

Abstract
Kv1.1 channels cluster at juxtaparanodes of myelinated axons in the vagus nerve, the primary conduit for parasympathetic innervation of the heart. Kcna1-null mice lacking these channels exhibit neurocardiac dysfunction manifested by atropine-sensitive atrioventricular conduction blocks and bradycardia that may culminate in sudden death. To evaluate whether loss of Kv1.1 channels alters electrogenic properties within the nerve, we compared the intrinsic excitability of single myelinated A- and Aδ-axons from excised cervical vagus nerves of young adult Kcna1-null mice and age-matched, wild-type littermate controls. Although action potential shapes and relative refractory periods varied little between genotypes, Kv1.1-deficient large myelinated A-axons showed a fivefold increase in susceptibility to 4-aminopyridine (4-AP)-induced spontaneous ectopic firing. Since the repolarizing currents of juxtaparanodal Kv1 channels and nodal KCNQ potassium channels both act to dampen repetitive activity, we examined whether augmenting nodal KCNQ activation could compensate for Kv1.1 loss and reverse the spontaneous hyperexcitability in Kv1.1-deficient A-axons. Application of the selective KCNQ opener flupirtine raised A-axon firing threshold while profoundly suppressing 4-AP-induced spontaneous firing, demonstrating a functional synergy between the two compartments. We conclude that juxtaparanodal Kv1.1-deficiency causes intrinsic hyperexcitability in large myelinated axons in vagus nerve which could contribute to autonomic dysfunction in Kcna1-null mice, and that KCNQ openers reveal a transcompartmental synergy between Kv1 and KCNQ channels in regulating axonal excitability.
Key points
Voltage-gated Kv1.1 potassium channels cluster at juxtaparanodes of myelinated axons in the vagus nerve, which provides parasympathetic innervation to the heart.
Kcna1 knockout mice lacking Kv1.1 channels exhibit frequent atrioventricular cardiac conduction blocks that are abolished by atropine, suggestive of a vagal mechanism.
Electrophysiological analysis of single myelinated axons from wild-type and Kv1.1-deficient mouse vagus nerves revealed that the absence of Kv1.1 channels rendered large myelinated vagal axons far more susceptible to spontaneous ectopic firing in the presence of 4-aminopyridine.
KCNQ2 potassium channels are present within vagal nodes of Ranvier and their activation with flupirtine rescued single axon hyperexcitability mediated by juxtaparanodal Kv1.1-deficiency.
These results demonstrate a functional synergy between nodal and extranodal K+ channels and implicate KCNQ channels as potential targets for Kv1-related peripheral nerve hyperexcitability.
Introduction
Faithful propagation of action potentials (APs) along myelinated axons depends on the appropriate repertoire and spatial arrangement of ion channels (Rasband, 2011). At nodes of Ranvier, Na+ channels (Nav1.6) form dense clusters conducting the inward Na+ current that underlies impulse electrogenesis and saltatory conduction. Slowly activating and deactivating K+ channels (formed by KCNQ2 homoteteromers and KCNQ2/KCNQ3 heterotetramers) also cluster at nodes where they mediate the outward IKs current that prevents aberrant repetitive firing (Schwarz et al. 2006). At juxtaparanodes, fast voltage-gated Shaker-type K+ channels composed of heterotetramers of Kv1.1 and Kv1.2 potassium channel α-subunits are concealed beneath the myelin sheath where they prevent re-entrant excitation of the nodes following single impulses (Wang et al. 1993; Vabnick et al. 1999).
Disruption of nodal or juxtaparanodal potassium channel function leads to peripheral nerve hyperexcitability. In humans, inherited mutations in the KCNA1 gene, which encodes juxtaparanodal Kv1.1 channels, show clinical evidence of peripheral nerve hyperexcitability manifested as neuromyotonia or myokymia, and acquired neuromyotonia may arise from abnormal autoantibodies targeted against juxtaparanodal Kv1 subunits (Kleopa et al. 2006; Tomlinson et al. 2010). In some patients with KCNA1 mutations, generalized myokymia occurs in combination with epilepsy or as part of a syndrome known as episodic ataxia type 1 (Zuberi et al. 1999; Liguori et al. 2001; Demos et al. 2009). Inherited mutations in the KCNQ2 gene, which encodes nodal KCNQ2 channels, can also cause myokymia, as well as neonatal epilepsy (Dedek et al. 2001).
Mice lacking Kv1.1 channels due to targeted deletion of the Kcna1 gene exhibit neuronal excitability phenotypes similar to patients with defective Kv1.1 channels, including cold-induced neuromyotonia and severe epilepsy (Smart et al. 1998; Zhou et al. 1998). Studies of sciatic nerves from Kcna1-null mice reveal only minor alterations in compound action potential (CAP) shape and refractory period while Kv1.1-deficient phrenic nerve–diaphragm preparations show cooling-induced hyperexcitability emanating from nerve terminals (Smart et al. 1998; Zhou et al. 1999). Kv1.1-deficient mice die prematurely and exhibit neurogenic cardiac abnormalities, including atropine-sensitive atrioventricular conduction blocks and lethal seizure-associated bradyarrhythmias suggestive of a vagus nerve-mediated mechanism in this model; however, Kv1.1 proteins are also expressed at low levels in wild-type mouse heart where their absence could potentially impair intrinsic cardiac rhythmicity (Glasscock et al. 2010).
Since Kv1.1 channels are present in wild-type brain, nerve and heart, all three components of the neurocardiac axis could contribute to pathological brain–heart interactions in Kcna1-null mice. Here we examine the consequences of Kv1.1 deficiency in mouse vagus nerve. We hypothesize that the absence of Kv1.1 channels at vagal juxtaparanodes alters electrogenic properties along the nerve, predisposing it to hyperexcitability. To test this hypothesis, we characterized the whole nerve conduction profiles and performed single axon recordings from wild-type and Kv1.1-deficient mouse vagus nerves. Despite the clinical importance of vagus nerve excitability, the physiology of these fibres has not been previously explored in the mouse. Our experiments show that absence of Kv1.1 channels renders large myelinated vagal axons far more susceptible to spontaneous ectopic firing. We also demonstrate that activation of nodal KCNQ channels with flupirtine can rescue single axon hyperexcitability mediated by Kv1.1 deficiency, suggesting a functional synergy between the nodal and extranodal potassium channel compartments.
Methods
Ethical approval
We performed all procedures in accordance with the guidelines of the National Institutes of Health, as approved by the Animal Care and Use Committee of Baylor College of Medicine.
Animals
The Kcna1−/− mice carry a null mutation of the Kcna1 gene on chromosome 6 as a result of gene targeted deletion which removed the open reading frame, as previously described (Smart et al. 1998). For experiments involving wild-type nerves, we used the genotyped Kcna1+/+ siblings of the mutant animals. The average age of mice used in electrophysiology experiments was 35 ± 3 days and 33 ± 4 days (mean ± SD) for Kcna1+/+ and Kcna1−/− genotypes, respectively. Mice were housed at 22°C, fed ad libitum, and submitted to a 12 h light–dark cycle.
Genotyping
We isolated genomic DNA by digesting tail clips in Direct-PCR Lysis Reagent (Viagen Biotech, Los Angeles, CA, USA). To determine the genotypes of Kcna1 mice, we PCR amplified specific alleles using three unique primers: a mutant specific primer (5′-CCTTCTATCGCCTTCTTGACG-3′), a wild-type specific primer (5′-GCCTCTGACAGTGACCTCAGC-3′), and a common primer (5′-GCTTCAGGTTCGCCACTCCCC-3′). The PCR yielded amplicons of ∼337 bp for the wild-type allele and ∼475 bp for the mutant allele.
