

Abstract
The chemokine receptor CXCR4 is one of two principal coreceptors for HIV-1 entry into target cells. CXCR4 is known to form homodimers. We previously demonstrated that the amino (N)-terminus of viral macrophage protein (vMIP)-II is the major determinant for CXCR4 recognition, and that V1 peptide derived from the N-terminus of vMIP-II (1-21 residues) showed significant CXCR4 binding. Interestingly, an all-D-amino acid analog of V1 peptide, DV1 peptide, displayed even higher binding affinity and strong antiviral activity in inhibiting the replication of CXCR4-dependent HIV-1 strains. In the present study, we synthetically linked two DV1 peptides with the formation of a disulfide bond between the two cysteine residues present in the peptide sequence to generate a dimeric molecule potentially capable of interacting with two CXCR4 receptors. DV1 dimer showed enhanced binding affinity and antiviral activity compared with DV1 monomer. Ligand binding site mapping experiments showed that DV1 dimer overlaps with HIV-1 gp120 on CXCR4 binding sites, including several transmembrane (TM) residues located close to the extracellular side and the N-terminus of CXCR4. This finding was supported by the molecular modeling of CXCR4 dimer–DV1 dimer interaction based on the crystal structure of CXCR4, which showed that DV1 dimer is capable of interacting with the CXCR4 dimeric structure by allowing the N-terminus of each DV1 monomer to reach into the binding pocket of CXCR4 monomer. The development of this bivalent ligand provides a tool to further probe the functions of CXCR4 dimerization and to study CXCR4 heterodimerization with other receptors.
INTRODUCTION
Chemokine receptors are a group of TM proteins that belong to the superfamily of G-protein-coupled receptors (GPCRs) (1-3). Many chemokine receptors, such as CXCR4, are important targets for drug discovery (4). The natural ligands of chemokine receptors, the chemokines, act as chemoattractants that direct various types of leukocytes to sites of inflammation and to secondary lymphoid organs. Chemokines and their receptors are also involved in a wide range of human diseases, most notably Acquired Immune Deficiency Syndrome (AIDS) (1, 5-7). The entry of human immunodeficiency virus type 1 (HIV-1) into host cells requires two principal coreceptors, CXCR4 and CCR5, in addition to its main receptor, CD4 (8-11). During the asymptomatic stage of disease, macrophage (M)-tropic strains of HIV-1 primarily use CCR5 as an entry coreceptor (12-14). However, in 40 to 50% of HIV-1 infected individuals, T-cell (T)-tropic strains that predominantly use CXCR4 eventually replace M-tropic strains and lead to rapid disease progression (15-17). Natural ligands of CXCR4 or CCR5 can inhibit HIV-1 infection (18, 19) by blocking HIV-1 glycoprotein gp120 binding sites on CXCR4 or CCR5 (20, 21) and/or by inducing receptor internalization (22, 23).
The GPCRs were initially considered to exist as monomeric entities, but many are now known to be organized in constitutive dimeric or oligomeric complexes (24, 25). Co-expression studies with the GABAb-R2 receptor demonstrated that efficient surface expression and function are dependent on the association of this protein with the GABAb-R1 protein (26-28), suggesting that hetero-oligomerization is necessary for receptor function. Also co-immunoprecipitation and cross-linking studies by many research groups have confirmed either constitutive (29) or ligand-induced multimerization of CCR5 (30). Similarly, CXCR4 was shown to form constitutive homodimers, and it can associate with itself far more efficiently than its association with other GPCRs, such as CCR5 or C5a receptor (31, 32). In fact, the nuclear magnetic resonance structure of a constitutively dimeric stromal cell-derived factor (SDF)-1α in complex with a CXCR4 fragment showed that sulfotyrosines sTyr7 and sTyr12 of CXCR4 occupy positively charged clefts on opposing chemokine subunits (33). Another piece of evidence for CXCR4 dimerization involves the “warts, hypogammaglobulinemia, infections and myelokathexis” (WHIM) syndrome (34). This syndrome has been linked to mutations in the carboxyl (C)-terminus of CXCR4, which results in truncated variants that exhibit enhanced signaling but fail to desensitize and internalize following the stimulation by SDF-1α. WHIM is primarily a heterozygous disease in which truncated CXCR4 is co-expressed with the wild-type receptor. Dimerization has been proposed as the most likely mechanism to explain the dominance of mutant CXCR4 over the wild-type receptor (35).
The roles of CXCR4 dimerization in normal physiological functions, human diseases, and therapeutic development need to be further explored. Our previous studies on synthetic peptides derived from the N-terminus of vMIP-II encoded by the Kaposi’s sarcoma-associated herpes virus demonstrated that the N-terminus of vMIP-II is the major binding determinant for CXCR4 (36). A peptide named V1 derived from the N-terminus (1-21 residues) of vMIP-II showed CXCR4 binding and selectively prevented CXCR4 signal transduction and coreceptor function by blocking the entry of T- and dual-tropic HIV-1 isolates. Both interestingly and surprisingly, DV1 peptide, an all-D-amino acid analog of V1 peptide, displayed higher binding affinity toward CXCR4 than V1 and showed significant antiviral activity in inhibiting the replication of CXCR4-dependent HIV-1 strains (37), despite the dramatically different conformations of DV1 from that of V1. In the present study, we exploited the observation that an increase in binding affinity or potency for GPCR homodimers usually occurs with “bivalent ligands” containing two identical pharmacophores linked by a spacer in a single ligand (38-41). Specifically, we sought to investigate the possible effects of ligand dimerization on CXCR4 binding. Compared with its parent monomeric DV1, DV1 dimer demonstrated much improved CXCR4 binding activity and showed 2-3 fold increase in its antiviral activity. Ligand binding site mapping experiments illustrated that CXCR4–DV1 dimer interaction requires several TM residues (i.e., Tyr45, Phe87, Tyr121, Asp171, Trp252, Tyr255, Glu288, and Phe292) as well as the N-terminus of CXCR4, and that many of these residues overlap with HIV-1 gp120 binding sites. The molecular modeling of CXCR4 dimer–DV1 dimer interactions also showed that DV1 dimer could span two CXCR4 receptors, with the N-terminus of each DV1 monomer reaching into the binding pocket of CXCR4 monomer.
