

Abstract
During the last half-century, incidences of breast cancer have increased globally. Various factors—genetic and environmental— have been implicated in the initiation and progression of this disease. One potential environmental risk factor that has not received a lot of attention is the exposure to heavy metals. While several mechanisms have been put forth describing how high concentrations of heavy metals play a role in carcinogenesis, it is unclear whether chronic, low-level exposure to certain heavy metals (i.e. cadmium and nickel), can directly result in the development and progression of cancer. Cadmium and nickel have been hypothesized to play a role in breast cancer development by acting as metalloestrogens— metals that bind to estrogen receptors and mimic the actions of estrogen. Since the lifetime exposure to estrogen is a well-established risk factor for breast cancer, anything that mimics its activity will likely contribute to the etiology of the disease. However, heavy metals, depending on their concentration, are capable of binding to a variety of proteins and may exert their toxicities by disrupting multiple cellular functions, complicating the analysis of whether heavy metal-induced carcinogenesis is mediated by the estrogen receptor. The purpose of this review is to discuss the various epidemiological, in vivo, and in vitro studies that show a link between the heavy metals, cadmium and nickel, and breast cancer development. We will particularly focus on the studies that test whether or not these two metals act as metalloestrogens in order to assess the strength of the data supporting this hypothesis.
Keywords: Breast cancer, metalloestrogen, estrogen receptor, cadmium, nickel
Introduction
Breast cancer is one of the most common malignancies in the United States. Approximately 1 in 8 women in the U.S. will develop invasive breast cancer (1), underscoring the importance of understanding the factors that may contribute to the development of this disease. Multiple studies suggest that both genetic and environmental factors contribute to breast cancer development (2–4). Some of the environmental factors that have been recognized as suspected risk factors for breast cancer include ionizing radiation, hormone disruptors such as organohalogens, pesticides and environmental pollutants such as heavy metals. Regarding the latter, increasing epidemiological evidence suggests a strong association between exposure to heavy metals and the initiation, promotion and progression of breast cancer (5–8).
Heavy metals— which include arsenic, lead, mercury, cadmium, and nickel— are present naturally in the environment in minute concentrations; however with the increased usage in certain industrial processes such as smelting and electroplating, heavy metals have emerged as an environmental contaminant of growing concern. These heavy metals tend to accumulate in the body— a phenomenon called bioaccumulation (9). Bioaccumulation of heavy metals in soft tissues interferes with normal physiological functions. Generally, these heavy metals exert their toxic effects by forming complexes with organic compounds. When heavy metals bind to nitrogen-, oxygen- or sulfur-containing groups on enzymes, for example, they disrupt proper protein folding and thus can inactivate enzymes that function in key metabolic processes (10). Increased exposure to heavy metals is associated with impaired mitochondrial function, oxidative stress, DNA damage, deregulated cell growth and cell death (11, 12).
Recent studies have suggested that certain heavy metals such as cadmium and nickel can function as endocrine disruptors by mimicking the action of estrogens. As a result, these metals are often referred to as metalloestrogens (11–13). Since estrogen itself plays an important role in the development and progression of the disease, the ability of metalloestrogens to bind to and activate the estrogen receptors suggests that these compounds may also contribute to the development of breast cancer (5, 13). It is hypothesized that metal-induced estrogen receptor activation is a crucial step in the carcinogenic process (14). Therefore, the goal of this review is to examine the association between cadmium and nickel exposure and the development and progression of breast cancer. Specifically, we wish to determine (1) the carcinogenic potential of chronic, low-level exposure to these heavy metals and (2) whether or not the carcinogenic potential of cadmium and nickel is due, at least in part, to their ability to bind to estrogen receptors and act as metalloestrogens.
The role of estrogen receptor α, β, and GPER in breast cancer progression
Estrogens, produced by the female ovaries, play major roles in regulating the developmental processes of both normal and neoplastic breast epithelium. Estrogens are primarily synthesized during a female’s reproductive years to promote the growth and differentiation of sex tissues and organs in the reproductive system. They also play a role in brain function, bone maintenance and the accumulation of adipose tissue (15). The effects of estrogen are mediated by two types of estrogen receptor (ER)-regulated pathways – (1) the nuclear estrogen receptors (nER), which when activated translocate to the nucleus to function as transcription factors, and (2) the membrane estrogen receptors (mER), which are located on the plasma membrane or within the membrane of the endoplasmic reticulum (16). The deregulation of estrogenic pathways can lead to elevated transcriptional activity that may contribute to the development of cancer. Removal of both ovaries has been shown to reduce breast cancer risk, underscoring the importance of estrogen in the development of breast cancers, especially in cancers that express the estrogen receptor (ER+). The factors that contribute to increased cell proliferation of ER+ breast cancer include elevated levels of endogenous estrogen, increased exposure to pharmaceutical and environmental estrogens (17), and deregulated expression of ERs in the cell, which can lead to abnormal expression of genes associated with cell growth. Therefore, overexposure to estrogen and the overexpression of ER can both contribute to the etiology of breast cancer (18).
More than 70% of primary breast cancers in women are nERα+ and show estrogen receptor-dependent growth (19–21). ERα and ERβ are nuclear receptors that display considerable homology in the DNA-binding and ligand-binding domains but vary greatly in the NH2-terminal transactivation AF-1(activation function-1) domains (Fig. 1) (22, 23). Both isoforms regulate transcription through classical and nonclassical pathways. Through the classical mode of ER signaling, AF-1 functions in a cell- and promoter-specific manner to enhance the overall transcriptional response of ER (24). ERα and/or ERβ then are activated by their ligand, estradiol, with similar affinities and bind directly to the the same consensus estrogen response element (ERE) (Fig 2) in promoter or enhancer regions of target genes, which is followed by recruitment of the coregulators (25). Target genes regulated by this classical pathway include breast cancer marker gene trefoil factor 1 (TFF1 or pS2), cathepsin D (CTD), cyclin D1, insulin-like growth factor-binding protein 1 (IGFBP1), lactoferrin (LTF) and prolactin (PRL). Additionally, ER can also function through a nonclassical pathway by cross-talking with other transcription factors (26), such as activator protein-1 (AP-1), Sp1 and NF-κB. This cross-talk allows estrogen receptor to regulate genes that do not contain an ERE, thereby increasing the number of genes that ER modulates (23, 27–33). Many of these transcription factors regulate expression of genes crucial for cell cycle progression, migration, proliferation and apoptosis; and therefore all can contribute to breast carcinogenesis. Examples of target genes studied extensively in breast cancer research include cyclin D1, c-myc and IGF-1.
Figure 1. ERα and ERβ are homologous in their functional domains.

The two ER isoforms display a high degree of homology in the DNA binding domain (DBD) and ligand binding domain (LBD), but are highly variable in the NH2-terminal transactivation AF-1 domain, also referred to as the A/B hypervariable domain. Percentages indicate percent identity between the two receptors. Figure is adapted from the review by Gustafsson (179, 180).
Figure 2. Cross-talking between ERα/ERβ and GPR30 signaling pathways.
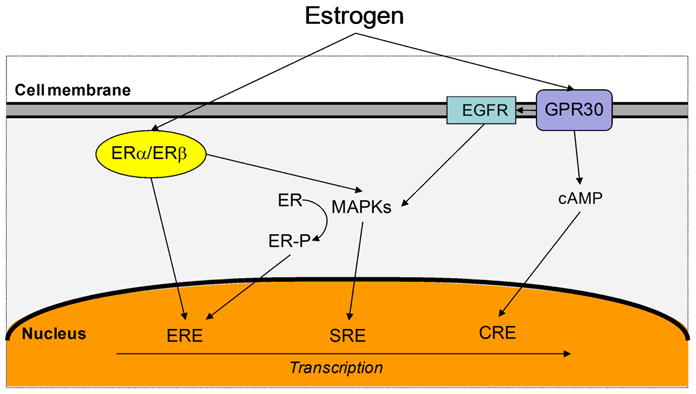
Estrogen can activate both long-term genomic (left) and rapid nongenomic (right) pathways leading to the transcription of downstream genes necessary for cell growth and development. ERα/ERβ activation can lead to both direct transcription activation in the nucleus or via rapid signaling of mitogen-activated protein kinases (MAPKs). In contrast, GPR30 cannot directly activate transcription processes but can rapidily activate nongenomic signaling including the activation of MAPKs resulting in the expression of transcription factors such as c-fos. cAMP is also produced via GPR30 activation. ERα/ERβ nd GPR30 signaling can induce both positive and negative effects on one another, depending upon the signaling components in the cell at a given time. Several other regulatory pathways are possible but not shown. Figure adapted from Prossnitz 2008 (181). ERE, estrogen response element; SRE, serum response element; CRE, cAMP response element.
ERα and ERβ seem to display similar ligand-dependent transcriptional functions and yet appear to play different roles in breast cancer progression. Although ERα has long been established as an important player in promoting the development and progression of breast cancer (34, 35), the link between ERβ and breast cancer is unclear due to conflicting data and the complexity of the ERβ isoforms. While some studies have suggested that the expression of ERβ is indicative of more advanced tumors (36–38), data from other studies have indicated that ERβ may play a protective role in breast cancer by increasing apoptosis in the presence of antiestrogens (39–41). Thus, much of what we know about ER-related breast cancer has focused largely on ERα. In a study performed by Holst et al., 20.6% of 2,000 clinical breast cancer samples showed an amplification of the ERα gene (ESR1) (42). Of those tumor samples, 99% showed an overexpression of the ERα protein, suggesting the importance of ERα in breast cancer development. Consequently, reducing estrogen levels or estrogen receptor activity through the administration of anti-estrogen compounds has been used to treat breast cancer patients.
In addition to the genomic actions mediated by nuclear estrogen receptors, estrogen also stimulates nongenomic signaling through the activation of one membrane form of estrogen receptor, GPR30 (GPER). With mERs, estrogens can act without directly binding onto DNA or altering gene expression. Filardo et al. was the first to show a function of GPR30 in estrogen signaling (43); and in 2005, GPR30 was finally identified as a bona fide estogen receptor (44, 45). GPR30 is a seven-transmembrane-spanning receptor reported to be located in the plasma membrane (45–47) and the endoplasmic reticulum (44). GPR30 functions by specifically binding to estrogen and causing rapid intracellular signaling and an activation of a downstream cascade, which includes epidermal growth factor release (EGFR) activation and increased intracellular cyclic AMP, leading to activation of transcriptional activities necessary for proliferation (Fig 2) (43, 47–49). Thus, EGFR plays a criticial role in regulating normal cell growth and physiology (50). Other cellular responses mediated by EGFR include activation of mitogen-activate protein kinsases MAPKs (such as extracellular signal-regulated kinase Erk-1 and Erk-2) which in turn phosphorylate numerous proteins that alter cell structure and regulate cell cycle checkpoints and gene transcription (51).
In 2009, Liu et al. showed that GPR30 was expressed in 37 of 74 cases of invasive ductal breast carcinomas. The binding of estrogen to GPR30 has also been shown to induce breast cancer cell proliferation and migration in vitro (52, 53). This increased cell proliferation has been attributed to the rapid activation of Erk-1/2, a downstream effector of growth factor signaling (54, 55). The dysregulation of the EGFR to MAPK pathway may have particular significance to breast carcinogenesis. Both Filardo et al. and Sivaraman et al. hypothesized that MAPKs may provide a mechanism whereby hyperactive growth factor signaling may activate estrogen-dependent breast tumor growth (56, 57). However, other researchers have contradicted this hypothesis by implicating GPR30 in apoptosis and cell cycle arrest (58–60). Although the role of GPR30 in breast cancer remains unclear, the combined effects of nER and mER activation can possibly lead to increases in cell cycle progression and cell proliferation that are associated with breast carciogenesis (Fig 2) (61).
Heavy metals— cadmium and nickel
Cadmium exists naturally in the earth’s crust. However, the most common forms of cadmium found in the environment are salts, comprised of sulfides, chlorides or oxides. These compounds are found in soil, water, rock sediments and the atmosphere at varying concentrations. Cadmium is found in ocean waters at 0.1μg/liter or less, in river water between <1 and 13.5ng/liter, and in the soil of non-polluted areas ranging from 0.2 to 0.4 mg/kg (62, 63). Atmospheric levels of cadmium range up to 5ng/m3 in rural areas, from 0.005 to 0.015 μg/m3 in urban areas and up to 0.06μg/m3in industrial areas (62, 63). Cadmium is released as a byproduct of various industrial activities, including the mining, galvanizing, and smelting of other metals like zinc, lead and copper. Cadmium is used to produce batteries, fertilizers, and paint pigments. Roughly 15,000 tons of cadmium are produced worldwide (64). Human exposure to cadmium is generally due to consumption of contaminated water or food or inhalation of cigarette smoke or fumes from smoldering metal. In 1989 and 1993, the World Health Organization (WHO) set the safe intake limit to 7μg cadmium/week/kg body weight.
Nickel is also a heavy metal and is widely distributed in the environment. It is found in agricultural soil at concentrations ranging from 3 to 1000 mg/kg and at concentrations of 1.5 to 8.5 mg/kg in forest floor samples collected from the northeastern United States (65). A small amount of nickel occurs naturally in water at levels of 0.228–0.693 μg/liter in ocean water and generally less than 2 μg/liter in fresh water (65). Nickel enters the human body through inhalation, ingestion and absorption. For the general population, the most common exposure to nickel is through the consumption of certain foods such as cacao products and nuts, which contain 10 and 3 mg nickel/kg, respectively (66). According to the WHO, the safe dietary intake of nickel is 4.2 μg Ni/kg/day (67).
