immobilization of single beads by shrinking the microwell diameter. When the microwells had 1 μm diameter and depth, they could accommodate one or two SSLBs, as shown in Figure S-1 in the supporting information.
Figure 2.

Scanning electron micrographs of SSLB arrays. (a) A SEM image of 700 nm diameter SSLBs immobilized in microwells. (b) A cross-sectional SEM image of a single SSLB immobilized in a microwell. The lighter coating on the substrate is the Al2O3 layer.
The SSLB arrays were characterized by fluorescence microscopy. Figure 3a shows a SSLB array covering a 100 μm × 100 μm area with 936 SSLBs. Each SSLB in the array is in effect a discrete SLB. The distribution of fluorescence intensities from individual SSLBs can be plotted in histograms as shown in Figure S-2 in the supporting information. Analyzing large groups of individual SSLBs by their intensity distribution makes it possible to acquire large sample numbers from single images. In general, SSLBs occupy greater than 75% of the available microwells. Figure 2b shows an overlay of brightfield and fluorescence images for an array that had 550 microwells and 416 immobilized SSLBs, giving an occupancy rate of 76 %.
Figure 3.

High density SSLB arrays and 2-component arrays. (a) Rhodamine-labeled SSLBs assembled into a 100 μm × 100 μm array. The area contains 936 SSLBs. (b) An overlay of bright field and fluorescence images showing a rhodamine-labeled SSLB array with 76 % occupancy. (c–e) A 2-component SSLB array where all SSLBs contain rhodamine-PE (c), but only a subset contain GM1 and are able to bind CTX (d). (e) Merger of rhodamine and CTX fluorescence showing fluorescence colocalization for SSLBs that contain GM1. The blue circles surround three SSLBs that display fluorescence in the rhodamine channel but not the CTX channel, indicating that they do not contain GM1.
Immobilization of multiple populations of beads in microwells has been demonstrated for multiplex optical biochemical assays, including single molecule studies.34–36 In an analogous manner, random arrays of two types of SSLBs can be immobilized in microwells by sequential deposition. One set of SSLBs contained egg PC and 1 % Rho-PE, while the other contained egg PC, 1 % Rho-PE and 2 % GM1. The SSLBs were deposited with 100× lower initial concentration (1.7 × 107 SSLBs/mL) which resulted in somewhat decreased occupancy. The array was then exposed to a solution containing Alexa 488-tagged B-subunit of cholera toxin (CTX), which binds GM1. (Figure 3c–e) All of the SSLBs fluoresce in the red channel due to the presence of Rho-PE on all SSLBs. However, after CTX incubation only a subset of the SSLBs in the array (those containing GM1) bound the CTX, indicated by spots of red and green overlap in the merged image shown in Figure 3e. The blue circles in Figure 3c-e surround three SSLBs that fluoresce in the red rhodamine channel, but not in the green CTX channel. This indicates that these SSLBs do not contain GM1, demonstrating that negative control binding sites can be co-immobilized with positive binders. Moreover, there are very few spots that appear in the green channel (CTX) only, indicating that CTX has minimal nonspecific adsorption to the substrate due to BSA passivation.
An alternative to mixing bead populations on individual arrays is to separately array different SSLBs on the same chip. To demonstrate this experimental approach, SSLBs containing Rho-PE and NBD-PE were immobilized in adjacent arrays on a single chip. This allows SSLBs to be encoded optically as well as spatially. After 5 μL droplets containing the different SSLB types were deposited on 8 2 × 2 mm arrays by hand with an Eppendorf pipette, the PDMS squeegee was passed over all of the arrays in a continuous motion to remove excess SSLBs from the chip surface. Figure 4a shows the positions of the Rho-PE and NBD-PE arrays, while Figure 4b shows fluorescence images from 8 separate arrays on a single 10 × 20 mm chip. Even though the PDMS squeegee was pulled along the long axis of the chip, there was no fluorescence overlap due to Rho-PE SSLBs being redeposited in arrays that were initially filled with NBD-PE SSLBs, and vice-versa. With this sample preparation method, it may be possible to perform array imaging with a high-resolution microarray scanner to increase imaging throughput. To increase throughput, the array size could be reduced which may require automated spotting equipment for sample deposition. The key parameter, however, would be spacing the arrays such that there is no redeposition of SSLBs during the squeegee process. In the current chip configuration the arrays are spaced by 3 mm, but this could probably be reduced to 1 mm with minimal cross-talk between arrays. Reducing the array size to 1 mm with 1 mm spacing between the arrays would allow for a 4-fold increase of the number of arrays on a chip.