EXPERIMENTAL PROCEDURES
Total Chemical Synthesis of DV1 and DV1 Dimer
DV1 or [12A]DV1 was assembled on a CLEAR amide resin (Peptides International, Louisville, KY) using the Applied Biosystems 433A peptide synthesizer (Applied Biosystems, Foster city, CA). Fmoc-chemistry and FastMoc protocol were employed for the synthesis. The replacement of cysteine residue at position 12 with alanine residue, as in [12A]DV1, was to promote a disulfide bond formation only at position 11 (Table 1). The protecting groups for Fmoc amino acids are Arg, Pmc; Asp, OtBu; Gln, Trt; Cys, Trt; His, Trt; Lys, Boc; Ser, Tyr, tBu; Trp, Boc. HBTU and HOBt were used as coupling reagents and additives. Single coupling was used for all the amino acids. After washing with dichloromethane and methanol, the peptide was dried and cleaved from the resin with the cool mixture of trifluoroacetic acid/water/thioanisole/ethanedithiol/triisopropylsilane/phenol 82:5:5:3:2:3 (v/v) for 4 hrs at room temperature with occasionally gentle shaking. The crude peptide was precipitated in ice-cold tert-butyl methyl ether, washed several times, freeze dried and purified using semi-preparative HPLC on a Vydac 218TP1010 column. The eluent A is 0.1% TFA in water, while the eluent B is acetonitrile/water 80:20 (v/v) with 0.08% TFA. The absorbance was monitored at 215 nm. The major peak containing the expected peptide was collected and lyophilized. For the disulfide bond formation of [12A]DV1, the crude peptide was dissolved in ammonium acetate buffer at pH 8.0 to which 5-10% dimethyl sulfoxide was added. The peptide concentration was kept at 5 mmol/L. The reaction was monitored using HPLC. After 48 hrs, the mixture was filtered and applied to a semi-preparative column for purification. The final products were checked by analysis HPLC and confirmed by MALDI-TOF mass spectrometry. We also conducted HPLC analysis to examine whether DV1 was stable and remained monomeric under the cell media condition and during the time course used for the antiviral assays (data not shown). It was found that DV1 remained monomeric after 48 hrs as no dimerization of DV1 was identified by RP-HPLC. In addition, DV1 remain stable as approximately 77% of DV1 remained intact after 48 hrs.
Table 1.
Amino Acid Sequences of DV1 and DV1 Dimer.
| Analog Designation |
Amino Acid Sequencesa |
|---|---|
| DV1 |
LGASWHRPDKCCLGYQ
KRPLP |
| DV1 Dimer |
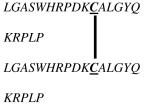
|
D-amino acids are shown in italic. The replacement of cysteine residue at position 12 with alanine residue, as in [12A]DV1, was to promote a disulfide bond formation only at position 11.
Materials
Plasmid pcDNA3-CXCR4, CXCR4 monoclonal (MAb) antibody 12G5, and human kidney cell line 293 were obtained through the AIDS Research and Reference Reagent Program (Division of AIDS, NIAID, National Institute of Health, Bethesda, MD). Cell culture media and G418 were purchased from CAMBREX (Walkersville, MD). Dulbecco’s Modified Eagle’s Medium (DMEM) plus 10% fetal bovine serum and 5% penicillin-streptomycin was used to maintain 293 cells.
Site-Directed Mutagenesis and Transfection
All the CXCR4 mutants were previously prepared with the QuikChange site-directed mutagenesis kit (Stratagene, La Jolla, CA) and stably transfected into 293 cells using Tfx-50 reagents (Promega, Madison, WI) or Lipofectamine 2000 (Invitrogen, Carlsbad, CA) according to the manufacturer’s instructions (42, 43). The selective medium containing G418 (800 μg/ml) was used to isolate stably transfected cells that were subsequently single-cloned.
Competition Receptor Binding Assays Using Receptor Specific Antibody
Competition binding experiments (44, 45) were performed using a single concentration of 12G5 (50 ng/ml), in a final volume of 100 μl FACS buffer containing 5 × 105 cells in 96-well plates in the presence of various concentrations of unlabeled DV1 or DV1 dimer. Samples were incubated on ice for 40 mins. The cells were washed with 200 μl FACS buffer and stained with 10 μg FITC-conjugated goat anti-mouse IgG for 30 mins at 4°C. As a negative control, cells were stained only with the secondary antibody. The cells were washed with FACS buffer and resuspended in 100 μl FACS buffer before being analyzed on the Wallac Victor2 1420 Multilabel counter (Turku, Finland). At least three independent experiments were performed.
Intracellular Calcium Measurements
Sup T1 cells (107 cells/ml) were loaded with 2 μM fura-2/AM (Molecular Probes, Eugene, OR) and 0.01% Pluronic F-127 (Sigma) in Hank’s balanced salt saline (140 mM NaCl, 5 mM KCl, 10 mM Hepes, pH 7.4, 1 mM CaCl2, 1 mM MgCl2, 1 mg/ml glucose, and 0.025% BSA) for 20 mins at room temperature (42, 44, 46). The cells were washed and resuspended in the same buffer to 106 cells/ml. Fura-2 fluorescence was measured on the fluorescence spectrophotometer (ISA SPEX FluoroMax-2) using excitation wavelengths of 340 nm and 380 nm, and an emission wavelength of 510 nm.
Assay for Inhibition of HIV-1 Infection
HeLa CD4 LTR β-gal cells were cultured with the required selection antibiotics (47). 24 hrs prior to the initiation of the assay, the cells were plated into 96-well flat bottom microtiter plates in media without selection antibiotics. At 24 hrs, media were removed. The compound diluted in media was placed on the cells and incubated for 15 mins at 37°C. A known titer of the IIIB strain of HIV-1 was added to the wells. The incubation was continued at 37°C for 1 to 2 hrs. At the end of the incubation, the wells were washed 2 times with media. The culture was continued for 40 to 48 hrs. At the termination of the assay, media were removed. β–galactosidase enzyme expression was determined by chemiluminescence according to the manufacturer’s instructions (Tropix Gal-screen™, Bedford, MA). Compound toxicity was monitored on a replica plate by MTS dye reduction. All determinations were performed in triplicate with serial Log10 dilutions of the test materials.
Focal Infectivity Assay
Single-cycle infectivity assays were performed as described previously (48). Briefly, HeLa cells expressing large quantities of CD4 were seeded at the concentrations of 5000 cells/well into 1 cm2 wells of a 48-well culture plate. The next day, the cells were incubated with 8 μg/mL of DEAE-dextran for 20 mins at room temp. 10,000 RT units of HXBc2 viruses at final volume of 0.1 mL were added to the cells and incubated for 2 to 4 hrs, at which point 0.5 mL of fresh complete DMEM medium was added to the cells and incubated for 48 hrs at 37 C. The cells were then fixed with 95% ethanol for 3 to 6 mins and rinsed with saline solution containing 1 mM EDTA. Immunoperoxidase staining was done as previously described (48) using 0.1 mL/well of 1:5 diluted supernatant from the anti-HIV p24 hybridoma 183-H12-5C obtained from the NIH AIDS Research and Reference Reagent Program. The stained foci of infection were counted using a dissecting microscope.
