

Abstract
Dendritic spine morphology is modulated by protein kinase p38, a mitogen-activated protein (MAPK), in the hippocampus. Protein p38MAPK is a substrate of wip1, a protein phosphatase. The role of wip1 in the central nervous system (CNS) has never been explored. Here, we report a novel function of wip1 in dendritic spine morphology and memory processes. Wip1 deficiency decreases dendritic spine size and density in pyramidal neurons of the hippocampal CA1 region. Simultaneously, impairments in object recognition tasks and contextual memory occur in wip1 deficient mice, but are reversed in wip1/p38 double mutant mice. Thus, our findings demonstrate that wip1 modulates dendritic morphology and memory processes through the p38MAPK signaling pathway. In addition to the well-characterized role of the wip1/p38MAPK in cell death and differentiation, we revealed the novel contribution of wip1 to cognition and dendritic spine morphology, which may suggest new approaches to treating neurodegenerative disorders.
Keywords: Wip1 phosphatase, p38MAPK, memory, dendritic spine morphology, hippocampus, signaling pathway
Introduction
Dendritic spines are the main excitatory sites of the neurons in brain, undergoing morphological changes in response to neuronal activity.1-3 Because of this continual dynamic, an increase in the size and number of dendritic spines might represent a physical substrate for learning and memory processes.1,3 However, these molecular mechanisms underlying learning and memory processes and modulation of dendritic spine structure, are not fully understood.
Several molecular families, including scaffold proteins, and regulators of the cytoskeleton, have been found involved in the regulation of dendritic spine morphology and density. In particular, several protein kinases, such as mitogen-activated protein kinase (MAPK), calcium/calmodulin-dependent protein kinase and protein phosphatases (PPs) (PP1 and PP2) present in the post-synaptic compartment, are essential factors in the regulation of dendritic spine morphology4-6 and synaptic plasticity.4,7,8 In addition, PPs have been previously associated with Alzheimer disease (AD), which is the most common cause of memory deficits and dementia.9,10
Wip1, encoded by the PPM1D gene, is part of the PP2C family of protein serine-threonine phosphatases.11 To date, three dephosphorylation targets of wip1 [p53, p38MAPK and ataxia-telangiectasia mutated (ATM)] have been reported.12-15 For p38MAPK, wip1 dephosphorylates an activating threonine residue that is essential for p38MAPK activity.16 Wip1 expression is induced in a p53-dependent manner following gamma and UV radiation.11,16 Although wip1 mRNA was found in mouse brain,12 the roles of wip1 in the CNS, particularly in dendritic spine morphology and/or memory processes, have never been explored.
p38MAPK, located in postsynaptic dendrites, has been implicated in cytoskeleton reorganisation.17-19 Inhibition of p38MAPK activity leads to an increase in the size of dendritic spines,20 which implicates p38MAPK in the regulation of the morphology of dendritic spines. Given that p38MAPK is a downstream element of wip1 signaling, it might be hypothesized that wip1 would have functions in CNS neurons. Here, we have studied novel roles of wip1 in the modulation of dendritic spine morphology and learning and memory processes through the p38MAPK pathway in the CNS.
Results
Role of wip1 protein in modulating dendritic spine morphology
Dendritic spine morphology was examined by Golgi staining, performed on hippocampal sections from adult wip1+/+ and wip1–/– mice. The staining revealed significant differences between wip1+/+ and wip1–/– mice in terms of dendritic morphology (Fig. 1A and B). Measurements of spine length, spine head width and spine density were performed on Golgi stained neurons in CA1 of the hippocampus. Our results showed that spine length was significantly reduced in wip1–/– (0.79 ± 0.02 μm) vs. wip1+/+ neurons (0.89 ± 0.05 μm; p = 0.04; Fig. 1Ba). Spine head widths were also significantly reduced in wip1–/–hippocampal neurons (0.41 ± 0.03 μm) compared with the ones measured in wip1+/+ hippocampal neurons (0.5 ± 0.07 μm; p = 0.023; Fig. 1Bb). The spine density measured in CA1 of wip1+/+ mice (17.7 ± 3.77) was more than 70% greater than in wip1–/– mice (6.7 ± 0.5; p = 0.0012; Fig. 1Bc). The spine length (0.8 ± 0.01 µm) and the spine head width (0.38 ± 0.05 µm) were significantly reduced in wip1+/– mice compared with wip1+/+ mice (p = 0.048 and p = 0.02, respectively) (Fig. 1Ba and b). Regarding spine density (number of spines per 10μm length of dendrites), the significant decrease (35%) reported in wip1+/– mice compared with control mice (11.4 ± 2.06 µm, p = 0 0.012) was lower than that observed in the wip1–/– mice (62%, p = 0.0012) (Fig. 1Bc). These data were confirmed by the immunostaining of the actin filaments with rhodamine-phalloidin in the hippocampal neurons from wip1+/+, wip1+/– and wip1–/– mice (Fig. S1). Quantification showed that spine lengths in wip1−/− (1.35 ± 0.06 μm) and in wip1+/− (1.84 ± 0.16 μm) hippocampal cells were significantly reduced compared with the spine lengths in wip1+/+ neurons (2.23 ± 0.15 μm). The spine densities of wip1+/− (1.7 ± 0.2) and wip1−/− (1.9 ± 0.5) neurons were also significantly decreased compared with the spine density measured in wip1+/+ cells (2.4 ± 0.8). However, spine head width did not differ significantly. Interestingly, branching indexes (number of branch points divided by average dendritic length) in wip1+/− (0.54 ± 0.07) and wip1−/− (0.44 ± 0.05) neurons were significantly reduced, compared with wip1+/+ mice (1.02 ± 0.05). Altogether, these data demonstrate, for the first time, that wip1 modulates dendritic spine morphology in hippocampus.

Figure 1. Morphological analysis of the dendrites in CA1 of the hippocampus in wip1 wt and wip1 ko mice. (A) Golgi staining of dendrites of adult pyramidal neurons in CA1 of the hippocampus of wip wt, wip1 het and wip1 ko mice. Representative images of dendritic fragments from wip1 wt, wip1 het and wip1 ko mice. (B) Quantification of spine length (a), spine head width (b) and spine density (c) of the Golgi stained neurons in wip1 wt (n = 9), wip1 het (n = 4) and wip1 ko mice (n = 8). *p < 0.05, **: p < 0.01. Bars = 4 ?m.