Vagus nerve isolation
Mice were killed with isoflurane and their left cervical vagus nerves excised by tying off the proximal end with a 5–0 silk suture at the level of the carotid bifurcation, severing the distal end where it enters the thorax, and transecting the nerve above the knot on the proximal end to free the entire cervical length. We then carefully removed the nerve (3–5 mm length) by the suture and immersed it in an oxygenated modified Krebs solution containing (in mm): 124 NaCl, 3 KCl, 2 CaCl2, 2 MgSO4, 26 NaHCO3, 10 glucose and 3 Hepes. The excised nerve was then positioned on a 2% agarose gel block and placed in a submerged recording chamber constantly perfused with the oxygenated modified Krebs solution. To isolate single axons, the nerves were briefly (2–5 min) incubated in a very low concentration of trypsin (0.0625%; Gibco) in the recording chamber to loosen the distal perineurium and allow access to individual fibre endings. This treatment produced no visible structural changes in the myelin of single axons. Furthermore, action potentials recorded from single axons did not exhibit broadening in the presence of 4-aminopyridine suggesting that the functional integrity of the myelin was preserved.
Compound nerve and single axon action potential recordings
A bipolar tungsten electrode was inserted into the proximal end of the nerve for stimulation. For CAP recording, a sharp glass electrode filled with 1 m NaCl (5–6 MΩ) was inserted into the distal nerve trunk for field potential recording. Conduction speeds were calculated by dividing the poststimulus AP latency (in ms) for each fibre population by the interelectrode distance. For single axon action potential recording, glass pipette suction electrodes were pulled with a 2–5 μm inner tip diameter. The input resistance was about 1 MΩ. A single axon was drawn into the electrode by gentle suction with a final seal resistance between 3 and 5 MΩ. Stimulation strength was increased until it reliably evoked compound nerve or single fibre action potentials, and the stimulation threshold defined as the minimal effective stimulation strength. Unless noted otherwise, we stimulated with 150–200% of the minimal stimulation strength. Signals were recorded with an Axopatch 1D amplifier (Molecular Devices, Sunnyvale, CA, USA) in current-clamp mode sampled at 40 kHz. In some cases, variation in seal resistance during the recording affected the single axon action potential amplitude but not its shape. Experiments were carried out at a bath temperature of 22°C unless specified otherwise. To block Kv1 potassium channels, 4-aminopyridine (4-AP; Sigma-Aldrich, St Louise, MO, USA) was added to the recording solution at a final concentration of either 100 μm or 1 mm. To activate Kcnq2 channels, a 100× stock solution of flupirtine maleate (Sigma-Aldrich) dissolved in dimethyl sulfoxide (DMSO; Sigma-Aldrich) was added to the recording solution to achieve a final concentration of 30 μm flupirtine, a concentration previously shown to alter clinical signs of excitability in myelinated axons of rat sural nerve (Sittl et al. 2010). Nerves showed no significant changes in excitability when recorded in solutions containing 1% DMSO without flupirtine.
Antibodies
For detection of Kv1.1 subunits, we used mouse monoclonal anti-Kv1.1 antibodies (clone K20/78; 1:1000 dilution; UC Davis/NIH NeuroMab Facility) and rabbit polyclonal anti-Kv1.1 antibodies (1:400; gift of Dr J. Trimmer, University of California, Davis, CA, USA). For co-localization experiments, we used mouse monoclonal anti-Kv1.2 antibodies (clone K14/16; 1:1000 dilution; UC Davis/NIH NeuroMab Facility) and rabbit polyclonal anti-KCNQ2 antibodies (1:300; gift of Dr E. Cooper, Baylor College of Medicine, Houston, TX, USA). The anti-KCNQ2 antibodies target the N-terminal portion of the protein as previously described (Pan et al. 2006).
Immunofluorescence
Left cervical vagus nerves were excised from 5-week-old mice, immersed in cold phosphate-buffered saline (PBS), teased and mounted onto microscope slides without fixation. After drying overnight at room temperature, nerves were blocked and permeabilized for 1 h in antibody vehicle (5–10% BSA, 0.3% Triton X-100 in PBS), followed by incubation in primary antibody diluted in vehicle for 15–20 h at room temperature. Subsequently, sections were washed three times in PBS and incubated for 1 h in Alexa Fluor 488 goat anti-rabbit F(ab’)2 secondary antibody and/or Alexa Fluor 555 goat anti-mouse F(ab’)2 secondary antibody (1:1000 dilution in vehicle; Invitrogen). Finally, sections were rinsed three times in PBS and then air dried at room temperature for 30 min. Once dry, the slides were coverslipped and mounted using ProLong Gold anti-fade reagent with DAPI (Invitrogen). Images were captured using an Olympus IX71 microscope (Olympus America) fitted with a digital CCD camera (Hamamatsu), and they were later colourized and adjusted for lighting levels using Adobe Photoshop Elements software (Adobe Systems). External diameters of nerve fibres were measured from 40× magnification digital images using ImageJ software (NIH).
Results
Compound action potentials in mouse vagus nerve
Electrical stimulation of wild-type mouse vagus nerve gave rise to a complex compound action potential (CAP) composed of three distinct waves corresponding to A-, Aδ - and C-axons (Fig. 1A). The first sharp peak in the CAP correlates with larger diameter, myelinated A-axons, which are characterized by lower stimulation thresholds and shorter latencies, equivalent to faster conduction velocities of 5.1 and 8.3 m s−1 at 22°C and 30°C, respectively. The second peak was broader and lower in amplitude, corresponding to smaller diameter, myelinated Aδ-axons with longer latencies and slower conduction velocities of 1.5 and 2.5 m s−1 at 22°C and 30°C, respectively. The third high amplitude peak was the broadest and represents non-myelinated C-axons, which exhibit the longest latencies and conduct the slowest relative to the other populations (0.4 m s−1 at 22°C and 0.6 m s−1 at 30°C). Compared to A-axons, the Aδ- and C-axons required greater stimulation intensities for recruitment (Fig. 1A). The waveforms and conduction velocities of the mouse CAP were similar to those measured in rat vagus nerve recordings (Docherty et al. 2005). Vagus nerves from Kcna1-null mice exhibited qualitatively similar CAP recordings compared to wild-type animals and showed no obvious differences in waveform shapes or conduction velocities (Fig. 1B and C).
Figure 1. CAPs recorded from mouse vagus nerve reveal three distinct axon populations.

A, as the stimulation intensity is increased, three waves become visible in CAPs recorded from wild-type mouse vagus nerve. These peaks result from recruitment of A-, Aδ- and C-axons. The A- and Aδ-axons correspond to larger (>4 μm) and smaller (2–4 μm) diameter myelinated axons, respectively. The C wave corresponds to non-myelinated axons (<2 μm). A-axons exhibited the lowest firing thresholds, followed by the Aδ- and C-axons, which required higher stimulation intensities to fire APs. B, nerves lacking Kv1.1 channels showed similar waveforms for all three axon groups in CAP recordings compared to wild-type nerves. C, each axon population exhibited a characteristic conduction velocity (mean ± SD) that varied with temperature and showed no change in the absence of Kv1.1 channels. The larger, myelinated A-axons conducted APs about three times faster than the smaller, myelinated Aδ-axons, which conducted APs about three times faster than the non-myelinated C-axons.
Action potentials in single myelinated vagus nerve axons
CAPs can reveal evoked nerve conduction abnormalities, but mask more subtle excitability changes present in single axons (Kocsis et al. 1983). Following brief trypsin digestion to loosen the nerve sheath, we could isolate individual myelinated axons and record single APs using a suction microelectrode (Fig. 2A). We determined axon type by measuring diameters and differences in response latency. Larger diameter axons (>4 μm) produced short latency responses (<1 ms) corresponding to the A population, whereas smaller diameter fibres (2–4 μm) gave longer latency responses (>2 ms) consistent with the Aδ population (Fig. 2B). The amplitude of evoked action potentials varied between wild-type axons. The small size of C-fibres (≤1 μm) rendered them inaccessible for recording.