Mapping Experiments Using Receptor Specific Antibody
Binding experiments were performed using a single concentration (5 μg/ml) of 12G5 in a final volume of 100 μl FACS buffer containing 5 × 105 cells in 96-well plates in the presence of various concentrations of DV1 dimer (43). Cells were incubated on ice for 40 mins and washed with FACS buffer before being stained with 10 μg of FITC-conjugated goat anti-mouse IgG for 30 mins at 4°C. As a negative control, cells were stained only with the secondary antibody. The cells were washed with FACS buffer and resuspended in 100 μl FACS buffer before being analyzed on the Wallac Victor2 1420 Multilabel counter (Turku, Finland). The percent (%) bound 12G5 was calculated by taking the ratio of the mean fluorescence intensity in the presence of DV1 dimer over the fluorescence in the absence of DV1 dimer. Only when the point mutation of a CXCR4 residue increased the % bound 12G5 of DV1 dimer compared with that of the wild-type receptor by more than 10% was the residue considered important for ligand binding. Although the testing concentration can be reduced to as low as 5 nM to test the binding activity of high-affinity ligands, such as DV1 dimer, the high concentration of 300 nM was used to clearly demonstrate any differences between the wild-type receptor and CXCR4 mutants. At least three independent experiments were performed. Although only the binding data at one testing concentration are shown for simplicity, the data represent the mean values of three independent assays with the error bars indicating the standard deviations.
Molecular Modeling of CXCR4 Dimer–DV1 Dimer Interactions
DV1 dimer was built by MOE (Molecular Operating Environment) program, and CXCR4 dimer is from PDB crystal structure 3ODU. The molecular-dynamics (MD) simulation was performed using MOE and AMBER99 force field. During the MD simulation, only residues in the extracellular loops of CXCR4 and all residues in ligands were allowed to move, whereas the rest of CXCR4 residues were frozen to their positions. The MD simulation was performed for 100 ps while the temperature of the system was maintained at 300K. As control and comparison studies, we also performed MD simulations using a single CXCR4 and DV1 dimer to examine whether DV1 dimer may interact with a single CXCR4 (data not shown), using the methods similar to those described above.
RESULTS AND DISCUSSION
Enhanced Binding Affinity of DV1 Dimer
It has been previously demonstrated that the N-terminus of vMIP-II is the major determinant for CXCR4 recognition (36, 49). DV1 peptide, an all-D-amino acid analog of V1 peptide (1-21 residues) derived from the N-terminus of vMIP-II (36), displayed enhanced binding affinity toward CXCR4 and significant antiviral activity in inhibiting the replication of CXCR4-dependent HIV-1 strains (37). These results also illustrated the remarkable stereochemical flexibility of the CXCR4–peptide interface. Since the early 1980s, “bivalent ligands,” which combine two pharmacophores linked by a spacer in a single molecule, have been synthesized for many different GPCRs as specific pharmacological tools. Some of these bivalent molecules showed higher binding affinity, potency, or selectivity over their respective monomeric counterparts (38-41, 50). In the present study, we designed DV1 dimer to investigate possible effects of ligand dimerization on CXCR4 binding, as well as to develop novel CXCR4-binding molecules with enhanced binding affinity and antiviral activity. Antibody competition binding assays showed that the CXCR4 binding activity of DV1 dimer (IC50 = 3 nM) was fourteen times greater than that of DV1 (IC50 = 43 nM) (Fig. 1; Table 2). This observation is consistent with the design concept that DV1 dimer may be able to span two CXCR4 receptors and interact with the CXCR4 dimer structure, which leads to increased receptor binding affinity. Consistent with this notion, the native state of human CXCR4 may be a dimer, as suggested by the previous reports that CXCR4 sedimented in a manner consistent with a dimer in cell lysates prepared with non-denaturing detergents, whereas CCR5 sedimented as a monomer under these conditions (31, 32). This is further supported by the recently solved crystal structures of CXCR4 that reveal a consistent homodimer (51).
Fig. 1. CXCR4 binding activities of DV1 and DV1 dimer.
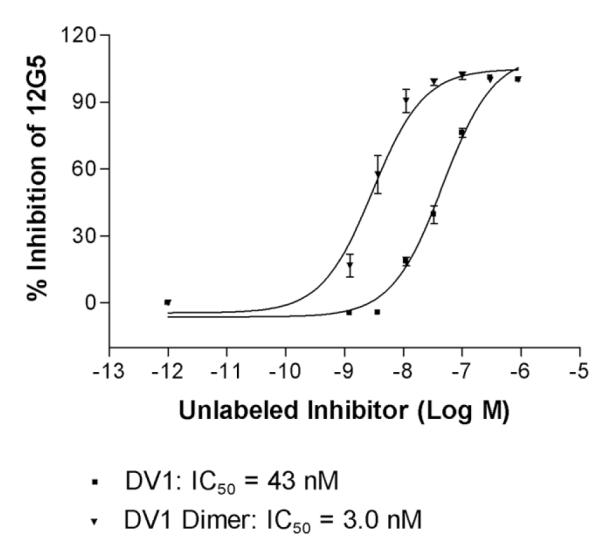
12G5 antibody competition binding assays were used to determine their IC50 values. Stably transfected 293 cells were used in the binding experiments. The binding data were analyzed using the PRISM program (GraphPad Inc., San Diego, CA). All data are shown as mean ± S.D. from at least three independent experiments.
Table 2.
Binding and Antiviral Activities of DV1 and DV1 Dimera
| Analog Designation |
CXCR4 Binding (nM) |
Antiviral Activity Using Infectious Virus Assays (μM) |
Antiviral Activity Using Single-Cycle Focal Infectivity Assays (μM) |
|---|---|---|---|
| DV1 | 43 ±5.01 | 12.1 ±3.26 | 24 ±0.00 |
| DV1 Dimer | 3.0 ± 0.52 | 4.4 ±3.10 | 10 ±0.49 |
The binding and antiviral activities of DV1 and DV1 dimer are shown by their IC50 values.
Signaling Activity of DV1 Dimer
We conducted biochemical studies on the signaling activity of DV1 dimer. Consistent with the antagonistic nature of DV1 monomer (37), DV1 dimer at 50 nM did not induce any significant mobilization of calcium (Ca2+) in Sup T1 cells expressing CXCR4. This contrasted with the rapid Ca2+ mobilization induced by the normal CXCR4 ligand, SDF-1α (Fig. 2). However, since CXCR4 can induce a variety of signaling events (including intracellular Ca2+ mobilization) following SDF-1α binding, the possibility that DV1 dimer may be able to induce other signaling events not investigated in this work cannot be completely excluded.
Fig. 2. Signaling activity of DV1 dimer.

All the data shown are representative of at least three independent experiments. Ca2+ influx in Sup T1 cells was measured in response to 50 nM SDF-1α (control) and DV1 dimer. DV1 dimer was also tested at higher concentrations and found to have no effect in activating Ca2+ influx.