Wip1 deficiency impairs object recognition task and contextual memory
As wip1 plays a role in the modulation of dendritic spine morphology, we hypothesized that wip1 might be involved in learning and memory processes. We therefore performed three behavioral tests: an object recognition test to evaluate associative memory,21 contextual fear conditioning22 and a T-maze spontaneous alternation task.23
Recognition memory tasks have previously been shown to be hippocampal-dependent in lesion studies in both rodents24-26 and primates.27,28 To assess whether wip1 affects associative memory, the object recognition test was performed with wip1+/+, wip1+/– and wip1–/– mice. During the training session of this test, the percentage of time spent exploring the identical objects in the open-field was not significantly different among wip1+/+ (19.4%), wip1+/– (17.8%) and wip1–/– (20.7%; n = 8–11) mice. Nevertheless, both wip1+/+ and wip1+/– mice showed a significantly higher recognition index (60.3% and 59.6%, respectively) compared with wip1–/– mice (49.6%, p = 0.0013 and p = 0.0186 respectively; Fig. 2). With a recognition index inferior to 50%, the mice deficient in wip1 showed an inability to remember the familiar object previously explored in the training session and they spent less time exploring the new object, suggesting that wip1 deficient mice have impaired memory in the object recognition test.
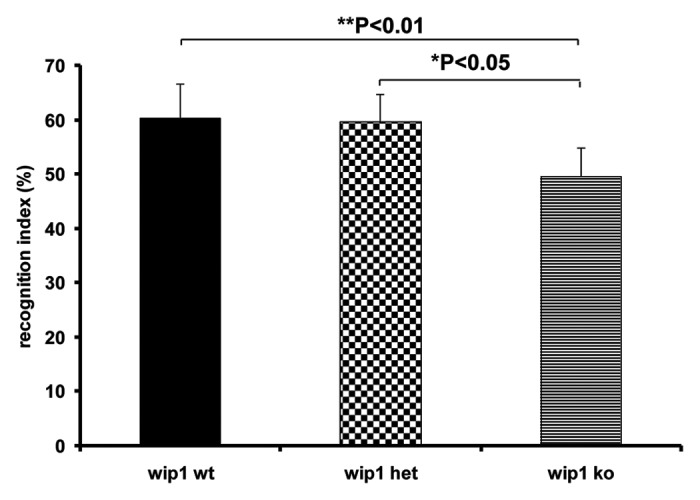
Figure 2. Evaluation of associative memory in wip1 wt, wip1 het and wip1 ko mice. Evaluation of memory by object recognition test in wip1 wt (n = 11), wip1 het (n = 8) and wip1 ko (n = 9) mice. Values of recognition index are means ? SEM *: p < 0.05, **: p < 0.01.
In order to further explore the differences in memory between wip1+/+ and wip1–/– mice, animals were tested on contextual fear conditioning and T-maze spontaneous alternation tasks (Fig. 3A--C). Both of these behavioral tasks have previously been reported involving the hippocampal region.29-32
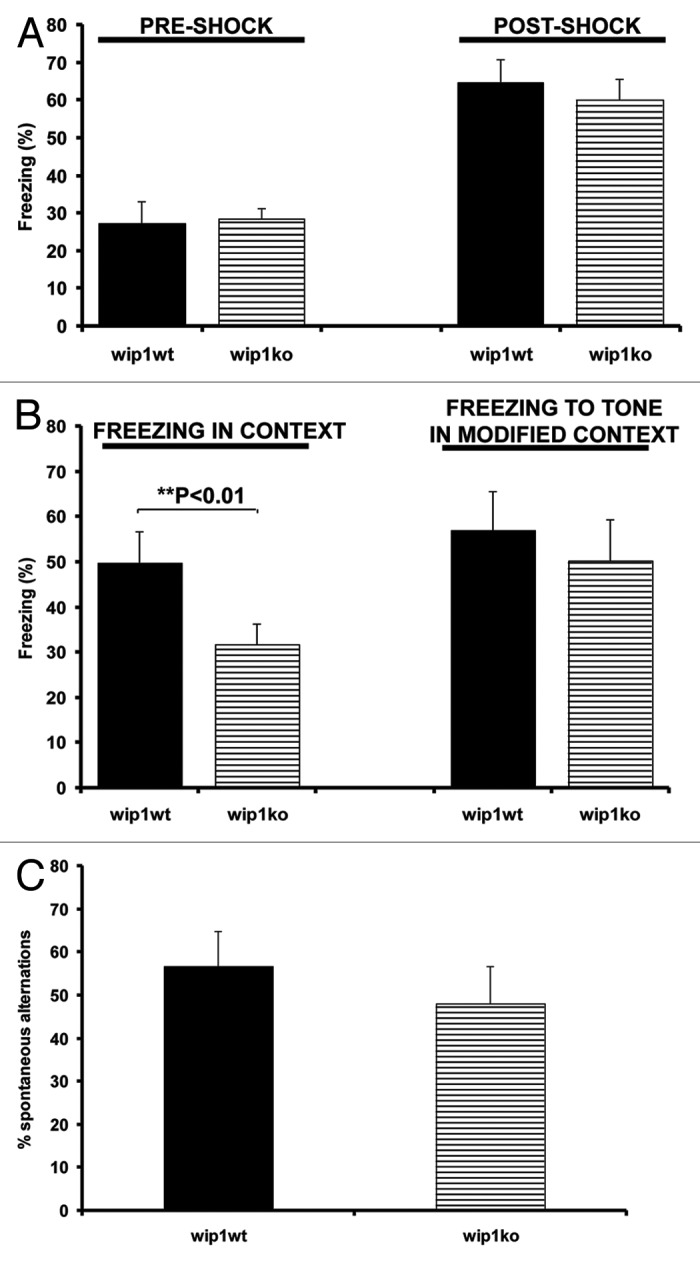
Figure 3. Evaluation of contextual memory and T-maze test in wip1 wt and wip1 ko mice. (A) Freezing response of wip1 wt (n = 11) and wip1 ko (n = 9) mice during fear conditioning trials (tone + shock conditioning). Values of freezing are expressed as mean ? SEM. (B) Freezing response of wip1 wt and wip1 ko mice to contextual exposure and to presentation of conditioned stimulus (tone) in a modified context 24 h following fear conditioning. Values of freezing are expressed as mean ? SEM **: p < 0.01. (C) Spontaneous alternation tasks on the T-maze for wip1 wt (n = 11) and wip1 ko (n = 9). Values of spontaneous alternation are expressed as mean ? SEM.
In the training trial of the contextual fear conditioning task, mice were placed in the test chamber and subsequently received two weak footshocks. Before the footshocks were delivered, the wip1+/+ and wip1–/– mice explored the conditioning chamber with no significant difference in their mobility (Fig. 3A, p > 0.05). Both groups of mice displayed a comparable increase in freezing while the footshocks were administered, demonstrating that these mice do not have a sensorimotor performance deficit, such as an inability to freeze (Fig. 3A, p > 0.05). When returned to the conditioning chamber on the next day, the wip1+/+ mice showed a greater conditioned freezing response (49.78 ± 6.9%) than wip1 deficient mice (31.6 ± 4.5%) (Fig. 3B, p = 0.0012). In contrast to this impaired contextual memory, wip1–/– mice were not significantly impaired in the different context of a modified chamber, where they displayed similar levels of conditioned freezing to the tone (CS) as the wip1+/+ mice (Fig. 3B, p > 0.05). Thus the deficit in contextual fear conditioning was not due to a general impairment in freezing ability but was more likely related to a cognitive deficit.33 Thus, in this hippocampal-dependent task, wip1 deficient mice showed impairment, as they did in the object recognition task.