Figure 2. Action potentials recorded from individual myelinated axons in mouse vagus nerve.

A, microscopy allowed selection of the larger A-axons (>4 μm diameter) and the smaller Aδ-axons (<4 μm diameter) for single axon recordings achieved via a suction recording electrode. B, AP latencies correlated with axon type. As demonstrated here in superimposed recordings from two individual axons belonging to the same wild-type nerve, larger diameter axons (>4 μm) gave shorter latency responses of <1 ms consistent with A-axons, while smaller diameter axons (<4 μm) fired after a latency of >2 ms as expected for an Aδ-axon. The stimulation artifact is indicated with an arrow.
Refractory periods in single vagus nerve axons
We evaluated the effects of Kv1.1-deficiency on impulse conduction by measuring the relative refractory period of single axons from wild-type and Kcna1-null vagus nerves. Using paired stimulus pulses, we measured the relative refractory period of individual myelinated axons. We first adjusted the stimulation intensity to the minimum required to reliably evoke a single AP, then determined the relative refractory period by stimulating the axon with paired pulses spaced at progressively shorter interstimulus intervals until the second response was no longer elicited. For wild-type A-axons, when the interval between two stimuli was shorter than 10 ms, the second response typically failed in an all-or-none fashion (Fig. 3A). However, as the stimulation intensity was doubled, the second stimulus successfully evoked APs even at interstimulus intervals as short as 5 ms (Fig. 3B). Since increased stimulation strength could overcome refractoriness, the failure to initiate a second action potential with minimal stimulation was probably due to a transient elevation in the firing threshold mediated by partial Na+ channel inactivation. Wild-type A-axons exhibited dramatically shorter recovery times than Aδ-axons. Whereas wild-type Aδ-axons required interstimulus intervals greater than 60 ms for full recovery, A-axons only required about 10 ms (Fig. 3C). Thus, as expected, the larger myelinated A-axons conduct APs faster and can fire at higher frequencies than the smaller myelinated Aδ-axons.
Figure 3. Recovery periods following a single action potential differ drastically between A- and Aδ-axons and can be prolonged by Kv1 channel blockade with 4-AP.

A, B and D, superimposed, successive wild-type A-fibre responses to paired pulses as the intersimulus interval was progressively shortened in 1 ms steps until the response failed. A, paired-pulse experiments using the minimum stimulation intensity required to reliably evoke APs allowed measurement of the relative refractory period for single axons. Wild-type A-axons usually fired APs until the interstimulus interval became less than 10 ms. As the interstimulus interval approached the relative refractory period, the second response would occasionally fail and then return before disappearing altogether as the interval was shortened further. The second response always failed in an all-or-none fashion. B, doubling the stimulation strength was sufficient to overcome the transient increase in firing threshold during the relative refractory period, allowing APs to be evoked at interstimulus intervals as short as 5 ms in wild-type A-axons. C, Aδ-axons exhibited a nearly sixfold longer relative refractory period than A-axons. Kv1.1-deficiency did not significantly alter the relative refractory periods. Values expressed as means ± SD. Number of axons measured shown in the bars. D, Kv1 channel blockade with 1 mm 4-AP prolonged the recovery period in wild-type A-axons. When stimulated at twice the minimal stimulation strength in the presence of 4-AP, wild-type A-axons required twice as long (∼10 ms) to recover before firing another AP (compare with B). In addition, with 4-AP the second response no longer failed in an all-or-none fashion, but was instead delayed and reduced in amplitude before failing completely. E, paired stimulation in a wild-type A-axon at baseline (without 4-AP) and in the presence of 1 mm 4-AP. At an interstimulus interval of 5 ms, the second response to twice minimal stimulation in the presence of 4-AP was delayed, broader and reduced in amplitude.
In A-axons from Kcna1-null mice that lack juxtaparanodal Kv1.1 channels, the recovery time was slightly longer (∼1 ms) than in wild-type axons, but this difference was not statistically significant (Fig. 3C). Likewise, the refractory period of Kv1.1-deficient Aδ-axons did not show obvious deviation from wild-type axons. Therefore, the lack of Kv1.1 channels does not appear to significantly affect recovery between APs in paired-pulse experiments.
We also analysed the effects of potassium channel blockade with 4-AP, which blocks both Kv1.1 and Kv1.2 subunits. When we applied 1 mm 4-AP to wild-type axons, they exhibited spontaneous discharges, hindering our ability to perform reliable paired-pulse measurements. However, in a small number of axons that did not exhibit spontaneous activity, we found that 1 mm 4-AP greatly lengthened the refractory period in A-axons, even when double the minimum stimulation intensity was applied to overcome an increase in firing threshold. While doubling the stimulation strength allowed A-axons to conduct action potentials with interstimulus intervals as short as 5 ms, when Kv1.1 and Kv1.2 channels were antagonized with 4-AP, the second response failed even with intervals as long as 10 ms (Fig. 3D). Furthermore, the second response was delayed and reduced in amplitude before eventually failing, consistent with prolongation of the relative refractory period (Fig. 3E). Since 4-AP always evoked spontaneous APs in Aδ-axons (see below), we were unable to reliably carry out measurements similar to those in A-axons. Therefore, it is not clear if Aδ-axons behave the same way as A-axons at high frequency in the presence of 4-AP.
4-AP unmasks hyperexcitability in Kv1.1-deficient vagal axons
Pharmacological blockade of Kv1 channels produced both spontaneous and evoked repetitive activity. Addition of 1 mm 4-AP induced spontaneous APs in about 50% of wild-type A-axons (n = 15) in the absence of electrical stimulation (Fig. 4A). In all six of the wild-type A-axons examined, the majority of these spontaneous discharges occurred rhythmically at average intervals of about 250 ms (∼4 Hz; Fig. 4B). Although the amplitude of this 4 Hz activity varied slightly from one discharge to the next, most resembled normal stimulation-evoked APs in amplitude and appearance. In addition to the spontaneous APs, five of six axons also exhibited occasional smaller amplitude discharges that only occurred shortly after a full-size spontaneous AP (within 8 to 30 ms), suggesting they were activity-driven (Fig. 4B). These partial spikes were usually about 25–50% of the amplitude of the prior full-size spontaneous AP. As further evidence that these low amplitude, short interval potentials were activity-driven, when APs were evoked with electrical stimulation in the presence of 4-AP, they were often followed by repetitive activity of similar appearance and latency (Fig. 4A).
Figure 4. Kv1.1-deficient vagal axons exhibit increased susceptibility to spontaneous activity in the presence of 4-AP.
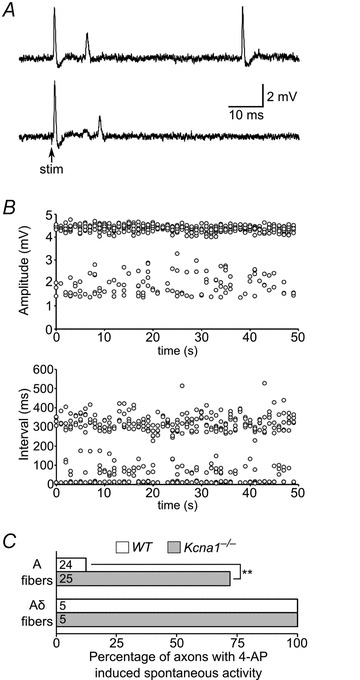
A, spontaneous APs (top trace) in a wild-type A-axon recorded in the presence of 1 mm 4-AP to block Kv1 channels. These spontaneous APs were usually high amplitude and similar in appearance to stimulation-evoked APs; however a subset were broader, reduced in amplitude, and usually occurred within ∼15 ms of a previous full-size spontaneous AP, suggesting they might be activity-driven. In the same axon, 4-AP also induced partial spikes during the ∼15 ms following stimulation evoked APs (bottom trace). B, the distribution of amplitudes and intervals between spontaneous APs in a wild-type A-axon in the presence of 1 mm 4-AP is plotted for a 50 s recording. Most APs exhibited amplitudes of about 4.5 mV, but some showed reduced amplitudes between about 1.5 and 3 mV. These smaller amplitude APs generally corresponded to the shorter interval APs occurring shortly after a prior full-size AP. C, summary of the percentage of axons displaying spontaneous activity in the presence of 100 μm 4-AP. A-axons lacking Kv1.1 channels exhibited a greater than fivefold increase in the incidence of spontaneous activity (**P < 0.0001 using two-tailed Fisher's exact test). Aδ-axons always displayed spontaneous activity regardless of genotype, suggesting they are especially susceptible to 4-AP-induced hyperexcitability. Numbers of axons analysed shown in the bars.
