

Abstract
The structural core of DNA, a continuous stack of aromatic heterocycles, the base pairs, which extends down the helical axis, gives rise to the fascinating electronic properties of this molecule that is so critical for life. Our laboratory and others have developed diverse experimental platforms to investigate the capacity of DNA to conduct charge, termed DNA-mediated charge transport (DNA CT). Here, we present an overview of DNA CT experiments in solution, on surfaces, and with single molecules that collectively provide a broad and consistent perspective on the essential characteristics of this chemistry. DNA CT can proceed over long molecular distances but is remarkably sensitive to perturbations in base pair stacking. We discuss how this foundation, built with data from diverse platforms, can be used both to inform a mechanistic description of DNA CT and to inspire the next platforms for its study: living organisms and molecular electronics.
1. Introduction
DNA holds great promise as a medium for charge transport (CT) in nanoscale electronic and biomedical devices due to its stability and structural programmability.1,2 Conductive properties of DNA were forecast in 1962 by Eley and Spivey when they observed similarities between stacked DNA base pairs and stacked graphene sheets: both are composed of planar, aromatic molecules, and both exhibit an inter-plane stacking distance of 3.4 Å.3 Evidence of DNA-mediated CT was presented in a 1993 experiment involving oxidative quenching of a DNA-bound metal complex through the DNA base stack.4 Since then, the ability of DNA to mediate CT reactions has been verified in many experimental systems, and the factors that affect the rate and efficiency of the reaction are for the most part well understood. Despite our knowledge of the fundamental characteristics of DNA CT, these systems remain quite challenging to model. Indeed, the nature of the CT bridge must be considered, and variations in the base sequence, the introduction of perturbations such as base mismatches between the donor and the acceptor, and dynamic motions of the bases, among other factors, can alter the rate of the reaction and the yield of CT products. It is clear that a mechanistic description must be informed by and consistent with the characteristics of DNA CT that have been observed and validated across diverse experimental platforms (Fig. 1).
Fig. 1.

Platforms for the study of DNA CT. In solution (top), donor and acceptor molecules are covalently tethered or otherwise incorporated into opposite ends of a DNA duplex. DNA CT is initiated by photoexcitation of the donor and measured by spectroscopic or biochemical methods. On electrode surfaces (center), DNA is covalently tethered to the surface by one end and modified with a redox-active probe moiety on the distal end. An applied potential to the electrode results in DNA CT to the distal probe and produces a characteristic DNA-mediated redox signal. With single molecules (bottom), one DNA duplex is covalently attached by amide bonds across a gap that has been cut in a carbon nanotube within an electrical circuit. Current flow through the CNT-DNA device is a reflection of DNA CT through the single DNA duplex that bridges the gap and can be used to make fundamental measurements of DNA conductivity.
Here, we present an overview of DNA CT experiments conducted in solution, on surfaces, and with single molecules, focusing on studies in our laboratory but also highlighting others. We show that several characteristics of DNA CT appear to be general, irrespective of the experimental platform used to observe this process. We then discuss how these conserved characteristics can inform a mechanistic description of DNA CT.
2. DNA CT in solution
The majority of experiments that examine the nature of DNA CT have been conducted in solution. In general, solution-phase DNA CT systems involve a photoexcited charge donor separated from a charge acceptor by a DNA bridge (Fig. 1). By positioning the donor and acceptor at opposite ends of the duplex, it is possible to survey the base sequence between them in a systematic manner in order to gain information about the CT characteristics of the medium itself. A wide variety of donors and acceptors have been utilized in solution measurements of DNA CT, and some are illustrated in Scheme 1. In contrast to other experimental platforms that will be discussed in later sections, almost all solution studies involve CT from the excited state, so the values obtained depend on the photophysical characteristics of the charge donor. The measurements are also ensemble measurements, so the reaction parameters obtained in solution experiments represent average values. The solution state provides many measurement techniques, including steady-state and time-resolved luminescence and transient absorption spectroscopies, as well as biochemical DNA oxidation assays. In addition, any conclusions drawn from observations of DNA CT in aqueous, solution-phase experiments are immediately applicable to biological systems.
Scheme 1.

Probes used for the study of DNA CT.
2.1 Interactions between probes and DNA
Primary among the requirements for efficient DNA CT is the necessity for intimate electronic interaction between the donor–acceptor pair and the DNA base stack. The effect of varying the strength of DNA association is nicely illustrated in an experiment involving naphthalimide (NI) derivatives bearing substituents that render them positively-charged, negatively-charged, or neutral without greatly modifying the structure of the probe.5 The cationic and neutral derivatives bind, as evidenced by hypochromicity in their absorbance spectra upon the addition of DNA and by luminescence quenching, while the anionic derivative does not. The lack of binding by the anionic derivative is attributed to electrostatic repulsion by the negatively-charged DNA phosphate backbone. Importantly, the electrostatic association between NI and DNA is not the only interaction that allows for the observed spectroscopic changes upon binding; the planar, aromatic character of the probe lends itself to intercalative binding as well. Indeed, for the NI derivatives, the changes in the absorption and luminescence intensities are comparable for the neutral and cationic species, despite their difference in charge. It is the intimacy of the intercalative binding mode, rather than the promiscuity of the electrostatic one, that influences the photophysics of these molecules. These different modes of interaction between probes and DNA are illustrated in Fig. 2.
Fig. 2.

Binding modes of donors and acceptors influence their participation in DNA CT. Δ-[Rh(phi)2(phen)]3+ (top left), intercalates between the DNA bases and is thus well coupled to the DNA π-stack and can participate in DNA CT. In contrast, [Ru(NH3)6]3+ (center right) which electrostatically binds the negatively charged phosphate backbone and Λ-[Ru(phen)3]2+ (bottom right) which binds within the major groove, are not well coupled and do not participate in DNA CT.
Intercalation involves accommodation of a planar, aromatic molecule into the DNA base stack through a slight unwinding and lengthening of the duplex. Crystallography has shown that an intercalated probe, sandwiched between two existing base pairs, therefore becomes incorporated into the π-stack and behaves as an additional base. Binding in this manner is driven by the increased stability afforded by π-stacking and hydrophobic interactions. Octahedral metal complexes bearing aromatic, planar ligands intercalate particularly strongly due to the combination of intercalation of the ligand and electrostatic attraction between such positively-charged complexes and the phosphate backbone of DNA. For this reason, metallointercalators have been used extensively in studies of DNA CT.
Several factors influence the strength of intercalation by metal complexes. Chief among these is steric complementarity between the bound complex and the DNA backbone. For example, a large differential in binding strength is observed between Δ and Λ octahedral enantiomers.6 The basis of this enantiomeric preference was verified in NMR and X-ray crystallography studies: while Δ-α-[Rh[(R,R)-Me2trien](phi)]3+ ((R,R)-Me2trien = 2R,9R-diamino-4,7-diazadecane; phi = 9,10-phenanthrenequinone diimine) binds DNA by intercalation from the major groove, intercalation of the Λ isomer is hindered by steric clashes between the (R,R)-Me2trien ancillary ligands and the DNA phosphate backbone.7,8 In fact, noncovalent interactions between the ancillary ligands of metal complexes and the DNA backbone are so influential that they can be employed to induce binding selectivity toward particular DNA sequences.9-13 Besides steric interactions between the DNA backbone and the ancillary ligands of metal complexes, the size, shape, and hydrophobicity of the intercalating ligand also affects binding affinity.14,15 Finally, although intercalative binding by metal complexes is quite strong, the stabilization gained through π-stacking and hydrophobic effects does not necessarily lead to a single binding conformation. Differences in the orientation of the bound complex, which may arise upon binding to different sequences, can influence the solvent accessibility of the complex or the electronic coupling between the complex and the base stack.16-18
2.2 DNA CT between metallointercalators
Early experiments involving metallointercalators proved that CT between well coupled probes can occur through the base stack. The first experimental verification of long range DNA CT involved oxidative quenching of photoexcited [Ru(phen)2(dppz)]2+ (phen = 1,10-phenanthroline; dppz = dipyrido[3,2-a:2′,3′-c]phenazine) by [Rh(phi)2(phen)]3+.4 The two probes were appended to opposite ends of a DNA 15-mer via short, flexible covalent linkers, and intercalation was verified by luminescence and photocleavage experiments. When the donor and acceptor were instead appended to different duplexes, no quenching was observed, proving that quenching occurs intraduplex. This control also showed that quenching was occurring through the base stack. In a similar experiment, DNA-mediated oxidative quenching of [Os(phen)2(dppz)]2+* by [Rh(phi)2(bpy)]3+ indicated that no portion of the observed quenching in these types of systems occurs by energy transfer, since the luminescence of the Os complex does not overlap spectrally with the absorption of the Rh complex.19 Finally, quenching of DNA-bound [Ru(phen)2(dppz)]2+* by the intercalator [Rh(phi)2(phen)]3+ was much more effective than quenching by [Ru(NH3)6]3+, which does not intercalate, underscoring that electronic coupling between the CT probes and the DNA base stack is necessary for DNA CT to take place.20
2.3 DNA bases as charge acceptors
Besides comprising the CT medium, DNA bases and base analogs can also function as charge acceptors. Using DNA bases as charge acceptors offers the advantage that they are inherently coupled electronically to the rest of the base stack. Importantly, the oxidation potentials of the bases vary as G (Eox = 1.3 V vs. NHE) < A (1.4 V) < C (1.6 V) < T (1.7 V);21 note that these values are taken from studies with individual nucleosides, rather than a DNA duplex. Depending on the excited state energy of a particular photooxidant, it is possible (and is indeed common) that only a subset of the bases undergo oxidation in hole transfer (HT) experiments. In addition, ab initio molecular orbital calculations have shown that the oxidation potential of each base depends on its sequence context,22 and that stacked guanines at GG and GGG sites have lower oxidation potentials than isolated guanines.23-25 In HT experiments, the mobile cation is therefore expected to localize at low potential guanine sites.
2.3.1 Metallointercalators as base oxidants
Many experiments have shown that G can be oxidized via DNA CT. For example, while high-energy (313 nm) irradiation of DNA-bound [Rh(phi)2(bpy)]3+ results in direct strand cleavage at the site of Rh binding through C3′-hydrogen abstraction, lower energy (365 nm) irradiation leads to oxidative damage only at GG sites.26 Guanine can also be oxidized over long distances by complexes such as [Ru(phen)2(dppz)]3+. This is accomplished following oxidation of the Ru2+ complex using the “flash-quench” technique, which involves preliminary oxidation of the excited Ru intercalator to the 3+ state by a diffusing quencher.27 The G radical cation generated in this manner can persist in solution for over one millisecond, and has been observed by transient absorption spectroscopy, EPR, and time-resolved infrared spectroscopy.28-30 Notably, strong electronic coupling between the oxidant and the base stack is necessary for efficient CT in these systems as well. This is shown in an experiment involving a family of [Ru(bpy)2(dppz)]2+ (bpy = 2,2′-bipyridine) derivatives, where the observed amount of DNA-mediated G oxidation correlates directly with the intercalation ability of the Ru oxidant.31
In DNA duplexes containing multiple acceptor sites, the injected charge equilibrates between them. This effect is observed in Ru flash-quench systems, where each of several guanines in the DNA sequence is oxidized to the same extent, no matter the distance between the Ru oxidant and the guanine site.27 Hole equilibration between G and GGG sites has also been observed by Giese following photoinduced hole injection to the G site.32,33 The property of charge equilibration between low potential sites can be used advantageously for the characterization of DNA as a medium for CT. In particular, comparing the yield of oxidation at two GG sites, one proximal and one distal to the oxidant binding site, can provide information about the conductivity of the base stack intervening between them.
The distal/proximal guanine oxidation assay is especially useful in understanding how DNA sequence affects the efficiency of CT. For example, in systems utilizing a Rh photooxidant, the relative amount of guanine oxidation at a distal GG site compared to oxidation at a proximal GG site is several-fold greater when the base sequence between the two GG sites is comprised of only A than when it is comprised of only T or of an alternating (TA)n sequence.34 Differences in transport efficiency through various base sequences are related to differences in static disorder, or variations in local DNA conformations and energetics, within the bridge.35 But this equilibration really occurs because the guanine radical is not an effective irreversible hole trap; its lifetime is ~10−4 s in DNA.28 Experiments utilizing very fast charge traps can be used to more precisely understand the effects of sequence on the efficiency of DNA CT. The cyclopropyl rings of N6-cyclopropyldeoxyadenosine (CPA), N4-cyclopropyldeoxycytosine (CPC) and N2-cyclopropyldeoxyguanine (CPG) open within 2 ps of cation localization on those bases.36 When incorporated into a DNA base stack, cyclopropyl-modified bases are therefore sensitive probes of charge occupation along the CT bridge. Interestingly, in Rh photooxidation experiments, CPC is oxidized as efficiently as CPG, suggesting that the mobile charge occupies high-energy bases as well as low-energy bases.37 Similar experiments also show that the yield of charge trapping depends on the sequence beyond the hole trap as well as the sequence intervening between the photooxidant and the trap.38 Both of these results suggest that the charge is delocalized among several bases during transport.