Figure 4.
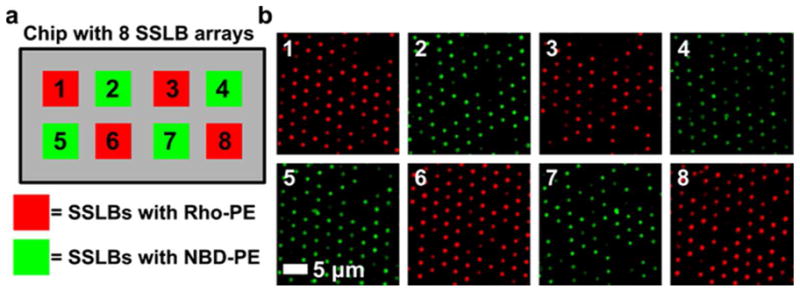
Two populations of SSLBs arrayed separately on a single chip. (a) Schematic illustration of a chip with eight arrays showing the position of SSLB arrays that contain Rho-PE lipids (red) and NBD-PE lipids (green). The actual dimensions of the chips were approximately 10 mm × 20 mm. (b) Fluorescence images of eight SSLB arrays from a single chip that were laid out as shown in the illustration in (a). The numbers in (b) correspond to the positions indicated by the numbers in (a).
Each SSLB in the array can function as a discrete sensing element for detecting the binding of CTX to GM1 on SSLBs. This allows us to collect large samples numbers (N > 1000) in a single image. To investigate the sensing capabilities of the SSLB array we made SSLBs with 0, 0.5, 1 and 2 % GM1. SSLBs with each different concentration of GM1 were immobilized on separate arrays. The SSLB arrays were then incubated with 50 nM CTX and the fluorescence from individual array spots was analyzed. The intensities from individual SSLBs in the arrays were plotted as histograms, shown in Figure 5a. There is a clear shift in the intensity distribution as the amount of GM1 increases, and the increase is linear as a function of GM1 concentration. (Figure 5b) The images corresponding to these data can be seen in Figure S-3 in the supporting information. Analyzing each SSLB as a discrete supported lipid bilayer, then compiling the results into histogram form provides a convenient method to analyze binding data from thousands of discrete binding sites.
Figure 5.

Binding assays with SSLB arrays and long-term array stability. (a) Histograms showing the distribution of fluorescence intensity from individual SSLBs containing 0.5, 1 and 2 % GM1 after incubation with 50 nM CTX. The solid lines are Gaussian fits to the data. (b) Plot of mean fluorescence intensity from all SSLBs in arrays exposed to 50 nM CTX as a function of GM1 concentration. (c) Binding curves showing mean fluorescence intensity from all SSLBs in arrays containing 2 % GM1 SSLBs after incubation with varying concentrations of CTX. Red and blue lines are fits to Langmuir isotherm and Hill-Waud binding models, respectively. (d) Mean array occupancy (black circles) and fluorescence intensity (red squares) for a SSLB array monitored over the course of one week. The error bars in (c–d) represent the standard deviations of the distributions of fluorescence intensity corresponding to each data point.