When we assessed performance in a T-maze spontaneous alternation task measuring exploratory behavior as well as working memory, there was no significant difference between wip1+/+ and wip1−/− mice for the measurement of the percentage of spontaneous alternation (Fig. 3C, p = 0.06). Nevertheless, wip1 deficient mice had a smaller percentage of spontaneous alternation (48.06 ± 8.6%) than wip1 wild-type mice (56.66 ± 8.3%) (Fig. 3C). This result was not due to a difference in completed trials, wip1−/− mice having completed on average 8.38 ± 1.2 trials of the 10 trials compared with the 8.63 ± 0.9 trials completed by the control mice.
Together with the impairments in dendritic spine morphology, the behavioral data further demonstrates that wip1 has functions in the CNS.
p38 deficiency maintains novel object recognition and contextual fear conditioning memory and normal dendritic spine morphology
To investigate the potential role of p38MAPK in memory, we measured novel object recognition test performance, contextual fear conditioning and the dendritic spine morphology in p38Kl/+ and p38+/+ littermates.
In the novel object recognition task, the training session revealed a comparable percentage of time spent exploring the object in both groups of mice (n = 8; 20.4% and 17.5% for p38+/+ and p38Kl/+ mice respectively). During the test, the p38+/+ mice (60.1%) showed no difference in recognition index compared with the p38Kl/+ mice (59.9%, p > 0.05; Fig. 4A). Both groups of mice tested presented a recognition index superior to 50%, suggesting that the memory in these mice was not affected by the deficiency in the p38MAPK gene. In the contextual fear conditioning test there was significant difference in the mobility of p38+/+ and p38Kl/+ mice before and after the footshocks during the conditioning phase (Fig. 4B, p > 0.05). The p38+/+ and p38Kl/+ mice displayed a similar percentage of freezing in context (49.8% and 50.7% respectively) and freezing to tone in a modified context (56.9% and 55% respectively) (Fig. 4C, p > 0.05). Together these data show that memory in the novel object recognition test, contextual fear conditioning test and T-maze spontaneous alternation test (Fig. 4D) was similar in p38Kl/+ mice and control mice.

Figure 4. Behavioral tests and dendrite spine morphology in p38 wt and p38Kl/+ mice. (A) Evaluation of memory by object recognition test in p38 wt (n = 11) and p38KI/+ (n = 9) mice. Recognition index values are shown as mean ? SEM. (B) Freezing response of p38 wt (n = 11) and p38KI/+ (n = 9) mice during fear conditioning trials (tone + shock conditioning). Values of freezing are expressed as mean ? SEM. (C) Freezing response of p38 wt and p38KI/+ mice to contextual exposure and to presentation of conditioned stimulus (tone) in a modified context 24 h following fear conditioning. Values of freezing are expressed as mean ? SEM. (D) T-maze spontaneous alternation for p38 wt (n = 11) and p38KI/+ (n = 9). Values of spontaneous alternation are expressed as mean ? SEM. (E) Golgi staining of dendrites of adult pyramidal neurons in CA1 hippocampus p38 wt and p38KI/+ mice. Representative images of dendritic fragments from p38 wt and p38KI/+ mice. (F) Quantification of spine length (a), spine head width (b), and spine density (c) of Golgi stained neurons in p38 wt (n = 8) and p38KI/+ mice (n = 7). Bars = 4 ?m.
Next, we further analyzed spine morphology in CA1 of the hippocampus. Golgi staining did not reveal any significant differences between p38Kl/+ and p38+/+ mice in terms of dendritic spine morphology as measured by the three parameters analyzed (p > 0.05; Fig. 4E and F). These results indicate that, in contrast to wip1, mutant p38MAPK deficient mice showed no impairment in ability to acquire associative and contextual memory tasks and also a preservation of normal dendritic spine morphology in the hippocampus.
Wip1 deficiency associated impairments in dendritic spine morphology, and memory are reversed in wip1–/–p38Kl/+ double mutant mice
The previous results indicate that p38MAPK is a downstream element of wip1 signaling in modulation of dendritic spine morphology and memory processes. To further confirm this notion, we investigated dendritic spines in CA1 of the hippocampus and memory in the novel object recognition test, contextual fear conditioning test and T-maze spontaneous alternation procedure in wip1–/–p38Kl/+ and wip1+/+p38+/+ littermates. Analysis of spine morphology (Fig. 5A and B) showed no significant differences in spine length and spine head widths between neurons from wip1+/+p38+/+ (1.17 ± 0.10 μm and 0.56 ± 0.08 μm, respectively) and wip1–/–p38Kl/+ (0.99 ± 0.08 μm and 0.58 ± 0.06 μm, respectively) mice (p > 0.05; Fig. 5Ba and b). However, the spine density in wip1–/–p38Kl/+ mice (7.4 ± 0.67) was significantly decreased by about 50% compared with their wip1+/+p38+/+ littermates (15.6 ± 1.7; p = 0.002; Fig. 5Bc).

Figure 5. Wip1 deficiency-associated changes in dendritic spine morphology and behavioral tests are reversed in the wip1kop38KI/+ double mutant mice. (A) Golgi staining of dendrites of adult pyramidal neurons in CA1 hippocampus in wip wt p38wt and wip1kop38KI/+ mice. Representative pictures of dendritic fragments from wip1wtp38wt and wip1kop38KI/+ mice. (B) Quantification of spine length (a), spine head width (b), and spine density (c) of the Golgi stained neurons in wip1wtp38wt (n = 7) and wip1ko p38KI/+ mice (n = 6). Bars = 4 ?m. (C) Evaluation of memory by object recognition test in wip1wtp38wt (n = 11) and wip1kop38Kl/+ (n = 8) mice. Recognition index values are reported as mean ? SEM. (D) Freezing response of wip1wtp38wt (n = 11) and wip1kop38Kl/+ (n = 8) mice during fear conditioning trials (tone + shock conditioning). Values of freezing are expressed as mean ? SEM. (E) Freezing response of wip1wtp38wt and wip1kop38Kl/+ mice to contextual exposure and to presentation of conditioned stimulus (tone) in a modified context 24 h following fear conditioning. Values of freezing are expressed as mean ? SEM. (F) T-maze spontaneous alternation in wip1wtp38wt (n = 11) and wip1kop38Kl/+ (n = 8). Values of spontaneous alternation are expressed as mean ? SEM.
The reduction in dendritic spine density was higher in wip1 deficient mice (about 70%) than in double mutant mice (about 50%), suggesting that wip1 modulates spine morphology through the p38MAPK signaling pathway but not necessarily exclusively through this pathway.