Epidemiological studies linking cadmium and nickel exposure to breast cancer development
The International Agency for Research on Cancer (IARC) and the United States National Toxicology Program (NTP) designate cadmium as a human carcinogen (68–72). Cadmium has been associated with cancers of the pulmonary system, prostate, liver, hemapoetic system, urinary bladder and stomach and is also a multi-tissue animal carcinogen (23, 73–75). Particularly, cadmium exposure has also been associated with increased incidences of breast cancer development (13, 76). In a recent study carried out by Julin et al., dietary cadmium exposure was positively linked to breast cancer development in postmenopausal women (8). Antila et al. found high concentrations of cadmium (ranging from 3.2 to 86.9 μg/g) in breast tissue from breast cancer patients (6). Additionally, a case study published by McElroy and colleagues recorded the cadmium levels in urine samples of 246 women diagnosed with breast cancer and of 254 controls (7). The study demonstrated a positive correlation between cadmium urine levels and breast cancer risk. Another case study carried by Strumylaite and colleagues found significantly higher levels of cadmium in malignant breast tumor tissue (0.053 μg/g) than in normal breast tissue (0.02 μg/g) (77). Other studies have confirmed this finding (78, 79). Although these data demonstrate a correlation between cadmium levels and breast cancer, they fall short at implicating cadmium as the etiological agent.
Minute amounts of nickel are considered nontoxic. In general, the average nickel concentration in urine ranges from 1 to 3 μg/g creatinine (80). However, increased exposure to nickel compounds due to its increased usage in industrial processes has been shown to negatively impact the development of mammalian cells, leading to increased incidences of breast cancer development (13, 81). In fact, certain nickel compounds have been deemed carcinogenic by the IARC since the 1970s (82, 83). A recent study comparing 20 breast cancer patients with 8 healthy individuals showed higher levels of cadmium, chromium, lead, and nickel in the 20 breast cancer tissue biopsies than in the 8 healthy biopsies (78). These data supported an earlier study by Sherif and colleagues that reported a significant (albeit small) increase in nickel concentration in breast tumor tissue compared to normal breast tissue (84) indicating a positive correlation between increased nickel levels and breast cancer development.
Evidence of cadmium and nickel acting as metalloestrogens during breast cancer development
As mentioned previously, metalloestrogens are defined as a group of heavy metals that mimic the physiological actions of estrogen. The precise mechanism behind this mimicry is uncertain. Recent studies have suggested that metalloestrogens may function as endocrine disruptors, perturbing the normal hormonal cycle and altering the development of the mammary gland through both the classical and nonclassical ER binding pathways aforementioned (14, 85). However, most cadmium studies have largely focused on acute cadmium exposures, and little is known about the effects of chronic, low-level cadmium exposure on human breast cancer development and/or progression (Table 1). Because the half-life of cadmium ranges from 12 to 30 years (86–88) and the body does not possess an active mechanism for cadmium elimination, it remains in the body. Benbrahim-Tallaa and colleagues have shown that more than 40 weeks of exposure to 2.5μM cadmium transformed normal human breast epithelial cells MCF-10A into cells displaying a basal-like phenotype. The cells showed an increase in colony formation and invasive potential, and a loss of contact inhibition (89). Animal studies (Table 1) have shown that acute cadmium exposure increases uterine weight, induces changes in the uterine lining, and increases the density of epithelial cells in the mammary glands, all of which are early signs of breast tumorigenesis (85, 90–92). Additionally, in vitro experiments have shown that cadmium can promote ER+ breast cancer cell growth, and this is found to be dependent on ERα (14, 93–96). According to Stoica et al. cadmium binds to ERα— with a KD of 4.5 × 10−10 M—and blocks the binding of 17β-estradiol (14). While the presence of cadmium does not alter the estrogen receptors’ KD for estrogen (~2.9 × 10−10 M), it does decrease the total number of available estrogen binding sites. Thus, since cadmium does not alter the binding affinity of estradiol to the receptor, it is thought to interfere with estradiol binding in a noncompetitive manner. In a study carried out by our lab, concentrations as low as 1μM Cd induced signficant cell proliferation in three ERα-positive breast cancer cell lines (MCF-7, T-47D and ZR-75-1) after 2, 4 and 6 days of exposure (96). In this same study, we also deduced that the nonclassical ER target genes— CycD1, c-myc and CTD— were up-regulated by Cd. Subsequent silencing of ERα or blocking the receptor with anti-estrogens mitigated the stimulatory affect of cadmium on ER+ breast cancer cells, thus showing requirement of ERα in mediating the cellular effects of cadmium (96). Similarly, several other studies have demonstrated that cadmium-induced gene expression is also dependent on the estrogen recpetor (14, 93, 97). Recent evidence also suggests that cadmium can initiate mitogenic actions through the binding of the membrane-bound form of estrogen receptor, GPR30, via the Erk-1/-2 cascading pathway (Fig. 2) (98, 99). Yu et al. showed that cadmium induces a proliferative response at concentrations ranging from 50 to 500nM in ER-negative/GPR30-positive SKBR3 breast cancer cells, but no such response was observed in a GPR30-mutant cell line. The lowest exposure of 50nM is comparable to the blood Cd level (140nM) reported by Fell et al. in occupationally-exposed people (100). These studies provide another mechanism for how cadmium may promote mammary gland carcinogenesis via an estrogen receptor.
Table 1.
Summary of experiments examining the effects of acute and chronic cadmium exposure on gene expression in breast cancer models
| Type of Exposure | Experimental System (ER status of cells) | Cd Concentration & Exposure Time | Response | Mechanism: Is it mediated through ER? | Gene Expression | References |
|---|---|---|---|---|---|---|
|
| ||||||
|
Acute Cells |
MCF-7 cells (ERα+/ERβ+/GPR30+) | 10−12 M CdCl2 for 1 hr | Blocked estradiol binding in a noncompetitive manner | Yes | KD of cadmium is 2.96×10−10M | Stoica (14) |
| 10−6M CdCl2 for 6 days | Increased ER-regulated gene expression and cell growth | Yes | ↑ PR and pS2 | Martin (13) | ||
| 10−6M CdCl2 for 24 and 48 hr | Increased ER-regulated gene expression and cell growth of MCF-7 and other ERa+ cell lines: T47D and ZR-75-1 | Yes | ↑ cyclin D, CTD, c-myc, ↓ p21 | Siewit (96) | ||
| 10−6 M for 24 hr | Increased ER-regulated gene expression and cell growth | Yes | ↑ PR and pS2 | Garcia-Morales (93) | ||
| 20 or 50μM CdCl2 for 52.5 hr | Cadmium activated ERα mediated transcription; Hsp22 exhibited interaction with Hsp27 | Yes | ↑ Hsp22 | Sun (97) | ||
| 10μM CdCl2 for 24 hr | Decreased levels of ERα and increased cell proliferation | Yes | ↑ ERK1/2, Akt, PDGFRα, c-jun, c-fos | Brama (55) | ||
| 5μM for 1 to 48 hr | Induced transcription of Cholinephosphotransferase (CPT) | No | ↑ CPT | Sinha Roy (194) | ||
|
| ||||||
| T47D cells (ERα+/ERβ+) | 10−10 to 10−6 M | Activated mER signaling leading to ERK1/2 activation; activated ERE-dependent transcription | Yes | ↑ ERK1/2 MAPK response | Zang (193) | |
|
| ||||||
| SKBR-3 cells (ERα-/ERβ-/GPR30+) | 5.0×10−7 M CdCl2 for 24 hr | Rapidly increased mER gene expression; Increased cell growth | Yes GPR-30 | ↑ cAMP, Raf-1, MEK-1, Erk-1/2 | Yu (98) | |
|
| ||||||
| COS-1 cells transfected with ERα | 10−12-10−6 M CdCl2 for 24 hr | Cadmium activates ERα mediated transcription (↑ CAT1) | Yes | ↑ CAT | Stoica (14) | |
|
| ||||||
| MDA-MB231 cells (ERα-/GPR30−) | 5.0×10−6 M CdCl2 for 96hr | Decreased transcriptional activity and intracellular signaling of AEG-12 and NF-kB | No | ↓ AEG-1, ↓ c-fos, c-jun, p63 | Luparello (182) | |
| 5.0×10−6 M CdCl2 | Stimulates p38 transcriptional activity | No | ↑ p38 | Casano (183) | ||
| 5 or 50 μM CdCl2 for 24 or 96 hr | Respiration activity increased after 96 hr exposure; increased reactive oxygen species; expression of hsp60, hsp70, Cytochrome oxidase II and IV were dependent on duration of Cd exposure | No | ↑ hsc/hsp70 ↓ Cytochrome oxidase II & IV |
Cannino (184) | ||
| 5 μM for 96 hr | Cells exhibited protective role after exposure | No | ↑ hsp70 and metallothioneins | Sirchia (185) | ||
|
| ||||||
| Non-tumoral HB2 cells | 50 μM for 24 hr | Induced cell death via non-apoptotic pathways | Not identified | Sirchia (186) | ||
|
| ||||||
|
Acute Animals |
Ovarectomized Rat | Single injection 5μg CdCl2 kg−1 b.w. | Increased uterine weight, thickness of uterine epithelium, and density of mammary gland | Not identified | ↑ PgR & C2 in mammary gland | Höfer (90) |
| Ovarectomized Rat | Single injection 0.05–2000 μg CdCl2 kg−1 b.w. | Increased uterine weight and thickness of uterine epithelium; increased pre-malignant characteristics | Not Identified | Johnson (85) | ||
| Transgenic ERE-Luc reporter mouse | 5 and 50μg CdCl2 kg−1 b.w. for 3 d | Increased uterine epithelium height | Not identified | ↑ Erk1/2 in liver | Ali (91) | |
|
| ||||||
|
Chronic1 Cells |
MCF-10A (ERα-) | 2.5×10−6 M CdCl2 for 40 wk | Increased invasion and colony formation; decreased contact inhibition; displayed basal-like phenotype | Not identified | ↑c-myc and k-ras | Benbrahim-Tallaa (89) |
|
| ||||||
|
Chronic1 Animal |
Mouse | 2–3 mg CdCl2 kg−1 b.w. for 2 or 7 wk | Increased labuloalveolar development of mammary gland | Not identified | Alonso-Gonzales (92) | |
Gray: Studies done with chronic cadmium exposure; White: Studies done with acute exposure
CAT: chloramphenicol acetyltransferase
AEG-1: astrocyte-elevated gene 1
Although animal studies have revealed that over-exposure to nickel compounds can lead to tumor formation in multiple tissue sites such studies have failed to link nickel with breast cancer development in experimental animals (Table 2) (101–103). Inhalation of metallic nickel (at a concentration of 15 mg/m3 for six hours/day, four to five days per week for 21 months) revealed neoplastic growth within the lungs of Wistar and Behesda black rats (104). Additionally, Ivankovic et al. observed fibrosacromas, mesotheliomas, and/or rhabdomyosarcomas in 10% of Syrian golden hamsters exposed to a single high dose of nickel powder (105). No tumors were found in the negative control group. Such studies provide evidence that nickel exposure increases the incidence of tumors in several species and at multiple tissue sites, but not specifically breast tissue.
Table 2.