In an additional assay, we held the GM1 concentration in the SSLBs constant at 2 %, then incubated the arrays with CTX concentrations ranging from 16 pM to 158 nM. After subtraction of the fluorescence background, we fit the data to Langmuir isotherm and Hill-Waud binding models. The equations for the Langmuir and Hill-Waud binding models are shown in Eq. 1 and 2, respectively:
| (1) |
| (2) |
where F is the measured fluorescence intensity, Fmax is the fluorescence intensity when the binding is saturated, [CTX] is the concentration of CTX, KD and KH are the equilibrium dissociation constants from the Langmuir and Hill-Waud binding models, respectively, and n is the Hill cooperativity coefficient in the Hill-Waud model. The Langmuir isotherm resulted in an apparent dissociation constant (KD) of 1.6 ± 0.2 nM (mean ± standard error of the fit), while the Hill-Waud fit gave an apparent dissociation constant (KH) of 1.4 ± 0.2 nM. (Figure 5c) The dissociation constants for GM1-CTX binding reported in the literature vary widely depending on the nature of the immobilized GM1, the buffer conditions and method of analysis. Some groups have reported KD as low as 4.55 pM,37 while other groups have reported values as high as 370 nM.38 Shi and coworkers formed supported lipid bilayers with 2 % GM1 in a microfluidic chip, analyzed CTX binding with total internal reflectance fluorescence (TIRF) microscopy and calculated a KD of 0.46 nM and a KH of 0.39 nM.39 Winter et al. determined a KD value of approximately 30 nM using colloidal phase transition analysis of lipid-coated beads.40 In general, the dissociation constants determined with the SSLB arrays agree well with literature values. The Hill-Waud binding model also tests for binding cooperativity, which is commonly observed in CTX/GM1 interactions because CTX is pentavalent. From the Hill-Waud fit, we determined a Hill coefficient of cooperativity of 1.3. A Hill coefficient of greater than 1 indicates cooperative binding, which agrees with results from other groups, although it is somewhat lower than the 1.9 measured by Shi and coworkers.39,41 It has been suggested that for multivalent CTX/GM1 binding to occur the GM1 must be freely diffusible.42 Our observation of cooperative binding suggests that lipid bilayers of the SSLB are in a fluid state, which has been shown in other reports where fluorescence recovery after photobleaching (FRAP) was used to analyze bilayer fluidity on larger (~5 μm) beads.26 However, the lower Hill coefficient in this study suggests that the lipid diffusion may be somewhat hindered. This may be due to the interactions of the outer leaflet of the SSLB with the walls of the surrounding microwell. Alternatively, the apparent hindrance of lipid diffusion may be due to the spherical nature of the bead substrate. While the SSLBs in this study are too small to investigate with standard FRAP techniques, it would be possible to determine the lipid diffusion coefficients with other methods. For applications where lipid diffusion is an important parameter, the lipid diffusion coefficient could be determined by NMR. In one study Köchy and Bayerl used 2H-NMR to determine that the lipid diffusion coefficient on 640 nm diameter SiO2 beads was quite similar to the diffusion coefficient on flat surfaces determined by FRAP.43 Thus it appears that the microwell walls may influence lipid diffusion somewhat. The influence of the microwell substrate on lipid diffusion could be probed by altering the surface chemistry of the walls. Making the walls more repulsive to lipid interactions, for example by coating them with PEG, could make the lipids more freely diffusible. The increased diffusion of lipids may result in more cooperative binding in studies where multivalent interactions are investigated.
We also investigated the long-term stability of the SSLB arrays. SSLB arrays were prepared as usual then imaged immediately after preparation. Then the arrays were submerged in PBS and stored in a refrigerator between subsequent daily image acquisitions. Images of the arrays were acquired for one week after initial preparation. After one week there was no significant degradation of the arrays in terms of occupancy and fluorescence intensity. (Figure 5d) The long-term stability of the SSLB arrays suggests that the arrays may be prepared days in advance and used on-demand for binding assays.