The efficiency of DNA CT depends not only on the degree of electronic coupling between the redox pair and the base stack, but also on the degree of coupling between the bases themselves. It is this heterogeneous and dynamic nature of the base stack that differentiates it from bridges used in other molecular wire systems. Due to the necessity for coupling between the bases, any influence or modification that compromises the integrity of the base stack decreases the efficiency of CT. This principle is illustrated effectively by systems in which gross structural aberrations intervene between the charge donor and the charge acceptor (Fig. 3). For example, the yield of DNA-mediated oxidation at GG or GGG sites distal to a Rh photooxidant decreases when a bulge is placed between the donor and the acceptor.39,40 Double crossover assemblies represent the extreme of complete electronic isolation between the bases of adjoining duplexes. In such systems, no DNA-mediated oxidation is observed in the duplex to which the oxidant is not bound.41 While structural perturbations imposed by base pair mismatches are more subtle than those presented by bulges, mismatches intervening between the donor and the acceptor can also decrease the efficiency of DNA CT.27,42 Importantly, pyrimidine·pyrimidine mismatches, which are the most thermodynamically destabilized, are most effective at attenuating CT to distal guanine doublets.43 Finally, protein binding can introduce both large-scale and small-scale structural perturbations in the DNA base stack. Binding of the methylase M. HhaI, which removes its target base from the base stack, between a hole donor and a GG acceptor decreases the yield of oxidative damage at the guanine sink.44 Binding of proteins that severely bend the duplex, such as TATA-binding protein, decreases the yield of long-range DNA CT, while binding of proteins that do not distort the duplex, but instead rigidify it, such as the restriction endonuclease R. PvuII or the transcription factor Antennapedia homeodomain protein, actually enhance the yield of DNA CT.45 From these many examples, it is clear that the detrimental effects of structural perturbations to the stacked duplex on the yield of DNA CT are quite general.
Fig. 3.
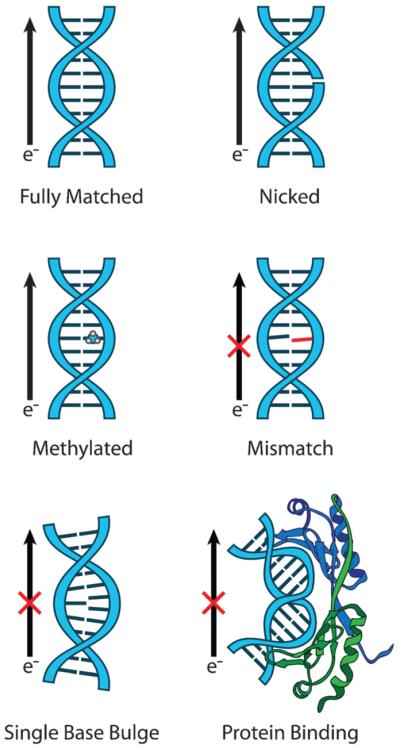
Structural perturbations to the base π-stack inhibit DNA CT. For efficient DNA CT, bases in the duplex must be well stacked with each other to achieve proper π-orbital overlap and electronic coupling. This occurs naturally in the case of fully matched DNA (top left). Nicks in the sugar phosphate backbone (top right) and methylation of the DNA bases (center left) do not interfere with the base stack and thus do not inhibit DNA CT. However, attenuation of DNA CT is observed for perturbations that disrupt the base stack including single base mismatches (center right), bound proteins that severely kink the DNA (bottom right), and single base bulges (bottom left). The attenuation in DNA CT caused by these structural perturbations, and others, has been measured with solution, surface, and single molecule platforms.
The distance of CT also affects the yield of oxidation, but the distance dependence is itself determined by other factors. For instance, [Ru(phen)2(dppz)]2+ * quenching by [Rh(phi)2(phen)]3+ is nearly quantitative across a 15-mer (40 Å) duplex, indicating a very shallow distance dependence in these constructs.4 This conclusion is echoed in CPA-opening experiments, where no statistical difference is observed in the yield of the ring-opened product with increasing distance from a Rh photooxidant.46 Similarly, guanine oxidation is observed in solution-based systems with little change in the relative yield as the distance is increased from 28 to 63 base pairs (100 Å to 200 Å).26,35 The yield of HT from G to GGG has also been compared over distances ranging from 1 to 16 base pairs (3.4 to 54.4 Å).32
Interestingly, the distance dependence in these systems is strong when transfer occurs over 3 or fewer bases, but is weak over longer distances; whether transfer over short distances in these systems occurs through the DNA stack or not is unclear. The shallow distance dependence observed for DNA CT over very long distances contrasts with electron transfer (ET) through other media such as molecular wires and proteins, where the yield of ET generally decreases exponentially with increasing distance.47,48
2.3.2 Organic probes as base oxidants
Organic charge donors have also been used to oxidize DNA bases and base analogs. Organic probes often share many characteristics with intercalating ligands, including the structural requirements of planarity and aromaticity, as well as strong absorption and luminescence properties. One important difference between organic probes and metallointercalators involves the method of incorporation of organic probes into DNA systems. While most organic probes fulfill the appropriate structural requirements for intercalative binding, they are often assumed (or intentionally constructed) to interact with only the terminal base pair of the duplex. Such end-capping interactions are certainly enhanced by favorable base stacking and hydrophobic effects. However, the electronic coupling between end-capping probes and the DNA base stack is weaker than that between metallointercalators and the base stack. Indeed, molecular modeling of donor-linked hairpins has shown that chromophore/base pair π-stacking distances vary from 3.5 to 4.2 Å (compared to the 3.4 Å interbase spacing of natural DNA), and melting of such hairpins results in very little change in the absorbance of the chromophore, indicating a lack of excitonic interactions with the DNA bases.49 Despite these limitations, such systems have provided valuable insights into the dynamics of DNA CT.
The first step in any proposed mechanism for DNA CT is charge injection by the excited donor. The products of this process are the donor radical anion and a radical cation within the bridge, which is usually assumed to lie at a low potential site. Following charge separation, charge recombination may occur to reform the initial state. The rates of these processes are conveniently determined by monitoring the lifetime of the excited state with time-resolved luminescence and transient absorption (TA) spectroscopies. These techniques have been used to observe charge injection and charge recombination in DNA hairpins linked by a number of different excited-state hole donors, including phenanthrene and naphthalene derivatives, stilbene-4,4′-dicarboxamide (Sa), and diphenylacetylene (DPA).50-53 The rates of charge injection by these donors vary from <1 × 107 to 1 × 1012 s−1, and the rates of charge recombination vary from 1.1 × 108 to 5.0 × 1011 s−1. Plotting the free energy change of charge injection and charge recombination against the measured rates yields two Marcus curves: one for constructs in which a G·C base pair has been placed adjacent to the donor, and one for constructs in which the nearest G·C base pair is separated from the donor by two T·A steps. The appearance of two independent Marcus curves in these systems suggests that hole injection to guanine at short distances occurs in a single step.
Single-step CT of this type has been verified over short distances in Sa-linked hairpin constructs. The Sa* lifetime decreases exponentially as the length of the (A·T)n bridge separating the donor from a G or GG site increases. Such an exponential distance dependence is predicted by Marcus theory, and can be quantified using a simplified form of the Marcus–Levich–Jortner equation for non-adiabatic ET,
where kCT is the rate of the CT process under study, k0 is a preexponential factor, β is a distance decay parameter characteristic of the bridge, and d is the bridge length. In Sa-hairpin systems, β = 0.7 Å−1 for charge separation and β = 0.9 Å −1 for charge recombination.51 These values are slightly lower than those observed for protein-mediated electron transport (β ≈ 0.9 Å−1), but are much higher than values obtained for DNA CT in other systems (β ≈ 0.1 Å−1).46,54 This discrepancy may be explained by differences in coupling between the donor and the π-stack in these various systems.
Another feature of these assemblies is the high rigidity of the base with Sa fixed in the hairpin arrangement. This rigidity may be important to keep in mind, given that base dynamics also appear to regulate DNA CT. For instance, femtosecond spectroscopic experiments have shown that reorientation of an intercalated ethidium donor into a CT-active conformation precedes CT.55 In those studies, the ethidium moiety was attached to the DNA duplex through an extended covalent tether, allowing for a high degree of conformational flexibility. Rigidifying the ethidium donor by incorporating it directly into the backbone decreases the rate of CT by two orders of magnitude, signifying the importance of conformational flexibility for efficient CT.56
The rates of charge injection and charge recombination can also be modulated by incorporating modified bases into the duplex. For example, replacing G by 7-deazaguanine (Z) or 8-oxo-7,8-dihydro-2′-deoxyguanosine, both of which have lower oxidation potentials than guanine, increases the lifetime of the charge-separated state.52,57 Modification of the base opposite low potential sites can also alter the lifetime of the charge-separated state. This effect is observed in systems utilizing a NI donor, where addition of 5-bromocytosine opposite G or 5-bromouracil opposite A increases the lifetime of the NI•− intermediate.58,59 Presumably, halogen incorporation into the structures of C and U lowers the oxidation potential of complementary G and A sites, increasing the lifetime of the charge-separated state.
In duplexes containing more than one low potential site, charge injection may be followed by charge equilibration. In the absence of a spectroscopic reporter for this process, the rate of CT between low potential sites can only be deduced with the assistance of elaborate kinetics models in which the charge is assumed to hop via superexchange from one low potential site to another without occupying intervening bases.49 This model has been applied in various contexts to gain insight into how variations in sequence affect the rate of charge equilibration. In one example, rates of 5 × 107 s−1 and 5 × 106 s−1 were extracted for HT and reverse HT, respectively, from G to GG.60 In another example, the rate of hopping from G to GG over a single A step was calculated to be faster than hopping over AA or T steps.61 It should be noted that such kinetic models greatly simplify the mechanics of DNA; the effects of charge delocalization, base dynamics, and charge occupation at higher potential sites are ignored. Still, the values obtained from these models are informative as to the range of reactivity.
2.4 DNA CT between organic probes
The incorporation of strongly absorbing charge acceptors such as stilbenediether (Sd) and phenothiazine (PTZ) into DNA CT systems allows one to follow charge arrival at the acceptor by TA spectroscopy. Such probes complete the temporal landscape from charge injection to charge acceptance, enabling researchers to accurately time the duration of charge occupation on the bridge. The additional kinetic information obtained from measurements of the formation and decay of acceptor-based transient intermediates also increases the accuracy of estimations for rates of charge hopping within the bridge. While the use of strong chromophores as charge acceptors undoubtedly improves the quality of information gained in DNA CT studies, these systems still hold limitations: diagnostic absorption bands in the visible region often overlap, making it difficult to deconvolute the donor and acceptor signals; TA spectroscopy is inherently less sensitive than luminescence methods, so the rates determined using TA spectroscopy are not as precise as those obtained using time-resolved luminescence; and poor interactions between the donor–acceptor pair and the DNA bridge can decrease the apparent rate of CT. Nonetheless, the use of low-potential chromophores as terminal charge acceptors has been beneficial to the study of DNA CT.
Just as in donor-linked hairpins, the rate of charge injection in stilbene-capped hairpin constructs is sensitive to sequence and hairpin length. For example, while the rate of charge injection from Sa into bridges consisting of (T·A)n steps are invariant to distance, the introduction of G·C steps into the sequence imparts a slight distance dependence.62 This was observed in carefully conducted ultrafast spectroscopic studies where the TA profile corresponding to charge injection was fit to a biexponential model. For injection into a (T·A)6 bridge, the faster component with a rate of 1.0 × 1012 s−1 was assigned to hole injection into the bridge, while the slower component with a rate of 2.6 × 1010 s−1 was assigned to solvent and nuclear relaxation of the contact ion pair.63 For charge injection into (T·A)n(G·C) sequences, these rates varied with bridge length from 5.3 × 1011 and 2.6 1010 s−1 for n = 1 to 1.2 × 1011 and 1.9 × 1010 s−1 for n = 4. The collected data for this series corresponds to β = 0.19 Å−1 for the fast component and β = 0.05 Å−1 for the slow component. Another sequence-dependent effect is seen upon the incorporation of AT repeats into the bridge. Similar to donor-linked hairpin systems, the rate of charge injection into Sd-capped hairpin bridges consisting of only A is several-fold higher than charge injection into bridges containing AT repeats of similar length.64 The sequence affects the yield of injection as well as the rate, as measured by the transient absorbance of the acceptor cation radical. This sequence dependence was observed in diblock oligomers, where the donor and acceptor were separated by bridges of the type (A·T)n(G·C)m. In such systems, the yield of charge separation is improved 5-fold over constructs consisting of only (A·T)n+m tracts65. Similar sequence and distance effects were observed in systems utilizing naphthaldiimide (NDI) or acridine as the hole donor and PTZ as the charge acceptor.66,67
With the additional kinetic information provided by the acceptor chromophore, calculations of the hopping rate within the bridge were revisited. Using an unbiased random walk model, the rate of G-to-G hopping through G tracts in Sa–Sd systems was determined to be 4.3 × 109 s−1, slightly higher than 1.2 × 109 s−1, the rate calculated for A-to-A hopping by the same method.68 In NDI–PTZ systems, the rate constant for hopping between neighboring A sites was 2 × 1010 s−1.69 These values are several orders of magnitude higher than the rates obtained for hopping from G to GG in donor-linked hairpin systems. It would be interesting to compare calculated hopping rates through identical bridges in the presence and absence of the Sd hole acceptor.