The novel object recognition test showed that there was no difference in the recognition index between wip1–/–p38Kl/+ (57.8%) and wip1+/+p38+/+ mice (60.1%, p > 0.05) (Fig. 5C). Likewise, the contextual fear test showed no significant difference in the percentage of freezing measured in wip1–/–p38Kl/+ and wip1+/+p38+/+ mice during the conditioning training phase (pre- and post-shock, Fig. 5D, p > 0.05), and during the tests undertaken the following day in the same context and in a modified environment (Fig. 5E, p > 0.05). When we evaluated performance on the T-maze spontaneous alternation task, there was no significant difference between the percentage of spontaneous alternation in wip1–/– p38Kl/+ mice compared with the controls (Fig. 5F, p > 0.05).
Together, these data demonstrate that wip1 modulates dendritic spine morphology, associative, contextual and working memory through a p38MAPK-dependent signaling pathway.
Discussion
There is currently a paucity of published information regarding the role(s) of wip1/p38MAPK in the CNS. In the present study, we have revealed novel functions of the wip1/p38MAPK signaling pathway in the CNS. Wip1 phosphatase likely dephosphorylates p38MAPK in the dendritic spines of hippocampal neurons, and can play a modulatory role in dendritic spine morphology, associative and contextual memory.
As reported previously, wip1 mRNA is expressed in the brain12 and p38MAPK is located in the post synaptic area.17 Interestingly, in situ analyses in CA1 of the hippocampus of wip1–/– mice have revealed a reduction in the dendritic spine head width and length as well as the dendritic spine density. The role of p38MAPK in cytoskeletal organization via its action on the mechanisms of polymerization/depolymerization has been well described.17,34 However, we have reported that spine size and density are unchanged in p38MAPK deficient mice. Alteration in the spine morphology caused by p38MAPK deficiency was evident in the wip1–/–p38Kl/+ mice, but was attenuated regarding the dendritic spine density in CA1 of the hippocampus. The reduction in dendritic spine density is higher in wip1 deficient mice (about 70%) than in double mutant mice (about 50%) compared with wild type mice, suggesting wip1 modulates spine morphology through the p38MAPK signaling pathway—but not completely. Interestingly, arcadlin, a protocadherin present in hippocampal neurons, has been shown to act as a “negative” regulator of the number of synaptic spines via p38MAPK and TAO2 β, a MAPK kinase kinase (MAPKKK),35 thus confirming the implied role of p38MAPK in the negative regulation of spine density. In addition, the elimination of wip1 and p38MAPK genes by knockout may also induce a reorganization of the elements involved in regulating the density of the spines in the hippocampus. In addition other pathways may be involved in this process. It is well established now that once wip1 is induced by p53, wip1 not only directly dephosphorylates p38MAPK (as mentioned above) but Mdm2, p53 and ATM as well. ATM and p-p53 (at serine 15) have been over found expressed in the hippocampus of wip1 deficient mice (compare Fig. S2). Interestingly, mice homozygous for a transgene encoding p44, a short and naturally occurring isoform of the p53, display cognitive decline and synaptic impairment early in life. Recent studies showed that in the absence of p53, wip1 expression can be induced by the JNK-c-Jun pathway, which is activated in Alzheimer disease brain.36,37 Activation of the JNK-c-Jun pathway has also been implicated in the decline of hippocampal long-term potentiation.38 Further studies are necessary to identify the molecular mechanisms responsible.
Many phosphatases have been previously involved in memory processes. Acquisition of memory is impaired in calcineurin, PP1 and PP2A deficient mice.39,40 In addition, the majority of PP has been found to be downregulated in the brains of Alzheimer disease (AD) sufferers,41 although activation of calcineurin has also been reported in AD brain.42,43 Because AD sufferers display a lack of initiative in addition to anterograde amnesia, it is interesting to examine how closely the mouse model resembles human symptomatology, using the T-maze spontaneous alternation test. Spontaneous alternation has been tested in several murine models of AD, with diverse results. Lower rate of spontaneous alternation have been reported in transgenic mice with the Swedish mutation of the 695 isoform (β APP695SWE), or the 770 isoform (β APP770SWE) combined with the M146L preselinin mutation compared with non-transgenic mice—and this even before mature Aβ protein plaques appear.44-46 However, segmental trisomic 16 (Ts65Dn) mice overexpressing βAPP and several other genes showed equivalent levels of spontaneous alternation to controls.47 Similarly, in our study, the spontaneous alternation rates for mutant wip1 ko mice were statistically comparable to the wild type mice, even if this result was close to the threshold of significance (p = 0.06), with an apparent lower (more than 15%) spontaneous alternation level for the mutant mice compared with controls (Fig. 2C). In addition, the results reported in two well established memory tests (contextual fear conditioning and novel object recognition tests) were highly significant for the mutant wip1 mice compared with the wild type (p = 0.0012 and p = 0.0013, respectively) demonstrating for the first time a role of wip1 in an associative memory task. Interestingly, the disturbance of PP1 anchoring by knockout of the gene encoding spinophilin alters not only learning and memory processes,48 but also the formation and the function of dendritic spines.49 Regarding wip1 heterozygote mice, the dendritic spine was affected in the hippocampus (reduction of 35% compared with controls; Fig. 1B); however, these mice did not show any disturbances of memory in the object recognition test (Fig. 2). Since wip1 protein is still present in the hippocampus region of the heterozygous mice during the object recognition test, wip1 can still dephosphorylate its downstream factors (including p38MAPK) and balance the memory deficit. Furthermore, despite experimental findings reporting a role for dendritic spine morphology in mediating learning and memory, the extent of the importance of structural plasticity remains debatable. For instance, it has been estimated that only a small portion of spines formed during training actually contribute to long-term memory.50 Thus, even although changes in spine morphology at specific synapses are likely to be important for learning and memory, it is not necessarily the case that all changes in spine morphology will lead to an impairment of memory.
Impairment of associative and contextual memories was observed in the wip1 deficient mice and reversed in wip1–/ p38Kl/+ double mutant mice compared with wild type mice, indicating that wip1 plays a role in the modulation of these memory processes, at least in part, through the p38MAPK pathway.
Dendritic spine morphology has been correlated with the processes of learning and memory.1,5,51 The direction of the causal connection between these events (if any) remains unresolved. Interestingly, alterations of spine density have been previously reported in hippocampus from an AD animal model, β APP695SWE transgenic mice.52 Substantial morphological changes in dendritic spine density have mainly been observed in the prefrontal cortex and the hippocampus from AD and other dementia sufferers.53 We report that deletion of wip1 not only leads to abnormal dendritic morphology but also leads to an impairment of learning and memory. The results described here, do not establish any direct causal connection between dendritic spine morphology and memory processes. However, our observations demonstrate novel functions of wip1 in the CNS in terms of modulating dendrite spine morphology and memory processes, which may provide a better understanding of the molecular mechanism underlying human cognitive disorders. Recent studies of p38MAPK and wip1 in old mice (22–25 mo old) showed a reduction of wip1 protein levels during the aging process, which contributes to p38MAPK activation.54 Downregulation of Ppm1d mRNA expression with aging has also been reported in neural stem cells in the subventricular zone.55 Interestingly, wip1 deficient mice and old (22–25 mo old) wild type mice showed a considerable increase in p38MAPK-dependent signaling, suggesting that wip1 may play a substantial role in the molecular basis for aging processes through p38MAPK signaling modulation.54,56 This finding is consistent with our result that when the expression of wip1 is decreased or nil there are disturbances in memory. This process seems to be partly p38MAPK-dependent as showed in our results. We have also reported a significant increase in the expression levels of p-p38MAPK in the hippocampus in wip1 deficient mice measured by immunoblotting (compare Fig. S2A and B). This upregulation of activated p38MAPK was also found when protein fractions (total, nuclear and cytoplasmic) from the hippocampus of wip1 deficient mice was analyzed. Wip1 deficient mice could be a new model of study for aging disorders since the expression of wip1 is decreased similarly in the hippocampus of the wild type old mice (while p-p38MAPK is increased). In addition, disturbances in memory tasks occurred in wip1 deficient mice seem to be similar to events occurring during aging). Although much remains to be done, there is promise that a better understanding of the physiological and pathophysiological roles of wip1 may lead to novel approaches for preventing and treating various disorders, such as aging disorders, neurological pathologies and cancer.