CONCLUSION
In conclusion, we have demonstrated a new method for forming high-density SLB arrays in size-tunable microwells. Random 2-component arrays can be prepared by serial immobilization of SSLBs and arrays of separate SSLB populations can be encoded optically and spatially on a single chip. Immobilization of single SSLBs in individual microwells prevents aggregation or removal during washing, which could occur if SSLBs were randomly dispersed on the surface without defined, recessed immobilization sites. The arrays can be used for sensing CTX-GM1 interactions and the large number of array spots per image results in large sample numbers from a single image acquisition. In the future, this platform will be used with optically encoded SSLBs with more than a one type of receptor to demonstrate truly multiplexed assays. Because the arrays patterns are defined by photolithography there is a great deal of design flexibility afforded by this method. Beyond their use in sensing, these arrays could be used as versatile cell culture substrates to investigate focal adhesion and spatiotemporal signaling properties of cells44,45 or curvature-dependent binding of proteins to lipid bilayers.46 Furthermore, use of porous beads47 could allow the inclusion of transmembrane proteins, such as G-protein coupled receptors48 onto the SSLBs for high-throughput drug screening studies.
Supplementary Material
Acknowledgments
This work was supported by grants to S.H.O. from the National Institutes of Health (R01 GM092993), the National Science Foundation (NSF CAREER Award and DBI 0964216), the Office of Naval Research (ONR) Young Investigator Program and the Minnesota Partnership Award for Biotechnology and Medical Genomics. Device fabrication was performed at the University of Minnesota Nanofabrication Center (NFC), which receives support from the NSF through the National Nanotechnology Infrastructure Network. S.H.O. also acknowledges support from the WCU Program # R31-10032 funded by the Ministry of Education, Science & Technology and the National Research Foundation of Korea. The authors wish to thank Hyungsoon Im for assistance with illustrations and Shailabh Kumar for assistance with scanning electron microscopy.
Footnotes
SUPPORTING INFORMATION AVAILABLE
Histogram of SSLB array fluorescence intensity, SEM image of multiple SSLBs in microwells with larger diameters, and fluorescence images of CTX binding to SSLB arrays. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Lingwood D, Simons K. Science. 2010;327:46–50. doi: 10.1126/science.1174621. [DOI] [PubMed] [Google Scholar]
- 2.Walde P, Cosentino K, Engel H, Stano P. Chembiochem. 2010;11:848–865. doi: 10.1002/cbic.201000010. [DOI] [PubMed] [Google Scholar]
- 3.Castellana ET, Cremer PS. Surf Sci Rep. 2006;61:429–444. doi: 10.1016/j.surfrep.2006.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tamm LK, McConnell HM. Biophys J. 1985;47:105–113. doi: 10.1016/S0006-3495(85)83882-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Keller C, Kasemo B. Biophys J. 1998;75:1397–1402. doi: 10.1016/S0006-3495(98)74057-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Reviakine I, Brisson A. Langmuir. 2000;16:1806–1815. [Google Scholar]