The rate of hole arrival at the acceptor is generally taken as the rate of formation of the acceptor radical cation as measured by TA spectroscopy. Takada et al. used NI as a donor and PTZ as an acceptor to observe this process over a distance of 100 Å.70 In these systems, HT rates decreased with increasing n in (GA)n sequences from 570 × 105 for n = 2 to 6.2 × 105 s−1 for n = 12. Transfer was retarded through (GT) n sequences over the same length regime, decreasing as 11 × 105 for n = 2 to 1.2 × 104 s−1 for n = 12. Importantly, the introduction of an A·C or G·T mismatch amidst (GA)n repeats decreased the rate of charge arrival 100-fold.
The rate of charge arrival at the acceptor depends on the length of the bridge. Interestingly, this distance dependence is not strictly exponential, as was observed for charge injection. In femtosecond TA measurements of DNA CT through A tracts of varying lengths in stilbene-capped hairpins, the strong, exponential distance dependence in rate and yield observed over 1 to 4 base pairs gave way to a very weak distance dependence from 5 to 7 base pairs.71 This change in behavior is presumably due to competition between charge recombination, which is efficient at short distances, and hole trapping. Importantly, for CT over a bridge of only 1 or 2 base pairs, charge injection and charge arrival are observed to occur simultaneously; that is, CT is accomplished in a single, coherent step. This transition from highly distance-dependent coherent exchange to distance-independent bridge-mediated hopping is similar to the pattern observed by Giese et al. for HT between G and GGG.32 Despite the decrease in CT yield with increasing distance, CT through long bridges can take place at remarkably fast rates. In NI–PTZ systems, PTZ•+ began forming within the laser lifetime (<10 ns), even over 32 base pairs (108.8 Å).66
The results of several experiments implicate the difference in HOMO energy levels (ΔHOMO) between bases in the DNA bridge as a primary factor influencing the rate of DNA CT. For example, replacing adenine with Z or diaminopurine (D) in mixed sequences increases the efficiency of CT several-fold.72,73 In a more extensive study, a good correlation was observed between the rate of formation of PTZ•+ in NI–PTZ systems and ΔHOMO between the low potential sites G, D, and 8-bromoguanine, and the high potential sites 2-aminopurine (Ap), A, 8-bromoadenine, and thymine when low and high potential sites were placed in alternating sequences.74 In this study, it was the difference in HOMO energies, rather than the absolute HOMO energies, that affected the rate of CT. This observation parallels that of Lewis et al. for donor-linked hairpins in which the distance dependence of CT correlated with the donor–base energy gap rather than with the absolute energies of the donor and the accepting base.75
The effect of mismatches on the rate of CT has been observed in several systems, some of which are mentioned above. A depression in CT rates upon the incorporation of mismatches has also been calculated using hopping models. Here, hopping to G-containing mismatches is slower than hopping to G·C sites, and hopping rates over T·T, A·A, and A·C mismatches roughly correlate with the thermodynamic stabilities of those mismatches.76 Additionally, in some NI–PTZ systems the formation of PTZ•+ was not observed when an A·A mismatch was placed within the bridge.66 Finally, in single molecule fluorescence correlation experiments employing bright fluorophores as charge donors, the fluorescence correlation lifetime increased 2- to 13-fold upon the introduction of a mismatch, depending on the identity and position of the mismatch, indicating a lack of DNA CT.77 Based on the large effects that mismatches have on the dynamics of DNA CT, kinetic measurements have been proposed as a method to discriminate well matched from mismatched DNA.76
2.5 DNA CT between bases
Although a large number of experiments have been conducted using chromophore-linked hairpins and end-capping probes, modest electronic coupling between probes bound by these methods and the DNA base stack may influence the measured rates and yields of CT. For this reason, it is desirable to use CT probes that are better integrated into the base stack. Luminescent DNA base analogs have proven especially useful in this respect. Unlike tethered intercalators, the positions of such probes are well defined, and in contrast to end-capping probes, the electronic coupling between these donors and neighboring bases is just as strong as coupling between the natural bases. Photoactive base analogs therefore serve as sensitive probes for DNA CT.
The prototypical photoactive base analog is Ap. In DNA, Ap forms a well stacked base pair with T, providing a stable fluorescent reporter of DNA dynamics and CT. Importantly, the redox potential of Ap* (E1[Ap*/Apred] ≈ 1.5 V vs. NHE) is high enough to oxidize guanine.78 This property is exemplified in a series of experiments involving Ap* fluorescence quenching by G, where biexponential fluorescence decay rates increase from 1.9 × 109 and 3.2 × 108 s−1 for unquenched Ap* to 5.6 × 109 and 7.7 × 108 s−1 for strands in which Ap and G are separated by 6.8 Å.78 Quenching over various distances has also been examined. Using the decrease in Ap* fluorescence quenching with increasing distance to the nearest G site as a proxy for charge separation, a β-value of 0.14 Å−1 is obtained. In contrast to the shallow distance dependence observed for CT between Ap* and G, for CT between excited 1-N6-ethenoadenine (Aε) and G, β = 1.0 Å−1. This large difference is attributed to the difference in stacking: Ap forms strong hydrogen bonds with its complementary T and strong π-stacking interactions with the bases surrounding it, while Aε is sterically bulky, does not pair with T, and adopts a poorly-stacked conformation. The low β-value observed for CT between Ap* and G also contrasts with the efficiency of CT between Ap* and Z, for which β = 0.36 Å−1. The distance dependence of CT therefore varies with the energy difference between the donor and the acceptor in Ap–G systems. The directionality of CT also affects the efficiency. The yield of damage induced by a charge moving in the 5′- to 3′-direction shows a shallower distance dependence than a charge moving in the 3′- to 5′-direction.79 Finally, differences in the efficiency of DNA CT are also observed depending on whether the donor and acceptor are placed on the same strand. When G is placed on the opposite strand to Ap, quenching is less efficient, showing that intrastrand DNA CT is preferred. This conclusion was also drawn in other experimental systems,61 and is corroborated by theory.80
The use of well stacked base analogs as donors has enabled the careful examination of the effect of temperature on DNA CT. While previous G oxidation assays had shown that an increase in temperature results in a higher yield of damage at distant sites, temperature-based kinetics experiments involving intercalating or end-capping probes are lacking.35 After all, an increase in temperature would likely affect the dynamics of an extrinsic probe differently than it would the dynamics of the base stack, leading to complications in data interpretation. Any changes in dynamic motion experienced by base analogs such as Ap, on the other hand, are expected to be identical to changes in dynamic motion occurring in the rest of the base stack. A series of luminescence quenching studies utilizing base analogs as intrinsic probes verified that increased dynamical motion facilitates DNA CT. In femtosecond TA spectroscopic experiments, the rate of CT between Ap* and G increases with increasing temperature to a maximum value of 1.2 × 1011 s−1 at the melting temperature of the duplex.81 This trend extends to low temperatures: DNA-mediated quenching of Ap* luminescence is strongly suppressed at 77 K.82 Similar results were also observed in steady-state fluorescence quenching studies.83 From these experiments, it has become clear not only that increased dynamics enhance CT, but that dynamic motion of the base stack is actually required for DNA CT to take place at all.
2.6 The generality of DNA CT in solution
These principles are expected to operate not only in HT, but also in DNA-mediated ET. Indeed, in experiments involving the charge donor [Ir(ppy)2(dppz)]+ (ppy = 2-phenylpyridine), a metallointercalator that is competent for DNA reduction as well as oxidation from the excited state, both processes occur with a similar, shallow distance dependence.84-86 A 5′- to 3′-direction preference is also observed for ET between photoexcited 5-(naphthalen-1-ylethynyl)uracil to 5-bromouracil, similar to the preference observed in HT from Ap* to G.87 In ET experiments involving excited tetrathiophene as an electron donor and DPA as an electron acceptor, the rate of hopping through T tracts was measured as 4.4 × 1010 s−1, which is faster than hopping through A or G tracts.88 Similarly, in terthiophene-linked hairpins, charge separation and recombination rates were estimated as ~1011 and 1010 s−1, respectively.89 In this work, for hairpins with sequences CnT5–n, increasing n led to faster charge separation, presumably due to charge delocalization within the C tract. It appears, therefore, that DNA HT and DNA ET are analogous processes. These similarities observed between DNA-mediated HT and ET in solution-based experiments strongly suggest that the conductive properties of DNA are general.
3. DNA CT on surfaces
In contrast to solution-based strategies for the study of electron transfer, surface platforms provide a solid handle that can be used to anchor study molecules into defined conformations.90 In the case of electrochemistry, this handle is the electrode surface which replaces the donor and allows for the controlled application of a potential.91 Importantly, while solution studies necessarily involve excited-state measurements, electrochemistry allows for the study of ground-state electron transfer processes. Additionally, electrochemical platforms are highly compatible with the aqueous, buffered environments that biomolecules require to maintain their native, biologically relevant structure. Many groups have taken advantage of these benefits for the study of electron transfer processes in diverse biomolecules and for the construction of electrochemical biosensors.92-97 Beyond electrochemistry, some measurements of DNA CT have also been performed on surfaces using photoelectrochemistry98-100 and fluorescence.101 Like solution studies, these experiments require photoexcitation and produce excited-state measurements.
In this section, we focus on experiments that involve ground-state, surface measurements of DNA CT. Specifically, we describe the extensive work by our group with DNA-modified electrodes using a variety of redox probes (Fig. 1, Scheme 1). The isolation of a DNA-mediated path as the only route for CT to the redox probe in these electrodes has allowed for our directed study of DNA CT and application of this property for electrochemical biosensing. In this work, we have observed the same characteristics for DNA CT that have been identified in solution studies. Namely, redox probes must be well coupled to the DNA π-stack to participate in DNA CT, and CT that is truly DNA-mediated is exquisitely sensitive to the structural integrity of the DNA path that extends from the electrode surface to the redox probe. Additionally, DNA CT can occur efficiently over very long distances.
3.1 DNA-modified electrodes
In our laboratory, we have developed and thoroughly characterized DNA-modified electrodes for the study of ground-state DNA CT and for use in biosensing applications that exploit this property.102,103 In our platform, DNA duplexes are modified with a linker on one end that allows for their self-assembly into monolayers on an electrode surface. Most commonly, we utilize alkanethiol linkers104 for the attachment to gold electrodes, but for experiments that require a wider potential window we employ pyrene linkers105 that allow for attachment to graphite electrodes. We have thoroughly characterized the structure of these assemblies with a variety of techniques including radioactive labeling,104-106 atomic force microscopy,105-108 and scanning tunneling microscopy.109 From these studies, we have confirmed that film density can be controlled by Mg2+, which promotes dense packing.104-106Additionally, the duplexes adopt an upright orientation with an angle relative to the surface that can be modulated by the applied potential; in the absence of a potential, duplexes are oriented at a ~45° angle to the surface and with the application of positive or negative potentials, the anchored duplexes are attracted to or repelled by the surface, respectively, from this set point.105,107 Thus, the DNA duplexes function as highly sensitive extensions of the electrode surface into solution.
In order to study CT that is mediated by these DNA extensions, a redox-active probe moiety is incorporated at or near the end of the DNA that is distal from the surface. For this purpose, we have utilized noncovalent104,105,110-116 and covalent106,117-125 redox probes as well as DNA-binding proteins that are redox-active.126-133 These different probes are illustrated in Fig. 4. After assembly of the DNA monolayer, the modified electrode is treated with a backfilling agent, such as mercaptohexanol, to passivate the surface against direct reduction of the redox probe and remove nonspecifically adsorbed DNA. In the completed DNA-modified electrode, CT is mediated from the electrode surface to the redox probe via the intervening path of well stacked DNA bases. Importantly, experiments with this platform are all performed in aqueous, buffered solution such that the DNA maintains a native, CT-active conformation.
Fig. 4.
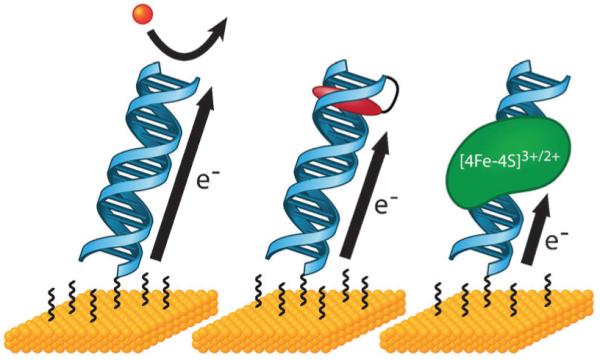
DNA-modified electrodes allow for the measurement of DNA CT to diverse redox-active species. As long as probe molecules are well coupled to the DNA π-stack, DNA-modified electrodes can be used to measure DNA-mediated redox processes of a variety of noncovalent (left) and covalent (center) redox probes, as well as proteins with redox-active cofactors (right). Attenuation of the redox signal by the incorporation of a mismatch or other structural perturbation to the π-stack can be used as a diagnostic to determine if the observed signal is indeed DNA-mediated.