We examined the dose dependence of 4-AP-induced spontaneous activity in A- and Aδ-axons and found a clear difference between these fibre types. Whereas about 50% of A-axons exhibited spontaneous repetitive activity with 1 mm 4-AP, when we recorded using low concentration 100 μm 4-AP, only 13% of wild-type A-axons (n = 24) exhibited spontaneous discharges (Fig. 4C). Interestingly, at this low 4-AP concentration, all wild-type Aδ-axons tested (n = 5) showed spontaneous activity, indicating increased susceptibility to spontaneous discharges in the presence of 4-AP, despite possessing higher firing thresholds with electrical stimulation compared to A-axons (Fig. 4C). Repetitive activity in Aδ-axons was usually uniform in amplitude and occurred at shorter interspike intervals of about 110–170 ms (∼7 Hz). Furthermore, Aδ-axons did not show the partial spike discharges seen in A-axons, at least at 100 μm 4-AP. Therefore, wild-type A-axons exhibited spontaneous activity when exposed to 4-AP, but required higher concentrations that more completely block Kv1.1 and Kv1.2 subtypes, whereas Aδ-axons are especially susceptible to hyperexcitability with even a small degree of Kv1 blockade.
We then analysed Kcna1-null axons for repetitive activity in the presence of 4-AP to determine whether they were more excitable. In contrast to wild-type axons, Kv1.1-deficient A-axons were far more sensitive to Kv1 channel blockade. In the presence of a low concentration (100 μm) of 4-AP, 72% of Kv1.1-deficient A-axons (n = 25) showed spontaneous discharges, a greater than fivefold increase compared to wild-type axons (Fig. 4C). The sustained spontaneous firing in Kv1.1-deficient axons occurred at intervals of about 90–280 ms, similar to wild-type axons. About 40% of Kv1.1-deficient axons with spontaneous APs also showed short-interval (10–25 ms), lower amplitude discharges that resembled the activity-driven partial spikes observed in wild-type axons. Repetitive activity following evoked APs (within ∼25 ms) was more rare, occurring in only about 20% of Kv1.1-deficient A-axons that exhibited spontaneous activity. The 4-AP induced spontaneous activity in axons lacking Kv1.1 channels was temperature dependent; at 30°C, 4-AP failed to induce spontaneous repetitive firing (data not shown). Like their wild-type counterparts, all Kv1.1-deficient Aδ-axons showed spontaneous activity in the presence of 100 μm 4-AP (n = 5; Fig. 4C). The frequency of spontaneous APs was similar in both genotypes occurring about five times per minute in the presence of 100 μm 4-AP. The greatly increased susceptibility of Kv1.1-deficient axons to 4-AP-induced hyperexcitability indicates that juxtaparanodal Kv1 channels are important for stabilizing the membrane excitability of myelinated axons in the vagus nerve.
Alteration of action potential shape by Kv1 channel inhibition
Next we asked whether Kv1.1-deficiency or Kv1 blockade with 4-AP alters AP shape in myelinated vagal axons. Kv1 channel blockade with 4-AP dramatically broadens APs when applied to myelinated axons in the central nervous system (CNS) or non-myelinated axons, but in the peripheral nervous system (PNS) 4-AP appears to affect AP shape only in immature myelinated axons and not in adults (Sherratt et al. 1980; Rasband et al. 1999; Vabnick et al. 1999). However, these studies of the relationship between 4-AP and peripheral AP shape were largely restricted to rat sciatic nerve or compound action potentials. Therefore we examined the normal variation in AP shape among single vagal A- and Aδ-axons of the young adult mouse and then investigated the consequences of Kv1.1-deficiency or Kv1 blockade with 4-AP.
Among wild-type A-axons, we observed a large degree of variation in AP shape that is likely to be attributable to subtle differences in axon diameter and ion channel composition. To quantify the AP shape, we plotted the hyperpolarization overshoot as a percentage of the peak amplitude versus the half-width of the AP (Fig. 5A). In general, A-axons exhibited APs with half-widths ranging from 0.2 to 0.6 ms followed by some degree of hyperpolarization overshoot. We expected that a stronger K+ current during repolarization would produce a larger hyperpolarization overshoot and narrower AP width. The resulting plot showed the expected inverse correlation between AP half-width and the amplitude of hyperpolarization overshoot. Out of 37 A-axons, only three axons showed no hyperpolarization overshoot (not plotted). However, the data clustered into three distinct groupings that could be approximately linearly fitted, suggesting the presence of at least three subcategories of A-axons that differ slightly in their electrical properties independent of technical issues, such as the distance of the recording electrode from the last node of Ranvier or seal resistance. The subtle intrinsic electrical variation among A-fibres suggests that functional differences exist even between axons belonging to the same general fibre population.
Figure 5. A- and Aδ-axons exhibit differences in AP waveform that are altered by Kv1 blockade with 4-AP.
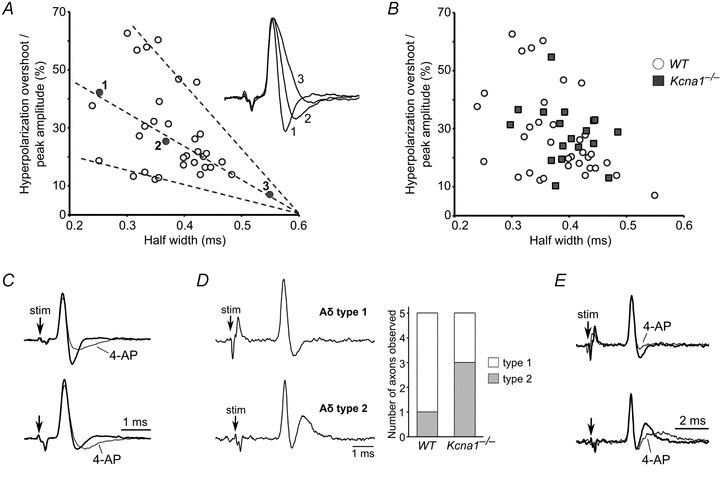
A, wild-type A-axons exhibited a large degree of variation in AP shape between axons, manifested by differences in half-width and degree of hyperpolarization overshoot. To quantify AP shape, we plotted the hyperpolarization overshoot of the AP as a percentage of the AP amplitude versus the half-width of the AP. The resulting plot showed that as the degree of overshoot increased, the AP half-width decreased. This trend can be clearly seen in the inset, which shows the superimposition of three AP waveforms with differing degrees of hyperpolarization overshoot; these three APs correspond to the three labelled points in the graph. The plot also revealed the presence of three putative subtypes of A-axons based on their different ratios of hyperpolarization overshoot to half-width; these three subcategories are indicated by the dashed lines. B, to determine whether Kv1.1 deficiency alters AP shape, we plotted the hyperpolarization overshoot amplitude versus half-width in Kcna1-null A-axons and overlaid the results with our wild-type distribution. We did not observe any obvious differences between the two distributions, suggesting that the absence of Kv1.1 channels does not drastically alter AP waveform in vagal A-axons. C, antagonism of both Kv1.1 and Kv1.2 channels with 100 μm 4-AP reduced the amplitude of the hyperpolarization overshoot of APs in A-axons (top trace) and slowed their recovery to baseline (top and bottom trace). D, Aδ-axons showed two distinct AP waveforms. The first type exhibited a typical AP morphology with hyperpolarization overshoot and recovery to baseline similar to A-axons. The second type exhibited hyperpolarization overshoot followed by a large depolarizing afterpotential. A histogram shows the relative incidence of these two types of APs in Aδ-axons from wild-type and Kv1.1-deficient nerves. E, application of 100 μm 4-AP to wild-type Aδ-axons reduced the hyperpolarization overshoot similar to A-axons, and reduced the amplitude of the depolarizing afterpotential while prolonging its recovery to baseline.