Antiviral Activity of DV1 Dimer
The ability of DV1 dimer to inhibit HIV-1 infection via CXCR4 was tested using both infectious virus and single-cycle infective assays. Consistent with the binding data, DV1 dimer was more potent in blocking HIV-1 entry (IIIB strain) than its parent DV1 peptide (Fig. 3A–B; Table 2). Infectious virus assays revealed that DV1 dimer has an IC50 value of 4.4 M for HIV-1 inhibition, which is a 3-fold lower than that of DV1 (IC50 = 12.1 M) (Fig. 3A). Similarly, using single-cycle virus (HXBc2) in focal infectivity assays, DV1 dimer (IC50 = 10 M) was found to be a more effective and potent blocker of HIV-1 entry than DV1 (IC50 = 24 M) (Fig. 3B). It should be noted that the degree of improvement (2-3 fold increase) in the antiviral activity of DV1 dimer is not the same as the increase in the binding assay (14 fold increase). This may be due to the insensitivity of the antiviral assays which has been observed by us and other groups in the past. When compared with the affinity of early isolates of HIV-1 for CCR5, late isolates of HIV-1 that utilize CXCR4 are known to achieve high replication efficiency despite the significantly lower affinity of HIV-1 gp120 for CXCR4 (52, 53). The HIV-1 envelope glycoproteins are trimers and contain three receptor-binding sites. As such, the interactions of envelope glycoprotein trimers with head-to-head dimers of CXCR4 could result in formation of hexameric arrays at the virus-cell interface. If this multimeric complex is involved in viral infection, it could compensate for low gp120-CXCR4 affinity (31, 32). Based on this mechanism, even though DV1 dimer has an increase in the affinity for CXCR4 receptor as compared to that of DV1 monomer, the increase in the ability of DV1 dimer to prevent viral infection which involves multimeric array interactions described above would be less as compared to that of CXCR4 binding and blocking monomeric gp120-CXCR4 interaction. This may explain the different degrees of increase in CXCR4 binding affinity and antiviral activity of DV1 dimer. However, the possibility that 2 to 3 fold increase in the antiviral activity of DV1 dimer arises from the bivalency of the ligand without the need for a true bivalent interaction with CXCR4 dimer cannot be completely ruled out, although our molecular modeling supports the true CXCR4 dimer–DV1 dimer interactions (Fig. 5).
Fig. 3. Antiviral activity of DV1 dimer.

The antiviral activity of DV1 dimer was compared to that of DV1 using both virus infection (A) and focal infectivity assays (B). HeLa CD4 LTR β-gal cells were used in infectious virus assays, and HeLa cells are used in single-cycle focal infectivity assays. %VC and %CC represent percent of virus control and percent of cell control, respectively. All the data shown are representative of at least three independent experiments with error bars indicating the standard deviations.
Fig. 5. Molecular modeling of CXCR4 dimer–DV1 dimer interactions.

A, Interactions between CXCR4 dimer and DV1 dimer after 100 ps molecular-dynamics (MD) simulation. DV1 monomer A is shown as red stick, DV1 monomer B as green stick, CXCR4-A as grey carton, and CXCR4-B as cyan carton, and CXCR4 binding site residues as gray sticks. B, Top view of DV1 dimer interacting with CXCR4 dimer. DV1 monomer A is shown as red line, and DV1 monomer B as green line. C, Side view of DV1 dimer interacting with CXCR4 dimer.
DV1 Dimer Overlaps with HIV-1 gp120 on CXCR4 Binding Sites
The mechanism of HIV-1 inhibition by DV1 dimer was examined by ligand binding site mapping experiments using a panel of site-directed mutants of CXCR4 (43). We previously demonstrated that all these mutants display stable expression levels comparable to or higher than wild-type CXCR4 (42, 43). Among the extracellular domain mutants, only DNX4 (a CXCR4 mutant with the entire N-terminus (codons 2-25) deleted) reduced the binding activity of DV1 dimer, as it increased the percentage (%) of 12G5 binding by 15% compared with the wild-type CXCR4 (Fig. 4). Unlike DNX4, none of the mutants of the second (ECL2) or third extracellular loop (ECL3) impaired the binding affinity of DV1 dimer (Fig. 4). Similarly, several TM mutants, including H79A, D97A, W161A, P163A, Y219A, Y256A, D262A, H294A and N298A, all failed to reduce the binding activity of DV1 dimer (Fig. 4). Only Tyr45, Phe87, Tyr121, Asp171, Trp252, Tyr255, Glu288, Phe292 and the N-terminus were involved in DV1 dimer binding (Table 3; Fig. 4). Specifically, Y45A, Y121A, W252A, Y255A, and F292A decreased DV1 dimer binding by 20%, whereas F87A and E288D reduced the binding affinity of DV1 dimer by 40% (Fig. 4), and D171A impaired DV1 dimer binding by 60% (Fig. 4). Despite several distinct binding sites required for DV1 dimer interactions with CXCR4, its binding site also overlaps with that of SDF-1α. Areas of potential overlap include three transmembrane residues (i.e., Phe87, Asp171, and Phe292) and the N-terminus of CXCR4 (Table 3) (43). These results are consistent with our previous report that several D-amino acid-containing analogs based on the full-length chemokine vMIP-II, especially RCP168, interact with many residues on CXCR4 TM and extracellular domains that are important for HIV-1 entry, but not SDF-1α binding or signaling (42, 43) (Table 3). In fact, the only difference between DV1 dimer and RCP168 with respect to CXCR4 binding sites is that RCP168 requires Asp97 for its interactions with CXCR4, whereas DV1 dimer does not.
Fig. 4. Binding activity of DV1 dimer to wild-type CXCR4 and mutants.

The data represent the mean values of three independent assays with the error bars indicating the standard deviations. The binding affinity of DV1 dimer was decreased by DNX4, Y45A, Y121A, W252A, Y255A, or E288D. F87A, D171A, and F292A also attenuated the binding activity of DV1 dimer. The other mutants had little effect on DV1 dimer binding.
Table 3.
Binding Sites of DV1 Dimer Versus RCP168, SDF-1α, and HIV-1 gp120
| CXCR4 Domains | DV1 Dimera | RCP168 | SDF-1α | HIV-1 gp120 |
|---|---|---|---|---|
| N-terminus | DNX4 | DNX4 | DNX4 | DNX4 |
| TM1 | Tyr45 | Tyr45 | Tyr45 | |
| TM2 | Phe87 | Phe87, Asp97 | Phe87 | |
| TM3 | Tyr121 | Tyr121 | ||
| TM4 | Asp171 | Asp171 | Asp171 | Asp171 |
| TM6 | Trp252, Tyr255 | Trp252, Tyr255 | Trp252, Tyr255 | |
| TM7 | Glu288, Phe292 | Glu288, Phe292 | Phe292 | Glu288 |
The residues highlighted in bold are also involved in HIV-1 coreceptor activity of CXCR4.