3.2 Noncovalent redox probes
In the development of this platform, noncovalent, redox-active small molecules were initially used as probes for DNA CT. For experiments with such probes, electrodes with densely packed DNA films are required in order to prevent access of the freely diffusing probe molecules to the surface. In initial work, we showed that for such films, noncovalent probes are restricted to binding the portion of the DNA duplex at the top of the monolayer, near the film-solution interface.104,110 Since single base mismatches had been shown to dramatically interfere with DNA CT in solution studies, despite the lack of disruption to the global duplex structure, we investigated this phenomenon with DNA-modified electrodes. The restricted binding of the noncovalent probe at the top of the duplex ensures that single base mismatches located in the middle of a 15-mer duplex will necessarily be on the DNA path between the surface and redox probe. We compared the mismatch sensitivity of the DNA groove binder [Ru(NH3)6]3+ to a diverse array of small molecule DNA intercalators including methylene blue (MB), [Ir(bpy)(phen)(phi)]3+, and daunomycin.110 As in solution studies, we observed that a close interaction between the probe and the DNA π-stack is essential for reduction that is DNA-mediated and thus sensitive to a mismatch; reduction of the DNA intercalators was significantly inhibited by the presence of a mismatch while reduction of the DNA groove binder [Ru(NH3)6]3+ was unaffected by mismatches.110
A more in-depth look at the behavior of [Ru(NH3)6]3+ as compared to MB in this platform clearly illustrates how the DNA functions as an extension of the electrode surface for molecules that can properly access the DNA CT pathway of the π-stacked bases.111 For both [Ru(NH3)6]3+ and MB, high salt conditions decreased the number of molecules that were reduced, reflecting the inhibition of both electrostatic and intercalative DNA binding modes, respectively, with increased ionic strength of the solution. This result indicates that the reduction of both probes is dependent on their tight binding to DNA. However, polymerization of 2-naphthol to completely passivate the electrode surface against direct probe interaction revealed fundamental differences in the pathways by which these probes are reduced. While reduction of MB was minimally affected by this total passivation (~3% signal reduction), reduction of [Ru(NH3)6]3+ was decreased significantly (~70% signal reduction).111 Thus, while [Ru(NH3)6]3+ is reduced directly at the electrode surface and the DNA functions as simply a charged guiderail that helps to facilitate its diffusion toward the surface, MB does not require access to the surface for its reduction. Instead, the ability of MB to intercalate into the DNA duplex and interact directly with the π-stacked bases allows it to access a DNA-mediated pathway for reduction that extends through the DNA, above the passivated surface. Unlike [Ru(NH3)6]3+ that cannot access DNA CT, for the well coupled MB, the DNA functions as an electrical conduit through which charge is conducted from the surface directly to the distally bound probe.
Additional work with this platform showed that not only must the redox probe be well coupled into the DNA π-stack, but the proper stacking of the bases themselves is also critical for DNA CT to occur. Further work with the MB probe showed that its DNA-mediated signal could be amplified in an electrocatalytic cycle with ferricyanide110,112 and used to detect all base mismatches.113 Notably, G·A mismatches, the most structurally stable type of mismatch, cannot be detected without electrocatalytic amplification.113 Additionally, we used this strategy to detect base lesions and found that while lesions that do not cause duplex destabilization or inhibit base stacking do not disrupt DNA CT, those lesions that do cause such structural perturbations are sensitively detected by an attenuation of DNA CT to the MB redox probe.114 We investigated other conformations of DNA with this platform and found that like B-form DNA, A-form DNA (DNA–RNA hybrid duplexes) can also efficiently mediate charge through stacked bases, and mismatches within its sequence are readily detected.115 In contrast, Z-form DNA, which is more rigid and has significantly less intrastrand base stacking than B- and A-form, shows significantly attenuated DNA CT.115 Incorporation of a 3′-endo-locked nucleotide into B-form DNA, which is known to disrupt base stacking, causes signal attenuation similar to that caused by the incorporation of a mismatch.116 However, incorporation of this modified nucleotide into A-form DNA, which can better accommodate its structure into the base stack, does not show attenuation in DNA CT.116
3.3 Covalent redox probes
The development of covalent redox probes for the DNA-modified electrode platform opened the door for more precise characterization of DNA CT, as we could define the location of the probe in the duplex. Initial work made it clear that, like noncovalent probes, electronic coupling to the π-stack was absolutely required for the DNA-mediated reduction of covalent probes. For MB that is covalently attached by a flexible alkane linkage to the distal end of the DNA, a redox signal is observed for low ionic strength conditions that permit intercalation of the tethered MB into the duplex.111 However, in a high salt environment that discourages intercalation, no redox signal for MB is observed despite its attachment to the DNA.111 This result mirrors the ionic dependence of the DNA-mediated reduction of freely diffusing MB and again highlights the importance of the direct electronic connection between the redox probe and the π-orbitals of the stacked DNA bases for DNA CT to occur; intercalation is still required for reduction of the covalent MB probe as the σ-bonds of the alkane linkage alone do not provide this electronic connection.
Intercalation is not the only mechanism to establish this electronic coupling to the π-stack. In a study that investigated different linkages to covalently attach the redox probes anthraquinone (AQ) or 2,2,6,6-tetramethylpiperidine 1-oxyl (TEMPO) directly to a modified uridine base, rigid acetylene linkages allowed for the DNA-mediated reduction of both probes that was sensitive to the incorporation of mismatches.117 In contrast, alkane linkages for both probes resulted in significantly weaker redox signals that were not influenced by mismatches. In these cases, although the acetylene holds the probe rigidly away from the DNA and prevents interaction directly with the base stack, this conjugated linkage still provides a connective path that electronically couples the probe to the base stack. The alkane linkages do not create this electronic conjugation and, in contrast to the previous example with MB, do not structurally allow for direct interaction between the redox probe and DNA π-stack. From these examples we see that, in an added level of complexity to noncovalent probes, the covalent linkage now functions as the critical gatekeeper that can either promote or prevent the redox probe from achieving a CT-active conformation. In addition to MB, AQ, and TEMPO, we characterized the linkages and conditions necessary for the DNA-mediated reduction of a variety of other covalent redoxactive moieties including daunomycin,106,118-120 disulfide bonds,121 Redmond Red,122 and Nile Blue.123-125
The use of covalent probes eliminates concern about the direct surface reduction of freely diffusing probes, making it possible to use low density DNA films and expanding this platform to include hybridization and protein binding studies. In developing this technology for the electrochemical detection of DNA-binding proteins and their activity, we found that proteins that perturb the DNA π-stack upon binding cause a dramatic attenuation in the DNA-mediated signal of the distally attached redox probe.106,123 We sensitively measured this effect for the DNA methyltransferase HhaI and uracil-DNA glycosylase, which both flip a base out of the DNA base stack upon binding, as well as the TATA-binding protein (TBP), which kinks the DNA by 90°. Proteins that do not bind DNA, such as BSA, or proteins that bind but do not distort the DNA such as the PvuII restriction enzyme bound to its methyl-protected restriction site do not cause this signal attenuation. As an important and informative control, we compared the effect of binding by wild-type HhaI and the mutant Q237W of HhaI.106 Wild-type HhaI inserts Gln237 into the DNA base stack to flip out the target base, and this disruption of the DNA CT path results in significant signal attenuation of the redox probe. However, binding by the Q237W mutant of HhaI results in only minimal attenuation of the redox signal. In this case, the Q237W mutant inserts Trp into the base stack instead of Gln, and this aromatic, heterocyclic amino acid fills the place of the flipped base in the π-stack, allowing for the DNA-mediated flow of charge to the probe. This control underscores the central role of the stacked DNA bases in forming a conduit for DNA CT.
Covalent tethering of the redox probe also makes it possible to use this platform for more detailed, fundamental characterization studies of DNA CT. The directionality of the mismatch effect on DNA CT was clearly established; incorporation of a C·A mismatch in the DNA between the gold surface and a covalent daunomycin probe causes significant signal attenuation, while the reverse case, in which the mismatch is located above the daunomycin relative to the surface, shows no signal attenuation.120 The incorporation of nicks in the sugar–phosphate backbone of the duplexes that make up the DNA-modified electrodes causes no signal attenuation of a covalent daunomycin probe.119 In contrast, the incorporation of a single base mismatch causes significant signal attenuation for both intact and nick-containing DNA, again illustrating that DNA CT propagates through the stacked base pairs, not the sugar–phosphate backbone.119
Most significant among these fundamental studies, covalent tethering opens the door for ground state distance dependence studies of DNA CT that are not possible in solution platforms or with noncovalent electrochemistry. Initial experiments in this area demonstrated that the covalent placement of daunomycin at different positions along a 15-mer duplex (50 Å) does not influence the signal intensity or splitting of the cathodic and anodic peaks as measured by cyclic voltammetry.120 For DNA CT over similar DNA distances, we investigated the contribution of the alkanethiol tether that attaches the DNA to the gold electrode on the rate of DNA CT and found that the number of methylene units in the tether absolutely dominates the observed CT rate.118 These results echoed the shallow distance dependence that had been observed in solution experiments, and we worked to push our measurements of DNA CT to longer distances. Our extension of these DNA-modified electrodes from single electrodes to multiplexed chips made it possible to run the more complex experiments and precise side-by-side controls that are needed for reliable measurements of DNA CT over very long distances.124 Using this multiplexed platform, we investigated the distance dependence of DNA CT over 100 base pairs (340 Å) to a covalent Nile Blue redox probe.125 Remarkably, we observed the same signal size and attenuation from a single base mismatch for both 100-mer and 17-mer DNA duplexes. Importantly, cyclic voltammetry of the 100-mer displayed the broad, split cathodic and anodic peaks that are characteristic of DNA-mediated processes and indicate that probe reduction does not occur by direct surface contact. Additionally, efficient cutting by the restriction enzyme RsaI, as reported by the near complete disappearance of the redox signal, indicates that the 100-mer adopts an upright, biologically active conformation that is readily recognized and accessed by the protein. Kinetics measurements of DNA CT through the 100-mer and 17-mer revealed the same rate, showing that DNA CT through the 100-mer is still limited by the C6 alkanethiol tether used for both DNA lengths. Assuming that the rate through the 100-mer is no faster than the rate through the alkanethiol tether, we made a conservative estimate for β, a parameter that describes the distance dependence of the CT rate through a bridge, of DNA CT to be 0.05 Å−1.125 This remarkably shallow distance dependence for DNA CT is on the order of our measurements in solution studies.78
3.4 Proteins as redox probes
Small molecules are not the only redox-active players that can participate in DNA CT. We have identified numerous proteins with redox-active cofactors, such as [Fe–S] clusters, whose biologically relevant, DNA-bound potentials may be observed with our DNA-modified electrode platform. For these experiments, electrodes are assembled with DNA that is modified only with a thiol tether; upon protein binding, redox-active cofactors that are well coupled to the DNA π-stack serve to report a DNA-mediated CT signal.126 Like small molecule redox probes, the integrity of the DNA π-stack is critical for strong electrochemical signals from these proteins; the incorporation of intervening abasic sites, lesions, or mismatches in the DNA electrode significantly attenuate the redox signal.126-129
Such biologically integrated redox probes can be used to monitor protein activity that involves the coupling of the redox cofactor to the π-stack. For example, the flavin cofactor of the light-activated DNA repair enzyme photolyase can be used to electrochemically monitor this protein as it binds and repairs pyrimidine dimer lesions.128 Upon initial binding of photolyase to thymine dimer-containing DNA, a weak signal from the flavin cofactor is observed due to the destacked lesion that disrupts DNA CT and inhibits proper coupling of the flavin cofactor to the π-stack. Upon photoactivation, the redox signal gradually increases as photolyase repairs the thymine dimer and restores efficient DNA CT to the flavin cofactor. Following this, the redox signal gradually disappears as the protein dissociates from the now repaired DNA.
Experiments with redox-active proteins on this platform illustrate the importance of using DNA-modified electrodes to measure relevant DNA-bound potentials. The transcription factor SoxR provides an informative case.130 This bacterial sensor of oxidative stress, which initiates the expression of an array of genes to respond to oxidative stress, is activated by oxidation of its [2Fe–2S] cluster. Using bare and DNA-modified highly ordered pyrolytic graphite (HOPG) electrode surfaces, we determined that the redox potential of the cluster in SoxR shifts by 490 mV when bound to DNA.130 This important shift maintains the protein in an inactive state during normal conditions such that it only becomes activated when the cell experiences conditions of high oxidative stress. Proper coupling to the DNA π-stack is additionally important for this protein, as solution experiments have shown that SoxR can be activated from a distance by DNA CT.134 Clearly, as in the case of small molecule redox probes, strong electronic coupling to the DNA π-stack is essential to produce measurable electrochemical signals. For proteins like SoxR, however, this coupling has an added level of significance, as it can actually modulate the redox character of the protein itself, significantly impacting the function of the protein in a biological context. Furthermore, through this coupling, the protein is granted access to the DNA CT pathway and can thereby participate in the efficient, coordinated activities that can be achieved through long-distance signaling.