To determine whether Kv1.1-deficiency modifies AP shape in A-axons, we analysed Kcna1-null APs by again plotting hyperpolarization overshoot as a percentage of the peak amplitude versus the half-width of the AP (Fig. 5B). If the absence of Kv1.1 channels significantly alters AP shape, then the plot should show a different pattern from wild-type axons. Instead we found that the distribution of overshoot percentage versus half-width for Kv1.1-deficient A-axons was indistinguishable from wild-type axons, suggesting minimal, if any, alteration of AP waveform in Kv1.1 knockouts.
We next investigated the effects of partially blocking Kv1 channels with 4-AP on the AP waveform. Blockade of both Kv1.1 and Kv1.2 channels with 100 μm 4-AP in wild-type A-axons exhibited a reduction in the amplitude of the hyperpolarization overshoot, without changes in AP width (Fig. 5C). 4-AP significantly slowed the membrane potential recovery in axons whether they displayed large or less prominent hyperpolarization in control solutions (Fig. 5C). Our results suggest that Kv1.1 and Kv1.2 channels in wild-type A-axons influence the hyperpolarization phase of the AP without exerting obvious effects on repolarization during the falling phase.
We next analysed variation in the AP shape between Aδ-axons. The small diameter and resulting fragility of Aδ-axons renders them less amenable to recording, but we were still able to detect two types of Aδ-waveforms. The majority of wild-type Aδ-axons (4/5) exhibited APs similar to A-axons with hyperpolarization overshoot followed by recovery to baseline (Fig. 5D). However, in one instance, we observed an Aδ-axon with hyperpolarization overshoot followed by a significant depolarizing afterpotential (Fig. 5D). This type of depolarization was not seen in A-axons, suggesting it is unique to the smaller, myelinated Aδ-axons.
Kv1.1-deficient Aδ-axons also displayed these two predominant AP shapes, but the presence of a depolarizing afterpotential appeared to be more common (3/5) suggesting a potential difference in AP shape due to the absence of Kv1.1 channels in a subset of Aδ-axons (Fig. 5D). Following application of 100 μm 4-AP, wild-type Aδ-axons exhibited a reduction in hyperpolarization overshoot (Fig. 5E). In Aδ-axons with prominent depolarizing afterpotentials, 4-AP reduced the initial amplitude of the depolarizing overshoot while prolonging its recovery to baseline (Fig. 5E). Similar to our observations in A-axons, 4-AP never altered the width of APs in Aδ-axons. Therefore, Kv1 channels appear to play similar roles in both A- and Aδ-axons by mediating the hyperpolarization phase of the AP.
Activation of KCNQ channels with flupirtine increases firing threshold and inhibits hyperexcitability in single vagal axons
Since the slow nodal IKs current mediated by KCNQ/Kv7 potassium channels can act as a damper on aberrant repetitive activity, we tested whether activation of nodal KCNQ channels with flupirtine might rescue hyperexcitability caused by Kv1.1 deficiency. Although KCNQ2 channels have been reported at nodes in both CNS and PNS, their presence in vagus nerve has never been examined. Using immunofluorescence we detected KCNQ2 channels at nodes of Ranvier along myelinated axons in mouse vagus nerve. Co-immunolabelling showed that Kv1.1-positive juxtaparanodes always flanked a KCNQ2-positive node (Fig. 6A). However, Kv1.1 immunoreactivity was either absent or weak at some KCNQ2-positive nodes in smaller diameter axons (<5 μm), probably belonging to the Aδ population (Fig. 6B). Since Kv1.1 usually colocalizes with Kv1.2 subunits in peripheral nerve, we also analysed Kv1.2-immunoreactivity and found that Kv1.1-positive juxtaparanodes were always Kv1.2-positive as well (Fig. 6C). Nerves from Kcna1-null animals showed an absence of Kv1.1 labelling, but Kcnq2 and Kv1.2 immunoreactivity persisted (Fig. 6D and E).
Figure 6. KCNQ2 channels localize to nodes of Ranvier in mouse vagus nerve.
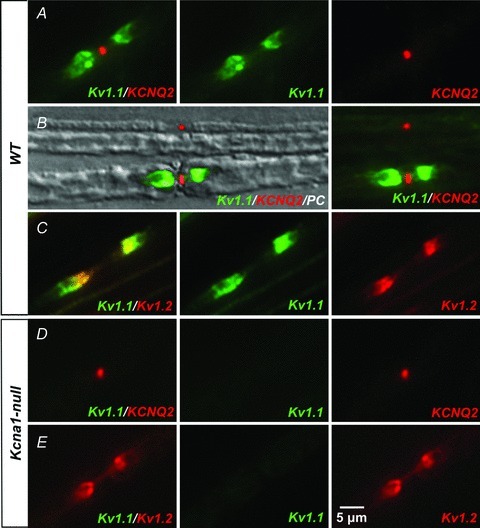
A, we detected KCNQ2 channels at nodes of myelinated vagal axons of 5-week-old wild-type mice using immunofluorescence. Co-labelling with anti-Kv1.1 antibodies showed Kv1.1-immunoreactivity at juxtaparanodes flanking the Kcnq2-positive nodes. B, Kv1.1-immunoreactivity appeared either weak or absent at many KCNQ2-positive nodes belonging to smaller fibres (<5 μm), as demonstrated in this phase contrast (PC) image superimposing Kv1.1- and KCNQ2-immunofluorescence. The small diameter (∼2 μm) fibre at the top shows only KCNQ2-positive nodal labelling, while the large diameter (∼7 μm) fibre at the bottom shows both Kv1.1-positive juxtaparanodal and KCNQ2-positive nodal immunoreactivity. C, Kv1.1-positive juxtaparanodes always exhibited Kv1.2-immunoreactivity. D and E, Kcna1-null fibres lack Kv1.1 labelling, but KCNQ2- and Kv1.2-immunoreactivity is preserved.
We next examined the effects of flupirtine on axonal excitability to determine whether augmenting KCNQ channel activation can functionally compensate for Kv1.1 deficiency in vagus nerve. Flupirtine is a synthetic KCNQ channel opener that causes an increase in KCNQ-mediated IKs, a slow activating, non-inactivating outward potassium current that hyperpolarizes axons, keeping them further from their firing threshold (Gunthorpe et al. 2012). Upon exposure to 30 μm flupirtine, wild-type A- and Aδ-axons exhibited an increase in AP firing threshold. When axons were stimulated at the minimum intensity required to reliably evoke action potentials, flupirtine abolished firing until the stimulation intensity was increased by about 25–50% (Fig. 7A and B). Furthermore, flupirtine increased the amplitude of the hyperpolarization overshoot in APs recorded from A-axons (Fig. 7C). These results are consistent with an activation of nodal KCNQ channels, which apparently hyperpolarizes the nodal resting membrane potential, as demonstrated by an increase in the axonal AP threshold.