- 7.Potma EO, Xie XS. J Raman Spectrosc. 2003;34:642–650. [Google Scholar]
- 8.Kraft ML, Weber PK, Longo ML, Hutcheon ID, Boxer SG. Science. 2006;313:1948–1951. doi: 10.1126/science.1130279. [DOI] [PubMed] [Google Scholar]
- 9.Dahlin A, Zäch M, Rindzevicius T, Käll M, Sutherland DS, Höök F. J Am Chem Soc. 2005;127:5043–5048. doi: 10.1021/ja043672o. [DOI] [PubMed] [Google Scholar]
- 10.Im H, Wittenberg NJ, Lesuffleur A, Lindquist NC, Oh SH. Chem Sci. 2010;1:688–696. doi: 10.1039/C0SC00365D. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.White RJ, Zhang B, Daniel S, Tang JM, Ervin ES, Cremer PS, White HS. Langmuir. 2006;22:10777–10783. doi: 10.1021/la061457a. [DOI] [PubMed] [Google Scholar]
- 12.Steinem C, Janshoff A, Ulrich WP, Sieber M, Galla HJ. Biochim Biophys Acta. 1996;1279:169–180. doi: 10.1016/0005-2736(95)00274-x. [DOI] [PubMed] [Google Scholar]
- 13.Lang H, Duschl C, Vogel H. Langmuir. 1994;10:197–210. [Google Scholar]
- 14.Yamazaki V, Sirenko O, Schafer RJ, Nguyen L, Gutsmann T, Brade L, Groves JT. BMC Biotechnol. 2005;5:18. doi: 10.1186/1472-6750-5-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Groves JT, Dustin ML. J Immunol Meth. 2003;278:19–32. doi: 10.1016/s0022-1759(03)00193-5. [DOI] [PubMed] [Google Scholar]
- 16.Salaita K, Nair PM, Petit RS, Neve RM, Das D, Gray JW, Groves JT. Science. 2010;327:1380–1385. doi: 10.1126/science.1181729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Groves JT, Boxer SG. Acc Chem Res. 2002;35:149–157. doi: 10.1021/ar950039m. [DOI] [PubMed] [Google Scholar]
- 18.Bally M, Bailey K, Sugihara K, Grieshaber D, Voros J, Stadler B. Small. 2010;6:2481–2497. doi: 10.1002/smll.201000644. [DOI] [PubMed] [Google Scholar]
- 19.Yang TL, Jung SY, Mao HB, Cremer PS. Anal Chem. 2001;73:165–169. doi: 10.1021/ac000997o. [DOI] [PubMed] [Google Scholar]
- 20.Groves JT, Ulman N, Boxer SG. Science. 1997;275:651–653. doi: 10.1126/science.275.5300.651. [DOI] [PubMed] [Google Scholar]
- 21.Morigaki K, Baumgart T, Offenhausser A, Knoll W. Angew Chem Int Ed. 2001;40:172–174. [PubMed] [Google Scholar]
- 22.Yee CK, Amweg ML, Parikh AN. J Am Chem Soc. 2004;126:13962–13972. doi: 10.1021/ja047714k. [DOI] [PubMed] [Google Scholar]
- 23.Hovis JS, Boxer SG. Langmuir. 2001;17:3400–3405. [Google Scholar]
- 24.Kaufmann S, Sobek J, Textor M, Reimhult E. Lab Chip. 2011;11:2403–2410. doi: 10.1039/c1lc20073a. [DOI] [PubMed] [Google Scholar]
- 25.Bieri C, Ernst OP, Heyse S, Hofmann KP, Vogel H. Nat Biotechnol. 1999;17:1105–1108. doi: 10.1038/15090. [DOI] [PubMed] [Google Scholar]
- 26.Baksh MM, Jaros M, Groves JT. Nature. 2004;427:139–141. doi: 10.1038/nature02209. [DOI] [PubMed] [Google Scholar]
- 27.Goodey A, Lavigne JJ, Savoy SM, Rodriguez MD, Curey T, Tsao A, Simmons G, Wright J, Yoo SJ, Sohn Y, Anslyn EV, Shear JB, Neikirk DP, McDevitt JT. J Am Chem Soc. 2001;123:2559–2570. doi: 10.1021/ja003341l. [DOI] [PubMed] [Google Scholar]
- 28.Walt DR. Chem Soc Rev. 2010;39:38–50. doi: 10.1039/b809339n. [DOI] [PubMed] [Google Scholar]
- 29.Smith AM, Vinchurkar M, Gronbech-Jensen N, Parikh AN. J Am Chem Soc. 2010;132:9320–9327. doi: 10.1021/ja100294k. [DOI] [PubMed] [Google Scholar]