Among the residues discussed above, Tyr45, Asp171, Trp252, Tyr255, Glu288 and the N-terminus are known to play key roles in the coreceptor activity of CXCR4 (42, 43, 54-57). Since these residues are located on the upper part of the TM barrel of CXCR4, close to the extracellular side, they are likely to be involved with direct interactions with DV1 dimer and/or HIV-1 gp120. Recently, the long-awaited crystal structure of CXCR4 was published by Wu et al who reported five independent crystal structures of CXCR4 bound to an antagonist small molecule IT1t and to a cyclic peptide CVX15 at 2.5 to 3.2 angstrom resolution (51). These structures provide new clues about the interactions between CXCR4 and SDF-1α as well as with HIV-1 gp120. All structures have revealed consistent CXCR4 homodimers with an interface, including TM helices V and VI of CXCR4, which may be involved in regulating signaling. Compared with previous GPCR structures, the binding pocket of CXCR4 is larger, more open, and located closer to the extracellular surface, and includes acidic residues Asp187, Glu288, and Asp97 that are important for SDF-1α binding. This suggests that Lys1, the most critical residue in SDF-1α for receptor activation, could reach into the CXCR4 pocket and interact with one of these acidic residues. The importance of Glu288 for SDF-1α signaling was previously demonstrated by our laboratory (42), and its critical role in ligand binding, including that of DV1 dimer, was further illustrated in the present study. Similarly, the basic character of the gp120 V3 loop, which becomes exposed upon CD4 binding, could potentially penetrate the CXCR4 binding pocket, thereby interacting with one of these acidic residues. As such, one can hypothesize that the gp120 V3 loop, which can reach into CXCR4 TM domains, may be blocked by DV1 dimer that may directly interact with some of these important TM residues, including Glu288. Furthermore, the potential ability to span two CXCR4 receptors with the N-terminus of each DV1 monomer reaching into the binding pocket of CXCR4 monomer may explain its increased binding affinity and antiviral activity of DV1 dimer. Indeed, based on our molecular modeling of CXCR4 dimer–DV1 dimer interactions (Fig. 5) and schematic illustration of the locations of residues important for DV1 dimer (Fig. 6), the binding pocket of DV1 dimer appears to be much larger and more open compared with that of SDF-1α which seems to be narrower. This wide binding pocket of DV1 dimer can be advantageous in blocking the formation of hexameric arrays between HIV glycoprotein trimers and CXCR4 dimers more efficiently.
Fig. 6. Schematic illustration of the locations of residues important for DV1 dimer binding on CXCR4 TM domains.

This model was drawn based on the crystal structure of CXCR4 (51), and it accurately represents the locations of TM helices of CXCR4. DV1 monomer A is shown as red stick, DV1 monomer B as green stick, CXCR4-A as pink carton, and CXCR4-B as cyan carton. The residues involved in the binding activities of both SDF-1α and DV1 dimer are highlighted with red spots, whereas those selectively involved in DV1 dimer binding (most of which overlap with HIV-1 binding) are highlighted with black spots. Black spots also include additional residues important for DV1 dimer binding as predicted by the molecular modeling of CXCR4 dimer–DV1 dimer interactions. Such overlapping sites between HIV-1 and DV1 dimer may serve as a potential target recognized by new selective anti-HIV inhibitors. It should be noted that all the residues highlighted with red/black spots are pointing inside toward the TM pocket, not pointing out toward the lipid bilayer.
Molecular Modeling of CXCR4 Dimer–DV1 Dimer Interactions
Further support for this view of CXCR4 dimer–DV1 dimer interactions was provided by molecular-dynamics (MD) simulation, whereby the N-terminus of DV1 dimer was manually docked into the groove of CXCR4 dimer, based on the recently solved crystal structures of CXCR4 (51). This computer docking study was performed based on the experimental findings of important CXCR4 residues as guides. The complex was energy minimized and subjected to a 100 ps MD simulation. During the simulation, the C-terminus of DV1 dimer showed flexibility toward CXCR4 dimer. For DV1 monomer A, Leu1A formed a hydrogen (H) bond with CXCR4 A chain Asp171 (Fig. 5). Ser4A also H-bonded with Arg188, while His6A and Arg7A of DV1 monomer A formed H-bonds with Asp193 and Glu32 of CXCR4 A chain, respectively. Cys28 of CXCR4 A chain also interacted with Asp9A of DV1 monomer A via a H-bond. Leu1B of DV1 monomer B formed a H-bond with CXCR4 B chain Glu288. Similar to the DV1 monomer A, His6B of DV1 monomer B exhibited a H-bond with Asp193, while Trp5B interacted with Arg188 via a H-bond. It should be noted that CXCR4 monomer–DV1 dimer interaction is unfavorable under the same constraints, as MD simulations showed that the binding pocket of a single CXCR4 could only accommodate the binding interactions of one monomer chain of DV1 dimer (data not shown). Taken together, the modeling data support the notion that the N-terminus of each DV1 monomer can reach into the binding pocket of each CXCR4 monomer, which is formed by several TM residues, including Asp171 and Glu288. These results are also consistent with our CXCR4 mutational studies.
Our results also provide interesting insights into the stereochemical tolerance of CXCR4–peptide interactions. We previously demonstrated that DV1 monomer binds to CXCR4 with potency and selectivity comparable with or higher than their L-peptide counterparts (37). This was surprising because of the profoundly different side chain topologies between D- and L-enantiomers with mirror image conformations. The high binding affinity of DV1 dimer also demonstrates the remarkable stereochemical flexibility of the CXCR4–peptide interface. These observations are consistent with the recent CXCR4 crystal structures that show a larger, more flexible, and open ligand binding pocket located closer to the extracellular surface as compared with the binding sites of other GPCR structures noted in the past (51). These features of CXCR4–ligand binding sites differ from those observed in other GPCRs, and help to explain the stereochemical flexibility demonstrated by our previous and present studies of D-peptide ligands. One may further speculate that the flexibility of the CXCR4–ligand interface might be a feature of CXCR4 that HIV-1 capitalizes on. This might allow sequence and conformational variations to occur in the V3 loop of HIV-1 envelope glycoprotein gp120. If the resulting changes in the gp120 V3 loop are accommodated by the flexible binding surface of CXCR4 but not by that of an antibody, this may be a potential mechanism for the virus to evade neutralizing antibodies while maintaining high affinity for the coreceptor that is necessary for viral entry into the cell.
Implications for the Study of CXCR4 Dimerization and Design of New HIV-1 Inhibitors
We report here that, as compared with its parent DV1 peptide, DV1 dimer shows enhanced CXCR4 binding and antiviral activity in inhibiting the replication of CXCR4-dependent HIV-1 strains. The residues on CXCR4 required for the binding activity of DV1 dimer overlap extensively with those involved in the HIV-1 coreceptor activity of the receptor. While one cannot rule out the possibility that the increase in the antiviral activity of DV1 dimer may arise from the bivalency of the ligand, molecular modeling of CXCR4 dimer–DV1 dimer interactions shows that DV1 dimer is capable of interacting with CXCR4 dimer with higher affinity than the monomeric interaction, which can lead to increased antiviral activity. As such, we have demonstrated a facile synthetic approach of using peptide disulfide bonds to generate bivalent peptide ligands such as DV1 dimeric peptide that can be used to study and analyze the functional roles of CXCR4 homodimerization. It is conceivable that this strategy can be used to generate bivalent ligands linking a CXCR4 peptide and the peptide of another receptor for the study of CXCR4 heterodimerization with other receptors. It is noted that a different chemical linker strategy was also recently reported to generate bivalent ligand probes of CXCR4 dimerization (58). Finally, an increase in the antiviral activity of the bivalent DV1 ligand as compared to the monovalent DV1 suggests that new and more effective HIV-1 inhibitors may be developed by using bivalent ligands.