Summary of experiments examining the effects of acute and chronic nickel exposure on gene expression in various cancer models
| Type of Exposure | Experimental System | Ni Concentration and Exposure Time | Response | Mechanism: Is it mediated through ER? | Gene Expression | References |
|---|---|---|---|---|---|---|
|
| ||||||
|
Acute Cell |
Breast cancer cells (MCF-7)1 | 1 μM NiCl2 for 6 days | Increased breast cancer cell proliferation | Yes | ↑ PR and pS2 | Martin (13) |
| 1mM NiCl2 for 6 and 24 hr | Increased HIF-α and p53 protein levels, accompanied by MDM-2 protein induction | No | ↑ HIF-α and p53 | Salnikow (106) | ||
|
| ||||||
| Lung cancer cells (A549) | 0.25 μM to 1 mM NiCl2 | Inhibited cell growth through IKKα-dependent manner; increased cells in G1/G0 phase | No | ↓ cyclin D1 ↑ p21 |
Ouyang (112) | |
| 0.5 and 0.75 mM NiCl2 at 24 hr; 1mM NiCl2 for 12–72 hr | ↑ H3K9 mono-and dimethylation, critical for long-term gene silencing | Epigenetic Modification; No | ↓ H3K9 methyltransferase | Chen (127) | ||
| 1mM NiCl2 for 24 hr | ↑ H3K4 trimethylation in promoter and coding regions for CA9 and NDRG1 | Epigenetic Modification; No | ↑ CA9 and NDRG1 | Tchou-Wong (108) | ||
| 0.25 to1.0 mM NiCl2 or Ni3S2 for 24 hr | Induced histone modification; alters epigenetic homeostasis in cells | Epigenetic Modification; No | Ke (124, 125) | |||
| 1mM NiCl2 for 6 and 24 hr | Increased HIF-α and p53 protein levels | No | ↑ HIF-α & p53 | Salnikow (106) | ||
|
| ||||||
| Lung epithelial cells (Beas-2B) | 1 to 4 μg/cm2 Ni3S2 for 48 hr | Induced M phase arrest; inhibited cell growth | No | ↑ cyclin B1, cyclin D, cyclin E | Ding (111) | |
| 0.25 to 2 μg/cm2 NiCl2 for 48 hr | Increased cell apoptosis, induced growth arrest through Akt/ASK1/p38 signaling pathway | No | ↓ Bcl-2, Bcl-xL ↑ p38, MAPK |
Pan (187) | ||
| 0.5 to 1.0 mM NiCl2 | Increased COX-2 expression via NFAT and NF-kB | No | ↑ COX-2 | Cai (188) | ||
| Mouse fibroblast cells (L-929) | 200 μM Ni2+ for 24, 48 and 72 hr | Identified 20 up-regulated genes and 19 down-regulated genes | No | Lü (113) | ||
| Mouse fibroblasts (HIF-1α+) | 1 mM NiCl2 for 20 hr | Activated hypoxia-inducible transcription factor-1 (HIF-1) and induced tumor marker Cap43 gene. | No | ↑ Nip3 ↑ p21, HSP70, p53, GADD45 |
Salnikow (107) | |
|
| ||||||
| NER proficient human fibroblast (GM00637) | 50 to 300 μM NiCl2 for 24 hr | Enhanced BPDE-induced mutation frequency by inhibiting nucleotide excision repair (NER) pathway | No | Hu (189) | ||
|
| ||||||
| Human osteoblast cells (HOS TE-85) | 2–10 mg/mL NiS, for 24 hr | Decreased retinoblastoma (pRB) phosphorylation; acquired anchorage-independent growth | No | ↓pRb phosphorylation | Lin (190) | |
|
| ||||||
| Human hepatoma cells (HepG2) | 150 μM NiCl2 hexahydrate for 48 hr | Up-regulation of genes associated with cell cycle progression-similar to other carcinogenic chemicals | No | Induced gene expression | Kawata (109) | |
|
| ||||||
| Human keratinocytes | 0.5mM to 2.0 mM NiCl2 for up to 48 hr | Decreased ΔNp63 activity; activation of NF-κB | No | ↓ΔNp63, IRF3, IRF7, and↑p21 | Zhang (191) | |
|
| ||||||
| Human lymphocytes | 15uM NiCH and NiO for 2hr | Exhibited higher DNA single-strand breaks | No | M’Bemba-Meka (192) | ||
|
| ||||||
|
Acute Animal |
Rats | 0.9mg Ni/100g Ni(CO)4 i.v. at 2 and 4 wk intervals | Resulted in undifferentiated sarcomas, fibrosarcomas, carcinomas, hemangioenthelioma, leukemia, and lymphoma | Not identified | Lau (101) | |
|
| ||||||
|
Chronic Cells |
Transformed mouse fibroblast cells | Cells resistant up to 200 μM Ni, but not treated with Ni. | ↑GSH, GSTA4, GSTT2 suggest enforcement of antioxidant defenses | No | ↑ β-catenin, c-myc, cyclin D1 | Kowara (110) |
|
| ||||||
|
Chronic Animal |
Rat and Mice | 0.15 to 1 mg/m3 Ni3S2; 1.25 to 2.5 mg/m3 NiO for2 yr | Increased alveolar/bronchiolar and adrenal medulla neoplasms in male and female rats. | No | Dunnick (102) | |
Gray: Studies done in breast cancer models; White: Studies done in other cancer models
GSH: glutathione synthetase; GSTA4: glutathione-S-transferase A4; GSTT: glutathione-S-transferase theta
In spite of the lack of evidence from animal studies, other studies have suggested that nickel (like cadmium) may function as a metalloestrogen and alter estrogen receptor activity (Table 2) (13, 106–108). In vitro studies have shown the ability of nickel to mimic the effects of estradiol on cell proliferation and block estradiol binding to ERα (13). MCF-7 cells treated with either 10−9 M estradiol or 10−6 M Ni shows 2–5 fold increase in cell growth. Additionally, as with cadmium, the presence of nickel does not affect the receptor’s KD yet does decrease the number of estradiol binding sites, suggesting that nickel also interferes with estradiol binding in a noncompetitive manner (13). As with cadmium, the binding of nickel to the estrogen receptor appears to induce the expression of genes associated with cell growth (13). Although few studies have analyzed the effects of nickel on gene expression in mammary cells (Table 2), the effects of nickel has been widely studied in other cell types (109–113). Data from microarray analysis of nickel-transformed mouse fibroblasts revealed an overexpression of cyclin D1 (110), a gene that has been shown to play an important role in breast cancer cell growth (114–116). Furthermore, studies with human pulmonary cells suggest that nickel has the ability to increase cyclin D1, cyclin E and cyclin B1 expression (111). Despite the increased expression of cyclin D1 and cyclin E, which has been shown to promote the G1/S transition, the induction of cyclin B1 resulted in the induction of M-phase arrest and inhibited cell growth (111). Similarly, Ouyang et al. also demonstrated that nickel inhibits cell growth, this time by decreasing the expression of cyclin D1. The discrepancy in these studies may reflect both the different model systems (breast and fibroblast vs. lung) and concentrations of nickel used in these studies.
Molecular interactions between estrogen receptors and the heavy metals, cadmium and nickel
Several researchers have attempted to map out the cadmium interaction domain within the estrogen receptor in order to better understand how cadmium functions as an estrogen receptor modulator. However, a consensus of where cadmium binds has not been determined. The two possibilities are that (1) cadmium binds to the ligand binding domain (LBD), and (2) cadmium may replace the zinc in the DNA binding domain. In one study, specific amino acids of the LBD, including C381, C447, E523, H524 and D538, were identified as possible cadmium interaction sites (14). However, further analysis using chemical modification and mass spectroscopy identified several other cysteine (C) residues as having high affinities for the divalent form of cadmium, and these amino acids were not the same as those previously identified (14, 117). Additionally, Glu (E), His (H) and Asp (D) residues were not protected from chemical modification when the ER was combined with cadmium, thus suggesting these residues actually have low affinity for Cd2+, a finding that has been confirmed in studies of other metallo-proteins (117–119). Much less is known about the binding between nickel and the estrogen receptors. An analysis by Martin and colleagues proposed that nickel activates the estrogen receptor through the LBD of ERα (13). The LBD of ERα includes C381, C417, C447, and C530, and nickel is believed to interact with C381 and C447 to activate ERα (13).
Another prospective cadmium-binding site is the DNA binding domain, which is logical since the coordination chemistry of Cd2+ is similar to that Zn2+. Cd2+ has the ability to replace Zn2+ because they have similar chemical properties (120). In vitro studies have shown that the replacement of Zn with Cd slightly increases the DNA binding affinity of ERα (95, 121–123). This may translate to changes in transcriptional activity. However, further studies are necessary to map the actual cadmium binding site and this will offer further insights into how cadmium functions as an estrogen receptor modulator. Similar in vitro studies have indicated that nickel can also replace Zn2+ in the DNA binding domains of the estrogen receptors (95, 123), but this replacement results in a decreased DNA binding affinity, likely due to the difference in the coordination chemistry of zinc (tetrahedral) and nickel (octahedral), which may in turn cause a conformational change that may decrease DNA binding. This suggests that the mechanisms of how nickel and cadmium function as metalloestrogens are likely different.
Other mechanisms of cadmium- or nickel-induced carcinogenesis
Chromatin modifications
Attempting to elucidate whether or not certain heavy metals contribute to breast cancer progression via estrogen receptor binding is complicated by the fact that heavy metals are capable of binding to a plethora of macromolecules and cellular structures. Above a certain concentration threshold, many heavy metals, such as copper, mercury, and silver, are very effective antimicrobial agents, as they bind to and inactivate various proteins and enzymes. Similarly, heavy metal-induced carcinogenesis can result from the binding of heavy metals to proteins other than the estrogen receptor. Nickel, for example, plays significant roles in chromatin remodeling, which is a proposed mechanism for nickel-induced carcinogenicity (124–130). Specifically, exposure of cells to nickel has been reported to alter the acetylation, methylation and ubiquitination of histone proteins (124, 125, 131), which in turn affects transcription. In general, histone acetylation is associated with gene activation and histone deacetylation results in gene repression (132–134). Methylation at histones H3K4, H3K36, and H3K79 has been linked to transcription activation, whereas methylation at H3K9, H3K27, and H4K20 has been associated with gene repression (94, 134–136). Studies describing nickel-induced epigenetic changes have reported a global loss of both histone acetylation and H3K4 methylation and a global increase in H3K9 methylation, all of which are associated with gene silencing. These findings, however, contradict the observation that exposure to low concentrations of nickel results in not only gene repression but also gene activation (124–127, 134). Furthermore, pre-treatment of cells with trichostatin A, a histone deacetylase inhibitor, significantly reduces nickel-induced cell transformation, suggesting that histone deacetylation is an important step in nickel-mediated carcinogenesis.
In addition to nickel-induced histone modifications, nickel has been shown to preferentially bind to heterochromatin by replacing the Mg2+ ions that are naturally found in high concentrations and thus play an important role in the condensation of heterochromatin (126). It is surmised that nickel may lead to greater chromatin condensation, perhaps even promoting heterochromatin formation in critical parts of the DNA, such as those regions containing tumor suppressor genes (137). The nickel accumulated in the heterochromatin regions may also induce oxidative damage in the DNA via a Fenton-like reaction, further disrupting gene expression and perhaps contributing to carcinogenesis (138).
Nickel has also been shown to increase DNA methylation, which is also associated with gene silencing (126, 139). More specifically, nickel exposure has been shown to alter the methylation patterns of the p53 and p16 promoters (139). Silencing such key tumor suppressor genes likely serves as an important mechanism of nickel-induced carcinogenesis (139, 140). While many of these studies were carried out in other cancer types (i.e. lung), similar mechanisms of nickel-induced carcinogenesis are expected to be involved in breast cancer cells (140, 141). Furthermore, some of the mechanisms of nickel-induced chromatin modification described above (such as histone deacetylation) may actually involve the estrogen receptor and thus lend further support to the hypothesis that nickel does indeed act as a metalloestrogen in breast cancer development. Multiple histone acetylases (HATs) and deacetylases (HDACs) are known to interact with the estrogen receptor (CBP, p300, p/CAF, p160 SRC family of coactivators, and HDAC 1and 6) (142–144), so it is possible that the interaction between nickel and ERα mediates the recruitment of chromatin-modifying proteins to the promoters of nickel-induced/repressed genes.
Unlike nickel, few studies have linked cadmium to epigenetic modifications, and such modifications are not likely a major mechanism of cadmium-induced carcinogenesis. Only a few studies have suggested that exposure to cadmium alters the global DNA methylation patterns (89, 145, 146). More specifically, acute cadmium exposure has been shown to induce hypomethylation, while chronic cadmium exposure results in hypermethylation (129). Changes in the methylation patterns of cells exposed to cadmium were mediated by altering the activity of DNA methyltransferase (145). In support of these observations, Benbrahim-Tallaa et al. showed that a 10-week exposure to 10μM cadmium was able to increase global DNA methylation and induce malignant transformation in prostate epithelial cells. Contrary to these observations, Huang et al. reported that chronic exposure of cadmium leads to global hypomethylation and cell proliferation in K562 leukemia cells (146). While these differences may be attributed to different model systems, it does suggest that further studies on cadmium’s affect on DNA methylation are necessary, especially in breast cancer cells. Furthermore, there appear to be no studies linking cadmium exposure to histone modifications, underscoring the need for further studies in this area of metal-induced carcinogenesis.
Aneuploidy
Aneuploidy— the phenomenon in which a cell possesses an abnormal number of chromosomes— is found in most cancerous lesions and is believed to play a significant role in cancer progression (147–150). In fact, some researchers have found evidence supporting the contention that aneuploidy is involved in the early stages— if not the actual initiation— of cancer cell development (151–153). Several studies have demonstrated the aneugenic potential of heavy metals (154–158). In one particular study carried out by Seoane and Dulout, cadmium and nickel salts such as CdCl2 (1–4μM), CdSO4 (0.033–0.134μM), NiCl2 (13–54μM), and NiSO4 (200–800μM) all induced aneuploidy in human fibroblast cells (159). The precise molecular mechanism behind this aneugenic effect, however, has not been determined.
Interestingly, estrogen itself has been shown to induce aneuploidy. In a study published in 2002 by Li et al., estrogen-induced mammary gland tumors (MGTs) in female ACT rats were found to have a significantly higher degree of aneuploidy than those MGTs induced by chemical carcinogens (153). Additionally, this same study revealed that these estrogen-induced MGTs greatly resembled invasive human ductal carcinoma in situ (DCIS) breast cancer in terms of degree of aneuploidy and increased amplification of the c-myc gene. An earlier in vitro study by Epe and colleagues demonstrated that peroxidative estrogen metabolites could directly interact with the tubulin protein and thus interfere with the assembly of microtubules, potentially affecting proper mitotic spindle formation (160). More relevant to this review, however, is a recent study by Hontz et al. which showed that estrogen (more specifically, estradiol-17β) increased expression of the mitotic kinases Aurora A and B via an ERα-dependent pathway (161). High levels of Aurora A and B lead to aneuploidy and consequently are believed to contribute to cancer progression (162–164). Specifically, Aurora A overexpression and aneuploidy have been found in primary invasive ductal breast cancer (161). Although more studies need to be done to determine exactly how cadmium and nickel lead to aneuploidy, the discovery that estrogen can induce aneuploidy through the estrogen receptor suggests that the aneugenic effect of cadmium and nickel could at least partially be due to their ability to bind to the estrogen receptor and act as metalloestrogens.