Figure 7. Flupirtine rescues 4-AP induced hyperexcitability in single vagal axons.
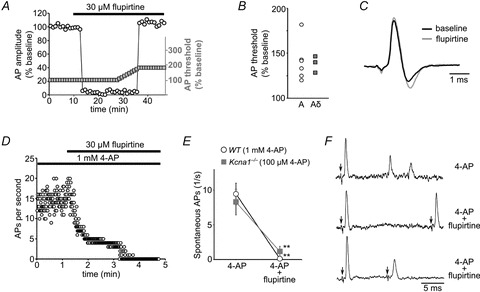
A, when we administered 30 μm flupirtine to wild-type A-axons to activate nodal KCNQ channels, we observed an increase in AP firing threshold that caused stimulation-evoked APs (left Y-axis, open circles) to disappear until the stimulation intensity was increased sufficiently to reach the new firing threshold (right Y-axis, grey boxes). B, flupirtine (30 μm) increased the AP firing threshold by about 25–50% in both wild-type A- and Aδ-axons. C, flupirtine increased the hyperpolarization overshoot phase of the AP waveform indicative of an increase in inward K+ current. D, raw data showing the elimination of 4-AP-induced spontaneous activity in a wild-type A-axon by application of flupirtine. E, flupirtine-mediated KCNQ channel activation suppressed 4-AP induced spontaneous activity in wild-type A-axons completely (n = 6) and in Kv1.1-deficient A-axons by 85% (n = 6). Values expressed as means ± SEM. **P < 0.001 using two-tailed, paired t test. F, flupirtine failed to rescue the 4-AP-induced prolongation of refractory period in wild-type A-axons. The top trace shows smaller partial spikes following a stimulation-evoked AP in the presence of 1 mm 4-AP. With the addition of flupirtine (middle trace), paired stimulation evokes a full-size action potential when the interstimulus interval is long (∼20 ms), but at shorter intervals (∼10 ms; bottom trace), the second response triggers only a lower amplitude impulse. Compare to Fig. 3B and D which shows that A-axons normally only require about 5 ms for recovery at baseline and about 10 ms in the presence of 1 mm 4-AP.
Since KCNQ channels can be powerful regulators of nodal membrane excitability, we examined the effects of flupirtine on abnormal spontaneous activity in the presence of 4-AP. In wild-type A-axons, 30 μm flupirtine almost completely abolished spontaneous firing in the presence of a high concentration of 4-AP (1 mm; Fig. 7D and E). In Kv1.1-deficient A-axons, which are more sensitive to 4-AP-induced spontaneous activity, flupirtine also profoundly inhibited spontaneous repetitive firing, reducing it by about 85% (Fig. 7E). In addition, flupirtine dramatically suppressed repetitive activity following APs evoked in the presence of 4-AP (Fig. 7F). These results demonstrate a functional synergism between juxtaparanodal Kv1 and nodal KCNQ channels that allows excessive activation of one to compensate for loss of the other despite their separate subcellular localization within distinct axonal compartments.
However, the functional rescue of Kv1-channel deficiency by activation of KCNQ channels was not complete. Although flupirtine suppressed 4-AP induced spontaneous activity, it did not reverse the 4-AP-mediated lengthening of the refractory period in paired stimulation protocols (Fig. 3D and E). When we stimulated wild-type A-axons with paired pulses at double the minimal stimulation strength in the presence of 1 mm 4-AP, the second response still failed at intervals that were about twice as long (∼10 ms) as wild-type A-axons (∼5 ms; Fig. 7F). The partial rescue of Kv1-mediated excitability defects by a KCNQ opener demonstrates that although Kv1 and KCNQ channels exhibit synergy, they are not functionally interchangeable.
Discussion
Kv1.1 channels and vagus nerve hyperexcitability
This work provides the first detailed electrophysiological description of single myelinated axons from mouse vagus nerve. This cranial nerve carries the parasympathetic autonomic input between the brainstem and heart to regulate heart rate and cardiac conduction. The composition of the vagus nerve is complex, being composed of both motor and sensory components carried by combinations of myelinated A- and Aδ-axons and non-myelinated C-axons (Agostoni et al. 1957; Docherty et al. 2005). Here we showed that extensive electrophysiological heterogeneity exists among myelinated vagal axons. In addition to differences in conduction velocity, paired-stimulation recordings showed that A-axons can follow much faster stimulation frequencies than Aδ-axons, requiring only one-sixth of the time to recover between APs compared to Aδ-axons. Interestingly, we also observed three potential subclasses of A-axons based on AP shape, which exhibited different degrees of hyperpolarization overshoot and half-widths. Aδ-axons may also demonstrate AP shape heterogeneity, since a subset displayed unique depolarizing afterpotentials following an initial depolarization. Smaller Aδ-axons also exhibited an extreme predisposition to spontaneous firing in response to 4-AP despite requiring higher electrical stimulation thresholds to evoke an action potential. The difference in relative refractory periods and 4-AP susceptibility between A- and Aδ-axons may reflect variation in ion channel composition as well as stem from increased electrotonic coupling between node and internode in smaller diameter fibres (Chiu & Ritchie, 1984). Although we have not directly investigated it here, our findings raise the possibility that axonal hyperexcitability in vagus nerves in Kcna1-null mice in vivo may potentiate parasympathetic drive to the heart, and trigger bradycardia and atrioventricular conduction failure. Kv1.1-deficient mice exhibit a fivefold increase in the occurrence of interictal AV blocks compared to wild-types; during seizures the incidence of AV blocks trends higher, suggesting ictal recruitment of autonomic cardiac brainstem centers (Glasscock et al. 2010). Treatment with the parasympathetic blocker atropine abolishes the interictal AV blocks, implicating a vagally mediated cholinergic receptor origin (Glasscock et al. 2010).
Kv1 channels and the stabilization of membrane excitability at nodes of Ranvier
Our results provide two lines of evidence that juxtaparanodal Kv1 channels stabilize resting membrane excitability and AP electrogenesis in myelinated axons of mouse vagus nerve. First, 4-AP mediated Kv1-channel blockade induced spontaneous action potentials, demonstrating that reduction of juxtaparanodal Kv1 currents elevates axon excitability. The stabilizing effect of juxtaparanodal Kv1 channels is further underscored by the increased susceptibility of genetically deficient Kcna1−/− axons to generate spontaneous APs in the presence of 4-AP. Second, activation of nodal KCNQ channels with flupirtine elevated the firing threshold of myelinated axons, and abolished spontaneous APs in the presence of 4-AP, linking their origin to threshold changes controlled by Kv1 channels.
Previous studies examining the origin of ectopic impulse electrogenesis in the absence of Kv1.1 channels have utilized recordings from sciatic and phrenic nerve preparations. CAP recordings of Kv1.1-deficient sciatic nerve revealed only very subtle AP broadening and minor increases in refractory period, both of which could be exacerbated at a low temperature of 22°C (Smart et al. 1998). Cooling to 18°C also led to spontaneous discharges in Kv1.1-deficient phrenic nerves (Zhou et al. 1999). This spontaneous activity originated at the nerve terminal region, since lidocaine applied between the terminal and recording site abolished it. Computer modelling showed that the generation of spontaneous activity relies on the unique geometry of the distal axon as the internodes progressively shorten in the smaller calibre preterminal zone (Zhou et al. 1999). However, this mechanism appears distinct from the spontaneous APs we observed in the presence of 4-AP, since the vagus nerve preparations in our study only included axon trunks. Thus our finding that Kv1.1-deficient axons have a dramatically low theshold for ectopic impulse generation in the presence of 4-AP demonstrates an important role of Kv1 channels in controlling nodal excitability along the length of myelinated axon shafts in addition to their effects on the preterminal membrane.