Acknowledgments
This work was supported by grants from the National Institutes of Health.
ACKNOWLEDGEMENTS
HeLa cells expressing high levels of CD4 (H1-J cells) were generous gift of David Kabat.
ABBREVIATIONS
- SDF-1α
stromal cell-derived factor-1α
- vMIP-II
viral macrophage inflammatory protein-II
- HIV
human immunodeficiency virus
- AIDS
acquired immune deficiency syndrome
REFERENCES
- 1.Murphy PM. The molecular biology of leukocyte chemoattractant receptors. Annu Rev Immunol. 1994;12:593–633. doi: 10.1146/annurev.iy.12.040194.003113. [DOI] [PubMed] [Google Scholar]
- 2.Kobilka B. Adrenergic receptors as models for G Protein-coupled receptors. Annu Rev Neurosci. 1992;15:87–114. doi: 10.1146/annurev.ne.15.030192.000511. [DOI] [PubMed] [Google Scholar]
- 3.Strader CD, Fong TM, Tota MR, Underwood D. Structure and function of G protein-coupled receptors. Annu Rev Biochem. 1994;63:101–132. doi: 10.1146/annurev.bi.63.070194.000533. [DOI] [PubMed] [Google Scholar]
- 4.Choi WT, Duggineni S, Xu Y, Huang Z, An J. Drug discovery research targeting the CXC chemokine receptor 4 (CXCR4) J Med Chem. 2012;55:977–994. doi: 10.1021/jm200568c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Baggiolini M, Dewald B, Moser B. Human chemokines: an update. Annu Rev Immunol. 1997;15:675–705. doi: 10.1146/annurev.immunol.15.1.675. [DOI] [PubMed] [Google Scholar]
- 6.Berger EA, Murphy PM, Farber JM. Chemokine receptors as HIV-1 coreceptors: roles in viral entry, tropism, and disease. Annu Rev Immunol. 1999;17:657–700. doi: 10.1146/annurev.immunol.17.1.657. [DOI] [PubMed] [Google Scholar]
- 7.Proudfoot A. Chemokine receptors: multifaceted therapeutic targets. Nature Reviews/Immunology. 2002;2:106–115. doi: 10.1038/nri722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Feng Y, Broder CC, Kennedy PE, Berger EA. HIV-1 entry cofactor: functional cDNA cloning of a seven-transmembrane, G protein-coupled receptor. Science. 1996;272:872–877. doi: 10.1126/science.272.5263.872. [DOI] [PubMed] [Google Scholar]
- 9.Hill CM, Littman DR. Natural resistance to HIV? Nature. 1996;382:668–669. doi: 10.1038/382668a0. [DOI] [PubMed] [Google Scholar]
- 10.Trkola A, Dragic T, Arthos J, Binley JM, Olson WC, Allaway GP, Cheng-Mayer C, Robinson J, Maddon PJ, Moore JP. CD4-dependent, antibody-sensitive interactions between HIV-1 and its co-receptor CCR5. Nature. 1996;384:184–187. doi: 10.1038/384184a0. [DOI] [PubMed] [Google Scholar]
- 11.Wu L, Gerard NP, Wyatt R, Choe H, Parolin C, Ruffing N, Borsetti A, Cardoso AA, Desjardin E, Newman W, Gerard C, Sodroski J. CD4-induced interaction of primary HIV-1 gp120 glycoproteins with the chemokine receptor CCR5. Nature. 1996;6605:179–183. doi: 10.1038/384179a0. [DOI] [PubMed] [Google Scholar]
- 12.Alkhatib G, Combadiere C, Broder CC, Feng Y, Kennedy PE, Murphy PM. CC CKR5: a RANTES, MIP-1alpha, MIP-1beta receptor as a fusion cofactor for macrophage-tropic HIV-1. Science. 1996;272:1955–1958. doi: 10.1126/science.272.5270.1955. [DOI] [PubMed] [Google Scholar]
- 13.Deng H, Liu R, Ellmeier W, Choe S, Unutmaz D, Burkhart M, Marzio PD, Marmon S, Sutton RE, Hill CM, Davis CB, Peiper SC, Schall TJ, Littman DR, Landau NR. Identification of a major co-receptor for primary isolates of HIV-1. Nature. 1996;381:661–666. doi: 10.1038/381661a0. [DOI] [PubMed] [Google Scholar]
- 14.Dragic T, Litwin V, Allaway GP, Martin SR, Huang Y, Nagashima KA, Cayanan C, Maddon PJ, Koup RA, Moore JP, Paxton WA. HIV-1 entry into CD4+ cells is mediated by the chemokine receptor CC-CKR-5. Nature. 1996;381:667–673. doi: 10.1038/381667a0. [DOI] [PubMed] [Google Scholar]
- 15.Cheng-Mayer C, Seto D, Tateno M, Levy JA. Biologic features of HIV-1 that correlate with virulence in the host. Science. 1988;240:80–82. doi: 10.1126/science.2832945. [DOI] [PubMed] [Google Scholar]
- 16.Tersmette M, Lange JM, de Goede RE, de Wolf F, Eeftink-Schattenkerk JK, Schellekens PT, Coutinho RA, Huisman JG, Goudsmit J, Miedema F. Association between biological properties of human immunodeficiency virus variants and risk for AIDS and AIDS mortality. Lancet. 1989;1:983–985. doi: 10.1016/s0140-6736(89)92628-7. [DOI] [PubMed] [Google Scholar]
- 17.Schellekens PT, Tersmette M, Roos MT, Keet RP, de Wolf F, Coutinho RA, Miedema F. Biphasic rate of CD4+ cell count decline during progression to AIDS correlates with HIV-1 phenotype. AIDS. 1992;6:665–669. doi: 10.1097/00002030-199207000-00008. [DOI] [PubMed] [Google Scholar]
- 18.Bleul CC, Farzan M, Choe H, Parolin C, Clark-Lewis I, Sodroski J. The lymphocyte chemoattractant SDF-1 is a ligand for LESTR/fusin and blocks HIV-1 entry. Nature. 1996;382:829–833. doi: 10.1038/382829a0. [DOI] [PubMed] [Google Scholar]
- 19.Oberlin E, Amara A, Bachelerie F, Bessia C, Virelizier JL, Arenzana-Seisdedos F, Schwartz O, Heard JM, Clark-Lewis I, Legler DF, Loetscher M, Baggiolini M, Moser B. The CXC chemokine SDF-1 is the ligand for LESTR/fusin and prevents infection by T-cell-line-adapted HIV-1. Nature. 1996;382:833–835. doi: 10.1038/382833a0. [DOI] [PubMed] [Google Scholar]
- 20.Oravecz T, Pall M, Norcross MA. Beta-chemokine inhibition of monocytotropic HIV-1 infection. Interference with a postbinding fusion step. J Immunol. 1996;157:1329–1332. [PubMed] [Google Scholar]
- 21.Arenzana-Seisdedos F, Virelizier JL, Rousset D, Clark-Lewis I, Loetscher P, Moser B, Baggiolini M. HIV blocked by chemokine antagonist. Nature. 1996;383:400. doi: 10.1038/383400a0. [DOI] [PubMed] [Google Scholar]