Conclusion
Breast cancer— like all cancers— is a complex disease, from its initiation to its progression. No two cases are alike, and the factors that contribute to its development can vary significantly between patients. Certain risk factors, such as increased exposure to estrogen, are well-established as integral to the development of most types of breast cancer. However, the rising incidence of breast cancer cases (currently 25% of all female cancers in the U.S. are breast cancers) has driven scientists to search for other factors that may explain this alarming increase.
Environmental contaminants such as heavy metals have emerged as a possible risk factor due to their increased usage in certain industrial processes, as mentioned previously. There is actually little debate as to whether or not heavy metals can cause cancer. Certain cadmium, nickel, and even arsenic compounds have been deemed carcinogenic by the IARC since the late 1970’s and 1980’s (68, 82, 165, 166). Of these heavy metals, however, we found that it was cadmium which has the strongest correlation with breast cancer development. Most of the epidemiological, in vivo, and in vitro studies we analyzed solidly supported the link between increased cadmium exposure and breast cancer development. For nickel, the evidence, though supportive, was much smaller. Epidemiological studies linking nickel to breast cancer were positive but few in number, and there were virtually no animal studies testing the effect of increased nickel exposure to breast cancer development. There were some very compelling in vitro studies involving nickel, particularly those carried out by Martin and colleagues, which showed that nickel can bind to the estrogen receptor in breast cancer cells and induce the expression of genes associated with cell growth (13). However, such studies are also small in number (Table 2). Even sketchier is the evidence linking arsenic to breast cancer. Although an Australian case study conducted by Hinwood et al. reported that 40% of the cancers caused by arsenic-contaminated drinking water were breast cancers (167, 168), in vivo and in vitro studies indicate that arsenic disrupts ER function and actually suppresses estrogen signaling pathway (169, 170)— findings that, in our view, effectively argue against arsenic as a potential metalloestrogen.
The main goal of this review was to determine if there is enough evidence to support the hypothesis that the heavy metals, cadmium and nickel at chronic, low-level concentrations can induce breast cancer by mimicking estrogen in the estrogen receptor signaling pathway, thus acting as metalloestrogens. We found that most studies analyzed the effects of acute heavy metal exposure on breast cancer development and progression (Tables 1 and 2). There is a general consensus that exposure to cadmium or nickel at levels greatly exceeding the concentration limits dictated by the World Health Organization (WHO) is exceedingly dangerous due to the promiscuous protein-binding patterns of heavy metals when they are at high enough concentrations. Our contention, however, is that chronic exposure to cadmium and nickel at concentrations well below WHO limits is still dangerous due to bioaccumulation and the peculiarly high affinity for specific proteins like the estrogen receptors.
Evidence obtained from in vivo and in vitro studies strongly suggests that cadmium can behave as a metalloestrogen. Cadmium has been shown to bind to ERα (with a KD nearly equivalent to that of estradiol), activate it, and induce expression of certain ER target genes. In addition, cadmium induces other estrogen-like effects which include: increased uterine weight; changes in uterine lining; increased epithelial cell density in mammary glands; increased cell proliferation; and increased aneuploidy. As strong as these data are, however, we feel that more experiments testing chronic, low-level cadmium exposure are needed to help confirm that cadmium-induced breast carcinogenesis is due, at least in part, to cadmium’s estrogenic potential. Currently, there are only a few studies on the effects of prolonged exposure to low levels of cadmium on breast cancer development and progression (Table 1). An additional weakness in the literature is the fact that the cadmium-binding site on ERα has not truly been determined yet. Additional structural studies— perhaps involving NMR or X-ray crystallography combined with protein modeling and computional chemistry— could potentially aid in the future development of therapeutics that might counteract cadmium’s effects.
Though we did not find a single study arguing against nickel serving as a metalloestrogen in breast cancer development, the number of studies providing support for this hypothesis was scant (Table 2). As with cadmium, nickel has been shown to bind to ERα, promote cell proliferation, and induce aneuploidy. However, many more studies at the animal, cellular, and molecular levels need to be carried out to effectively determine if and how low-dose, chronic nickel exposure can lead to breast cancer. Since much less is known about the nickel-binding site than the cadmium-binding site on ERα, more structural studies are required as well to confirm nickel’s role as a metalloestrogen.
Finally, it is also worth noting that both cadmium and nickel were found in various human samples including urine, hair, blood and breast tumor tissues (Table 3) (79, 84, 171–173). While healthy individuals had detectable levels of heavy metals, significantly higher levels of cadmium and nickel were found in patients with breast cancer (Table 3). The studies presented in Table 3 further argue the need for additional studies on chronic exposures to these metals at low concentrations. Also lacking in the literature are any studies evaluating whether heavy metal exposure during a critical developmental window— e.g. prenatal, puberty, or postmenopausal— would lead to an increased risk of breast cancer. Many reports do indicate a positive correlation between certain childhood cancers and prenatal exposure to endocrine disruptors such as diethylstilbestrol (DES) and 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) (174, 175). Animal studies have shown that rats exposed to tamoxifen during late gestation produce offspring with a greater sensitivity to DMBA-induced breast cancer (176). Epidemiological analyses have reported a positive correlation between prenatal exposure to elevated levels of natural estrogens and breast cancer (177); and strong evidence has implicated hormone replacement therapy (HRT) in increasing the breast cancer risk in peri- and postmenopausal women (178). Although a recently published study by Julin et al. indicated a correlation between dietary cadmium exposure and breast cancer risk in postmenopausal women, no other age group was analyzed (8). Thus, until more comprehensive analyses are carried out, it is not clear that exposure to cadmium or nickel during certain critical developmental periods increases breast cancer risk. However, despite the fact that more studies need to be done, there is sufficient evidence to warrant great concern over the increasing emission of heavy metals like cadmium and nickel into the environment. Acute exposures aside, the data suggest that even minimal levels of cadmium and nickel are potentially hazardous and could negatively impact the health of thousands of people.
Table 3.
Summary of experiments comparing the bioaccumulation of cadmium and nickel in biological samples from breast cancer patients and healthy individuals
| Biological Sample | Metal | Sample Size | Experimental Findings | Reference |
|---|---|---|---|---|
| Breast Tissue | Cd | 19 benign tissue samples and 21 malignant tissue samples | Cadmium (Cd) levels were higher in malignant than benign tissues (p = 0.009). | Strumylaite (77) |
| Cd | 32 healthy and 43 breast cancer subjects | A positive correlation was observed between cadmium concentration and smoking rate (p<0.05). | Antila (6) | |
| Cd, Ni, Al | 16 non-cancerous tissue samples, and 67 breast cancer tissue samples | Cd levels were higher in cancerous versus non-cancerous tissue (p<0.05). No significant difference was observed in Ni levels (p=0.057). |
Romanowicz-Makowska (171) | |
| Fe, Ni, Cr, Cu, Pb, Zn, Hg, Ag, Sn, Au, Pd | 20 breast cancer biopses and | Ni, Cr (p<0.00005), Zn (p<0.00001), Fe (p<0.0001), and Cd, Hg (p<0.005) levels were higher in breast cancer biopsies. | Ionescu (78) | |
| Ni, Ca, Cu, Zn, Fe, Cr, Mn, Se, Br, Rb, Sr, Hg, As, Pb, V, Se, Mo | Normal and neoplastic breast tissue from 25 patients | Levels of Ca, V, Cu, Zn, Se, Rb (P<0.001) and Ni (p<0.05) were higher in breast tumor tissue | Rizk (84) | |
| Breast tissue, urine and blood | Cd | 51 benign tumor patients and 57 breast cancer patients | Cd levels were significantly higher in malignant vs. benign tumor tissues p<0.01. Additionally, higher levels of Cd were found in the urine samples of patients with breast cancer p<0.001. A positive correlation exists between blood cadmium levels and tumorigenicity. | Strumylaite (173) |
| Urine | Cd | 254 healthy subjects and 246 breast cancer subjects | Breast cancer risk increases with cadmium concentration (p= 0.01) | McElroy (7) |
| Hair Sample | 36 elements including Cd and Ni | 52 stage III breast cancer subjects and 52 healthy subjects | Higher levels of Ag, As, Au, Bo, Ba, Be, Ca, Cd, Ce, Co, Cs, Ga, Mn, Ni, Pb, Sb, Sc, Se and Zn were found in breast cancer patients in comparison to healthy subjects (p<0.05). | Benderli Cihan (172) |
Acknowledgments
We thank Dr. Sibdas Ghosh and the Department of Natural Sciences and Mathematics at Dominican Univeristy of California for their support. This work was partially supported by the NIH-R15CA121983 to MCL.
Footnotes
The authors have no conflict of interest to disclose.
References
- 1.ACS. Cancer Facts & Figures 2010. American Cancer Society, Inc; 2010. [Google Scholar]
- 2.Parkin DM, Bray F, Ferlay J, Pisani P. Global cancer statistics, 2002. CA Cancer J Clin. 2005;55(2):74–108. doi: 10.3322/canjclin.55.2.74. [DOI] [PubMed] [Google Scholar]
- 3.Lichtenstein P, Holm NV, Verkasalo PK, Iliadou A, Kaprio J, Koskenvuo M, Pukkala E, Skytthe A, Hemminki K. Environmental and heritable factors in the causation of cancer--analyses of cohorts of twins from Sweden, Denmark, and Finland. N Engl J Med. 2000;343(2):78–85. doi: 10.1056/NEJM200007133430201. [DOI] [PubMed] [Google Scholar]
- 4.Nathanson KL, Wooster R, Weber BL. Breast cancer genetics: what we know and what we need. Nat Med. 2001;7(5):552–556. doi: 10.1038/87876. [DOI] [PubMed] [Google Scholar]
- 5.Darbre PD. Metalloestrogens: an emerging class of inorganic xenoestrogens with potential to add to the oestrogenic burden of the human breast. J Appl Toxicol. 2006;26(3):191–197. doi: 10.1002/jat.1135. [DOI] [PubMed] [Google Scholar]
- 6.Antila E, Mussalo-Rauhamaa H, Kantola M, Atroshi F, Westermarck T. Association of cadmium with human breast cancer. Sci Total Environ. 1996;186(3):251–256. doi: 10.1016/0048-9697(96)05119-4. [DOI] [PubMed] [Google Scholar]
- 7.McElroy JA, Shafer MM, Trentham-Dietz A, Hampton JM, Newcomb PA. Cadmium exposure and breast cancer risk. J Natl Cancer Inst. 2006;98(12):869–873. doi: 10.1093/jnci/djj233. [DOI] [PubMed] [Google Scholar]
- 8.Julin B, Wolk A, Bergkvist L, Bottai M, Akesson A. Dietary cadmium exposure and risk of postmenopausal breast cancer: a population-based prospective cohort study. Cancer research. 2012;72(6):1459–1466. doi: 10.1158/0008-5472.CAN-11-0735. [DOI] [PubMed] [Google Scholar]