The profound ramifications of strong coupling to the DNA π-stack are perhaps best illustrated by our extensive work with the [4Fe–4S] cluster-containing base excision repair enzymes MutY, EndoIII, and AfUDG.126,127,131,132 In electrochemical studies of these proteins, we found that DNA-modified electrodes are essential for measuring relevant DNA-bound potentials.131 While stable, quasi-reversible redox signals at ~90 mV vs. NHE are observed for these repair proteins on DNA-modified electrodes, no redox signal is observed in the same potential window when the proteins are applied directly to a bare gold electrode. In fact, HOPG electrode surfaces, which have a wider potential window than gold, are required to measure the redox chemistry of these proteins in the absence of DNA because the potentials of the unbound proteins are shifted beyond the potential window of gold.131 Electrochemical studies of EndoIII on bare HOPG and DNA-modified HOPG reveal that DNA binding shifts the redox potential of the [4Fe–4S]3+/2+ couple by >200 mV and stabilizes the [4Fe–4S]3+ state, thereby converting the cluster into a form that is redox-active under physiologically relevant conditions. Importantly, the redox state of the cluster influences the binding affinity of EndoIII for DNA, and in this way, long-distance, DNA-mediated redox of the cluster can influence protein binding and localization.133,135
Multiple experiments following this observation support a model in which this DNA-mediated redox activity is essential for the search process that these proteins undertake to efficiently locate and repair damage in the genome.133,135 Proteins that are well coupled to the DNA π-stack and that can access DNA CT have the capacity to participate in long distance, DNA-mediated signaling to coordinate search efforts and promote the localization of repair proteins around sites of DNA damage. This model was clearly illustrated in an electrochemical study of EndoIII variants with mutations around the [4Fe–4S] cluster.136 Mutants that exhibited large electrochemical redox signals and were thus well coupled to the DNA were also proficient at localizing near DNA damage, as observed by AFM. On the other hand, EndoIII mutants that exhibited small electrochemical signals, and thus poor coupling to the DNA, did not localize near mismatches. Thus, the degree of coupling of the [4Fe–4S] cluster to the π-stack as measured with DNA-modified electrodes relates directly to the ability of the protein to utilize DNA CT for long-distance signaling to locate and respond to DNA damage.
We are just beginning to understand the role of signaling via DNA CT in the coordination of complex biological processes. Studies with the DNA helicase XPD provide some perspective on the potential magnitude of the coupling of biological redox moieties to DNA CT.129 XPD contains a [4Fe–4S] cluster and is a critical participant in nucleotide excision repair and transcription. When bound to a DNA-modified electrode, the [4Fe–4S] cluster in XPD produces a strong signal that is highly sensitive to the presence of an intervening mismatch, indicating its DNA-mediated nature. Upon subsequent addition of ATP, which is required for helicase activity, this signal increases in a manner that is both substrate-specific and not observed for a mutant that is deficient in ATPase and helicase activity. Thus, this signal increase is reporting ATP hydrolysis by XPD and reflects conformational changes in the protein that improve coupling of the [4Fe–4S] cluster to the DNA π-stack during this reaction (Fig. 5). Importantly, the potential of DNA-bound XPD is 80 mV vs. NHE, which is near the potential of base excision repair proteins and which makes it redox-active under physiological conditions. Remarkably, when its localization near DNA damage is studied by AFM, XPD is found to cooperate with EndoIII to promote redistribution around mismatches, despite the fact that these proteins are derived from entirely different organisms.136 This result highlights the universal nature of DNA CT as a mechanism for long-distance biological signaling and coordination of protein activities even, between proteins of diverse structure and function.
Fig. 5.
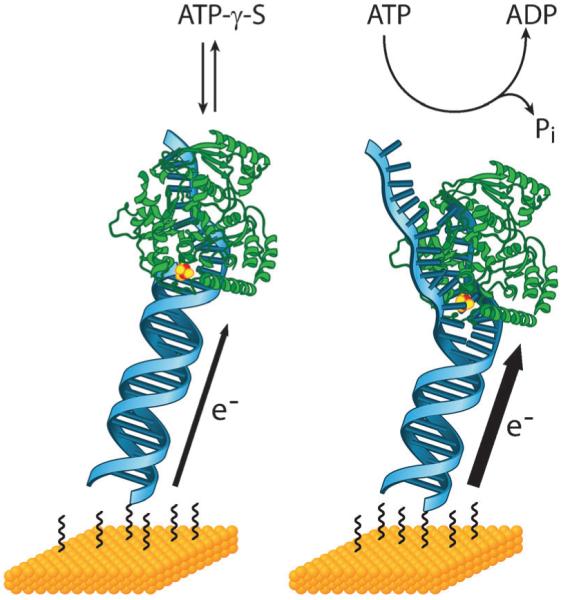
DNA-modified electrodes can be used to monitor proteins with redox-active cofactors. Here, the DNA helicase XPD which contains a [4Fe–4S] cluster is shown bound to a DNA-modified electrode. In the absence of any ATP or with the non-hydrolyzable ATP analog ATP-γ-S, a steady DNA-mediated signal from the cluster is measured (left). Upon addition of ATP, this signal increases significantly. This result reflects a conformational change in XPD during ATP hydrolysis that improves the coupling of the [4Fe–4S] cluster to the π-stack. Thus, DNA-modified electrodes report on how DNA coupling changes during protein activity and this information can provide insight into how DNA CT might be involved in the regulation and coordination of these activities.
4. DNA CT with single molecules
Conductivity measurements of single molecules allow for an understanding of the fundamental electronic properties of these molecules that cannot be achieved through bulk platforms.137 However, the study of single molecule, ground state, DNA conductivity presents a significant challenge that is clearly fore-shadowed by observations from bulk solution and electrochemical experiments; the nature of the electrical connection to the DNA and the integrity of the π-stacked duplex structure are critical to make meaningful conductivity measurements. Most initial experiments of single or few molecules of DNA by others utilized platforms with inconsistent or poorly defined electrical connections, and the DNA was not maintained in an aqueous, undamaged state.138-141 Not surprisingly, these experiments measured a full range of electrical behavior for DNA from insulating to semiconducting to conducting.138-141
Experiments by our group and others using AFM142 and STM109,143,144 of DNA films provided more consistent measurements, including low resistances for well matched DNA and increased resistance with a single base mismatch, as these methods involve better defined electrical connections for the DNA duplexes and allow measurements to be taken under aqueous conditions. However, as at least dozens of molecules can make contact with the probing tip in these experiments, these platforms did not allow for truly single molecule measurements of DNA conductivity. In this section, we describe our work with carbon nanotube (CNT)-based devices that have allowed for measurements of DNA CT in single molecules of DNA (Fig. 1). We also describe how we can use the sensitivity of this CT to detect single molecule protein binding and activity.
4.1 Carbon nanotube–DNA devices
Carbon nanotube (CNT)-based devices provide a platform that has well defined electrical contacts and has previously been used to measure the electrical properties of a variety of small molecule molecular bridges.137,145 In these devices, the CNT is connected into an electrical circuit, a gap is cut in the CNT, and a molecule of interest is covalently attached by amide bonds within the gap (Fig. 6). Importantly, these devices are compatible with an aqueous environment for DNA, and the similarity between the diameter of the CNTs and diameter of the DNA duplexes restricts covalent attachment to only a single duplex in the CNT gap. Working closely in collaboration with Colin Nuckolls’ laboratory, we have established this platform for effective, consistent measurements of single molecule DNA conductivity. To fabricate these devices, CNTs are grown on silicon wafers, and metal electrodes are patterned onto the wafer surface. After passivating the entire wafer with a layer of polymethylmethacrylate, ultra high resolution electron beam lithography followed by oxygen ion plasma is then used to cut gaps of defined width in the CNTs. This method of cutting results in carboxylic acid functionalization at each terminus of the CNT gap such that amine-modified DNA can be applied to covalently bridge the gap. The width of the gap can be tuned to precisely fit different lengths of DNA. Using these DNA-bridged devices, the source-drain current can be measured as a function of gating voltage to characterize the conductivity of the DNA spanning the gap relative to that of the carbon nanotube.
Fig. 6.

Carbon nanotube (CNT) devices allow for conductivity measurements in single molecules of DNA. In this platform, a CNT is connected into an electrical circuit (top). Then, high resolution electron beam lithography and oxygen ion plasma are used to cut a gap in the CNT that has a defined width and carboxylic acid end functionalization (center). A single, amine-modified DNA duplex of compatible length is then added and made to covalently bridge the gap by peptide coupling (bottom). Importantly, the DNA is functionalized with amines on both the 5′ and 3′ ends of just one of the strands in the duplex (shown here in blue) such that the noncovalent strand (green) may be easily exchanged for fully complementary or mismatched strands. DNA-mediated current can then be measured across this reconnected DNA–CNT device and compared to the current across the uncut CNT.
4.2 Single molecule measurements of DNA conductivity
Using these DNA–CNT devices, we determined that the resistance of DNA duplexes 6 nm in length lies in the range of 0.1–5 MΩ.146 Importantly, this range encompasses the resistance value (~1 MΩ) that would be expected through layered sheets of graphite of similar dimensions.146 Thus, with this platform, it is possible to validate the initial postulate of DNA conductivity made by Eley and Spivey in 1962 that the π-stacked bases of DNA might conduct charge in the same way as the structurally similar stacked π orbitals of graphite sheets.3 The connection of a single strand of DNA through functionalization of both the 5′ and 3′ ends of a duplex, to opposite sides of the CNT gap yields a robust device in which different duplexes, matched or mismatched, can be measured in the same device by introduction of various complementary or partially complementary strands noncovalently. From such experiments, we observed that charge flow through these devices is exquisitely sensitive to the introduction of a variety of single base mismatches, with resistances increasing ~300-fold upon hybridization of a mismatched strand.146 Additionally, these devices are readily recognized and cut by a restriction enzyme, which also shuts off the conductivity of the device. This sensitivity to mismatches and restriction activity, which mirror results seen in solution and electrochemical platforms, shows that we are indeed measuring DNA CT through single duplexes that adopt biologically relevant conformations (Fig. 7).
Fig. 7.

Single molecule measurements of DNA CT reflect perturbations to the DNA π-stack. (From left to right) Prior to cutting a gap, current flows through the CNT device. After the gap is cut, reconnection with a single DNA strand does not allow current flow; reconnection with duplex DNA is necessary to restore current flow. Incorporation of a mismatch, cutting with a restriction enzyme, and base flipping by a bound methyltransferase all shut off current flow through the device. This sensitivity to structural perturbations of the DNA π-stack is also observed in measurements of DNA CT in solution and on surfaces and indicates that these single molecule conductivity measurements are likewise DNA-mediated.
We have used this platform also to detect the single molecule binding and activity of SssI methylase.147 Like electrochemical experiments with methyltransferases,106 we showed that the binding and base-flipping action of SssI shuts off current flow through the device in a manner that is sequence-specific, cofactor-dependent, and reversible (Fig. 7).147 After initial treatment of the device with SssI, however, a subsequent treatment does not result in the same signal decrease due to the decreased binding affinity of SssI for methylated DNA. Thus, these devices sensitively report the structural changes to DNA associated with single molecule protein binding and maintain the DNA in its native form so that even the subtle addition of a methyl group can be recognized.
5. Characteristics of DNA CT inform a mechanistic description
From these complementary studies in solution, on surfaces, and with single molecules, several fundamental characteristics of DNA CT emerge: (i) the rate and yield of these reactions are heavily influenced by the base sequence and the extent of electronic coupling between the donor and the base stack, within the DNA bridge, and between the acceptor and the base stack; (ii) DNA CT occurs on the picosecond time scale, and the rate is gated by the dynamic motions of the donor–bridge–acceptor system as they move in and out of CT-active conformations; (iii) DNA CT is highly sensitive to the structural integrity of the stacked path of bases between the donor and acceptor; and (iv) the distance dependence for long-range CT is very shallow, and efforts to quantify this distance dependence are often limited by the platform used to make measurements rather than by the length of the DNA strand. A mechanistic description of DNA CT must reflect characteristics that are conserved for all measurements of DNA CT, regardless of platform type.
Two kinetic models that have been considered to describe DNA CT are superexchange (coherent) and localized hopping (incoherent). In superexchange, the orbitals of the DNA bridge are higher in energy than the donor and acceptor such that charge must tunnel through and only virtually occupy the DNA bridge.47 In localized hopping, the orbitals of the DNA bridge are close in energy to the donor and acceptor, so charge hops between low-potential sites and transiently occupies discrete base orbitals. We have recently reviewed the issues surrounding a mechanistic description of DNA CT in great detail.148 Here, we summarize the inconsistencies that arise in an attempt to reconcile the characteristics of DNA CT that have been observed experimentally with proposed CT mechanisms. We also identify additional mechanistic components that must be included in a suitable description of DNA CT.