- 30.Wittenberg NJ, Im H, Johnson TW, Xu XH, Warrington AE, Rodriguez M, Oh SH. ACS Nano. 2011;5:7555–7564. doi: 10.1021/nn202554t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hope MJ, Bally MB, Webb G, Cullis PR. Biochim Biophys Acta. 1985;812:55–65. doi: 10.1016/0005-2736(85)90521-8. [DOI] [PubMed] [Google Scholar]
- 32.Sweeney CM, Hasan W, Nehl CL, Odom TW. J Phys Chem A. 2009;113:4265–4268. doi: 10.1021/jp810837u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bayerl TM, Bloom M. Biophys J. 1990;58:357–362. doi: 10.1016/S0006-3495(90)82382-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Michael KL, Taylor LC, Schultz SL, Walt DR. Anal Chem. 1998;70:1242–1248. doi: 10.1021/ac971343r. [DOI] [PubMed] [Google Scholar]
- 35.Walt DR. Science. 2000;287:451–452. doi: 10.1126/science.287.5452.451. [DOI] [PubMed] [Google Scholar]
- 36.Rissin DM, Kan CW, Campbell TG, Howes SC, Fournier DR, Song L, Piech T, Patel PP, Chang L, Rivnak AJ, Ferrell EP, Randall JD, Provuncher GK, Walt DR, Duffy DC. Nat Biotechnol. 2010;28:595–599. doi: 10.1038/nbt.1641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kuziemko GM, Stroh M, Stevens RC. Biochemistry. 1996;35:6375–6384. doi: 10.1021/bi952314i. [DOI] [PubMed] [Google Scholar]
- 38.Moran-Mirabal JM, Edel JB, Meyer GD, Throckmorton D, Singh AK, Craighead HG. Biophys J. 2005;89:296–305. doi: 10.1529/biophysj.104.054346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Shi JJ, Yang TL, Kataoka S, Zhang YJ, Diaz AJ, Cremer PS. J Am Chem Soc. 2007;129:5954–5961. doi: 10.1021/ja069375w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Winter EM, Groves JT. Anal Chem. 2006;78:174–180. doi: 10.1021/ac0514927. [DOI] [PubMed] [Google Scholar]
- 41.Goins B, Freire E. Biochemistry. 1988;27:2046–2052. doi: 10.1021/bi00406a035. [DOI] [PubMed] [Google Scholar]
- 42.Lauer S, Goldstein B, Nolan RL, Nolan JP. Biochemistry. 2002;41:1742–1751. doi: 10.1021/bi0112816. [DOI] [PubMed] [Google Scholar]
- 43.Köchy T, Bayerl TM. Phys Rev E. 1993;47:2109–2116. doi: 10.1103/physreve.47.2109. [DOI] [PubMed] [Google Scholar]
- 44.Wu M, Holowka D, Craighead HG, Baird B. Proc Natl Acad Sci USA. 2004;101:13798–13803. doi: 10.1073/pnas.0403835101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Nair PM, Salaita K, Petit RS, Groves JT. Nat Protoc. 2011;6:523–539. doi: 10.1038/nprot.2011.302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hatzakis NS, Bhatia VK, Larsen J, Madsen KL, Bolinger PY, Kunding AH, Castillo J, Gether U, Hedegard P, Stamou D. Nat Chem Biol. 2009;5:835–841. doi: 10.1038/nchembio.213. [DOI] [PubMed] [Google Scholar]
- 47.Ashley CE, Carnes EC, Phillips GK, Padilla D, Durfee PN, Brown PA, Hanna TN, Liu JW, Phillips B, Carter MB, Carroll NJ, Jiang XM, Dunphy DR, Willman CL, Petsev DN, Evans DG, Parikh AN, Chackerian B, Wharton W, Peabody DS, Brinker CJ. Nat Mater. 2011;10:389–397. doi: 10.1038/nmat2992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Roizard S, Danelon C, Hassaine G, Piguett J, Schulze K, Hovius R, Tampe R, Vogel H. J Am Chem Soc. 2011;133:16868–16874. doi: 10.1021/ja205302g. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.