- 9.Islam E, Yang XE, He ZL, Mahmood Q. Assessing potential dietary toxicity of heavy metals in selected vegetables and food crops. J Zhejiang Univ Sci B. 2007;8(1):1–13. doi: 10.1631/jzus.2007.B0001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nikolic J, Sokolovic D. Lespeflan, a bioflavonoid, and amidinotransferase interaction in mercury chloride intoxication. Ren Fail. 2004;26(6):607–611. doi: 10.1081/jdi-200037149. [DOI] [PubMed] [Google Scholar]
- 11.Rollerova E, Urbancikova M. Intracellular estrogen receptors, their characterization and function (Review) Endocr Regul. 2000;34(4):203–218. [PubMed] [Google Scholar]
- 12.Misra UK, Gawdi G, Akabani G, Pizzo SV. Cadmium-induced DNA synthesis and cell proliferation in macrophages: the role of intracellular calcium and signal transduction mechanisms. Cell Signal. 2002;14(4):327–340. doi: 10.1016/s0898-6568(01)00268-6. [DOI] [PubMed] [Google Scholar]
- 13.Martin MB, Reiter R, Pham T, Avellanet YR, Camara J, Lahm M, Pentecost E, Pratap K, Gilmore BA, Divekar S, et al. Estrogen-like activity of metals in MCF-7 breast cancer cells. Endocrinology. 2003;144(6):2425–2436. doi: 10.1210/en.2002-221054. [DOI] [PubMed] [Google Scholar]
- 14.Stoica A, Katzenellenbogen BS, Martin MB. Activation of estrogen receptor-alpha by the heavy metal cadmium. Mol Endocrinol. 2000;14(4):545–553. doi: 10.1210/mend.14.4.0441. [DOI] [PubMed] [Google Scholar]
- 15.Byrne C, Divekar SD, Storchan GB, Parodi DA, Martin MB. Cadmium--a metallohormone? Toxicol Appl Pharmacol. 2009;238(3):266–271. doi: 10.1016/j.taap.2009.03.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ikeda K, Inoue S. Estrogen receptors and their downstream targets in cancer. Arch Histol Cytol. 2004;67(5):435–442. doi: 10.1679/aohc.67.435. [DOI] [PubMed] [Google Scholar]
- 17.Henderson BE, Feigelson HS. Hormonal carcinogenesis. Carcinogenesis. 2000;21(3):427–433. doi: 10.1093/carcin/21.3.427. [DOI] [PubMed] [Google Scholar]
- 18.Henderson BE, Ross R, Bernstein L. Estrogens as a cause of human cancer: the Richard and Hinda Rosenthal Foundation award lecture. Cancer research. 1988;48(2):246–253. [PubMed] [Google Scholar]
- 19.Clark GM, McGuire WL. Steroid receptors and other prognostic factors in primary breast cancer. Semin Oncol. 1988;15(2 Suppl 1):20–25. [PubMed] [Google Scholar]
- 20.Allred DC, Brown P, Medina D. The origins of estrogen receptor alpha-positive and estrogen receptor alpha-negative human breast cancer. Breast Cancer Res. 2004;6(6):240–245. doi: 10.1186/bcr938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Masood S. Estrogen and progesterone receptors in cytology: a comprehensive review. Diagn Cytopathol. 1992;8(5):475–491. doi: 10.1002/dc.2840080508. [DOI] [PubMed] [Google Scholar]
- 22.Mosselman S, Polman J, Dijkema R. ER beta: identification and characterization of a novel human estrogen receptor. FEBS Lett. 1996;392(1):49–53. doi: 10.1016/0014-5793(96)00782-x. [DOI] [PubMed] [Google Scholar]
- 23.Paech K, Webb P, Kuiper GG, Nilsson S, Gustafsson J, Kushner PJ, Scanlan TS. Differential ligand activation of estrogen receptors ERalpha and ERbeta at AP1 sites. Science. 1997;277(5331):1508–1510. doi: 10.1126/science.277.5331.1508. [DOI] [PubMed] [Google Scholar]
- 24.Hall JM, McDonnell DP. The estrogen receptor beta-isoform (ERbeta) of the human estrogen receptor modulates ERalpha transcriptional activity and is a key regulator of the cellular response to estrogens and antiestrogens. Endocrinology. 1999;140(12):5566–5578. doi: 10.1210/endo.140.12.7179. [DOI] [PubMed] [Google Scholar]
- 25.Huang HJ, Norris JD, McDonnell DP. Identification of a negative regulatory surface within estrogen receptor alpha provides evidence in support of a role for corepressors in regulating cellular responses to agonists and antagonists. Mol Endocrinol. 2002;16(8):1778–1792. doi: 10.1210/me.2002-0089. [DOI] [PubMed] [Google Scholar]
- 26.DeNardo DG, Kim HT, Hilsenbeck S, Cuba V, Tsimelzon A, Brown PH. Global gene expression analysis of estrogen receptor transcription factor cross talk in breast cancer: identification of estrogen-induced/activator protein-1-dependent genes. Mol Endocrinol. 2005;19(2):362–378. doi: 10.1210/me.2004-0267. [DOI] [PubMed] [Google Scholar]
- 27.Wang W, Dong L, Saville B, Safe S. Transcriptional activation of E2F1 gene expression by 17beta-estradiol in MCF-7 cells is regulated by NF-Y-Sp1/estrogen receptor interactions. Mol Endocrinol. 1999;13(8):1373–1387. doi: 10.1210/mend.13.8.0323. [DOI] [PubMed] [Google Scholar]
- 28.Safe S. Transcriptional activation of genes by 17 beta-estradiol through estrogen receptor-Sp1 interactions. Vitam Horm. 2001;62:231–252. doi: 10.1016/s0083-6729(01)62006-5. [DOI] [PubMed] [Google Scholar]
- 29.Kushner PJ, Agard DA, Greene GL, Scanlan TS, Shiau AK, Uht RM, Webb P. Estrogen receptor pathways to AP-1. The Journal of steroid biochemistry and molecular biology. 2000;74(5):311–317. doi: 10.1016/s0960-0760(00)00108-4. [DOI] [PubMed] [Google Scholar]
- 30.Porter W, Saville B, Hoivik D, Safe S. Functional synergy between the transcription factor Sp1 and the estrogen receptor. Mol Endocrinol. 1997;11(11):1569–1580. doi: 10.1210/mend.11.11.9916. [DOI] [PubMed] [Google Scholar]
- 31.Cerillo G, Rees A, Manchanda N, Reilly C, Brogan I, White A, Needham M. The oestrogen receptor regulates NFkappaB and AP-1 activity in a cell-specific manner. The Journal of steroid biochemistry and molecular biology. 1998;67(2):79–88. doi: 10.1016/s0960-0760(98)00078-8. [DOI] [PubMed] [Google Scholar]
- 32.Webb P, Nguyen P, Valentine C, Lopez GN, Kwok GR, McInerney E, Katzenellenbogen BS, Enmark E, Gustafsson JA, Nilsson S, et al. The estrogen receptor enhances AP-1 activity by two distinct mechanisms with different requirements for receptor transactivation functions. Mol Endocrinol. 1999;13(10):1672–1685. doi: 10.1210/mend.13.10.0357. [DOI] [PubMed] [Google Scholar]
- 33.Ray P, Ghosh SK, Zhang DH, Ray A. Repression of interleukin-6 gene expression by 17 beta-estradiol: inhibition of the DNA-binding activity of the transcription factors NF-IL6 and NF-kappa B by the estrogen receptor. FEBS Lett. 1997;409(1):79–85. doi: 10.1016/s0014-5793(97)00487-0. [DOI] [PubMed] [Google Scholar]
- 34.Sommer S, Fuqua SA. Estrogen receptor and breast cancer. Semin Cancer Biol. 2001;11(5):339–352. doi: 10.1006/scbi.2001.0389. [DOI] [PubMed] [Google Scholar]
- 35.Barone I, Brusco L, Fuqua SA. Estrogen receptor mutations and changes in downstream gene expression and signaling. Clinical cancer research: an official journal of the American Association for Cancer Research. 2010;16(10):2702–2708. doi: 10.1158/1078-0432.CCR-09-1753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Clarke R, Liu MC, Bouker KB, Gu Z, Lee RY, Zhu Y, Skaar TC, Gomez B, O’Brien K, Wang Y, et al. Antiestrogen resistance in breast cancer and the role of estrogen receptor signaling. Oncogene. 2003;22(47):7316–7339. doi: 10.1038/sj.onc.1206937. [DOI] [PubMed] [Google Scholar]
- 37.Bardin A, Boulle N, Lazennec G, Vignon F, Pujol P. Loss of ERbeta expression as a common step in estrogen-dependent tumor progression. Endocr Relat Cancer. 2004;11(3):537–551. doi: 10.1677/erc.1.00800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lazennec G, Bresson D, Lucas A, Chauveau C, Vignon F. ER beta inhibits proliferation and invasion of breast cancer cells. Endocrinology. 2001;142(9):4120–4130. doi: 10.1210/endo.142.9.8395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hodges-Gallagher L, Valentine CD, El Bader S, Kushner PJ. Estrogen receptor beta increases the efficacy of antiestrogens by effects on apoptosis and cell cycling in breast cancer cells. Breast Cancer Res Treat. 2008;109(2):241–250. doi: 10.1007/s10549-007-9640-6. [DOI] [PubMed] [Google Scholar]
- 40.Jarvinen TA, Pelto-Huikko M, Holli K, Isola J. Estrogen receptor beta is coexpressed with ERalpha and PR and associated with nodal status, grade, and proliferation rate in breast cancer. Am J Pathol. 2000;156(1):29–35. doi: 10.1016/s0002-9440(10)64702-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Nair HB, Perla RP, Kirma NB, Krishnegowda NK, Ganapathy M, Rajhans R, Nair SS, Saikumar P, Vadlamudi RK, Tekmal RR. Estrogen Receptor-beta Mediates the Protective Effects of Aromatase Induction in the MMTV-Her-2/neu x Aromatase Double Transgenic Mice. Horm Cancer. 2012;3(1–2):26–36. doi: 10.1007/s12672-011-0083-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Holst F, Stahl PR, Ruiz C, Hellwinkel O, Jehan Z, Wendland M, Lebeau A, Terracciano L, Al-Kuraya K, Janicke F, et al. Estrogen receptor alpha (ESR1) gene amplification is frequent in breast cancer. Nat Genet. 2007;39(5):655–660. doi: 10.1038/ng2006. [DOI] [PubMed] [Google Scholar]
- 43.Filardo EJ, Quinn JA, Bland KI, Frackelton AR., Jr Estrogen-induced activation of Erk-1 and Erk-2 requires the G protein-coupled receptor homolog, GPR30, and occurs via trans-activation of the epidermal growth factor receptor through release of HB-EGF. Mol Endocrinol. 2000;14(10):1649–1660. doi: 10.1210/mend.14.10.0532. [DOI] [PubMed] [Google Scholar]
- 44.Revankar CM, Cimino DF, Sklar LA, Arterburn JB, Prossnitz ER. A transmembrane intracellular estrogen receptor mediates rapid cell signaling. Science. 2005;307(5715):1625–1630. doi: 10.1126/science.1106943. [DOI] [PubMed] [Google Scholar]
- 45.Thomas P, Pang Y, Filardo EJ, Dong J. Identity of an estrogen membrane receptor coupled to a G protein in human breast cancer cells. Endocrinology. 2005;146(2):624–632. doi: 10.1210/en.2004-1064. [DOI] [PubMed] [Google Scholar]
- 46.Funakoshi T, Yanai A, Shinoda K, Kawano MM, Mizukami Y. G protein-coupled receptor 30 is an estrogen receptor in the plasma membrane. Biochem Biophys Res Commun. 2006;346(3):904–910. doi: 10.1016/j.bbrc.2006.05.191. [DOI] [PubMed] [Google Scholar]
- 47.Filardo EJ, Graeber CT, Quinn JA, Resnick MB, Giri D, DeLellis RA, Steinhoff MM, Sabo E. Distribution of GPR30, a seven membrane-spanning estrogen receptor, in primary breast cancer and its association with clinicopathologic determinants of tumor progression. Clinical cancer research: an official journal of the American Association for Cancer Research. 2006;12(21):6359–6366. doi: 10.1158/1078-0432.CCR-06-0860. [DOI] [PubMed] [Google Scholar]
- 48.Maggiolini M, Vivacqua A, Fasanella G, Recchia AG, Sisci D, Pezzi V, Montanaro D, Musti AM, Picard D, Ando S. The G protein-coupled receptor GPR30 mediates c-fos up-regulation by 17beta-estradiol and phytoestrogens in breast cancer cells. J Biol Chem. 2004;279(26):27008–27016. doi: 10.1074/jbc.M403588200. [DOI] [PubMed] [Google Scholar]
- 49.Prossnitz ER, Arterburn JB, Sklar LA. GPR30: A G protein-coupled receptor for estrogen. Molecular and cellular endocrinology. 2007;265–266:138–142. doi: 10.1016/j.mce.2006.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Stern DF. Tyrosine kinase signalling in breast cancer: ErbB family receptor tyrosine kinases. Breast Cancer Res. 2000;2(3):176–183. doi: 10.1186/bcr51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Chang L, Karin M. Mammalian MAP kinase signalling cascades. Nature. 2001;410(6824):37–40. doi: 10.1038/35065000. [DOI] [PubMed] [Google Scholar]
- 52.Pandey DP, Lappano R, Albanito L, Madeo A, Maggiolini M, Picard D. Estrogenic GPR30 signalling induces proliferation and migration of breast cancer cells through CTGF. The EMBO journal. 2009;28(5):523–532. doi: 10.1038/emboj.2008.304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Madeo A, Maggiolini M. Nuclear alternate estrogen receptor GPR30 mediates 17beta-estradiol-induced gene expression and migration in breast cancer-associated fibroblasts. Cancer research. 2010;70(14):6036–6046. doi: 10.1158/0008-5472.CAN-10-0408. [DOI] [PubMed] [Google Scholar]