5.1 Inconsistencies with superexchange and localized hopping models
In superexchange processes, the probability of tunneling and virtual occupation of the bridge becomes less and less favorable with increasing distance between the donor and acceptor. Thus, the rate of CT by superexchange is predicted to decrease exponentially as this donor–acceptor separation distance increases.47 Such an exponential distance dependence is only observed for DNA CT over short distances. In fact, both solution and surface experiments indicate that the distance dependence of DNA CT becomes very shallow for CT over long distances. Remarkably, measurements of DNA CT to a covalent redox probe over 34 nm on DNA-modified electrodes exhibit signals with the same magnitude and degree of attenuation from an intervening mismatch as DNA CT measurements over 5 nm, indicating that even over a distance of 34 nm, the rate of CT is still limited by the rate of tunneling through the alkanethiol linker that connects the DNA to the electrode surface.125 A conservative estimation for β of 0.05 Å−1 derived from these and other experiments is wholly incompatible with superexchange in DNA.125,148
By replacing the long, steeply distance dependent steps of superexchange with short hops that actually occupy the bridge, the distance dependence of a charge hopping mechanism becomes much more shallow.47,149 In this model, short hops occur between low-potential G sites along the DNA.150 Several lines of evidence highlight the deficiencies in such hopping models, however. For example, limiting transfer to hopping between guanine sites alone is not sufficient to explain CT through adenine tracts151 and several lines of experimentation have suggested instead that DNA CT occurs through a combination of hopping between G sites and tunneling through A·T base pairs.149,151-156 In support of this mechanism, Giese and co-workers noted a decrease in the yield of G oxidation with increasing lengths of A-T mixed sequences separating guanines.153-156 Importantly, in experiments involving G oxidation over various lengths of T, A, and mixed A-T bridges, it was the flexibility of the intervening sequence, not the length of the tract, that was the dominant factor in determining the oxidation yield.34 Additionally, the introduction of a GC segment within TA tracts actually decreases the yield of DNA CT.34 The exquisite sensitivity of DNA CT to single base mismatches that has been observed for all platforms is also poorly explained by hopping mechanisms. The suggestion that G-containing mismatches interrupt DNA CT by proton abstraction156 are inconsistent with experimental observations in which the degree of CT attenuation correlates with the thermodynamic destabilization of the mismatch.113 Instead, the idea that these structural distortions decrease the likelihood for DNA to adopt a well stacked conformation can be used to more universally explain the attenuation in DNA CT that is observed for a variety of base lesions114 and protein binding events.106,123
An additional inconsistency with the localized hopping mechanism is routinely observed in our electrochemistry experiments where the potentials that are applied for reduction of the small molecule redox probe or [Fe–S] cluster-containing protein are far below the reduction potentials of the bases (up to 1 V difference).103 Given this discrepancy, charge injection to an isolated base for localized hopping would be extraordinarily slow, at least 4–5 orders of magnitude slower148 than the linker-limited rate of 30 s−1 that is measured through our DNA-modified electrodes.117,120,125 Thus, to account for the consistent observation of DNA CT with our electrochemical platform, the mechanism of the CT process must necessarily involve a decrease in the energy gap between the Fermi level of the electrode surface and the oxidation potential of the individual bases. Together, the experimental results observed across several platforms demonstrate that localized hopping models are insufficient to accurately describe the dynamics of DNA CT.
5.2 Considering base dynamics in a mechanism for DNA CT
Neither the superexchange nor the localized hopping models include provisions for the profound impact that structural dynamics have on mediating DNA CT. From our early observations on the effect of sequence-dependent flexibilities of DNA CT, we proposed that long distance CT might occur through hopping between delocalized domains.34 The extent of delocalization, and the resulting physical definition of a domain, would clearly depend heavily on the dynamic motions that allow segments of the assembly to achieve CT-active conformations. Femtosecond spectroscopy experiments in solution provided strong support that dynamic motions within the DNA structure actually gate DNA CT; the CT rate is determined not by individual bases but by the simultaneous alignment of multiple components of the assembly, including the bases, the donor, and the acceptor, into CT-active conformations.55
The formation of discrete domains of delocalized charge was illustrated in greater detail by solution fluorescence studies in which we observed a periodic length dependence for the quenching yield of photoexcited Ap by G across A-tracts.83 That CT through the DNA duplex essentially encounters a gate at each sequential length of 3–4 base pairs indicates that this length is ideal for the formation of a CT-active, delocalized domain.83 A similar periodic length dependence was also measured for CT between photoexcited [Rh(phi)2(bpy’)]3+ and CPG.157 In a recent study we observed that the contribution of coherent and incoherent mechanisms to DNA CT is actually dictated by the capacity of different lengths of DNA to form delocalized domains. In this work, we studied CPG oxidation by photoexcited Ap across A-tracts and showed that for DNA CT over a full turn of the DNA helix, 7–8 base pairs when 2-aminopurine is included, a coherent CT mechanism is favored. For segments that are not integer multiples of the 3–4 base pair length that is ideal for the formation of a delocalized domain, the CPG oxidation yield decreases, indicating that CT must switch from a coherent to incoherent mechanism.158
In addition to clarifying the role of base dynamics in DNA CT, charge delocalization over multi-base domains helps explain why such severe attenuation of DNA CT is observed for lesions, mismatches, and protein-binding events that distort the DNA π-stack. These perturbations to the natural stacking within DNA cause the affected bases to preferentially adopt un-stacked conformations. When it comes to the formation of delocalized domains to facilitate CT, a primarily un-stacked base functions like a rotating disc on a multi-disk combination lock that is stuck on the wrong number. Although the other disks might turn fluidly to the correct combination the disk that is stuck in the wrong orientation will still prevent the lock from opening. Likewise, the bases surrounding a lesion, mismatch, or bound protein may be free to move into CT-active conformations, but the un-stacked base disrupts the formation of a domain over which charge could delocalize, dramatically shutting off DNA CT. A delocalized domain component to the mechanism additionally provides an explanation for the fast CT rates that are measured electrochemically; states in which the charge is delocalized over large domains would necessarily be lower in energy than the states in which the charge is localized on an individual base, thus enabling charge injection at the applied potentials used in electrochemistry experiments.
6. Conclusions and the next platforms for DNA CT
Studies of DNA CT with photooxidation, spectroscopy, and electrochemistry, in solution, on surfaces, and with single molecule techniques, and using a variety of donors and acceptors have established a solid, experimentally-based foundation to understand this process. These complementary vantage points are collectively valuable as they serve to validate shared characteristics and cast light on pieces of the DNA CT puzzle that are only visible from a particular experimental platform. Despite the diversity of platforms that we have explored, we consistently observe a common set of characteristics for CT processes that are mediated by DNA. Namely, the electronic coupling of the donor and acceptor to the π-stack of DNA is required to access DNA CT. The CT itself is highly sensitive to the structural integrity of the stack of bases between the donor and acceptor pair. For donors and acceptors that are well coupled to structurally undisturbed, undamaged DNA duplexes, the DNA can mediate CT that is rapid and has an extremely shallow distance dependence which allows this process to efficiently occur over very long distances. Additionally, this rapid rate is gated by the dynamic motions of the bases, donor, and acceptor as they move in and out of CT-active conformations.
Despite these consistent observables, a mechanism for DNA CT is still not well defined. DNA CT clearly does not fit within the bounds of either superexchange or hopping models. Our experiments suggest that transient delocalization of charge across multi-base domains must necessarily play an important role. Given the dynamic and structural complexity of the DNA molecule and the variability introduced by different sequences, donors, and acceptors, it is likely overly restrictive to confine this process to a single mechanistic description. Instead, as we continue to shed light on DNA CT from diverse experimental viewpoints it will be important to validate known characteristics and integrate new observations into a mechanistic understanding that is consistent with the complexity of this process.
Standing upon the strong foundation that was built by these solution, surface, and single molecule platforms in the pursuit of a mechanistic understanding, we are now well equipped to study and utilize DNA CT in two exciting new platforms: living organisms and molecular electronics. Indeed, our efforts to answer the question “How does DNA CT work?” has not only yielded a multifaceted understanding of this complex process but also inspired the fascinating questions that propel us toward these bigger platforms: How does biology utilize DNA CT to coordinate proteins in their efforts to meet complex cellular challenges? How can we utilize the unique conductive properties of DNA CT in the development of nanoelectronic devices? Although more work is clearly still needed to construct a fuller mechanistic understanding, what we already know about DNA CT indicates that these new platforms are promising investments in elucidating and applying this chemistry.
Acknowledgements
We are grateful to the NIH (GM49216 and GM61077) for their support of this work. We thank also our coworkers for their great efforts, sometimes against the tide, in carrying out their experiments.
Biography

Natalie B. Muren
Natalie B. Muren received her B.A. in chemistry from Willamette University in 2006, where she studied aminoglycoside antibiotics in the laboratory of Professor Sarah R. Kirk. Natalie’s graduate work in Professor Jacqueline K. Barton’s group involves both fundamental studies of DNA charge transport and the use of this sensitive chemistry for the electrochemical detection of clinically relevant DNA-binding proteins.

Eric D. Olmon