Materials and Methods
Mice
Wip1–/– mice (C57BL/6 background) were generated using a targeting construct from a murine genomic Wip1 clone derived from a 129/Sv phage genomic library as described previously.57p38Kl/+ mutant mice were generated as previously decribed by targeted mutation of p38MAPK in ES cells.54 Briefly, the original BAC clone RP23–83F4 of Mus musculus strain C57BL6/J (NCI Intramural Sequencing center) was used to generate targeting vectors. The targeting vectors contained the equivalent of 4.5 kb of mouse DNA with an exon containing the Thr180/Tyr182 sites of p38 α. These sites were subsequently mutated to alanine and phenylalanine by site-directed mutagenesis, leading to an inactivation of phosphorylation of these sites. The entire sequence was placed into the pTK plasmid with two flanking thymidine kinase genes and electroporated into B6 ES cells. The resulting mutated ES cells were used to generate mutant mice.
Wip1–/–p38Kl/+ mice were bred by crossing the wip1–/– and p38Kl/+ mice, and their wild type littermates were used as controls. All animal work was conducted under the institutional guidelines of the Animal Ethics Committees of the Singapore General Hospital and the Institute of Molecular and Cellular Biology in Singapore.
Golgi staining
Two- to three-month-old brains of littermate mice were processed and stained according to the protocol provided by the manufacture (FD Neuro Technologies) with some modification. In brief, 100 µm thick brain sections were cut on a vibratome after impregnation with sodium dichromate buffer. Hippocampus tissue sections were stained with silver nitrate substrate. Reflectance confocal micrographs were obtained using a laser scanning confocal microscope (LSM510 META, Zeiss; 63x objective for dendrite measurements and 40x for the hippocampal neuron images). Each dendrite analyzed was quantified through a z-series stack of 30–60 pictures (depending on the depth of the dendrite) taken at 0.35 to 0.7 µm intervals. Measurements of dendritic morphology were performed on neurons randomly chosen in the CA1 region of the hippocampus for analysis following previously published criteria.58 Briefly, fully impregnated cells were selected and, after identification of the apical shaft, basal dendrites were chosen for analysis. The spine quantification commenced on dendrites starting at more than 85 µm distal to the soma, and after the first branch point. Measurements were made out to a maximum of 200 µm from the soma and 20–30 µm dendritic segments were analyzed (n = 18–27 neurons for each studied genotype) and protrusions were measured as described previously.59 No distinction was made between the different spine types. The analysis of dendritic morphology was performed using Zeiss LSM Image Browser software (Version 4.2) by research assistants who were blind to genotypes and experimental manipulations.
Object recognition test
Memory was assessed by using a modified version of the object recognition test described previously.60 The experimental apparatus consisted of a Plexiglas open-field box (55 cm × 41 cm × 35 cm high). The apparatus was located in a sound-attenuated room and was illuminated with a 40 W bulb. The procedure consisted of three sessions: habituation, training, and retention. The animals (3–4 mo old) were videotaped in both training and retention sessions. Each mouse was individually habituated to the box, with 10 min of exploration in the absence of objects for three consecutive days (habituation session, days 1–3). During the training session, two identical objects (A) were placed in two opposite corners of the apparatus 5 cm from the side wall. Each animal was placed in the middle of the apparatus and allowed to explore the objects for 10 min (day 4). An animal was considered to be exploring the object when its head was facing the object (with the distance between the head and object approximately 2 cm or less) or when it was touching or sniffing the object. Ninety minutes after the training session, the animal explored the open field for 10 min in the presence of one familiar (A, the same one used during the training session) and one novel (B) object. Objects were chosen after determining in preliminary experiments that they were equally preferred. Between each trial both the open-field arena and the objects were washed with 70% ethanol solution. All sessions were videotaped, and an experimenter blind to the genotype and experimental conditions analyzed the object recognition behavior. Object placement was counterbalanced so that half of the animals in each treatment group saw the novel object on the left side (relative to the animal's start position) of the open-field arena, and the other half saw the novel object on the right side of the arena. A preference index in the retention session (a ratio of the amount of time spent exploring the novel object over the total time spent exploring both objects) was used to measure cognitive function. In the training session, the recognition index was calculated as a ratio of the time spent exploring the object that was replaced by the novel object in the retention session over the total exploring time.
Contextual fear test
The training chamber was a rectangular box 30 cm × 24 cm × 21 cm in size, equipped with a grid floor composed of stainless steel rods. The grid floor was connected to a shock generator, which delivered a 2 sec, 0.15 mA footshock. The stimulus light and generator were controlled by computer software (TSE Systems). The chamber was cleaned with 70% ethanol between animals. Mice (4 mo old, n = 8–11 per genotype) were trained and tested on two consecutive days. A 2 min pre-training observation period allowed for measurement of basal locomotor activity. Training consisted of placing a subject in the chamber, and allowing exploration for 2 min. Afterwards, an auditory cue (85 dB, Conditioned CS) was presented for 30 sec. The 2 sec footshock (0.15 mA, unconditioned stimulus, US) was administered for the final 2 sec of the CS. This procedure was repeated, and mice were removed from the chamber 30 sec later.
Twenty four hours after training, mice were returned to the same chamber in which training occurred (context), and freezing behavior was recorded by digital video tracking (Noldus Ethovision). Freezing was defined as lack of movement except that required for respiration. At the end of the 4 min context test, mice were returned to their home cage.
Approximately 2 h later, freezing was recorded in a novel environment context and in response to the cue. The novel environmental context consisted of visual modifications in the chamber, including black bands (3 cm) around the walls of the chamber and plexiglass on the floor. Mice were placed in the novel environment and freezing was recorded for 4 min. The auditory cue (CS) was then presented during the last 2 min. Freezing scores for each subject were expressed as percentage time spent freezing during each portion of the test.