- 54.Zhang W, Liu HT. MAPK signal pathways in the regulation of cell proliferation in mammalian cells. Cell Res. 2002;12(1):9–18. doi: 10.1038/sj.cr.7290105. [DOI] [PubMed] [Google Scholar]
- 55.Brama M, Gnessi L, Basciani S, Cerulli N, Politi L, Spera G, Mariani S, Cherubini S, d’Abusco AS, Scandurra R, et al. Cadmium induces mitogenic signaling in breast cancer cell by an ERalpha-dependent mechanism. Molecular and cellular endocrinology. 2007;264(1–2):102–108. doi: 10.1016/j.mce.2006.10.013. [DOI] [PubMed] [Google Scholar]
- 56.Filardo EJ, Quinn JA, Frackelton AR, Jr, Bland KI. Estrogen action via the G protein-coupled receptor, GPR30: stimulation of adenylyl cyclase and cAMP-mediated attenuation of the epidermal growth factor receptor-to-MAPK signaling axis. Mol Endocrinol. 2002;16(1):70–84. doi: 10.1210/mend.16.1.0758. [DOI] [PubMed] [Google Scholar]
- 57.Sivaraman VS, Wang H, Nuovo GJ, Malbon CC. Hyperexpression of mitogen-activated protein kinase in human breast cancer. J Clin Invest. 1997;99(7):1478–1483. doi: 10.1172/JCI119309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Teng J, Wang ZY, Prossnitz ER, Bjorling DE. The G protein-coupled receptor GPR30 inhibits human urothelial cell proliferation. Endocrinology. 2008;149(8):4024–4034. doi: 10.1210/en.2007-1669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Wang C, Dehghani B, Magrisso IJ, Rick EA, Bonhomme E, Cody DB, Elenich LA, Subramanian S, Murphy SJ, Kelly MJ, et al. GPR30 contributes to estrogen-induced thymic atrophy. Mol Endocrinol. 2008;22(3):636–648. doi: 10.1210/me.2007-0359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ariazi EA, Brailoiu E, Yerrum S, Shupp HA, Slifker MJ, Cunliffe HE, Black MA, Donato AL, Arterburn JB, Oprea TI, et al. The G protein-coupled receptor GPR30 inhibits proliferation of estrogen receptor-positive breast cancer cells. Cancer research. 2010;70(3):1184–1194. doi: 10.1158/0008-5472.CAN-09-3068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Prossnitz ER, Maggiolini M. Mechanisms of estrogen signaling and gene expression via GPR30. Molecular and cellular endocrinology. 2009;308(1–2):32–38. doi: 10.1016/j.mce.2009.03.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.IPCS. Cadmium. Environmental Health Criteria. Vol. 134. Geneva: World Health Organization; 1992. International Programme on Chemical Safety. [Google Scholar]
- 63.IPCS. Cadmium—Environmental aspects. Environmental Health Criteria. Vol. 135. Geneva: World Health Organization; 1992. International Programme on Chemical Safety. [Google Scholar]
- 64.Hayes RB. The carcinogenicity of metals in humans. Cancer Causes Control. 1997;8(3):371–385. doi: 10.1023/a:1018457305212. [DOI] [PubMed] [Google Scholar]
- 65.IPCS. Nickel. Environmental Health Criteria. Vol. 108. Geneva: World Health Organization; 1991. International Programme on Chemical Safety. [Google Scholar]
- 66.Nielsen GD, Soderberg U, Jorgensen PJ, Templeton DM, Rasmussen SN, Andersen KE, Grandjean P. Absorption and retention of nickel from drinking water in relation to food intake and nickel sensitivity. Toxicol Appl Pharmacol. 1999;154(1):67–75. doi: 10.1006/taap.1998.8577. [DOI] [PubMed] [Google Scholar]
- 67.World Health Organization (WHO) Guidelines for Drinking-water Quality. Nickel in Drinking-water. [ http://www.who.int/water_sanitation_health/gdwqrevision/nickel2ndadd.pdf]
- 68.IARC. Cadmium and cadmium compounds. IARC Monogr Eval Carcinog Risk Chem Man. 1976;11:39–74. [PubMed] [Google Scholar]
- 69.IARC. Cadmium and cadmium compounds. IARC Monogr Eval Carcinog Risks Hum. 1993;58:119–237. [PMC free article] [PubMed] [Google Scholar]
- 70.IARC. Beryllium, cadmium, mercury, and exposures in the glass manufacturing industry. IARC Monogr Eval Carcinog Risks Hum; Working Group views and expert opinions; Lyon. 9–16 February 1993; 1993. pp. 1–415. [PMC free article] [PubMed] [Google Scholar]
- 71.NTP. Cadmium and cadmium compounds. Rep Carcinog. 2002;10:42–44. [PubMed] [Google Scholar]
- 72.NTP. Cadmium and cadmium compounds. Rep Carcinog. 2011;12:80–83. [PubMed] [Google Scholar]
- 73.Hu J, Mao Y, White K. Renal cell carcinoma and occupational exposure to chemicals in Canada. Occup Med (Lond) 2002;52(3):157–164. doi: 10.1093/occmed/52.3.157. [DOI] [PubMed] [Google Scholar]
- 74.Waalkes MP, Rehm S, Cherian MG. Repeated cadmium exposures enhance the malignant progression of ensuing tumors in rats. Toxicol Sci. 2000;54(1):110–120. doi: 10.1093/toxsci/54.1.110. [DOI] [PubMed] [Google Scholar]
- 75.Waalkes MP, Anver MR, Diwan BA. Chronic toxic and carcinogenic effects of oral cadmium in the Noble (NBL/Cr) rat: induction of neoplastic and proliferative lesions of the adrenal, kidney, prostate, and testes. J Toxicol Environ Health A. 1999;58(4):199–214. doi: 10.1080/009841099157296. [DOI] [PubMed] [Google Scholar]
- 76.Choe SY, Kim SJ, Kim HG, Lee JH, Choi Y, Lee H, Kim Y. Evaluation of estrogenicity of major heavy metals. Sci Total Environ. 2003;312(1–3):15–21. doi: 10.1016/S0048-9697(03)00190-6. [DOI] [PubMed] [Google Scholar]
- 77.Strumylaite L, Mechonosina K, Tamasauskas S. Environmental factors and breast cancer. Medicina (Kaunas) 2010;46(12):867–873. [PubMed] [Google Scholar]
- 78.Ionescu JG, Novotny J, Stejskal V, Latsch A, Blaurock-Busch E, Eisenmann-Klein M. Increased levels of transition metals in breast cancer tissue. Neuro Endocrinol Lett. 2006;27 (Suppl 1):36–39. [PubMed] [Google Scholar]
- 79.Strumylaite L, Bogusevicius A, Ryselis S, Pranys D, Poskiene L, Kregzdyte R, Abdrachmanovas O, Asadauskaite R. Association between cadmium and breast cancer. Medicina (Kaunas) 2008;44(6):415–420. [PubMed] [Google Scholar]
- 80.Agency for Toxic Substances and Disease Registry. Toxicological Profile for Cadmium. U.S. Department of Health and Human Services; [ http://www.atsdr.cdc.gov/toxprofiles/tp5-p.pdf.] [PubMed] [Google Scholar]
- 81.Beyersmann D. Effects of carcinogenic metals on gene expression. Toxicology letters. 2002;127(1–3):63–68. doi: 10.1016/s0378-4274(01)00484-2. [DOI] [PubMed] [Google Scholar]
- 82.IARC. Nickel and nickel compounds. IARC Monogr Eval Carcinog Risk Chem Man. 1976;11:75–112. [PubMed] [Google Scholar]
- 83.IARC. Chromium, nickel and welding. IARC Monogr Eval Carcinog Risks Hum. 1990;49:1–648. [PMC free article] [PubMed] [Google Scholar]
- 84.Rizk SL, Sky-Peck HH. Comparison between concentrations of trace elements in normal and neoplastic human breast tissue. Cancer research. 1984;44(11):5390–5394. [PubMed] [Google Scholar]
- 85.Johnson MD, Kenney N, Stoica A, Hilakivi-Clarke L, Singh B, Chepko G, Clarke R, Sholler PF, Lirio AA, Foss C, et al. Cadmium mimics the in vivo effects of estrogen in the uterus and mammary gland. Nat Med. 2003;9(8):1081–1084. doi: 10.1038/nm902. [DOI] [PubMed] [Google Scholar]
- 86.Jin T, Lu J, Nordberg M. Toxicokinetics and biochemistry of cadmium with special emphasis on the role of metallothionein. Neurotoxicology. 1998;19(4–5):529–535. [PubMed] [Google Scholar]
- 87.Nawrot T, Plusquin M, Hogervorst J, Roels HA, Celis H, Thijs L, Vangronsveld J, Van Hecke E, Staessen JA. Environmental exposure to cadmium and risk of cancer: a prospective population-based study. Lancet Oncol. 2006;7(2):119–126. doi: 10.1016/S1470-2045(06)70545-9. [DOI] [PubMed] [Google Scholar]
- 88.Amzal B, Julin B, Vahter M, Wolk A, Johanson G, Akesson A. Population toxicokinetic modeling of cadmium for health risk assessment. Environmental health perspectives. 2009;117(8):1293–1301. doi: 10.1289/ehp.0800317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Benbrahim-Tallaa L, Tokar EJ, Diwan BA, Dill AL, Coppin JF, Waalkes MP. Cadmium malignantly transforms normal human breast epithelial cells into a basal-like phenotype. Environmental health perspectives. 2009;117(12):1847–1852. doi: 10.1289/ehp.0900999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Hofer N, Diel P, Wittsiepe J, Wilhelm M, Degen GH. Dose- and route-dependent hormonal activity of the metalloestrogen cadmium in the rat uterus. Toxicology letters. 2009;191(2–3):123–131. doi: 10.1016/j.toxlet.2009.08.014. [DOI] [PubMed] [Google Scholar]
- 91.Ali I, Penttinen-Damdimopoulou PE, Makela SI, Berglund M, Stenius U, Akesson A, Hakansson H, Halldin K. Estrogen-like effects of cadmium in vivo do not appear to be mediated via the classical estrogen receptor transcriptional pathway. Environmental health perspectives. 2010;118(10):1389–1394. doi: 10.1289/ehp.1001967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Alonso-Gonzalez C, Gonzalez A, Mazarrasa O, Guezmes A, Sanchez-Mateos S, Martinez-Campa C, Cos S, Sanchez-Barcelo EJ, Mediavilla MD. Melatonin prevents the estrogenic effects of sub-chronic administration of cadmium on mice mammary glands and uterus. J Pineal Res. 2007;42(4):403–410. doi: 10.1111/j.1600-079X.2007.00434.x. [DOI] [PubMed] [Google Scholar]
- 93.Garcia-Morales P, Saceda M, Kenney N, Kim N, Salomon DS, Gottardis MM, Solomon HB, Sholler PF, Jordan VC, Martin MB. Effect of cadmium on estrogen receptor levels and estrogen-induced responses in human breast cancer cells. J Biol Chem. 1994;269(24):16896–16901. [PubMed] [Google Scholar]
- 94.Martin C, Zhang Y. The diverse functions of histone lysine methylation. Nat Rev Mol Cell Biol. 2005;6(11):838–849. doi: 10.1038/nrm1761. [DOI] [PubMed] [Google Scholar]
- 95.Predki PF, Sarkar B. Effect of replacement of “zinc finger” zinc on estrogen receptor DNA interactions. J Biol Chem. 1992;267(9):5842–5846. [PubMed] [Google Scholar]
- 96.Siewit CL, Gengler B, Vegas E, Puckett R, Louie MC. Cadmium promotes breast cancer cell proliferation by potentiating the interaction between ERalpha and c-Jun. Mol Endocrinol. 2010;24(5):981–992. doi: 10.1210/me.2009-0410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Sun X, Fontaine JM, Bartl I, Behnam B, Welsh MJ, Benndorf R. Induction of Hsp22 (HspB8) by estrogen and the metalloestrogen cadmium in estrogen receptor-positive breast cancer cells. Cell Stress Chaperones. 2007;12(4):307–319. doi: 10.1379/CSC-276.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Yu X, Filardo EJ, Shaikh ZA. The membrane estrogen receptor GPR30 mediates cadmium-induced proliferation of breast cancer cells. Toxicol Appl Pharmacol. 2010;245(1):83–90. doi: 10.1016/j.taap.2010.02.005. [DOI] [PubMed] [Google Scholar]
- 99.Liu Z, Yu X, Shaikh ZA. Rapid activation of ERK1/2 and AKT in human breast cancer cells by cadmium. Toxicol Appl Pharmacol. 2008;228(3):286–294. doi: 10.1016/j.taap.2007.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Fell GS, Ottaway JM, Hussein FE. Application of blood cadmium analysis to industry using an atomic fluorescence method. Br J Ind Med. 1977;34(2):106–109. doi: 10.1136/oem.34.2.106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Lau TJ, Hackett RL, Sunderman FW., Jr The carcinogenicity of intravenous nickel carbonyl in rats. Cancer research. 1972;32(10):2253–2258. [PubMed] [Google Scholar]
- 102.Dunnick JK, Elwell MR, Radovsky AE, Benson JM, Hahn FF, Nikula KJ, Barr EB, Hobbs CH. Comparative carcinogenic effects of nickel subsulfide, nickel oxide, or nickel sulfate hexahydrate chronic exposures in the lung. Cancer research. 1995;55(22):5251–5256. [PubMed] [Google Scholar]
- 103.IARC. Cancer Monographs on the Evaluation of the Carcinogenic Risk of Chemicals to Humans: Chromium, nickel and welding. Internation Agency for Research on Cancer. 1990;48:1–648. [PMC free article] [PubMed] [Google Scholar]
- 104.Hueper WC. Experimental studies in metal cancerigenesis. IX. Pulmonary lesions in guinea pigs and rats exposed to prolonged inhalation of powdered metallic nickel. AMA Arch Pathol. 1958;65(6):600–607. [PubMed] [Google Scholar]