- 22.Amara A, Gall SL, Schwartz O, Salamero J, Montes M, Loetscher P, Baggiolini M, Virelizier J-L, Arenzana-Seisdedos F. HIV coreceptor downregulation as antiviral principle: SDF-1α-dependent internalization of the chemokine receptor CXCR4 contributes to inhibition of HIV replication. J Exp Med. 1997;186:139–146. doi: 10.1084/jem.186.1.139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Förster R, Kremmer E, Schubel A, Breitfeld D, Kleinschmidt A, Nerl C, Bernhardt G, Lipp M. Intracellular and surface expression of the HIV-1 coreceptor CXCR4/fusin on various leukocyte subsets: rapid internalization and recycling upon activation. J Immunol. 1998;160:1522–1531. [PubMed] [Google Scholar]
- 24.Prinster SC, Hague C, Hall RA. Heterodimerization of G-protein-coupled receptors: specificity and functional significance. Pharmacol Rev. 2005;57:289–298. doi: 10.1124/pr.57.3.1. [DOI] [PubMed] [Google Scholar]
- 25.Bouvier M. Oligomerization of G-protein-coupled transmitter receptors. Nat Rev Neurosci. 2001;2:274–286. doi: 10.1038/35067575. [DOI] [PubMed] [Google Scholar]
- 26.White JH, Wise A, Main MJ, Green A, Fraser NJ, Disney GH, Barnes AA, Emson P, Foord SM, Marshall FH. Heterodimerization is required for the formation of a functional GABA(B) receptor. Nature. 1998;396:679–682. doi: 10.1038/25354. [DOI] [PubMed] [Google Scholar]
- 27.Kaupmann K, Malitschek B, Schuler V, Heid J, Froestl W, Beck P, Mosbacher J, Bischoff S, Kulik A, Shigemoto R, Karschin A, Bettler B. GABA(B)-receptor subtypes assemble into functional heteromeric complexes. Nature. 1998;396:683–687. doi: 10.1038/25360. [DOI] [PubMed] [Google Scholar]
- 28.Jones KA, Borowsky B, Tamm JA, Craig DA, Durkin MM, Dai M, Yao WJ, Johnson M, Gunwaldsen C, Huang LY, Tang C, Shen Q, Salon JA, Morse K, Laz T, Smith KE, Nagarathnam D, Noble SA, Branchek TA, Gerald C. GABA(B) receptors function as a heteromeric assembly of the subunits GABA(B)R1 and GABA(B)R2. Nature. 1998;396:674–679. doi: 10.1038/25348. [DOI] [PubMed] [Google Scholar]
- 29.Benkirane M, Jin DY, Chun RF, Koup RA, Jeang KT. Mechanism of transdominant inhibition of CCR5-mediated HIV-1 infection by ccr5delta32. J Biol Chem. 1997;272:30603–30606. doi: 10.1074/jbc.272.49.30603. [DOI] [PubMed] [Google Scholar]
- 30.Vila-Coro AJ, Mellado M, Martín de Ana A, Lucas P, del Real G, Martínez-A C, Rodríguez-Frade JM. HIV-1 infection through the CCR5 receptor is blocked by receptor dimerization. Proc Natl Acad Sci U S A. 2000;97:3388–3393. doi: 10.1073/pnas.050457797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Babcock GJ, Farzan M, Sodroski J. Ligand-independent dimerization of CXCR4, a principal HIV-1 coreceptor. J Biol Chem. 2003;278:3378–3385. doi: 10.1074/jbc.M210140200. [DOI] [PubMed] [Google Scholar]
- 32.Percherancier Y, Berchiche YA, Slight I, Volkmer-Engert R, Tamamura H, Fujii N, Bouvier M, Heveker N. Bioluminescence resonance energy transfer reveals ligand-induced conformational changes in CXCR4 homo- and heterodimers. J Biol Chem. 2005;280:9895–9903. doi: 10.1074/jbc.M411151200. [DOI] [PubMed] [Google Scholar]
- 33.Veldkamp CT, Seibert C, Peterson FC, De la Cruz NB, Haugner J. C. r., Basnet H, Sakmar TP, Volkman BF. Structural basis of CXCR4 sulfotyrosine recognition by the chemokine SDF-1/CXCL12. Sci Signal. 2008;16:37. doi: 10.1126/scisignal.1160755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hernandez PA, Gorlin RJ, Lukens JN, Taniuchi S, Bohinjec J, Francois F, Klotman ME, Diaz GA. Mutations in the chemokine receptor gene CXCR4 are associated with WHIM syndrome, a combined immunodeficiency disease. Nat Genet. 2003;34:70–74. doi: 10.1038/ng1149. [DOI] [PubMed] [Google Scholar]
- 35.Lagane B, Chow KY, Balabanian K, Levoye A, Harriague J, Planchenault T, Baleux F, Gunera-Saad N, Arenzana-Seisdedos F, Bachelerie F. CXCR4 dimerization and beta-arrestin-mediated signaling account for the enhanced chemotaxis to CXCL12 in WHIM syndrome. Blood. 2008;112:34–44. doi: 10.1182/blood-2007-07-102103. [DOI] [PubMed] [Google Scholar]
- 36.Luo Z, Fan X, Zhou N, Hiraoka M, Luo J, Kaji H, Huang Z. Structure-function study and anti-HIV activity of synthetic peptide analogues derived from viral chemokine vMIP-II. Biochemistry. 2000;39:13545–13550. doi: 10.1021/bi000633q. [DOI] [PubMed] [Google Scholar]
- 37.Zhou N, Luo Z, Luo J, Fan X, Cayabyab M, Hiraoka M, Liu D, Han X, Pesavento J, Dong CZ, Wang Y, An J, Kaji H, Sodroski JG, Huang Z. Exploring the stereochemistry of CXCR4-peptide recognition and inhibiting HIV-1 entry with D-peptides derived from chemokines. J Biol Chem. 2002;277:17476–17485. doi: 10.1074/jbc.M202063200. [DOI] [PubMed] [Google Scholar]
- 38.Daniels DJ, Lenard NR, Etienne CL, Law PY, Roerig SC, Portoghese PS. Opioid-induced tolerance and dependence in mice is modulated by the distance between pharmacophores in a bivalent ligand series. Proc Nat Acad Sci USA. 2005;102:19208–19213. doi: 10.1073/pnas.0506627102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.George SR, O’Dowd BF, Lee SP. G-protein-coupled receptor oligomerization and its potential for drug discovery. Nat Rev Drug Discov. 2002;1:808–820. doi: 10.1038/nrd913. [DOI] [PubMed] [Google Scholar]
- 40.Messer WSJ. Bivalent ligands for G protein-coupled receptors. Curr Pharm Des. 2004;10:2015–2020. doi: 10.2174/1381612043384213. [DOI] [PubMed] [Google Scholar]