Eric D. Olmon grew up in Troy, Ohio and earned his B.S. in chemistry from The Ohio State University, where he studied the effect of DNA conformation on the efficiency of thymine dimer formation in the laboratory of Bern Kohler. Eric recently earned his PhD in chemistry from Caltech for his work involving the study of DNA-mediated charge transport by time-resolved spectroscopy.

Jacqueline K. Barton
Jacqueline K. Barton is the Arthur and Marion Hanisch Memorial Professor of Chemistry. She also currently serves as the Chair of the Division of Chemistry and Chemical Engineering at Caltech. Barton obtained her PhD in Inorganic Chemistry at Columbia University and served as a member of the faculty at Hunter College and Columbia University before joining Caltech in 1989. Her work is focused on the chemistry of double helical DNA. She has received many awards for this research including most recently the National Medal of Science (2011).
References
- 1.Saccà B, Niemeyer CM. Angew. Chem., Int. Ed. 2012;51:58–66. doi: 10.1002/anie.201105846. [DOI] [PubMed] [Google Scholar]
- 2.Aldaye FA, Palmer AL, Sleiman HF. Science. 2008;321:1795–1799. doi: 10.1126/science.1154533. [DOI] [PubMed] [Google Scholar]
- 3.Eley DD, Spivey DI. Trans. Faraday Soc. 1962;58:411–415. [Google Scholar]
- 4.Murphy CJ, Arkin MR, Jenkins Y, Ghatlia ND, Bossmann SH, Turro NJ, Barton JK. Science. 1993;262:1025–1029. doi: 10.1126/science.7802858. [DOI] [PubMed] [Google Scholar]
- 5.Takada T, Kawai K, Tojo S, Majima T. J. Phys. Chem. B. 2004;108:761–766. [Google Scholar]
- 6.Pyle AM, Long EC, Barton JK. J. Am. Chem. Soc. 1989;111:4520–4522. [Google Scholar]
- 7.Hudson BP, Barton JK. J. Am. Chem. Soc. 1998;120:6877–6888. [Google Scholar]
- 8.Kielkopf CL, Erkkila KE, Hudson BP, Barton JK, Rees DC. Nat. Struct. Mol. Biol. 2000;7:117–121. doi: 10.1038/72385. [DOI] [PubMed] [Google Scholar]
- 9.Sitlani A, Dupureur CM, Barton JK. J. Am. Chem. Soc. 1993;115:12589–12590. [Google Scholar]
- 10.Sitlani A, Barton JK. Biochemistry. 1994;33:12100–12108. doi: 10.1021/bi00206a013. [DOI] [PubMed] [Google Scholar]
- 11.Krotz AH, Hudson BP, Barton JK. J. Am. Chem. Soc. 1993;115:12577–12578. [Google Scholar]
- 12.Shields TP, Barton JK. Biochemistry. 1995;34:15037–15048. doi: 10.1021/bi00046a009. [DOI] [PubMed] [Google Scholar]
- 13.Hudson BP, Dupureur CM, Barton JK. J. Am. Chem. Soc. 1995;117:9379–9380. [Google Scholar]
- 14.Pyle A, Rehmann J, Meshoyrer R, Kumar C, Turro N, Barton JK. J. Am. Chem. Soc. 1989;111:3051–3058. [Google Scholar]
- 15.Kirsch-De Mesmaeker A, Orellana G, Barton JK, Turro NJ. Photochem. Photobiol. 1990;52:461–472. doi: 10.1111/j.1751-1097.1990.tb01787.x. [DOI] [PubMed] [Google Scholar]
- 16.Jenkins Y, Friedman AE, Turro NJ, Barton JK. Biochemistry. 1992;31:10809–10816. doi: 10.1021/bi00159a023. [DOI] [PubMed] [Google Scholar]
- 17.Hartshorn RM, Barton JK. J. Am. Chem. Soc. 1992;114:5919–5925. [Google Scholar]
- 18.Stemp EDA, Holmlin RE, Barton JK. Inorg. Chim. Acta. 2000;297:88–97. [Google Scholar]
- 19.Holmlin RE, Stemp EDA, Barton JK. J. Am. Chem. Soc. 1996;118:5236–5244. [Google Scholar]
- 20.Murphy CJ, Arkin MR, Ghatlia ND, Bossmann SH, Turro NJ, Barton JK. Proc. Natl. Acad. Sci. U. S. A. 1994;91:5315–5319. doi: 10.1073/pnas.91.12.5315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Steenken S, Jovanovic SV. J. Am. Chem. Soc. 1997;119:617–618. [Google Scholar]
- 22.Senthilkumar K, Grozema FC, Guerra CF, Bickelhaupt FM, Siebbeles LDA. J. Am. Chem. Soc. 2003;125:13658–13659. doi: 10.1021/ja037027d. [DOI] [PubMed] [Google Scholar]
- 23.Saito I, Takayama M, Sugiyama H, Nakatani K, Tsuchida A, Yamamoto M. J. Am. Chem. Soc. 1995;117:6406–6407. [Google Scholar]
- 24.Sugiyama H, Saito I. J. Am. Chem. Soc. 1996;118:7063–7068. [Google Scholar]
- 25.Saito I, Nakamura T, Nakatani K, Yoshioka Y, Yamaguchi K, Sugiyama H. J. Am. Chem. Soc. 1998;120:12686–12687. [Google Scholar]
- 26.Hall DB, Holmlin RE, Barton JK. Nature. 1996;382:731–735. doi: 10.1038/382731a0. [DOI] [PubMed] [Google Scholar]
- 27.Arkin MR, Stemp EDA, Coates Pulver S, Barton JK. Chem. Biol. 1997;4:389–400. doi: 10.1016/s1074-5521(97)90129-0. [DOI] [PubMed] [Google Scholar]
- 28.Stemp EDA, Arkin MR, Barton JK. J. Am. Chem. Soc. 1997;119:2921–2925. [Google Scholar]
- 29.Schiemann O, Turro NJ, Barton JK. J. Phys. Chem. B. 2000;104:7214–7220. [Google Scholar]
- 30.Olmon ED, Sontz PA, Blanco-Rodríguez AM, Towrie M, Clark IP, Vlček A, Barton JK. J. Am. Chem. Soc. 2011;133:13718–13730. doi: 10.1021/ja205568r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Delaney S, Pascaly M, Bhattacharya PK, Han K, Barton JK. Inorg. Chem. 2002;41:1966–1974. doi: 10.1021/ic0111738. [DOI] [PubMed] [Google Scholar]
- 32.Giese B, Amaudrut J, Kohler A-K, Spormann M, Wessely S. Nature. 2001;412:3–5. doi: 10.1038/35085542. [DOI] [PubMed] [Google Scholar]
- 33.Kendrick T, Giese B. Chem. Commun. 2002:2016–2017. doi: 10.1039/b205928b. [DOI] [PubMed] [Google Scholar]
- 34.Williams TT, Odom DT, Barton JK. J. Am. Chem. Soc. 2000;122:9048–9049. [Google Scholar]
- 35.Núñez ME, Hall DB, Barton JK. Chem. Biol. 1999;6:85–97. doi: 10.1016/S1074-5521(99)80005-2. [DOI] [PubMed] [Google Scholar]
- 36.Musa OM, Horner JH, Shahin H, Newcomb M. J. Am. Chem. Soc. 1996;118:3862–3868. [Google Scholar]
- 37.Shao F, O’Neill MA, Barton JK. Proc. Natl. Acad. Sci. U. S. A. 2004;101:17914–17919. doi: 10.1073/pnas.0408128101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Shao F, Augustyn KE, Barton JK. J. Am. Chem. Soc. 2005;127:17445–17452. doi: 10.1021/ja0563399. [DOI] [PubMed] [Google Scholar]
- 39.Hall DB, Barton JK. J. Am. Chem. Soc. 1997;119:5045–5046. [Google Scholar]
- 40.Delaney S, Yoo J, Stemp EDA, Barton JK. Proc. Natl. Acad. Sci. U. S. A. 2004;101:10511–10516. doi: 10.1073/pnas.0403791101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Odom DT, Dill EA, Barton JK. Chem. Biol. 2000;7:475–481. doi: 10.1016/s1074-5521(00)00133-2. [DOI] [PubMed] [Google Scholar]
- 42.Yoo J, Delaney S, Stemp EDA, Barton JK. J. Am. Chem. Soc. 2003;125:6640–6641. doi: 10.1021/ja034326u. [DOI] [PubMed] [Google Scholar]
- 43.Bhattacharya PK, Barton JK. J. Am. Chem. Soc. 2001;123:8649–8656. doi: 10.1021/ja010996t. [DOI] [PubMed] [Google Scholar]
- 44.Rajski SR, Kumar S, Roberts RJ, Barton JK. J. Am. Chem. Soc. 1999;121:5615–5616. [Google Scholar]
- 45.Rajski SR, Barton JK. Biochemistry. 2001;40:5556–5564. doi: 10.1021/bi002684t. [DOI] [PubMed] [Google Scholar]
- 46.Augustyn KE, Genereux JC, Barton JK. Angew. Chem., Int. Ed. 2007;46:5731–5733. doi: 10.1002/anie.200701522. [DOI] [PubMed] [Google Scholar]
- 47.Marcus RA, Sutin N. Biochim. Biophys. Acta. 1985;811:265–322. [Google Scholar]
- 48.Gray HB, Winkler JR. Annu. Rev. Biochem. 1996;65:537–561. doi: 10.1146/annurev.bi.65.070196.002541. [DOI] [PubMed] [Google Scholar]
- 49.Lewis FD, Letsinger RL, Wasielewski MR. Acc. Chem. Res. 2001;34:159–170. doi: 10.1021/ar0000197. [DOI] [PubMed] [Google Scholar]
- 50.Lewis FD, Wu T, Zhang Y, Letsinger RL, Greenfield SR, Wasielewski MR. Science. 1997;277:673–676. doi: 10.1126/science.277.5326.673. [DOI] [PubMed] [Google Scholar]
- 51.Lewis FD, Wu T, Liu X, Letsinger RL, Greenfield SR, Miller SE, Wasielewski MR. J. Am. Chem. Soc. 2000;122:2889–2902. [Google Scholar]
- 52.Lewis FD, Kalgutkar RS, Wu Y, Liu X, Liu J, Hayes RT, Miller SE, Wasielewski MR. J. Am. Chem. Soc. 2000;122:12346–12351. [Google Scholar]
- 53.Lewis FD, Liu X, Miller SE, Hayes RT, Wasielewski MR. J. Am. Chem. Soc. 2002;124:14020–14026. doi: 10.1021/ja027108u. [DOI] [PubMed] [Google Scholar]
- 54.Page CC, Moser CC, Chen X, Dutton PL. Nature. 1999;402:47–52. doi: 10.1038/46972. [DOI] [PubMed] [Google Scholar]
- 55.Wan C, Fiebig T, Kelley SO, Treadway CR, Barton JK, Zewail AH. Proc. Natl. Acad. Sci. U. S. A. 1999;96:6014–6019. doi: 10.1073/pnas.96.11.6014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Valis L, Wang Q, Raytchev M, Buchvarov I, Wagenknecht H-A, Fiebig T. Proc. Natl. Acad. Sci. U. S. A. 2006;103:10192–10195. doi: 10.1073/pnas.0600957103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kawai K, Matsutani E, Majima T. Chem. Commun. 2010;46:3277–3279. doi: 10.1039/c002810j. [DOI] [PubMed] [Google Scholar]
- 58.Kawai K, Osakada Y, Takada T, Fujitsuka M, Majima T. J. Am. Chem. Soc. 2004;126:12843–12846. doi: 10.1021/ja0475813. [DOI] [PubMed] [Google Scholar]
- 59.Kawai K, Osakada Y, Fujitsuka M, Majima T. Chem.–Eur. J. 2008;14:3721–3726. doi: 10.1002/chem.200701835. [DOI] [PubMed] [Google Scholar]
- 60.Lewis FD, Liu X, Liu J, Miller SE, Hayes RT, Wasielewski MR. Nature. 2000;406:51–53. doi: 10.1038/35017524. [DOI] [PubMed] [Google Scholar]
- 61.Lewis FD, Zuo X, Liu J, Hayes RT, Wasielewski MR. J. Am. Chem. Soc. 2002;124:4568–4569. doi: 10.1021/ja0177859. [DOI] [PubMed] [Google Scholar]
- 62.Lewis FD, Zhu H, Daublain P, Fiebig T, Raytchev M, Wang Q, Shafirovich V. J. Am. Chem. Soc. 2006;128:791–800. doi: 10.1021/ja0540831. [DOI] [PubMed] [Google Scholar]
- 63.Lewis FD, Zhu H, Daublain P, Sigmund K, Fiebig T, Raytchev M, Wang Q, Shafirovich V. Photochem. Photobiol. Sci. 2008;7:534–539. doi: 10.1039/b719715b. [DOI] [PubMed] [Google Scholar]
- 64.Lewis FD, Daublain P, Cohen B, Vura-Weis J, Shafirovich V, Wasielewski MR. J. Am. Chem. Soc. 2007;129:15130–15131. doi: 10.1021/ja076876o. [DOI] [PubMed] [Google Scholar]
- 65.Vura-Weis J, Wasielewski MR, Thazhathveetil AK, Lewis FD. J. Am. Chem. Soc. 2009;131:9722–9727. doi: 10.1021/ja9015217. [DOI] [PubMed] [Google Scholar]
- 66.Takada T, Kawai K, Fujitsuka M, Majima T. J. Am. Chem. Soc. 2006;128:11012–11013. doi: 10.1021/ja0641554. [DOI] [PubMed] [Google Scholar]
- 67.Kawai K, Osakada Y, Fujitsuka M, Majima T. J. Phys. Chem. B. 2008;112:2144–2149. doi: 10.1021/jp075326+. [DOI] [PubMed] [Google Scholar]
- 68.Conron SMM, Thazhathveetil AK, Wasielewski MR, Burin AL, Lewis FD. J. Am. Chem. Soc. 2010;132:14388–14390. doi: 10.1021/ja106991f. [DOI] [PubMed] [Google Scholar]
- 69.Takada T, Kawai K, Cai X, Sugimoto A, Fujitsuka M, Majima T. J. Am. Chem. Soc. 2004;126:1125–1129. doi: 10.1021/ja035730w. [DOI] [PubMed] [Google Scholar]
- 70.Takada T, Kawai K, Cai X, Sugimoto A, Fujitsuka M, Majima T. J. Am. Chem. Soc. 2004;126:1125–1129. doi: 10.1021/ja035730w. [DOI] [PubMed] [Google Scholar]
- 71.Lewis FD, Zhu H, Daublain P, Cohen B, Wasielewski MR. Angew. Chem., Int. Ed. 2006;45:7982–7985. doi: 10.1002/anie.200603455. [DOI] [PubMed] [Google Scholar]
- 72.Kawai K, Kodera H, Osakada Y, Majima T. Nat. Chem. 2009;1:156–159. doi: 10.1038/nchem.171. [DOI] [PubMed] [Google Scholar]
- 73.Kawai K, Kodera H, Majima T. J. Am. Chem. Soc. 2010;132:627–630. doi: 10.1021/ja907409z. [DOI] [PubMed] [Google Scholar]
- 74.Kawai K, Hayashi M, Majima T. J. Am. Chem. Soc. 2012;134:4806–4811. doi: 10.1021/ja2109213. [DOI] [PubMed] [Google Scholar]
- 75.Lewis FD, Liu J, Weigel W, Rettig W, Kurnikov IV, Beratan DN. Proc. Natl. Acad. Sci. U. S. A. 2002;99:12536–12541. doi: 10.1073/pnas.192432899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Osakada Y, Kawai K, Fujitsuka M, Majima T. Nucleic Acids Res. 2008;36:5562–5570. doi: 10.1093/nar/gkn505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Kawai K, Matsutani E, Maruyama A, Majima T. J. Am. Chem. Soc. 2011;133:15568–15577. doi: 10.1021/ja206325m. [DOI] [PubMed] [Google Scholar]
- 78.Kelley SO, Barton JK. Science. 1999;283:375–381. doi: 10.1126/science.283.5400.375. [DOI] [PubMed] [Google Scholar]