- 105.Ivankovic S, Zeller WJ, Komitowski D, Edler L, Lehman E, Frohlich N. Carcinogenesis of nickel alloys in the hamster following intratracheal instillation. Schriftenreihe der Bundesanstalt fu r Arbeitsschutz, Dortmund. 1988:1–58. [Google Scholar]
- 106.Salnikow K, An WG, Melillo G, Blagosklonny MV, Costa M. Nickel-induced transformation shifts the balance between HIF-1 and p53 transcription factors. Carcinogenesis. 1999;20(9):1819–1823. doi: 10.1093/carcin/20.9.1819. [DOI] [PubMed] [Google Scholar]
- 107.Salnikow K, Davidson T, Costa M. The role of hypoxia-inducible signaling pathway in nickel carcinogenesis. Environmental health perspectives. 2002;110 (Suppl 5):831–834. doi: 10.1289/ehp.02110s5831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Tchou-Wong KM, Kiok K, Tang Z, Kluz T, Arita A, Smith PR, Brown S, Costa M. Effects of nickel treatment on H3K4 trimethylation and gene expression. PLoS One. 2011;6(3):e17728. doi: 10.1371/journal.pone.0017728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Kawata K, Shimazaki R, Okabe S. Comparison of gene expression profiles in HepG2 cells exposed to arsenic, cadmium, nickel, and three model carcinogens for investigating the mechanisms of metal carcinogenesis. Environ Mol Mutagen. 2009;50(1):46–59. doi: 10.1002/em.20438. [DOI] [PubMed] [Google Scholar]
- 110.Kowara R, Karaczyn A, Cheng RY, Salnikow K, Kasprzak KS. Microarray analysis of altered gene expression in murine fibroblasts transformed by nickel(II) to nickel(II)-resistant malignant phenotype. Toxicol Appl Pharmacol. 2005;205(1):1–10. doi: 10.1016/j.taap.2004.10.006. [DOI] [PubMed] [Google Scholar]
- 111.Ding J, He G, Gong W, Wen W, Sun W, Ning B, Huang S, Wu K, Huang C, Wu M, et al. Effects of nickel on cyclin expression, cell cycle progression and cell proliferation in human pulmonary cells. Cancer Epidemiol Biomarkers Prev. 2009;18(6):1720–1729. doi: 10.1158/1055-9965.EPI-09-0115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Ouyang W, Zhang D, Li J, Verma UN, Costa M, Huang C. Soluble and insoluble nickel compounds exert a differential inhibitory effect on cell growth through IKKalpha-dependent cyclin D1 down-regulation. J Cell Physiol. 2009;218(1):205–214. doi: 10.1002/jcp.21590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Lu X, Bao X, Huang Y, Qu Y, Lu H, Lu Z. Mechanisms of cytotoxicity of nickel ions based on gene expression profiles. Biomaterials. 2009;30(2):141–148. doi: 10.1016/j.biomaterials.2008.09.011. [DOI] [PubMed] [Google Scholar]
- 114.Weinstat-Saslow D, Merino MJ, Manrow RE, Lawrence JA, Bluth RF, Wittenbel KD, Simpson JF, Page DL, Steeg PS. Overexpression of cyclin D mRNA distinguishes invasive and in situ breast carcinomas from non-malignant lesions. Nat Med. 1995;1(12):1257–1260. doi: 10.1038/nm1295-1257. [DOI] [PubMed] [Google Scholar]
- 115.Foulkes WD, Brunet JS, Stefansson IM, Straume O, Chappuis PO, Begin LR, Hamel N, Goffin JR, Wong N, Trudel M, et al. The prognostic implication of the basal-like (cyclin E high/p27 low/p53+/glomeruloid-microvascular-proliferation+) phenotype of BRCA1-related breast cancer. Cancer research. 2004;64(3):830–835. doi: 10.1158/0008-5472.can-03-2970. [DOI] [PubMed] [Google Scholar]
- 116.Alao JP. The regulation of cyclin D1 degradation: roles in cancer development and the potential for therapeutic invention. Mol Cancer. 2007;6:24. doi: 10.1186/1476-4598-6-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Nesatyy VJ, Rutishauser BV, Eggen RI, Suter MJ. Identification of the estrogen receptor Cd-binding sites by chemical modification. Analyst. 2005;130(7):1087–1097. doi: 10.1039/b501192b. [DOI] [PubMed] [Google Scholar]
- 118.Satofuka H, Fukui T, Takagi M, Atomi H, Imanaka T. Metal-binding properties of phytochelatin-related peptides. J Inorg Biochem. 2001;86(2–3):595–602. doi: 10.1016/s0162-0134(01)00223-9. [DOI] [PubMed] [Google Scholar]
- 119.Maier T, Yu C, Kullertz G, Clemens S. Localization and functional characterization of metal-binding sites in phytochelatin synthases. Planta. 2003;218(2):300–308. doi: 10.1007/s00425-003-1091-7. [DOI] [PubMed] [Google Scholar]
- 120.Deegan BJ, Bona AM, Bhat V, Mikles DC, McDonald CB, Seldeen KL, Farooq A. Structural and thermodynamic consequences of the replacement of zinc with environmental metals on estrogen receptor alpha-DNA interactions. J Mol Recognit. 2011;24(6):1007–1017. doi: 10.1002/jmr.1148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Low LY, Hernandez H, Robinson CV, O’Brien R, Grossmann JG, Ladbury JE, Luisi B. Metal-dependent folding and stability of nuclear hormone receptor DNA-binding domains. J Mol Biol. 2002;319(1):87–106. doi: 10.1016/S0022-2836(02)00236-X. [DOI] [PubMed] [Google Scholar]
- 122.Freedman LP, Luisi BF, Korszun ZR, Basavappa R, Sigler PB, Yamamoto KR. The function and structure of the metal coordination sites within the glucocorticoid receptor DNA binding domain. Nature. 1988;334(6182):543–546. doi: 10.1038/334543a0. [DOI] [PubMed] [Google Scholar]
- 123.Predki PF, Sarkar B. Metal replacement in “zinc finger” and its effect on DNA binding. Environmental health perspectives. 1994;102 (Suppl 3):195–198. doi: 10.1289/ehp.94102s3195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Ke Q, Davidson T, Chen H, Kluz T, Costa M. Alterations of histone modifications and transgene silencing by nickel chloride. Carcinogenesis. 2006;27(7):1481–1488. doi: 10.1093/carcin/bgl004. [DOI] [PubMed] [Google Scholar]
- 125.Ke Q, Ellen TP, Costa M. Nickel compounds induce histone ubiquitination by inhibiting histone deubiquitinating enzyme activity. Toxicol Appl Pharmacol. 2008;228(2):190–199. doi: 10.1016/j.taap.2007.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Lee YW, Klein CB, Kargacin B, Salnikow K, Kitahara J, Dowjat K, Zhitkovich A, Christie NT, Costa M. Carcinogenic nickel silences gene expression by chromatin condensation and DNA methylation: a new model for epigenetic carcinogens. Mol Cell Biol. 1995;15(5):2547–2557. doi: 10.1128/mcb.15.5.2547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Chen H, Ke Q, Kluz T, Yan Y, Costa M. Nickel ions increase histone H3 lysine 9 dimethylation and induce transgene silencing. Mol Cell Biol. 2006;26(10):3728–3737. doi: 10.1128/MCB.26.10.3728-3737.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Ellen TP, Kluz T, Harder ME, Xiong J, Costa M. Heterochromatinization as a potential mechanism of nickel-induced carcinogenesis. Biochemistry. 2009;48(21):4626–4632. doi: 10.1021/bi900246h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Arita A, Costa M. Epigenetics in metal carcinogenesis: nickel, arsenic, chromium and cadmium. Metallomics. 2009;1(3):222–228. doi: 10.1039/b903049b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Lu H, Shi X, Costa M, Huang C. Carcinogenic effect of nickel compounds. Mol Cell Biochem. 2005;279(1–2):45–67. doi: 10.1007/s11010-005-8215-2. [DOI] [PubMed] [Google Scholar]
- 131.Karaczyn AA, Golebiowski F, Kasprzak KS. Ni(II) affects ubiquitination of core histones H2B and H2A. Exp Cell Res. 2006;312(17):3252–3259. doi: 10.1016/j.yexcr.2006.06.025. [DOI] [PubMed] [Google Scholar]
- 132.Eberharter A, Becker PB. Histone acetylation: a switch between repressive and permissive chromatin. Second in review series on chromatin dynamics. EMBO Rep. 2002;3(3):224–229. doi: 10.1093/embo-reports/kvf053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Chen H, Tini M, Evans RM. HATs on and beyond chromatin. Curr Opin Cell Biol. 2001;13(2):218–224. doi: 10.1016/s0955-0674(00)00200-3. [DOI] [PubMed] [Google Scholar]
- 134.Ellen TP, Kluz T, Harder ME, Xiong J, Costa M. Heterochromatinization as a Potential Mechanism of Nickel-Induced Carcinogenesis. Biochemistry. 2009 doi: 10.1021/bi900246h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Krogan NJ, Kim M, Tong A, Golshani A, Cagney G, Canadien V, Richards DP, Beattie BK, Emili A, Boone C, et al. Methylation of histone H3 by Set2 in Saccharomyces cerevisiae is linked to transcriptional elongation by RNA polymerase II. Mol Cell Biol. 2003;23(12):4207–4218. doi: 10.1128/MCB.23.12.4207-4218.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Schubeler D, MacAlpine DM, Scalzo D, Wirbelauer C, Kooperberg C, van Leeuwen F, Gottschling DE, O’Neill LP, Turner BM, Delrow J, et al. The histone modification pattern of active genes revealed through genome-wide chromatin analysis of a higher eukaryote. Genes Dev. 2004;18(11):1263–1271. doi: 10.1101/gad.1198204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.McGarvey KM, Fahrner JA, Greene E, Martens J, Jenuwein T, Baylin SB. Silenced tumor suppressor genes reactivated by DNA demethylation do not return to a fully euchromatic chromatin state. Cancer research. 2006;66(7):3541–3549. doi: 10.1158/0008-5472.CAN-05-2481. [DOI] [PubMed] [Google Scholar]
- 138.Kasprzak KS, Bal W, Karaczyn AA. The role of chromatin damage in nickel-induced carcinogenesis. A review of recent developments. J Environ Monit. 2003;5(2):183–187. doi: 10.1039/b210538c. [DOI] [PubMed] [Google Scholar]
- 139.Govindarajan B, Klafter R, Miller MS, Mansur C, Mizesko M, Bai X, LaMontagne K, Jr, Arbiser JL. Reactive oxygen-induced carcinogenesis causes hypermethylation of p16(Ink4a) and activation of MAP kinase. Mol Med. 2002;8(1):1–8. [PMC free article] [PubMed] [Google Scholar]
- 140.Kowara R, Salnikow K, Diwan BA, Bare RM, Waalkes MP, Kasprzak KS. Reduced Fhit protein expression in nickel-transformed mouse cells and in nickel-induced murine sarcomas. Mol Cell Biochem. 2004;255(1–2):195–202. doi: 10.1023/b:mcbi.0000007275.22785.91. [DOI] [PubMed] [Google Scholar]
- 141.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144(5):646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 142.Wei LH, Torng PL, Hsiao SM, Jeng YM, Chen MW, Chen CA. Histone deacetylase 6 regulates estrogen receptor alpha in uterine leiomyoma. Reprod Sci. 2011;18(8):755–762. doi: 10.1177/1933719111398147. [DOI] [PubMed] [Google Scholar]
- 143.Kawai H, Li H, Avraham S, Jiang S, Avraham HK. Overexpression of histone deacetylase HDAC1 modulates breast cancer progression by negative regulation of estrogen receptor alpha. Int J Cancer. 2003;107(3):353–358. doi: 10.1002/ijc.11403. [DOI] [PubMed] [Google Scholar]
- 144.Moggs JG, Orphanides G. Estrogen receptors: orchestrators of pleiotropic cellular responses. EMBO Rep. 2001;2(9):775–781. doi: 10.1093/embo-reports/kve185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Takiguchi M, Achanzar WE, Qu W, Li G, Waalkes MP. Effects of cadmium on DNA-(Cytosine-5) methyltransferase activity and DNA methylation status during cadmium-induced cellular transformation. Exp Cell Res. 2003;286(2):355–365. doi: 10.1016/s0014-4827(03)00062-4. [DOI] [PubMed] [Google Scholar]
- 146.Huang D, Zhang Y, Qi Y, Chen C, Ji W. Global DNA hypomethylation, rather than reactive oxygen species (ROS), a potential facilitator of cadmium-stimulated K562 cell proliferation. Toxicology letters. 2008;179(1):43–47. doi: 10.1016/j.toxlet.2008.03.018. [DOI] [PubMed] [Google Scholar]
- 147.Evans HJ. Neoplasia and cytogenetic abnormalities. Basic Life Sci. 1985;36:165–178. doi: 10.1007/978-1-4613-2127-9_11. [DOI] [PubMed] [Google Scholar]
- 148.Oshimura M, Barrett JC. Chemically induced aneuploidy in mammalian cells: mechanisms and biological significance in cancer. Environ Mutagen. 1986;8(1):129–159. doi: 10.1002/em.2860080112. [DOI] [PubMed] [Google Scholar]
- 149.Hill RP. Tumor progression: potential role of unstable genomic changes. Cancer Metastasis Rev. 1990;9(2):137–147. doi: 10.1007/BF00046340. [DOI] [PubMed] [Google Scholar]