Although we do not directly measure it, our data are consistent with a role for Kv1 channels in maintaining the internodal resting membrane potential, thereby preventing spontaneous ectopic firing at nodes. Compartmental modelling studies predict that myelinated axons must possess an internodal resting potential maintained in part by voltage-gated K+ channels, or the node would be depolarized by up to 20 mV depending on fibre diameter (Chiu & Ritchie, 1984), and simulations suggest that Kv1 channel clustering is specifically required at juxtaparanodes to prevent re-entrant excitation following single impulses (Vabnick et al. 1999). Other indirect evidence for a role of Kv1 channels in maintaining conduction properties has been provided by studies of sciatic nerve axons lacking TAG-1 and Caspr2, two proteins required for proper Kv1 channel juxtaparanodal localization (Poliak et al. 2003; Traka et al. 2003). Mice lacking either of these two proteins display Kv1 channels that no longer cluster at juxtaparanodes but instead spread out diffusely along the internode. Both TAG-1 and Caspr2 knockout mice fail to show obvious changes in myelinated axon excitability at the level of compound action potential conduction, which has been interpreted as evidence that Kv1 channels are important for maintaining resting potential and that overall Kv1 channel content along the internode matters more than specific clustering at juxtaparanodes (Poliak et al. 2003; Poliak & Peles, 2003). A more detailed study of single axons of defined diameters will be required to assess the individual repetitive firing properties of remodelled fibres in these models.
Functional synergism between juxtaparanodal Kv1 channels and nodal KCNQ channels
The abolition of 4-AP-induced spontaneous APs by flupirtine reveals a transcompartmental functional interaction between Kv1 and KCNQ channel-mediated repolarization. Nodes of myelinated axons have long been known to possess slow outward rectifier channels that are blocked by tetraethylammonium (TEA), while the internodes contain predominantly fast, outward rectifiers that are sensitive to 4-AP (Baker et al. 1987). The 4-AP- and TEA-sensitive components have now been identified as Kv1 and KCNQ channels, respectively (Christie et al. 1989; Devaux et al. 2004; Schwarz et al. 2006).
Despite their differing kinetics and subcellular localization, the two channel populations work together to dampen aberrant excitability, and when either is compromised, axonal hyperexcitability ensues. When myelinated rat sciatic nerve fibres are treated with 4-AP, single impulses cause abnormal bursts of action potentials that are potentiated by additional blockade of KCNQ channels with TEA (Kocsis et al. 1987). Studies using phrenic-nerve diaphragm preparations have shown that, in the absence of Kv1.1 channels, cooling induces spontaneous nerve activity emanating from the nerve terminal regions that is amplified by TEA; the authors suggested that the TEA-sensitive channels become critical in modulating repetitive discharges when Kv1.1 expression is compromised (Zhou et al. 1999). Direct or indirect impairment of nodal KCNQ function by selective KCNQ blockers or genetic disruption of the spectrin cytoskeleton, respectively, causes stimulus-evoked repetitive afterdischarge in mouse sciatic nerves resembling that in nerves with genetic or pharmacological Kv1.1 deficiency (Schwarz et al. 2006; Devaux, 2010). Interestingly, given their functional overlap, mutations in both the KCNA1 and KCNQ2 genes have been implicated not only in human epilepsy but also peripheral nerve dysfunction manifested as neuromyotonia and myokymia, suggesting similar functional consequences when the channels are disrupted (Browne et al. 1994; Dedek et al. 2001).
Flupirtine is a synthetic positive allosteric modulator of KCNQ channels that causes a hyperpolarizing shift in the voltage dependence of channel activation (Martire et al. 2004; Xiong et al. 2008; Gunthorpe et al. 2012). In vitro CAP recordings of rat sural nerve show that flupirtine increases the late subexcitability phase (Sittl et al. 2010). Our demonstration that KCNQ activation with flupirtine can rescue Kv1-mediated spontaneous activity suggests that KCNQ channels are potential targets for Kv1-related peripheral nerve hyperexcitability.
Acknowledgments
Thanks to Dr Ed Cooper (Baylor College of Medicine, Houston, TX, USA) for generously supplying KCNQ2 antibody, as well as providing critical input during manuscript preparation. This work was supported by National Institutes of Health grants K99HL107641 (E.G.) from the National Heart, Lung, and Blood Institute and NS29709 (J.L.N.) and NS076916 (J.L.N.) from the National Institute for Neurological Disorders and Stroke. Additional funding was provided by a postdoctoral fellowship from the American Heart Association (E.G.).
Glossary
- 4-AP
4-aminopyridine
- AP
action potential
- CAP
compound action potential
- CNS
central nervous system
- PNS
peripheral nervous system
- TEA
tetraethylammonium
Author contributions
Experiments were performed at Baylor College of Medicine, Houston, TX, USA. E.G. conceived the experiments. E.G., J.Q. and M.J.K. designed the experiments. E.G. and J.Q. collected, analysed and interpreted the data. E.G., J.Q. and J.L.N. drafted the article. All authors approved the final version of the manuscript. The authors have no financial interests or conflicts to disclose.
References
- Agostoni E, Chinnock JE, De Daly MB, Murray JG. Functional and histological studies of the vagus nerve and its branches to the heart, lungs and abdominal viscera in the cat. J Physiol. 1957;135:182–205. doi: 10.1113/jphysiol.1957.sp005703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker M, Bostock H, Grafe P, Martius P. Function and distribution of three types of rectifying channel in rat spinal root myelinated axons. J Physiol. 1987;383:45–67. doi: 10.1113/jphysiol.1987.sp016395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Browne DL, Gancher ST, Nutt JG, Brunt ER, Smith EA, Kramer P, Litt M. Episodic ataxia/myokymia syndrome is associated with point mutations in the human potassium channel gene, KCNA1. Nat Genet. 1994;8:136–140. doi: 10.1038/ng1094-136. [DOI] [PubMed] [Google Scholar]
- Chiu SY, Ritchie JM. On the physiological role of internodal potassium channels and the security of conduction in myelinated nerve fibres. Proc R Soc Lond B Biol Sci. 1984;220:415–422. doi: 10.1098/rspb.1984.0010. [DOI] [PubMed] [Google Scholar]
- Christie MJ, Adelman JP, Douglass J, North RA. Expression of a cloned rat brain potassium channel in Xenopus oocytes. Science. 1989;244:221–224. doi: 10.1126/science.2539643. [DOI] [PubMed] [Google Scholar]
- Dedek K, Kunath B, Kananura C, Reuner U, Jentsch TJ, Steinlein OK. Myokymia and neonatal epilepsy caused by a mutation in the voltage sensor of the KCNQ2 K+ channel. Proc Natl Acad Sci U S A. 2001;98:12272–12277. doi: 10.1073/pnas.211431298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demos MK, Macri V, Farrell K, Nelson TN, Chapman K, Accili E, Armstrong L. A novel KCNA1 mutation associated with global delay and persistent cerebellar dysfunction. Mov Disord. 2009;24:778–782. doi: 10.1002/mds.22467. [DOI] [PubMed] [Google Scholar]