T-maze spontaneous alternation
Performance in the spontaneous alternation task was assessed using a T-maze as previously described.61,62 This procedure was performed in an enclosed “T”-shaped maze in which the long arm of the T (40 cm × 10 cm) served as a start arm and the short arms of the T (29 cm × 10 cm) served as the goal arms. Guillotine doors could close the arm entrances from the choice area. Training involved a single forced-choice trial with one goal arm blocked. Testing was for 10 free-choice arm entries or 16 min, whichever occurred first. Mice (4 mo old, n = 8–11 per genotype) placed in the start arm must enter (whole body, including tail) the left or right open goal arm and return to the choice area, and then enter the opposite arm for each trial. At the conclusion of each trial, the maze was cleaned of urine and feces. The number of spontaneous alternations completed (entering the right goal arm after the left goal arm was entered and vice versa), was recorded and expressed as a percentage of total arm entries. To account for differences in the number of entries made, the percentage of alternations accomplished (number of alternating entries achieved/total number of entries completed × 100) was taken as an index of enhanced performance in this task.
Data analysis
Data were analyzed by one way ANOVA, followed by Student’s t-test for paired groups (two-tailed) with p < 0.05 indicating a significant difference. For the electrophysiological data, ANOVA analysis was performed and followed by Bonferroni corrected post hoc tests, with p < 0.01 as a significance threshold.
Supplementary Material
Acknowledgments
We would like to thank Miss Siying Chen, Dr Xiao-Wei Tan and Miss Hnin Ngwe Yee Lwin from the Institute of Molecular and Cell Biology, Singapore and Dr Alice Lim from the Centre for Life Sciences, National University of Singapore for technical support. We are very grateful for the assistance and service from staff at the Electron Microscopy Unit, NUS, Singapore, and would like to thank in particular Miss Micky Leong for her help. We would like to thank Mr Gavin Symonds and Mrs Susan Heart from Carl Zeiss Pty. Ltd. for their help in analyzing the confocal pictures of dendritic spine morphology. This work was supported by grants to Z.-C.X. from the Singapore Health Foundation, Department of Clinical Research, Singapore General Hospital, Singapore. The authors from Institute of Molecular and Cell Biology are supported by A*STAR, Singapore.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Footnotes
Previously published online: www.landesbioscience.com/journals/celladhesion/article/20892
References
- 1.Matsuzaki M, Honkura N, Ellis-Davies GC, Kasai H. Structural basis of long-term potentiation in single dendritic spines. Nature. 2004;429:761–6. doi: 10.1038/nature02617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Tada T, Sheng M. Molecular mechanisms of dendritic spine morphogenesis. Curr Opin Neurobiol. 2006;16:95–101. doi: 10.1016/j.conb.2005.12.001. [DOI] [PubMed] [Google Scholar]
- 3.Chen LY, Rex CS, Casale MS, Gall CM, Lynch G. Changes in synaptic morphology accompany actin signaling during LTP. J Neurosci. 2007;27:5363–72. doi: 10.1523/JNEUROSCI.0164-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Colbran RJ, Brown AM. Calcium/calmodulin-dependent protein kinase II and synaptic plasticity. Curr Opin Neurobiol. 2004;14:318–27. doi: 10.1016/j.conb.2004.05.008. [DOI] [PubMed] [Google Scholar]
- 5.Colicos MA, Collins BE, Sailor MJ, Goda Y. Remodeling of synaptic actin induced by photoconductive stimulation. Cell. 2001;107:605–16. doi: 10.1016/S0092-8674(01)00579-7. [DOI] [PubMed] [Google Scholar]
- 6.Carmody LC, Bauman PA, Bass MA, Mavila N, DePaoli-Roach AA, Colbran RJ. A protein phosphatase-1gamma1 isoform selectivity determinant in dendritic spine-associated neurabin. J Biol Chem. 2004;279:21714–23. doi: 10.1074/jbc.M402261200. [DOI] [PubMed] [Google Scholar]
- 7.Collins CA, Wairkar YP, Johnson SL, DiAntonio A. Highwire restrains synaptic growth by attenuating a MAP kinase signal. Neuron. 2006;51:57–69. doi: 10.1016/j.neuron.2006.05.026. [DOI] [PubMed] [Google Scholar]
- 8.Krapivinsky G, Medina I, Krapivinsky L, Gapon S, Clapham DE. SynGAP-MUPP1-CaMKII synaptic complexes regulate p38 MAP kinase activity and NMDA receptor-dependent synaptic AMPA receptor potentiation. Neuron. 2004;43:563–74. doi: 10.1016/j.neuron.2004.08.003. [DOI] [PubMed] [Google Scholar]
- 9.LaFerla FM. Calcium dyshomeostasis and intracellular signalling in Alzheimer’s disease. Nat Rev Neurosci. 2002;3:862–72. doi: 10.1038/nrn960. [DOI] [PubMed] [Google Scholar]
- 10.Mattson MP, Chan SL. Neuronal and glial calcium signaling in Alzheimer’s disease. Cell Calcium. 2003;34:385–97. doi: 10.1016/S0143-4160(03)00128-3. [DOI] [PubMed] [Google Scholar]
- 11.Fiscella M, Zhang H, Fan S, Sakaguchi K, Shen S, Mercer WE, et al. Wip1, a novel human protein phosphatase that is induced in response to ionizing radiation in a p53-dependent manner. Proc Natl Acad Sci U S A. 1997;94:6048–53. doi: 10.1073/pnas.94.12.6048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Choi J, Appella E, Donehower LA. The structure and expression of the murine wildtype p53-induced phosphatase 1 (Wip1) gene. Genomics. 2000;64:298–306. doi: 10.1006/geno.2000.6134. [DOI] [PubMed] [Google Scholar]
- 13.Bulavin DV, Phillips C, Nannenga B, Timofeev O, Donehower LA, Anderson CW, et al. Inactivation of the Wip1 phosphatase inhibits mammary tumorigenesis through p38 MAPK-mediated activation of the p16(Ink4a)-p19(Arf) pathway. Nat Genet. 2004;36:343–50. doi: 10.1038/ng1317. [DOI] [PubMed] [Google Scholar]