- 150.Cheng KC, Loeb LA. Genomic instability and tumor progression: mechanistic considerations. Adv Cancer Res. 1993;60:121–156. doi: 10.1016/s0065-230x(08)60824-6. [DOI] [PubMed] [Google Scholar]
- 151.Duesberg P, Rausch C, Rasnick D, Hehlmann R. Genetic instability of cancer cells is proportional to their degree of aneuploidy. Proc Natl Acad Sci U S A. 1998;95(23):13692–13697. doi: 10.1073/pnas.95.23.13692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Cahill DP, Kinzler KW, Vogelstein B, Lengauer C. Genetic instability and darwinian selection in tumours. Trends Cell Biol. 1999;9(12):M57–60. [PubMed] [Google Scholar]
- 153.Li R, Yerganian G, Duesberg P, Kraemer A, Willer A, Rausch C, Hehlmann R. Aneuploidy correlated 100% with chemical transformation of Chinese hamster cells. Proceedings of the National Academy of Sciences of the United States of America. 1997;94(26):14506–14511. doi: 10.1073/pnas.94.26.14506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Seoane A, CFD Contribution to the validation of the anaphase––telophase test: aneugenic and clastogenic effects of cadmium sulfate, potassium dichromate and nickel chloride in Chinese hamster ovary cells. Genet Mol Biol. 1999;22:551–555. [Google Scholar]
- 155.Andersen O. Evaluation of the spindle-inhibiting effect of Ni++ by quantitation of chromosomal super-contraction. Res Commun Chem Pathol Pharmacol. 1985;50(3):379–386. [PubMed] [Google Scholar]
- 156.Coogan TP, Bare RM, Waalkes MP. Cadmium-induced DNA strand damage in cultured liver cells: reduction in cadmium genotoxicity following zinc pretreatment. Toxicol Appl Pharmacol. 1992;113(2):227–233. doi: 10.1016/0041-008x(92)90118-c. [DOI] [PubMed] [Google Scholar]
- 157.Parry JM, Sors A. The detection and assessment of the aneugenic potential of environmental chemicals: the European Community Aneuploidy Project. Mutat Res. 1993;287(1):3–15. doi: 10.1016/0027-5107(93)90140-b. [DOI] [PubMed] [Google Scholar]
- 158.Vega L, Gonsebatt ME, Ostrosky-Wegman P. Aneugenic effect of sodium arsenite on human lymphocytes in vitro: an individual susceptibility effect detected. Mutat Res. 1995;334(3):365–373. doi: 10.1016/0165-1161(95)90074-8. [DOI] [PubMed] [Google Scholar]
- 159.Seoane AI, Dulout FN. Genotoxic ability of cadmium, chromium and nickel salts studied by kinetochore staining in the cytokinesis-blocked micronucleus assay. Mutat Res. 2001;490(2):99–106. doi: 10.1016/s1383-5718(00)00145-5. [DOI] [PubMed] [Google Scholar]
- 160.Epe B, Harttig U, Stopper H, Metzler M. Covalent binding of reactive estrogen metabolites to microtubular protein as a possible mechanism of aneuploidy induction and neoplastic cell transformation. Environmental health perspectives. 1990;88:123–127. doi: 10.1289/ehp.9088123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Hontz AE, Li SA, Lingle WL, Negron V, Bruzek A, Salisbury JL, Li JJ. Aurora a and B overexpression and centrosome amplification in early estrogen-induced tumor foci in the Syrian hamster kidney: implications for chromosomal instability, aneuploidy, and neoplasia. Cancer research. 2007;67(7):2957–2963. doi: 10.1158/0008-5472.CAN-06-3296. [DOI] [PubMed] [Google Scholar]
- 162.Katayama H, Brinkley WR, Sen S. The Aurora kinases: role in cell transformation and tumorigenesis. Cancer Metastasis Rev. 2003;22(4):451–464. doi: 10.1023/a:1023789416385. [DOI] [PubMed] [Google Scholar]
- 163.Bischoff JR, Anderson L, Zhu Y, Mossie K, Ng L, Souza B, Schryver B, Flanagan P, Clairvoyant F, Ginther C, et al. A homologue of Drosophila aurora kinase is oncogenic and amplified in human colorectal cancers. The EMBO journal. 1998;17(11):3052–3065. doi: 10.1093/emboj/17.11.3052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Tatsuka M, Katayama H, Ota T, Tanaka T, Odashima S, Suzuki F, Terada Y. Multinuclearity and increased ploidy caused by overexpression of the aurora- and Ipl1-like midbody-associated protein mitotic kinase in human cancer cells. Cancer research. 1998;58(21):4811–4816. [PubMed] [Google Scholar]
- 165.IARC. Arsenic and arsenic compounds. IARC Monogr Eval Carcinog Risk Chem Hum. 1980;23:39–141. [PubMed] [Google Scholar]
- 166.IARC. Some metals and metallic compounds. IARC Monogr Eval Carcinog Risk Chem Hum. 1980;23:1–415. [PubMed] [Google Scholar]
- 167.Hinwood AL, Sim MR, Jolley D, de Klerk N, Bastone EB, Gerostamoulos J, Drummer OH. Risk factors for increased urinary inorganic arsenic concentrations from low arsenic concentrations in drinking water. Int J Environ Health Res. 2003;13(3):271–284. doi: 10.1080/0960312031000122424. [DOI] [PubMed] [Google Scholar]
- 168.Hinwood AL, Sim MR, Jolley D, de Klerk N, Bastone EB, Gerostamoulos J, Drummer OH. Hair and toenail arsenic concentrations of residents living in areas with high environmental arsenic concentrations. Environmental health perspectives. 2003;111(2):187–193. doi: 10.1289/ehp.5455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Chatterjee A, Chatterji U. Arsenic abrogates the estrogen-signaling pathway in the rat uterus. Reprod Biol Endocrinol. 2010;8:80. doi: 10.1186/1477-7827-8-80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Davey JC, Bodwell JE, Gosse JA, Hamilton JW. Arsenic as an endocrine disruptor: effects of arsenic on estrogen receptor-mediated gene expression in vivo and in cell culture. Toxicol Sci. 2007;98(1):75–86. doi: 10.1093/toxsci/kfm013. [DOI] [PubMed] [Google Scholar]
- 171.Romanowicz-Makowska H, Forma E, Brys M, Krajewska WM, Smolarz B. Concentration of cadmium, nickel and aluminium in female breast cancer. Pol J Pathol. 2011;62(4):257–261. [PubMed] [Google Scholar]
- 172.Benderli Cihan Y, Sozen S, Ozturk Yildirim S. Trace elements and heavy metals in hair of stage III breast cancer patients. Biol Trace Elem Res. 2011;144(1–3):360–379. doi: 10.1007/s12011-011-9104-z. [DOI] [PubMed] [Google Scholar]
- 173.Strumylaite L, Bogusevicius A, Abdrachmanovas O, Baranauskiene D, Kregzdyte R, Pranys D, Poskiene L. Cadmium concentration in biological media of breast cancer patients. Breast Cancer Res Treat. 2011;125(2):511–517. doi: 10.1007/s10549-010-1007-8. [DOI] [PubMed] [Google Scholar]
- 174.Anderson LM, Diwan BA, Fear NT, Roman E. Critical windows of exposure for children’s health: cancer in human epidemiological studies and neoplasms in experimental animal models. Environmental health perspectives. 2000;108 (Suppl 3):573–594. doi: 10.1289/ehp.00108s3573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Birnbaum LS, Fenton SE. Cancer and developmental exposure to endocrine disruptors. Environmental health perspectives. 2003;111(4):389–394. doi: 10.1289/ehp.5686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Hilakivi-Clarke L. Estrogens, BRCA1, and breast cancer. Cancer research. 2000;60(18):4993–5001. [PubMed] [Google Scholar]
- 177.Weiss HA, Potischman NA, Brinton LA, Brogan D, Coates RJ, Gammon MD, Malone KE, Schoenberg JB. Prenatal and perinatal risk factors for breast cancer in young women. Epidemiology. 1997;8(2):181–187. doi: 10.1097/00001648-199703000-00010. [DOI] [PubMed] [Google Scholar]
- 178.Glasier A. HRT and Breast Cancer. Women’s Health Medicine. 2006;3(1):15–17. [Google Scholar]
- 179.Gustafsson JA. Estrogen receptor beta--a new dimension in estrogen mechanism of action. The Journal of endocrinology. 1999;163(3):379–383. doi: 10.1677/joe.0.1630379. [DOI] [PubMed] [Google Scholar]
- 180.Thomas C, Gustafsson JA. The different roles of ER subtypes in cancer biology and therapy. Nature reviews Cancer. 2011;11(8):597–608. doi: 10.1038/nrc3093. [DOI] [PubMed] [Google Scholar]
- 181.Prossnitz ER, Arterburn JB, Smith HO, Oprea TI, Sklar LA, Hathaway HJ. Estrogen signaling through the transmembrane G protein-coupled receptor GPR30. Annual review of physiology. 2008;70:165–190. doi: 10.1146/annurev.physiol.70.113006.100518. [DOI] [PubMed] [Google Scholar]
- 182.Luparello C, Longo A, Vetrano M. Exposure to cadmium chloride influences astrocyte-elevated gene-1 (AEG-1) expression in MDA-MB231 human breast cancer cells. Biochimie. 2012;94(1):207–213. doi: 10.1016/j.biochi.2011.10.009. [DOI] [PubMed] [Google Scholar]
- 183.Casano C, Agnello M, Sirchia R, Luparello C. Cadmium effects on p38/MAPK isoforms in MDA-MB231 breast cancer cells. Biometals: an international journal on the role of metal ions in biology, biochemistry, and medicine. 2010;23(1):83–92. doi: 10.1007/s10534-009-9268-6. [DOI] [PubMed] [Google Scholar]
- 184.Cannino G, Ferruggia E, Luparello C, Rinaldi AM. Effects of cadmium chloride on some mitochondria-related activity and gene expression of human MDA-MB231 breast tumor cells. Journal of inorganic biochemistry. 2008;102(8):1668–1676. doi: 10.1016/j.jinorgbio.2008.04.002. [DOI] [PubMed] [Google Scholar]
- 185.Sirchia R, Longo A, Luparello C. Cadmium regulation of apoptotic and stress response genes in tumoral and immortalized epithelial cells of the human breast. Biochimie. 2008;90(10):1578–1590. doi: 10.1016/j.biochi.2008.06.011. [DOI] [PubMed] [Google Scholar]
- 186.Sirchia R, Luparello C. Short-term exposure to cadmium affects the expression of stress response and apoptosis-related genes in immortalized epithelial cells from the human breast. Toxicology in vitro: an international journal published in association with BIBRA. 2009;23(5):943–949. doi: 10.1016/j.tiv.2009.04.016. [DOI] [PubMed] [Google Scholar]
- 187.Pan J, Chang Q, Wang X, Son Y, Zhang Z, Chen G, Luo J, Bi Y, Chen F, Shi X. Reactive oxygen species-activated Akt/ASK1/p38 signaling pathway in nickel compound-induced apoptosis in BEAS 2B cells. Chemical research in toxicology. 2010;23(3):568–577. doi: 10.1021/tx9003193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Cai T, Li X, Ding J, Luo W, Li J, Huang C. A cross-talk between NFAT and NF-kappaB pathways is crucial for nickel-induced COX-2 expression in Beas-2B cells. Current cancer drug targets. 2011;11(5):548–559. doi: 10.2174/156800911795656001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Hu W, Feng Z, Tang MS. Nickel (II) enhances benzo[a]pyrene diol epoxide-induced mutagenesis through inhibition of nucleotide excision repair in human cells: a possible mechanism for nickel (II)-induced carcinogenesis. Carcinogenesis. 2004;25(3):455–462. doi: 10.1093/carcin/bgh012. [DOI] [PubMed] [Google Scholar]
- 190.Lin X, Dowjat WK, Costa M. Nickel-induced transformation of human cells causes loss of the phosphorylation of the retinoblastoma protein. Cancer research. 1994;54(10):2751–2754. [PubMed] [Google Scholar]
- 191.Zhang Z, Li W, Cheng S, Yao H, Zhang F, Chang Q, Ke Z, Wang X, Son YO, Luo J, et al. Nickel-induced down-regulation of DeltaNp63 and its role in the proliferation of keratinocytes. Toxicology and applied pharmacology. 2011;253(3):235–243. doi: 10.1016/j.taap.2011.03.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.M’Bemba-Meka P, Lemieux N, Chakrabarti SK. Nickel compound-induced DNA single-strand breaks in chromosomal and nuclear chromatin in human blood lymphocytes in vitro: role of oxidative stress and intracellular calcium. Mutation research. 2005;586(2):124–137. doi: 10.1016/j.mrgentox.2005.06.001. [DOI] [PubMed] [Google Scholar]
- 193.Zang Y, Odwin-Dacosta S, Yager JD. Effects of cadmium on estrogen receptor mediated signaling and estrogen induced DNA synthesis in T47D human breast cancer cells. Toxicology letters. 2009;184(2):134–138. doi: 10.1016/j.toxlet.2008.10.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Sinha Roy S, Mukherjee S, Mukhopadhyay S, Das SK. Differential effect of cadmium on cholinephosphotransferase activity in normal and cancerous human mammary epithelial cell lines. Molecular cancer therapeutics. 2004;3(2):199–204. [PubMed] [Google Scholar]