- 41.Kiessling LL, Gestwicki JE, Strong LE. Synthetic multivalent ligands as probes of signal transduction. Angew Chem Int Ed Engl. 2006;45:2348–2368. doi: 10.1002/anie.200502794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tian S, Choi WT, Liu D, Pesavento J, Wang Y, An J, Sodroski JG, Huang Z. Distinct functional sites for human immunodeficiency virus type 1 and stromal cell-derived factor 1a on CXCR4 transmembrane helical domains. J Virol. 2005;79:12667–12673. doi: 10.1128/JVI.79.20.12667-12673.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Choi WT, Tian S, Dong CZ, Kumar S, Liu D, Madani N, An J, Sodroski JG, Huang Z. Unique ligand binding sites on CXCR4 probed by a chemical biology approach: implications for the design of selective human immunodeficiency virus type 1 inhibitors. J Virol. 2005;79:15398–15404. doi: 10.1128/JVI.79.24.15398-15404.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kumar S, Choi WT, Dong CZ, Madani N, Tian S, Liu D, Wang Y, Pesavento J, Wang J, Fan X, Yuan J, Fritzsche WR, An J, Sodroski JG, Richman DD, Huang Z. SMM-Chemokines: a class of unnatural synthetic molecules as chemical probes of chemical receptor biology and leads for therapeutic development. Chemistry & Biology. 2006;13:69–79. doi: 10.1016/j.chembiol.2005.10.012. [DOI] [PubMed] [Google Scholar]
- 45.Zhou NL, Luo Z, Fan J, Cayabyab X, Hiraoka M, Liu M, Han D, Pesavento X, Dong J, Wang CZ, An Y, Kaji J, Sodroski H, J.G., Huang Z. Exploring the stereochemistry of CXCR4-peptide recognition and inhibiting HIV-1 entry with D-peptides derived from chemokines. J Biol Chem. 2002;277:17476–17485. doi: 10.1074/jbc.M202063200. [DOI] [PubMed] [Google Scholar]
- 46.Dong CZ, Kumar S, Choi WT, Madani N, Tian S, An J, Sodroski JG, Huang Z. Different stereochemical requirements for CXCR4 binding and signaling functions as revealed by an anti-HIV, D-amino acid-containing SMM-chemokine ligand. J Med Chem. 2005;48:7923–7924. doi: 10.1021/jm050829u. [DOI] [PubMed] [Google Scholar]
- 47.Chen SS, Lee SF, Hao HJ, Chuang CK. Mutations in the leucine zipper-like heptad repeat sequence of human immunodeficiency virus type 1 gp41 dominantly interfere with wild-type virus infectivity. J Virol. 1998;72:4765–4774. doi: 10.1128/jvi.72.6.4765-4774.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Platt EJ, Wehrly K, Kuhmann SE, Chesebro B, Kabat D. Effects of CCR5 and CD4 cell surface concentrations on infections by macrophagetropic isolates of human immunodeficiency virus type 1. J Virol. 1998;72:2855–2864. doi: 10.1128/jvi.72.4.2855-2864.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.LiWang AC, Cao JJ, Zheng H, Lu Z, Peiper SC, LiWang PJ. Dynamics study on the anti-human immunodeficiency virus chemokine viral macrophage-inflammatory protein-II (VMIP-II) reveals a fully monomeric protein. Biochem. 1999;38:442–453. doi: 10.1021/bi9812726. [DOI] [PubMed] [Google Scholar]
- 50.Demmer O, Dijkgraaf I, Schumacher U, Marinelli L, Cosconati S, Wester HJ, Kessler H. Design, synthesis and functionalization of dimeric peptides targetting chemokine receptor CXCR4. J Med Chem. 2011;54:7648–7662. doi: 10.1021/jm2009716. [DOI] [PubMed] [Google Scholar]
- 51.Wu B, Chien EY, Mol CD, Fenalti G, Liu W, Katritch V, Abagyan R, Brooun A, Wells P, Bi FC, Hamel DJ, Kuhn P, Handel TM, Cherezov V, Stevens RC. Structures of the CXCR4 chemokine GPCR with small-molecule and cyclic peptide antagonists. Science. 2010;330:1066–1071. doi: 10.1126/science.1194396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Turner S, Tizard R, DeMarinis J, Pepinsky RB, Zullo J, Schooley R, Fisher R. Resistance of primary isolates of human immunodeficiency virus type 1 to neutralization by soluble CD4 is not due to lower affinity with the viral envelope glycoprotein gp120. Proc Nat Acad Sci USA. 1992;89:1335–1339. doi: 10.1073/pnas.89.4.1335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Peden K, Emerman M, Montagnier L. Changes in growth properties on passage in tissue culture of viruses derived from infectious molecular clones of HIV-1LAI, HIV-1MAL, and HIV-1ELI. Virology. 1991;185:661–672. doi: 10.1016/0042-6822(91)90537-l. [DOI] [PubMed] [Google Scholar]
- 54.Brelot A, Heveker N, Pleskoff O, Sol N, Alizon M. Role of the first and third extracellular domains of CXCR-4 in human immunodeficiency virus coreceptor activity. J Virol. 1997;71:4744–4751. doi: 10.1128/jvi.71.6.4744-4751.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Doranz B, Orsini M, Turner J, Hoffman T, Berson J, Hoxie J, Peiper S, Brass L, Doms R. Identification of CXCR4 domains that support coreceptor and chemokine receptor functions. J Virol. 1999;73:2752–2761. doi: 10.1128/jvi.73.4.2752-2761.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Chabot DJ, Zhang PF, Quinnan GV, Broder CC. Mutagenesis of CXCR4 identifies important domains for human immunodeficiency virus type 1 ×4 isolate envelope-mediated membrane fusion and virus entry and reveals cryptic coreceptor activity for R5 isolates. J Virol. 1999;73:6598–6609. doi: 10.1128/jvi.73.8.6598-6609.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zhou N, Luo Z, Luo J, Liu D, Hall JW, Pomerantz RJ, Huang Z. Structural and functional characterization of human CXCR4 as a chemokine receptor and HIV-1 co-receptor by mutagenesis and molecular modeling studies. J Biol Chem. 2001;276:42826–42833. doi: 10.1074/jbc.M106582200. [DOI] [PubMed] [Google Scholar]
- 58.Tanaka T, Nomura W, Narumi T, Masuda A, Tamamura H. Bivalent ligands of CXCR4 with rigid linkers for elucidation of the dimerization state in cells. J Am Chem Soc. 2010;132:15899–15901. doi: 10.1021/ja107447w. [DOI] [PubMed] [Google Scholar]