- 79.O’Neill MA, Barton JK. Proc. Natl. Acad. Sci. U. S. A. 2002;99:16543–16550. doi: 10.1073/pnas.012669599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Neill PO, Parker AW, Plumb MA, Siebbeles LDA. J. Phys. Chem. B. 2001;105:5283–5290. [Google Scholar]
- 81.O’Neill MA, Becker H-C, Wan C, Barton JK, Zewail AH. Angew. Chem., Int. Ed. 2003;42:5896–5900. doi: 10.1002/anie.200352831. [DOI] [PubMed] [Google Scholar]
- 82.O’Neill MA, Barton JK. J. Am. Chem. Soc. 2004;126:13234–13235. doi: 10.1021/ja0455897. [DOI] [PubMed] [Google Scholar]
- 83.O’Neill MA, Barton JK. J. Am. Chem. Soc. 2004;126:11471–11483. doi: 10.1021/ja048956n. [DOI] [PubMed] [Google Scholar]
- 84.Shao F, Barton JK. J. Am. Chem. Soc. 2007;129:14733–14738. doi: 10.1021/ja0752437. [DOI] [PubMed] [Google Scholar]
- 85.Elias B, Genereux JC, Barton JK. Angew. Chem., Int. Ed. 2008;47:9067–9070. doi: 10.1002/anie.200803556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Elias B, Shao F, Barton JK. J. Am. Chem. Soc. 2008;130:1152–1153. doi: 10.1021/ja710358p. [DOI] [PubMed] [Google Scholar]
- 87.Tanaka M, Elias B, Barton JK. J. Org. Chem. 2010;75:2423–2428. doi: 10.1021/jo1000862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Park MJ, Fujitsuka M, Kawai K, Majima T. J. Am. Chem. Soc. 2011;133:15320–15323. doi: 10.1021/ja2068017. [DOI] [PubMed] [Google Scholar]
- 89.Park MJ, Fujitsuka M, Kawai K, Majima T. Chem.–Eur. J. 2012;18:2056–2062. doi: 10.1002/chem.201103663. [DOI] [PubMed] [Google Scholar]
- 90.Love JC, Estroff LA, Kriebel JK, Nuzzo RG, Whitesides GM. Chem. Rev. 2002;105:1103–1169. doi: 10.1021/cr0300789. [DOI] [PubMed] [Google Scholar]
- 91.Bard AJ, Faulkner LR. Electrochemical Methods. 2nd edn John Wiley & Sons; New York: 2002. [Google Scholar]
- 92.Frasconi M, Mazzei F, Ferri T. Anal. Bioanal. Chem. 2010;398:1545–1564. doi: 10.1007/s00216-010-3708-6. [DOI] [PubMed] [Google Scholar]
- 93.Arya S, Solanki P, Datta M, Malhotra B. Biosens. Bioelectron. 2009;24:2810–2817. doi: 10.1016/j.bios.2009.02.008. [DOI] [PubMed] [Google Scholar]
- 94.Samanta D, Sarkar A. Chem. Soc. Rev. 2011;40:2567–2592. doi: 10.1039/c0cs00056f. [DOI] [PubMed] [Google Scholar]
- 95.Liu J, Cao Z, Lu Y. Chem. Rev. 2009;109:1948–1998. doi: 10.1021/cr030183i. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Ronkainen NJ, Halsall HB, Heineman WR. Chem. Soc. Rev. 2010;39:1747–1763. doi: 10.1039/b714449k. [DOI] [PubMed] [Google Scholar]
- 97.Sadik OA, Aluoch AO, Zhou A. Biosens. Bioelectron. 2009;24:2749–2765. doi: 10.1016/j.bios.2008.10.003. [DOI] [PubMed] [Google Scholar]
- 98.Okamoto A, Kamei T, Saito I. J. Am. Chem. Soc. 2006;128:658–662. doi: 10.1021/ja057040t. [DOI] [PubMed] [Google Scholar]
- 99.Okamoto A, Kamei T, Tanaka K, Saito I. J. Am. Chem. Soc. 2004;126:14732–14733. doi: 10.1021/ja0449067. [DOI] [PubMed] [Google Scholar]
- 100.Takada T, Lin C, Majima T. Angew. Chem., Int. Ed. 2007;46:6681–6683. doi: 10.1002/anie.200701525. [DOI] [PubMed] [Google Scholar]
- 101.Takada T, Takeda Y, Fujitsuka M, Majima T. J. Am. Chem. Soc. 2009;131:6656–6657. doi: 10.1021/ja9009919. [DOI] [PubMed] [Google Scholar]
- 102.Drummond TG, Hill MG, Barton JK. Nat. Biotechnol. 2003;21:1192–1199. doi: 10.1038/nbt873. [DOI] [PubMed] [Google Scholar]
- 103.Gorodetsky AA, Buzzeo MC, Barton JK. Bioconjugate Chem. 2008;19:2285–2296. doi: 10.1021/bc8003149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Kelley SO, Barton JK. Bioconjugate Chem. 1997;8:31–37. doi: 10.1021/bc960070o. [DOI] [PubMed] [Google Scholar]
- 105.Gorodetsky AA, Barton JK. Langmuir. 2006;22:7917–7922. doi: 10.1021/la0611054. [DOI] [PubMed] [Google Scholar]
- 106.Boon EM, Salas JE, Barton JK. Nat. Biotechnol. 2002;20:282–286. doi: 10.1038/nbt0302-282. [DOI] [PubMed] [Google Scholar]
- 107.Kelley SO, Barton JK, Jackson NM, McPherson LD, Potter AB, Spain EM, Allen MJ, Hill MG. Langmuir. 1998;14:6781–6784. [Google Scholar]
- 108.Boon EM, Sam M, Barton JK, Hill MG, Spain EM. Langmuir. 2001;17:5727–5730. [Google Scholar]
- 109.Ceres DM, Barton JK. J. Am. Chem. Soc. 2003;125:14964–14965. doi: 10.1021/ja0384476. [DOI] [PubMed] [Google Scholar]
- 110.Kelley SO, Boon EM, Barton JK, Jackson NM, Hill MG. Nucleic Acids Res. 1999;27:4830–4837. doi: 10.1093/nar/27.24.4830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Boon EM, Jackson NM, Wightman MD, Kelley SO, Barton JK, Hill MG. J. Phys. Chem. B. 2003;107:11805–11812. [Google Scholar]
- 112.Boon EM, Barton JK, Bhagat V, Nerissian M, Wang W. Langmuir. 2003;19:9255–9259. [Google Scholar]
- 113.Boon EM, Ceres DM, Drummond TG, Hill MG, Barton JK. Nat. Biotechnol. 2000;18:1096–1100. doi: 10.1038/80301. [DOI] [PubMed] [Google Scholar]
- 114.Boal AK, Barton JK. Bioconjugate Chem. 2005;16:312–321. doi: 10.1021/bc0497362. [DOI] [PubMed] [Google Scholar]
- 115.Boon EM, Barton JK. Bioconjugate Chem. 2003;14:1140–1147. doi: 10.1021/bc034139l. [DOI] [PubMed] [Google Scholar]
- 116.Boon EM, Barton JK, Pradeepkumar PI, Isaksson J, Petit C, Chattopadhyaya J. Angew. Chem., Int. Ed. 2002;41:3402–3405. doi: 10.1002/1521-3773(20020916)41:18<3402::AID-ANIE3402>3.0.CO;2-6. [DOI] [PubMed] [Google Scholar]
- 117.Gorodetsky AA, Green O, Yavin E, Barton JK. Bioconjugate Chem. 2007;18:1434–1441. doi: 10.1021/bc0700483. [DOI] [PubMed] [Google Scholar]
- 118.Drummond TG, Hill MG, Barton JK. J. Am. Chem. Soc. 2004;126:15010–15011. doi: 10.1021/ja044910i. [DOI] [PubMed] [Google Scholar]
- 119.Liu T, Barton JK. J. Am. Chem. Soc. 2005;127:10160–10161. doi: 10.1021/ja053025c. [DOI] [PubMed] [Google Scholar]
- 120.Kelley SO, Jackson NM, Hill MG, Barton JK. Angew. Chem., Int. Ed. 1999;38:941–945. doi: 10.1002/(SICI)1521-3773(19990401)38:7<941::AID-ANIE941>3.0.CO;2-7. [DOI] [PubMed] [Google Scholar]
- 121.Gorodetsky AA, Barton JK. J. Am. Chem. Soc. 2007;129:6074–6075. doi: 10.1021/ja071006s. [DOI] [PubMed] [Google Scholar]
- 122.Buzzeo MC, Barton JK. Bioconjugate Chem. 2008;19:2110–2112. doi: 10.1021/bc800339y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Gorodetsky AA, Ebrahim A, Barton JK. J. Am. Chem. Soc. 2008;130:2924–2925. doi: 10.1021/ja7106756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Slinker JD, Muren NB, Gorodetsky AA, Barton JK. J. Am. Chem. Soc. 2010;132:2769–2774. doi: 10.1021/ja909915m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Slinker JD, Muren NB, Renfrew SE, Barton JK. Nat. Chem. 2011;3:230–235. doi: 10.1038/nchem.982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Boon EM, Livingston AL, Chmiel NH, David SS, Barton JK. Proc. Natl. Acad. Sci. U. S. A. 2003;100:12543–12547. doi: 10.1073/pnas.2035257100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Boal AK, Yavin E, Jukianova OA, O’Shea VL, David SS, Barton JK. Biochemistry. 2005;44:8397–8407. doi: 10.1021/bi047494n. [DOI] [PubMed] [Google Scholar]
- 128.DeRosa MC, Sancar A, Barton JK. Proc. Natl. Acad. Sci. U. S. A. 2005;102:10788–10792. doi: 10.1073/pnas.0503527102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Mui TP, Fuss JO, Ishida JP, Tainer JA, Barton JK. J. Am. Chem. Soc. 2011;133:16378–16381. doi: 10.1021/ja207222t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Gorodetsky AA, Dietrich LEP, Lee PE, Demple B, Newman DK, Barton JK. Proc. Natl. Acad. Sci. U. S. A. 2008;105:3684–3689. doi: 10.1073/pnas.0800093105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Gorodetsky AA, Boal AK, Barton JK. J. Am. Chem. Soc. 2006;128:12082–12083. doi: 10.1021/ja064784d. [DOI] [PubMed] [Google Scholar]
- 132.Romano CA, Sontz PA, Barton JK. Biochemistry. 2011;50:6133–6145. doi: 10.1021/bi2003179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Boal AK, Yavin E, Barton JK. J. Inorg. Biochem. 2007;101:1913–1921. doi: 10.1016/j.jinorgbio.2007.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Lee PE, Demple B, Barton JK. Proc. Natl. Acad. Sci. U. S. A. 2009;106:13164–13168. doi: 10.1073/pnas.0906429106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Genereux JC, Boal AK, Barton JK. J. Am. Chem. Soc. 2010;132:891–905. doi: 10.1021/ja907669c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Sontz PA, Mui TP, Fuss JO, Tainer JA, Barton JK. Proc. Natl. Acad. Sci. U. S. A. 2012;109:1856–1861. doi: 10.1073/pnas.1120063109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Feldman AK, Steigerwald ML, Guo X, Nuckolls C. Acc. Chem. Res. 2008;41:1731–1741. doi: 10.1021/ar8000266. [DOI] [PubMed] [Google Scholar]
- 138.Fink HW, Schonenberger C. Nature. 1999;398:407–410. doi: 10.1038/18855. [DOI] [PubMed] [Google Scholar]
- 139.Porath D, Bezryadin A, de Vries S, Dekker C. Nature. 2000;403:635–638. doi: 10.1038/35001029. [DOI] [PubMed] [Google Scholar]
- 140.Storm AJ, van Noort J, de Vries S, Dekker CS. Appl. Phys. Lett. 2001;79:3881–3883. [Google Scholar]
- 141.Kasumov AY, Kociak M, Gueron S, Reulet B, Volkov VT, Klinov DV, Bouchiat H. Science. 2000;291:280–282. doi: 10.1126/science.291.5502.280. [DOI] [PubMed] [Google Scholar]
- 142.Gohen H, Nogues C, Naaman R, Porath D. Proc. Natl. Acad. Sci. U. S. A. 2005;102:11589–11593. doi: 10.1073/pnas.0505272102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Hihath J, Xu B, Zhang P, Tao N. Proc. Natl. Acad. Sci. U. S. A. 2005;102:16979–16983. doi: 10.1073/pnas.0505175102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Wierzbinski E, Arndt J, Hammond W, Slowinski K. Langmuir. 2006;22:2426–2429. doi: 10.1021/la053224+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Guo X, Small JP, Klare JE, Wang Y, Purewal MS, Tam IW, Hong BH, Caldwell R, Huang L, O’Brien S, Yan J, Breslow R, Wind SJ, Hone J, Kim P, Nuckolls C. Science. 2006;311:356–359. doi: 10.1126/science.1120986. [DOI] [PubMed] [Google Scholar]
- 146.Guo X, Gorodetsky AA, Hone J, Barton JK, Nuckolls C. Nat. Nanotechnol. 2008;3:163–167. doi: 10.1038/nnano.2008.4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Wang H, Muren NB, Ordinario D, Gorodetsky AA, Barton JK, Nuckolls C. Chem. Sci. 2012;3:62–65. doi: 10.1039/C1SC00772F. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Genereux JC, Barton JK. Chem. Rev. 2010;110:1642–1662. doi: 10.1021/cr900228f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Bixon M, Giese B, Wessely S, Langenbacher T, Michel-Beyerle ME, Jortner J. Proc. Natl. Acad. Sci. U. S. A. 1999;96:11713–11716. doi: 10.1073/pnas.96.21.11713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Jortner J, Bixon M, Langenbacher T, Michel-Beyerle ME. Proc. Natl. Acad. Sci. U. S. A. 1998;95:12759–12765. doi: 10.1073/pnas.95.22.12759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Jortner J, Bixon M, Voityuk AA, Rosch N. J. Phys. Chem. A. 2002;106:7599–7606. [Google Scholar]
- 152.Bixon M, Jortner J. Chem. Phys. 2002;281:393–408. [Google Scholar]
- 153.Meggers E, Michel-Beyerle ME, Giese B. J. Am. Chem. Soc. 1998;120:12950–12955. [Google Scholar]
- 154.Giese B, Wessely S, Spormann M, Lindemann U, Meggers E, Michel-Beyerle ME. Angew. Chem., Int. Ed. 1999;38:996–998. doi: 10.1002/(SICI)1521-3773(19990401)38:7<996::AID-ANIE996>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- 155.Giese B. Acc. Chem. Res. 2000;33:631–636. doi: 10.1021/ar990040b. [DOI] [PubMed] [Google Scholar]
- 156.Giese B. Annu. Rev. Biochem. 2002;71:51–70. doi: 10.1146/annurev.biochem.71.083101.134037. [DOI] [PubMed] [Google Scholar]
- 157.Genereux JC, Augustyn KE, Davis ML, Shao F, Barton JK. J. Am. Chem. Soc. 2008;130:15150–15156. doi: 10.1021/ja8052738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Genereux JC, Wuerth SM, Barton JK. J. Am. Chem. Soc. 2011;133:3863–3868. doi: 10.1021/ja107033v. [DOI] [PMC free article] [PubMed] [Google Scholar]