- Devaux JJ. The C-terminal domain of ßIV-spectrin is crucial for KCNQ2 aggregation and excitability at nodes of Ranvier. J Physiol. 2010;588:4719–4730. doi: 10.1113/jphysiol.2010.196022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devaux JJ, Kleopa KA, Cooper EC, Scherer SS. KCNQ2 is a nodal K+ channel. J Neurosci. 2004;24:1236–1244. doi: 10.1523/JNEUROSCI.4512-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Docherty RJ, Charlesworth G, Farrag K, Bhattacharjee A, Costa S. The use of the rat isolated vagus nerve for functional measurements of the effect of drugs in vitro. J Pharmacol Toxicol Methods. 2005;51:235–242. doi: 10.1016/j.vascn.2004.08.011. [DOI] [PubMed] [Google Scholar]
- Glasscock E, Yoo JW, Chen TT, Klassen TL, Noebels JL. Kv1.1 potassium channel deficiency reveals braindriven cardiac dysfunction as a candidate mechanism for sudden unexplained death in epilepsy. J Neurosci. 2010;30:5167–5175. doi: 10.1523/JNEUROSCI.5591-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gunthorpe MJ, Large CH, Sankar R. The mechanism of action of retigabine (ezogabine), a first-in-class K+ channel opener for the treatment of epilepsy. Epilepsia. 2012;53:412–424. doi: 10.1111/j.1528-1167.2011.03365.x. [DOI] [PubMed] [Google Scholar]
- Kleopa KA, Elman LB, Lang B, Vincent A, Scherer SS. Neuromyotonia and limbic encephalitis sera target mature Shaker-type K+ channels: subunit specificity correlates with clinical manifestations. Brain. 2006;129:1570–1584. doi: 10.1093/brain/awl084. [DOI] [PubMed] [Google Scholar]
- Kocsis JD, Eng DL, Gordon TR, Waxman SG. Functional differences between 4-aminopyridine and tetraethylammonium-sensitive potassium channels in myelinated axons. Neurosci Lett. 1987;75:193–198. doi: 10.1016/0304-3940(87)90296-5. [DOI] [PubMed] [Google Scholar]
- Kocsis JD, Ruiz JA, Waxman SG. Maturation of mammalian myelinated fibers: changes in action-potential characteristics following 4-aminopyridine application. J Neurophysiol. 1983;50:449–463. doi: 10.1152/jn.1983.50.2.449. [DOI] [PubMed] [Google Scholar]
- Liguori R, Avoni P, Baruzzi A, Di Stasi V, Montagna P. Familial continuous motor unit activity and epilepsy. Muscle Nerve. 2001;24:630–633. doi: 10.1002/mus.1048. [DOI] [PubMed] [Google Scholar]
- Martire M, Castaldo P, D’Amico M, Preziosi P, Annunziato L, Taglialatela M. M channels containing KCNQ2 subunits modulate norepinephrine, aspartate, and GABA release from hippocampal nerve terminals. J Neurosci. 2004;24:592–597. doi: 10.1523/JNEUROSCI.3143-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan Z, Kao T, Horvath Z, Lemos J, Sul J-Y, Cranstoun SD, Bennett V, Scherer SS, Cooper EC. A common ankyrin-G-based mechanism retains KCNQ and NaV channels at electrically active domains of the axon. J Neurosci. 2006;26:2599–2613. doi: 10.1523/JNEUROSCI.4314-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poliak S, Peles E. The local differentiation of myelinated axons at nodes of Ranvier. Nat Rev Neurosci. 2003;4:968–980. doi: 10.1038/nrn1253. [DOI] [PubMed] [Google Scholar]
- Poliak S, Salomon D, Elhanany H, Sabanay H, Kiernan B, Pevny L, Stewart CL, Xu X, Chiu S-Y, Shrager P, Furley AJW, Peles E. Juxtaparanodal clustering of Shaker-like K+ channels in myelinated axons depends on Caspr2 and TAG-1. J Cell Biol. 2003;162:1149–1160. doi: 10.1083/jcb.200305018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasband MN. Composition, assembly, and maintenance of excitable membrane domains in myelinated axons. Semin Cell Dev Biol. 2011;22:178–184. doi: 10.1016/j.semcdb.2010.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasband MN, Trimmer JS, Peles E, Levinson SR, Shrager P. K+ channel distribution and clustering in developing and hypomyelinated axons of the optic nerve. J Neurocytol. 1999;28:319–331. doi: 10.1023/a:1007057512576. [DOI] [PubMed] [Google Scholar]
- Schwarz JR, Glassmeier G, Cooper EC, Kao T-C, Nodera H, Tabuena D, Kaji R, Bostock H. KCNQ channels mediate IKs, a slow K+ current regulating excitability in the rat node of Ranvier. J Physiol. 2006;573:17–34. doi: 10.1113/jphysiol.2006.106815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherratt RM, Bostock H, Sears TA. Effects of 4-aminopyridine on normal and demyelinated mammalian nerve fibres. Nature. 1980;283:570–572. doi: 10.1038/283570a0. [DOI] [PubMed] [Google Scholar]
- Sittl R, Carr RW, Schwarz JR, Grafe P. The Kv7 potassium channel activator flupirtine affects clinical excitability parameters of myelinated axons in isolated rat sural nerve. J Peripher Nerv Syst. 2010;15:63–72. doi: 10.1111/j.1529-8027.2010.00253.x. [DOI] [PubMed] [Google Scholar]
- Smart SL, Lopantsev V, Zhang CL, Robbins CA, Wang H, Chiu SY, Schwartzkroin PA, Messing A, Tempel BL. Deletion of the KV1.1 potassium channel causes epilepsy in mice. Neuron. 1998;20:809–819. doi: 10.1016/s0896-6273(00)81018-1. [DOI] [PubMed] [Google Scholar]
- Tomlinson SE, Tan SV, Kullmann DM, Griggs RC, Burke D, Hanna MG, Bostock H. Nerve excitability studies characterize Kv1.1 fast potassium channel dysfunction in patients with episodic ataxia type 1. Brain. 2010;133:3530–3540. doi: 10.1093/brain/awq318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Traka M, Goutebroze L, Denisenko N, Bessa M, Nifli A, Havaki S, Iwakura Y, Fukamauchi F, Watanabe K, Soliven B, Girault J-A, Karagogeos D. Association of TAG-1 with Caspr2 is essential for the molecular organization of juxtaparanodal regions of myelinated fibers. J Cell Biol. 2003;162:1161–1172. doi: 10.1083/jcb.200305078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vabnick I, Trimmer JS, Schwarz TL, Levinson SR, Risal D, Shrager P. Dynamic potassium channel distributions during axonal development prevent aberrant firing patterns. J Neurosci. 1999;19:747–758. doi: 10.1523/JNEUROSCI.19-02-00747.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Kunkel DD, Martin TM, Schwartzkroin PA, Tempel BL. Heteromultimeric K+ channels in terminal and juxtaparanodal regions of neurons. Nature. 1993;365:75–79. doi: 10.1038/365075a0. [DOI] [PubMed] [Google Scholar]
- Xiong Q, Gao Z, Wang W, Li M. Activation of Kv7 (KCNQ) voltage-gated potassium channels by synthetic compounds. Trends Pharmacol Sci. 2008;29:99–107. doi: 10.1016/j.tips.2007.11.010. [DOI] [PubMed] [Google Scholar]
- Zhou L, Messing A, Chiu SY. Determinants of excitability at transition zones in Kv1.1-deficient myelinated nerves. J Neurosci. 1999;19:5768–5781. doi: 10.1523/JNEUROSCI.19-14-05768.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou L, Zhang CL, Messing A, Chiu SY. Temperature-sensitive neuromuscular transmission in Kv1.1 null mice: role of potassium channels under the myelin sheath in young nerves. J Neurosci. 1998;18:7200–7215. doi: 10.1523/JNEUROSCI.18-18-07200.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuberi SM, Eunson LH, Spauschus A, De Silva R, Tolmie J, Wood NW, McWilliam RC, Stephenson JB, Stephenson JP, Kullmann DM, Hanna MG. A novel mutation in the human voltage-gated potassium channel gene (Kv1.1) associates with episodic ataxia type 1 and sometimes with partial epilepsy. Brain. 1999;122:817–825. doi: 10.1093/brain/122.5.817. [DOI] [PubMed] [Google Scholar]