- 14.Lu X, Nannenga B, Donehower LA. PPM1D dephosphorylates Chk1 and p53 and abrogates cell cycle checkpoints. Genes Dev. 2005;19:1162–74. doi: 10.1101/gad.1291305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Shreeram S, Hee WK, Demidov ON, Kek C, Yamaguchi H, Fornace AJ, Jr., et al. Regulation of ATM/p53-dependent suppression of myc-induced lymphomas by Wip1 phosphatase. J Exp Med. 2006;203:2793–9. doi: 10.1084/jem.20061563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Takekawa M, Adachi M, Nakahata A, Nakayama I, Itoh F, Tsukuda H, et al. p53-inducible wip1 phosphatase mediates a negative feedback regulation of p38 MAPK-p53 signaling in response to UV radiation. EMBO J. 2000;19:6517–26. doi: 10.1093/emboj/19.23.6517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bolshakov VY, Carboni L, Cobb MH, Siegelbaum SA, Belardetti F. Dual MAP kinase pathways mediate opposing forms of long-term plasticity at CA3-CA1 synapses. Nat Neurosci. 2000;3:1107–12. doi: 10.1038/80624. [DOI] [PubMed] [Google Scholar]
- 18.Huang CC, You JL, Wu MY, Hsu KS. Rap1-induced p38 mitogen-activated protein kinase activation facilitates AMPA receptor trafficking via the GDI.Rab5 complex. Potential role in (S)-3,5-dihydroxyphenylglycene-induced long term depression. J Biol Chem. 2004;279:12286–92. doi: 10.1074/jbc.M312868200. [DOI] [PubMed] [Google Scholar]
- 19.Zhu JJ, Qin Y, Zhao M, Van Aelst L, Malinow R. Ras and Rap control AMPA receptor trafficking during synaptic plasticity. Cell. 2002;110:443–55. doi: 10.1016/S0092-8674(02)00897-8. [DOI] [PubMed] [Google Scholar]
- 20.Pak DT, Yang S, Rudolph-Correia S, Kim E, Sheng M. Regulation of dendritic spine morphology by SPAR, a PSD-95-associated RapGAP. Neuron. 2001;31:289–303. doi: 10.1016/S0896-6273(01)00355-5. [DOI] [PubMed] [Google Scholar]
- 21.Ennaceur A, Aggleton JP. The effects of neurotoxic lesions of the perirhinal cortex combined to fornix transection on object recognition memory in the rat. Behav Brain Res. 1997;88:181–93. doi: 10.1016/S0166-4328(97)02297-3. [DOI] [PubMed] [Google Scholar]
- 22.Rudy JW, Huff NC, Matus-Amat P. Understanding contextual fear conditioning: insights from a two-process model. Neurosci Biobehav Rev. 2004;28:675–85. doi: 10.1016/j.neubiorev.2004.09.004. [DOI] [PubMed] [Google Scholar]
- 23.Deacon RM, Rawlins JN. T-maze alternation in the rodent. Nat Protoc. 2006;1:7–12. doi: 10.1038/nprot.2006.2. [DOI] [PubMed] [Google Scholar]
- 24.Clark RE, Zola SM, Squire LR. Impaired recognition memory in rats after damage to the hippocampus. J Neurosci. 2000;20:8853–60. doi: 10.1523/JNEUROSCI.20-23-08853.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wood ER, Mumby DG, Pinel JP, Phillips AG. Impaired object recognition memory in rats following ischemia-induced damage to the hippocampus. Behav Neurosci. 1993;107:51–62. doi: 10.1037/0735-7044.107.1.51. [DOI] [PubMed] [Google Scholar]
- 26.Gaskin S, Tremblay A, Mumby DG. Retrograde and anterograde object recognition in rats with hippocampal lesions. Hippocampus. 2003;13:962–9. doi: 10.1002/hipo.10154. [DOI] [PubMed] [Google Scholar]
- 27.Reed JM, Squire LR. Impaired recognition memory in patients with lesions limited to the hippocampal formation. Behav Neurosci. 1997;111:667–75. doi: 10.1037/0735-7044.111.4.667. [DOI] [PubMed] [Google Scholar]
- 28.Stark CE, Bayley PJ, Squire LR. Recognition memory for single items and for associations is similarly impaired following damage to the hippocampal region. Learn Mem. 2002;9:238–42. doi: 10.1101/lm.51802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kirkby RJ, Stein DG, Kimble RJ, Kimble DP. Effects of hippocampal lesions and duration of sensory input on spontaneous alternation. J Comp Physiol Psychol. 1967;64:342–5. doi: 10.1037/h0088012. [DOI] [PubMed] [Google Scholar]
- 30.Stevens R, Cowey A. Effects of dorsal and ventral hippocampal lesions on spontaneous alternation, learned alternation and probability learning in rats. Brain Res. 1973;52:203–24. doi: 10.1016/0006-8993(73)90659-8. [DOI] [PubMed] [Google Scholar]
- 31.Johnson CT, Olton DS, Gage FH, 3rd, Jenko PG. Damage to hippocampus and hippocampal connections: effects on DRL and spontaneous alternation. J Comp Physiol Psychol. 1977;91:508–22. doi: 10.1037/h0077346. [DOI] [PubMed] [Google Scholar]
- 32.Phillips RG, LeDoux JE. Lesions of the dorsal hippocampal formation interfere with background but not foreground contextual fear conditioning. Learn Mem. 1994;1:34–44. [PubMed] [Google Scholar]
- 33.Cahill L, McGaugh JL, Weinberger NM. The neurobiology of learning and memory: some reminders to remember. Trends Neurosci. 2001;24:578–81. doi: 10.1016/S0166-2236(00)01885-3. [DOI] [PubMed] [Google Scholar]
- 34.Cingolani LA, Goda Y. Actin in action: the interplay between the actin cytoskeleton and synaptic efficacy. Nat Rev Neurosci. 2008;9:344–56. doi: 10.1038/nrn2373. [DOI] [PubMed] [Google Scholar]
- 35.Yasuda S, Tanaka H, Sugiura H, Okamura K, Sakaguchi T, Tran U, et al. Activity-induced protocadherin arcadlin regulates dendritic spine number by triggering N-cadherin endocytosis via TAO2beta and p38 MAP kinases. Neuron. 2007;56:456–71. doi: 10.1016/j.neuron.2007.08.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Anderson AJ, Su JH, Cotman CW. DNA damage and apoptosis in Alzheimer’s disease: colocalization with c-Jun immunoreactivity, relationship to brain area, and effect of postmortem delay. J Neurosci. 1996;16:1710–9. doi: 10.1523/JNEUROSCI.16-05-01710.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhu X, Raina AK, Rottkamp CA, Aliev G, Perry G, Boux H, et al. Activation and redistribution of c-jun N-terminal kinase/stress activated protein kinase in degenerating neurons in Alzheimer’s disease. J Neurochem. 2001;76:435–41. doi: 10.1046/j.1471-4159.2001.00046.x. [DOI] [PubMed] [Google Scholar]
- 38.Wang Q, Walsh DM, Rowan MJ, Selkoe DJ, Anwyl R. Block of long-term potentiation by naturally secreted and synthetic amyloid beta-peptide in hippocampal slices is mediated via activation of the kinases c-Jun N-terminal kinase, cyclin-dependent kinase 5, and p38 mitogen-activated protein kinase as well as metabotropic glutamate receptor type 5. J Neurosci. 2004;24:3370–8. doi: 10.1523/JNEUROSCI.1633-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bennett PC, Zhao W, Ng KT. Concentration-dependent effects of protein phosphatase (PP) inhibitors implicate PP1 and PP2A in different stages of memory formation. Neurobiol Learn Mem. 2001;75:91–110. doi: 10.1006/nlme.1999.3959. [DOI] [PubMed] [Google Scholar]
- 40.Zeng H, Chattarji S, Barbarosie M, Rondi-Reig L, Philpot BD, Miyakawa T, et al. Forebrain-specific calcineurin knockout selectively impairs bidirectional synaptic plasticity and working/episodic-like memory. Cell. 2001;107:617–29. doi: 10.1016/S0092-8674(01)00585-2. [DOI] [PubMed] [Google Scholar]
- 41.Tian Q, Wang J. Role of serine/threonine protein phosphatase in Alzheimer’s disease. Neurosignals. 2002;11:262–9. doi: 10.1159/000067425. [DOI] [PubMed] [Google Scholar]
- 42.Hata R, Masumura M, Akatsu H, Li F, Fujita H, Nagai Y, et al. Up-regulation of calcineurin Abeta mRNA in the Alzheimer’s disease brain: assessment by cDNA microarray. Biochem Biophys Res Commun. 2001;284:310–6. doi: 10.1006/bbrc.2001.4968. [DOI] [PubMed] [Google Scholar]
- 43.Liu F, Grundke-Iqbal I, Iqbal K, Oda Y, Tomizawa K, Gong CX. Truncation and activation of calcineurin A by calpain I in Alzheimer disease brain. J Biol Chem. 2005;280:37755–62. doi: 10.1074/jbc.M507475200. [DOI] [PubMed] [Google Scholar]
- 44.Hsiao K, Chapman P, Nilsen S, Eckman C, Harigaya Y, Younkin S, et al. Correlative memory deficits, Abeta elevation, and amyloid plaques in transgenic mice. Science. 1996;274:99–102. doi: 10.1126/science.274.5284.99. [DOI] [PubMed] [Google Scholar]
- 45.Holcomb LA, Gordon MN, Jantzen P, Hsiao K, Duff K, Morgan D. Behavioral changes in transgenic mice expressing both amyloid precursor protein and presenilin-1 mutations: lack of association with amyloid deposits. Behav Genet. 1999;29:177–85. doi: 10.1023/A:1021691918517. [DOI] [PubMed] [Google Scholar]
- 46.Chapman PF, White GL, Jones MW, Cooper-Blacketer D, Marshall VJ, Irizarry M, et al. Impaired synaptic plasticity and learning in aged amyloid precursor protein transgenic mice. Nat Neurosci. 1999;2:271–6. doi: 10.1038/6374. [DOI] [PubMed] [Google Scholar]
- 47.Demas GE, Nelson RJ, Krueger BK, Yarowsky PJ. Spatial memory deficits in segmental trisomic Ts65Dn mice. Behav Brain Res. 1996;82:85–92. doi: 10.1016/S0166-4328(97)81111-4. [DOI] [PubMed] [Google Scholar]
- 48.Stafstrom-Davis CA, Ouimet CC, Feng J, Allen PB, Greengard P, Houpt TA. Impaired conditioned taste aversion learning in spinophilin knockout mice. Learn Mem. 2001;8:272–8. doi: 10.1101/lm.42101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Feng J, Yan Z, Ferreira A, Tomizawa K, Liauw JA, Zhuo M, et al. Spinophilin regulates the formation and function of dendritic spines. Proc Natl Acad Sci U S A. 2000;97:9287–92. doi: 10.1073/pnas.97.16.9287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Yang G, Pan F, Gan WB. Stably maintained dendritic spines are associated with lifelong memories. Nature. 2009;462:920–4. doi: 10.1038/nature08577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Nägerl UV, Eberhorn N, Cambridge SB, Bonhoeffer T. Bidirectional activity-dependent morphological plasticity in hippocampal neurons. Neuron. 2004;44:759–67. doi: 10.1016/j.neuron.2004.11.016. [DOI] [PubMed] [Google Scholar]
- 52.Knafo S, Alonso-Nanclares L, Gonzalez-Soriano J, Merino-Serrais P, Fernaud-Espinosa I, Ferrer I, et al. Widespread changes in dendritic spines in a model of Alzheimer’s disease. Cereb Cortex. 2009;19:586–92. doi: 10.1093/cercor/bhn111. [DOI] [PubMed] [Google Scholar]
- 53.Knobloch M, Mansuy IM. Dendritic spine loss and synaptic alterations in Alzheimer’s disease. Mol Neurobiol. 2008;37:73–82. doi: 10.1007/s12035-008-8018-z. [DOI] [PubMed] [Google Scholar]
- 54.Wong ES, Le Guezennec X, Demidov ON, Marshall NT, Wang ST, Krishnamurthy J, et al. p38MAPK controls expression of multiple cell cycle inhibitors and islet proliferation with advancing age. Dev Cell. 2009;17:142–9. doi: 10.1016/j.devcel.2009.05.009. [DOI] [PubMed] [Google Scholar]
- 55.Zhu YH, Zhang CW, Lu L, Demidov ON, Sun L, Yang L, et al. Wip1 regulates the generation of new neural cells in the adult olfactory bulb through p53-dependent cell cycle control. Stem Cells. 2009;27:1433–42. doi: 10.1002/stem.65. [DOI] [PubMed] [Google Scholar]
- 56.Le Guezennec X, Bulavin DV. WIP1 phosphatase at the crossroads of cancer and aging. Trends Biochem Sci. 2010;35:109–14. doi: 10.1016/j.tibs.2009.09.005. [DOI] [PubMed] [Google Scholar]
- 57.Choi J, Nannenga B, Demidov ON, Bulavin DV, Cooney A, Brayton C, et al. Mice deficient for the wild-type p53-induced phosphatase gene (Wip1) exhibit defects in reproductive organs, immune function, and cell cycle control. Mol Cell Biol. 2002;22:1094–105. doi: 10.1128/MCB.22.4.1094-1105.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Jacobs B, Driscoll L, Schall M. Life-span dendritic and spine changes in areas 10 and 18 of human cortex: a quantitative Golgi study. J Comp Neurol. 1997;386:661–80. doi: 10.1002/(SICI)1096-9861(19971006)386:4<661::AID-CNE11>3.0.CO;2-N. [DOI] [PubMed] [Google Scholar]
- 59.Campaña AD, Sanchez F, Gamboa C, Gómez-Villalobos MdeJ, De La Cruz F, Zamudio S, et al. Dendritic morphology on neurons from prefrontal cortex, hippocampus, and nucleus accumbens is altered in adult male mice exposed to repeated low dose of malathion. Synapse. 2008;62:283–90. doi: 10.1002/syn.20494. [DOI] [PubMed] [Google Scholar]
- 60.Dodart JC, Mathis C, Ungerer A. Scopolamine-induced deficits in a two-trial object recognition task in mice. Neuroreport. 1997;8:1173–8. doi: 10.1097/00001756-199703240-00023. [DOI] [PubMed] [Google Scholar]
- 61.Hughes RN. The value of spontaneous alternation behavior (SAB) as a test of retention in pharmacological investigations of memory. Neurosci Biobehav Rev. 2004;28:497–505. doi: 10.1016/j.neubiorev.2004.06.006. [DOI] [PubMed] [Google Scholar]
- 62.Frye CA, Walf AA. Progesterone enhances performance of aged mice in cortical or hippocampal tasks. Neurosci Lett. 2008;437:116–20. doi: 10.1016/j.neulet.2008.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.